

Abstract
The type 2 ryanodine receptor (RyR2) plays a key role in the cardiac intracellular calcium (Ca2+) regulation. We have previously shown that oxidative stress activates RyR2 in rabbit cardiomyocytes by promoting the formation of disulfide bonds between neighboring RyR2 subunits. However, the functional significance of this redox modification for human RyR2 (hRyR2) remains largely unknown. Here, we studied the redox regulation of hRyR2 in HEK293 cells transiently expressing the ryr2 gene. Analysis of hRyR2 cross-linking and of the redox-GFP readout response to diamide oxidation revealed that hRyR2 cysteines involved in the intersubunit cross-linking are highly sensitive to oxidative stress. In parallel experiments, the effect of diamide on endoplasmic reticulum (ER) Ca2+ release was studied in cells co-transfected with hRyR2, ER Ca2+ pump (SERCA2a) and the ER-targeted Ca2+ sensor R-CEPIA1er. Expression of hRyR2 and SERCA2a produced “cardiac-like” Ca2+ waves due to spontaneous hRyR2 activation. Incubation with diamide caused a fast decline of the luminal ER Ca2+ (or ER Ca2+ load) followed by the cessation of Ca2+ waves. The maximal effect of diamide on ER Ca2+ load and Ca2+ waves positively correlates with the maximum level of hRyR2 cross-linking, indicating a functional significance of this redox modification. Furthermore, the level of hRyR2 cross-linking positively correlates with the degree of calmodulin (CaM) dissociation from the hRyR2 complex. In skeletal muscle RyR (RyR1), cysteine 3635 (C3635) is viewed as dominantly responsible for the redox regulation of the channel. Here, we showed that the corresponding cysteine 3602 (C3602) in hRyR2 does not participate in intersubunit cross-linking and plays a limited role in the hRyR2 regulation by CaM during oxidative stress. Collectively, these results suggest that redox-mediated intersubunit cross-linking is an important regulator of hRyR2 function under pathological conditions associated with oxidative stress.
Keywords: Ca2+ signaling, Ca2+ waves, Ryanodine receptor, Sarcoplasmic reticulum and oxidative stress
Graphical abstract

Highlights
-
•
Oxidative stress promotes cardiac ryanodine receptor (RyR2) intersubunit crosslinking.
-
•
Human RyR2 crosslinking promotes Ca leak and calmodulin dissociation.
-
•
RyR2 C3602 is not involved in crosslinking, slightly affects calmodulin binding.
-
•
RyR2 crosslinking is an important pathology related RyR2 regulator.
1. Introduction
The type 2 ryanodine receptor (RyR2) is the major sarcoplasmic reticulum (SR) Ca2+ release channel that plays an essential role in cardiac excitation-contraction (EC) coupling [1,2]. During systole, RyR2 is activated by an inward Ca2+ current via voltage gated L-type Ca2+ channels, resulting in global Ca2+-induced Ca2+ release (CICR) [3]. The increase in cytosolic Ca2+ triggers cellular contraction (systole) followed by a relaxation phase (diastole). During diastole, cytosolic Ca2+ is pumped back into the SR by the SR Ca2+-ATPase (SERCA) and removed outside the cell by Na+-Ca2+ exchanger [4]. Spontaneous RyR2 opening during diastole creates SR Ca2+ leak that counterbalances SERCA Ca2+ uptake and sets SR Ca2+ load [5,6]. Due to its central role in cardiac function, RyR2 activity is tightly regulated. RyR2 dysfunction, caused either by mutations or stress-induced post-translational modifications, has been linked to various cardiac pathologies, including arrhythmias and heart failure (HF) [[7], [8], [9]].
Among various factors that regulate RyR2 activity, redox modifications of the channel play a particularly important role under pathological conditions associated with oxidative stress [[10], [11], [12]]. Each subunit of the RyR2 homo-tetrameric complex comprises 90 cysteine residues, but only 21 of them are accessible for redox modifications [13,14]. Excessive accumulation of reactive oxygen species (ROS) can cause RyR2 oxidation and increase SR Ca2+ leak, leading to arrhythmias and contractile dysfunction [[15], [16], [17], [18]]. It has been shown that ROS activate RyR2-mediated Ca2+ release by promoting spontaneous Ca2+ sparks [19,20] and Ca2+ waves [21]. In failing heart, RyR2 oxidation and aberrant SR Ca2+ release have been linked to the dissociation of the RyR2 regulator calmodulin (CaM) but not FKBP12.6 [22,23]. Despite its clinical significance, the molecular mechanisms of redox regulation as well as the redox-sensitive sites within human RyR2 (hRyR2) remain largely unknown.
The effect of oxidative stress on RyR structure and function is better described for its skeletal muscle isoform (RyR1). It has been shown that only a few cysteines within the RyR1 complex undergo redox modifications [24,25]. Cysteine 3635 (C3635) is considered to be the most important residue responsible for redox regulation of RyR1. This cysteine is localized within the CaM binding domain and can be involved in the channel's regulation by CaM [[26], [27], [28]]. It has been shown that during oxidative stress C3635 can undergo S-nitrosylation, S-glutathionylation or can be oxidized to a disulfide leading to intersubunit cross-linking [26]. Similarly to RyR1, the cardiac RyR2 is also prone to cross-linking during oxidative stress [29]. We have shown previously that this post-translational modification has a profound effect on SR Ca2+ leak during oxidative stress in rabbit cardiomyocytes [18]. However, it remains unclear whether the corresponding cysteine 3602 (C3602) in RyR2 is involved in cross-linking and what the functional consequence of C3602 redox modification might be. It has been shown that the mutation of C3602 to alanine modulates store overload-induced Ca2+ release by increasing activation and decreasing termination thresholds [30]. Surprisingly, this mutation prevented neither the channel's activation by oxidative stress nor its regulation by CaM.
In the present study, we assessed the functional significance of intersubunit cross-linking in the regulation of hRyR2. We found that redox-mediated hRyR2 cross-linking is a highly effective post-translational modification associated with oxidative stress. The levels of hRyR2 cross-linking positively correlated with an increase in ER Ca2+ leak and CaM dissociation from the channel. We also re-evaluated the functional significance of C3602, and showed that this residue is not involved in hRyR2 cross-linking. Moreover, C3602 plays only a limited role in hRyR2 regulation by CaM under control conditions and during oxidative stress.
2. Materials and METHODS
2.1. R-CEPIA1er, mCer-SERCA2a and GFP-hRyR2 expression
Generation of the vector containing mCer-SERCA2a has been previously described [31]. pCMV R-CEPIA1er was a gift from Dr. Masamitsu Iino (Addgene plasmid 58,216; http://n2t.net/addgene:58,216; RRID: Addgene_58,216). The vector encoding hRyR2 cDNA fused to green fluorescent protein (GFP) at the N-terminus was kindly provided by Dr. Christopher George (University of Cardiff). The C3602A hRyR2 (hRyRC3602A) mutant was generated by cloning a fragment of wild type hRyR2 (hRyR2WT) cDNA containing C3602, in the pBlueScript II SK(+) cloning vector (kindly provided by Dr. Ruben Mestril, Loyola University Chicago). The mutation C3602A was generated using a site-directed mutagenesis kit (Q5 site-directed mutagenesis kit, NEB). Specific primers containing the mutation were used to amplify the whole plasmid containing the hRyR2 fragment. The mutated RyR2 fragment was cloned back into the hRyR2 backbone, yielding the C3602A mutant. After verification of the mutagenesis by single pass analysis, the plasmids were amplified and used for experimentation. For functional measurements, HEK293 cells were grown on 30 mm plastic dishes for 24 h, and co-transfected with three different plasmids containing GFP-hRyR2, mCer-SERCA2a and R-CEPIA1er genes respectively. Transfection was carried out using polyethylenimine (PEI). 24 h after transfection, cells were transferred to laminin-coated Lab-Tex chambers (ThermoFisher Scientific, USA) and grown for another 24 h. Experiments on transfected cells were carried out 48 h after transfection when expression of the exogenous genes was optimal.
2.2. Measurements of RyR2 cross-linking
HEK293 cells were grown on 100 mm plastic dishes for 24 h and then transfected with the GFP-hRyR2 plasmid using PEI. Cells were further grown for another 48 h. Cells were harvested and resuspended in the solution containing (in mM): 150 K-aspartate, 0.25 MgCl2, 0.1 EGTA, 10 HEPES, pH 7.2. Cell suspensions were permeabilized with 0.005% saponin for 5 min and incubated with different diamide concentrations for 10 min. The control group was further treated with 5 mM DTT. The treated cells were harvested and resuspended in lysis buffer (150 mM NaCl, 25 mM Hepes pH 7.4, 1% Triton X-100, phosphatases inhibitors) with 5 mM N-ethylmaleimide (NEM) to block free sulfhydryl groups. Lysate samples were incubated with non-reducing Laemmli buffer for 10 min, ran on 2–10% polyacrylamide gradient SDS-PAGE gels and blotted overnight onto the nitrocellulose membrane. Cross-linking was detected using F1 anti-RyR2 primary antibody (1:1000; Santa Cruz, USA) and anti-mouse secondary antibody (1:5000. Santa Cruz, USA). The amount of hRyR2 cross-linking was analyzed in ImageJ software (NIH, USA), based on the disappearance of the monomeric 560kD band.
2.3. Generation of the roGFP-PLB sensor
pLPCX redox GFP (roGFP)-ORP1 was provided by Dr. Tobias Dick (German Cancer Research Group, Heidelberg). roGFP cDNA without the STOP codon was amplified and cloned into pCDNA3.1 (ThermoFisher Scientific, USA), obtaining pcDNA3.1_roGFP. pCMV-phospholamban (PLB) was kindly donated by Dr. Seth Robia (Loyola University Chicago, USA). The PLB cDNA was amplified and cloned into the pcDNA3.1_roGFP, after the roGFP cDNA, yielding pcDNA3.1_roGFP_PLB. In this construct, roGFP is fused to the N-terminus domain of PLB. After verification of sequences by single pass analysis (AGCT inc., USA), the roGFP-PLB plasmid was used for experimentation.
2.4. Measurements of oxidative stress, [Ca2+]ER and CaM-hRyR2 binding
All experiments were conducted using a laser scanning confocal microscope (Radiance 2000 MP, BioRad) equipped with a 40x oil objective lens (N.A. = 1.3). The cells prepared for an experiment were washed with a solution containing (in mM): 150 K-aspartate, 0.25 MgCl2, 0.1 EGTA, 10 HEPES, pH 7.2. Then, the plasma membrane was permeabilized with 0.005% saponin dissolved in the experimental solution containing (in mM): K-aspartate 100; KCl 15; KH2PO4 5; MgATP 5; EGTA 0.35; CaCl2 0.22; MgCl2 0.75; HEPES 10; dextran (MW: 40,000) 2% and pH 7.2. Free [Ca2+] and [Mg2+] were 200 nM and 1 mM respectively. After the permeabilization, the cells were perfused with saponin-free solution during an experiment.
The level of oxidative stress at the cytosolic side of the ER membrane was measured with roGFP-PLB. For roGFP excitation, we used the 488-nm line of the argon laser, and signal was collected at >515 nm. 2D images (512 × 512 pixels) were collected every 10 s at a scanning speed of 6 ms/line. As the roGFP fluorescence decreases with the increase of roGFP oxidation, the recorded signal (F) was presented as background-subtracted (1 − F/FMax) values. The background fluorescence was recorded during the maximum roGFP oxidation with 100 μM diamide. The maximal signal (FMax) was recorded in the presence of 5 mM DTT.
Changes in [Ca2+]ER were recorded with the Ca2+ sensor R-CEPIA1er in HEK293 cells expressing GFP-hRyR2 and mCer-SERCA2a. R-CEPIA1er was excited with the 543-nm line of a He–Ne laser and fluorescence was measured at >580 nm. For Ca2+ wave measurements, 2D images (256 × 256 pixels) were collected every 0.5 s at a scanning speed of 2 ms/line. For measurements of ER Ca2+ load-leak balance, 2D images (512 × 512 pixels) were collected every 5 s at a scanning speed of 6 ms/line. At the end of each experiment, caffeine (10 mM) was applied to induce complete depletion of [Ca2+]ER. Then, Fmax was estimated by application of 2 μM ionomycin and 10 mM Ca2+. The changes in [Ca2+]ER were presented normalized according to [Ca2+]ER =(F-Fmin)/(Fmax-Fmin).
To measure the CaM dissociation from hRyR2, we used CaM labeled at position 34 with Alexa Fluor 568 C5 maleimide (F-CaM), which was prepared as we previously described [32]. Permeabilized cells expressing GFP-hRyR2 were saturated with 300 nM F-CaM for 10 min, and then were equilibrated in 60 nM F-CaM. The solution in the chamber was replaced by the experimental solution containing diamide (25 or 50 μM) with F-CaM (60 nM), followed by 10 min incubation. After the subtraction of background fluorescence, the bound F-CaM fluorescence was calculated according to: F (F-CaM) = (F-Fmin)/(F0-Fmin). Fmin was obtained after treating cells with suramin (100 μM) with no F-CaM.
2.5. Statistics
All data presented as means ± SEM of n measurements. Obtained 2D images were analyzed using ImageJ software (NIH, USA). Groups were compared using the Student's t-test for unpaired data sets and differences were considered statistically significant at P < 0.05. Peak analysis, statistical analysis, and graphical representation of averaged data was carried out on Origin 2016 SR2 software (OriginLab, USA).
3. Results
3.1. Sensitivity of hRyR2 cross-linking to oxidative stress
We have shown that the thiol-specific oxidant diamide causes disulfide cross-linking between RyR2 subunits in rabbit ventricular myocytes [29]. To characterize the sensitivity of hRyR2 to oxidative stress, cross-linking was measured upon application of increasing concentrations of diamide in permeabilized HEK293 cells expressing GFP-hRyR2. This oxidative treatment led to the formation of hRyR2 high molecular weight oligomeric forms (>1 MDa) together with a gradual disappearance of the monomeric 560 kDa RyR2 band (Fig. 1A). The application of 5 mM reducing agent DTT prevented hRyR2 cross-linking induced by diamide. Changes in the monomeric hRyR2 band density after oxidation was used to quantify the cross-linking level. We found that incubation with 25 μM diamide caused a full disappearance of the monomeric band, yielding 100% hRyR2 cross-linking (Fig. 1D). Next, we analyzed the level of roGFP oxidation at the cytosolic side of ER membrane. The redox sensor roGFP reports the level of oxidation of two cysteines within the GFP β-barrel [33]. To anchor roGFP to the ER membrane, we fused it to the cytosolic N-terminus of transmembrane protein PLB (Fig. 1B). This led to a similar cellular localization pattern of roGFP-PLB as that of hRyR2 (Fig. 3A), suggesting that the sensor was successfully targeted to the ER membrane. Dose-dependence of roGFP-PLB cysteine oxidation was measured during the application of increasing concentrations of diamide (Fig. 1C). These experiments revealed that 25 μM diamide that produced the maximal hRyR2 cross-linking caused only 69 ± 5% (n = 11) roGFP-PLB cysteine oxidation and >50 μM diamide was required to fully oxidize this biosensor. We determined that the KM of hRyR2 cross-linking and of the roGFP signal were 6.27 ± 1.72 (n = 5) and 16.54 ± 6.58 μM (n = 11), respectively (Fig. 1D). It appeared that the hRyR2 cysteines involved in the cross-linking are highly sensitive to oxidative stress. Since 25 and 50 μM diamide produced similar hRyR2 cross-linking but different extents of roGFP-PLB oxidation, we analyzed how these two concentrations affect the hRyR2-Ca2+ release and the hRyR2-CaM binding.
Fig. 1.

Effect of diamide on hRyR2WTcross-linking and roGFP-PLB oxidation. A, Western blot images of hRyR2 cross-linking induced by increasing concentrations of diamide. The control samples were treated with 5 mM DTT to prevent hRyR2 oxidation and cross-linking. B, a schematic diagram of the roGFP-PLB sensor. The redox sensor roGFP was fused to the N-terminus of PLB to measure oxidative stress at the cytosolic side of ER membrane (left). Expression pattern of the roGFP-PLB sensor in HEK293 cells (right). C, changes of the roGFP-PLB signal in response to increasing concentrations of diamide. As the roGFP-PLB signal decreases with an increase of roGFP oxidation, the recorded signal (F) was presented as background-subtracted (1−F/FMax) values. The background fluorescence was recorded during the maximum roGFP oxidation with 100 μM diamide. FMax was recorded in the presence of 5 mM DTT. D, dose-dependence of the hRyR2WT cross-linking (n = 5 western blots) and the roGFP-PLB signal (n = 11 cells) from diamide concentration. The data were fitted by the Hill equation using the built-in Origin 2016 SR Hill function, which was also used to determine the Michaelis constant (Km).
Fig. 3.

Effect of diamide on CaM dissociation from hRyR2WT. A, representative images of HEK293 cells expressing GFP-RyR2WT (left), the Alexa 568-labeled CaM (F-CaM) signal (middle) and the merged GFP-RyR2WT and F-CaM signals (right). B, representative traces of F-CaM dissociation from GFP-RyR2WT upon application of 25 μM and 50 μM diamide. At the end of the experiment, suramin was applied to completely remove the F-CaM from hRyR2. C, Effect of diamide on the CaM dissociation from the hRyR2WT complex. On average, levels of the CaM dissociation induced by 25 μM (n = 11 cells) and 50 μM (n = 10 cells) diamide were not significantly different (p = 0.57).
3.2. Effect of oxidative stress on Ca2+ waves and ER Ca2+ load
Permeabilized HEK293 cells expressing GFP-hRyR2 and mCer-SERCA2a together with the ER Ca2+ sensor R-CEPIA1er were used to evaluate the effect of diamide on ER Ca2+ release. Co-expression of hRyR2 and SERCA2a generated periodic Ca2+ waves due to spontaneous activation of hRyR2 followed by SERCA Ca2+ reuptake (Fig. 2A; Control), thus providing a cellular model to reproduce “cardiac-like” Ca2+ release events [31]. Application of 25 μM diamide caused a fast decline of the basal [Ca2+]ER (or ER Ca2+ load) by 29.5 ± 3.4% (n = 22) followed by the cessation of Ca2+ waves. 50 μM diamide resulted in similar functional response and did not produce further decline in [Ca2+]ER (Fig. 2A and B). In the presence of 50 μM diamide, ER Ca2+ load decreased by 30.7 ± 4.2% (n = 17). The high frequency of Ca2+ waves at the 200 nM cytosolic [Ca2+] did not allow an accurate measurement of ER Ca2+ load. Lowering cytosolic [Ca2+] to 100 nM abolished Ca2+ waves, but maintained ER Ca2+ load at a steady level. We found that 25 and 50 μM diamide produced similar declines in ER Ca2+ load (Fig. 1 Sup). Since SERCA function is unaffected by these diamide concentrations [31], the decline in [Ca2+]ER was likely mediated by an increase in ER Ca2+ leak via hRyR2. These results show that the maximal effect of 25 μM diamide on Ca2+ waves and ER Ca2+ leak correlates with the maximum level of hRyR2 cross-linking (Fig. 1A), which is consistent with a functional importance of this redox modification. Since these experiments were conducted without adding exogenous CaM to permeabilized cells, the observed effect of diamide on ER Ca2+ release did not require CaM-hRyR2 interaction.
Fig. 2.

Effects of diamide on hRyR2WT-mediated Ca2+waves and ER Ca2+load. A, representative recordings of Ca2+ waves from two different cells (top and bottom) expressing hRyR2WT and SERCA2a in the control conditions (left) and during 25 μM or 50 μM diamide application (right). The dashed lines indicate the level of ER Ca2+ load. B, the effect of diamide on ER Ca2+ load. ER Ca2+ load was measured between Ca2+ waves. On average, changes in ER Ca2+ load induced by 25 μM (n = 22 cells) and 50 μM (n = 17 cells) diamide were not significantly different (p = 0.82; NS).
3.3. Effect of oxidative stress on the CaM dissociation from hRyR2
It has been shown that oxidative stress reduces CaM binding to the RyR2 macromolecular complex in cardiomyocytes [23]. Here, we asked whether the level of cross-linking correlates with the degree of CaM dissociation from hRyR2. To measure the diamide effect on CaM-hRyR2 binding, GFP-hRyR2 expressed in HEK293 cells were saturated with 300 nM Alexa-568 labeled CaM (F-CaM) after membrane permeabilization. Cells were then equilibrated with 60 nM F-CaM, corresponding to the physiological concentration of free CaM in cardiomyocytes [34]. The F-CaM signal co-localized with GFP-hRyR2, suggesting a formation of the CaM-hRyR2 complex (Fig. 3A). The Alexa-568 signal was not detected in cells that did not express hRyR2, suggesting that hRyR2a represents the vast majority of CaM binding in our cell model. The application of diamide caused CaM dissociation from hRyR2, which was measured as a relative decline in F-CaM fluorescence over time (Fig. 3B). Overall, 25 and 50 μM diamide decreased the F-CaM signal by 43.7 ± 2.7% (n = 11) and 46.4 ± 4.1% (n = 10), respectively. Although 50 μM diamide dissociated F-CaM at a higher rate, the final level of F-CaM signal was the same for both diamide concentrations (Fig. 3C). These results indicate that the degree of oxidative stress producing the maximal hRyR2 cross-linking also resulted in the maximal CaM dissociation from hRyR2.
Role of cysteine 3602 in hRyR2 cross-linking and functional response to oxidative stress.
Cysteine 3635 (C3635) in RyR1 has been shown to undergo functionally important redox modifications, including intersubunit cross-linking [25,26]. Given the high level (~65%) of homology between RyR1 and RyR2 [35], we tested whether the corresponding cysteine 3602 (C3602) in hRyR2 is involved in intersubunit cross-linking. CaM binding site is highly conserved in RyR1 and RyR2 (Fig. 2A Sup). According to recent cryo-EM 3D reconstruction of Ca2+-CaM bound to RyR [36,37], cysteine residues that correspond to C3635 in RyR1 and C3602 in hRyR2 are oriented similarly in the CaM binding site (Fig. 2B Sup), suggesting similar functional properties. C3602 was mutated to alanine to generate GFP-hRyR2C3602A. Then, Western blot analysis was employed to determine whether the C3602A mutation prevents the diamide-induced cross-linking. Similar to hRyR2WT, 25 μM diamide produced the full disappearance of the monomeric form of hRyR2C3602A, indicating 100% channel cross-linking (Fig. 4A). The functional response of hRyR2C3602A to oxidative stress was tested in the HEK293 cell model. Cells expressing the hRyR2C3602A mutant together with SERCA2a generated regular Ca2+ waves (Fig. 4B) with similar properties as cells with hRyR2WT (Fig. 2A). Treatment of the hRyR2C3602A expressing cells with diamide decreased ER Ca2+ load and abolished Ca2+ waves (Fig. 4B). Overall, 25 and 50 μM diamide decreased ER Ca2+ load by 26.2 ± 3.5% (n = 22) and 32.6 ± 4.2% (n = 9) in cells expressing hRyR2C3602A, respectively. These diamide-induced changes in ER Ca2+ load were not significantly different for hRyR2C3602A vs hRyR2aWT (Fig. 4C). These results strongly suggest that unlike the equivalent cysteine in RyR1, C3602 participates neither in hRyR2 cross-linking nor in hRyR2 activation during oxidative stress.
Fig. 4.

Effect of diamide on the channel's cross-linking, Ca2+waves and ER Ca2+load in cells expressing hRyR2C3602A. A, Western blot images of the channel's cross-linking induced by increasing concentrations of diamide in cells expressing hRyR2WT and hRyR2C3602A. B, representative recordings of Ca2+ waves from two different cells expressing hRyR2C3602A (top and bottom) in the control conditions (left) and during 25 μM or 50 μM diamide application (right). The dashed lines indicate the level of ER Ca2+ load. C, diamide-induced depletion of ER Ca2+ load in cells expressing hRyR2WT and hRyR2C3602A. On average, changes in ER Ca2+ load induced by diamide in hRyR2WT and hRyR2C3602A cells were not significantly different (for 25 μM diamide p = 0.49, n = 26 and 22 cells and for 50 μM diamide p = 0.81, n = 17 and 9 cells).
3.4. Role of cysteine 3602 in the regulation of hRyR2 by CaM
Since C3602 is located within the CaM binding domain, we examined whether the C3602A mutation prevents CaM dissociation from hRyR2 during oxidative stress. CaM dissociation from the hRyR2C3602A mutant was measured in the presence of 50 μM diamide (Fig. 5A) using an approach similar to that described in Fig. 3. These experiments revealed that the C3602A mutation does not affect CaM binding to hRyR2C3602A in the control conditions (measured as the F-CaM signal before the diamide application) and prevents the diamide-induced CaM dissociation by ~24% (Fig. 5B). Then, we examined whether the C3602A mutation alters the inhibitory effect of CaM on hRyR2-mediated Ca2+ release. Ca2+ waves in cells expressing hRyR2WT or hRyR2C3602A were recorded under control conditions and after adding 1 μM of exogenous CaM (Fig. 6A). In both experimental groups, CaM decreased Ca wave frequency, increased Ca2+ wave termination level and increased ER Ca load (Fig. 6B). However, the effect of CaM on ER Ca2+ load and Ca2+ wave frequency was significantly stronger in hRyR2C3602A expressing cells, suggesting that the C3602A mutation enhances the CaM inhibitory action. The subsequent application of 50 μM diamide decreased ER Ca2+ load by 29 ± 3% (n = 23) in hRyR2WT and by 19 ± 3% (n = 23) in hRyR2C3602A expressing cells. These results indicate that C3602 can be involved in the regulation of hRyR2 by CaM in the control conditions and during oxidative stress. However, the limited protective effect (~34%) of the C3602A mutation suggests that other cysteines, outside the CaM binding site, play more important role in the redox-mediated CaM-hRyR2 dissociation.
Fig. 5.

Effect of diamide on the CaM dissociation from hRyR2C3602A. A, representative traces of F-CaM dissociation from GFP-RyR2 upon application of 50 μM diamide in cells expressing hRyR2WT and hRyR2C3602A. Suramin was applied to completely dissociate the F-CaM from hRyR2. B, effect of diamide on CaM dissociation from hRyR2WT and hRyR2C3602A. On average, the C3602A mutation reduced diamide-induced CaM dissociation from hRyR2 by 24% (p = 0.002; hRyR2WT n = 22 and hRyR2C3602A n = 19 cells).
Fig. 6.

Effects of CaM and diamide on Ca waves and ER Ca2+load in cells expressing hRyR2WTand hRyR2C3602A. A, representative recordings of Ca2+ waves in cells expressing hRyR2WT and hRyR2C3602A under control conditions, after 1 μM CaM application, and the subsequent application of 50 μM diamide (as indicated). The dashed lines indicate the level of ER Ca2+ load and Ca2+ wave termination. B, effect of CaM on Ca2+ wave frequency (p = 0.001 n = 22 and n = 33 cells), Ca2+ waves termination (p = 0.15, n = 22 and 33 cells) and ER Ca2+ load (p = 0.03, n = 36 and 40 cells) in hRyR2WT and hRyR2C3602A expressing cells. C, effect of 50 μM diamide on ER Ca load in the presence of CaM in hRyR2WT and hRyR2C3602A expressing cells. Overall, the C3602A mutation prevented diamide-induced depletion of ER Ca2+ load by 34% (p = 0.011, n = 23 hRyR2WT and 23 hRyR2C3602A cells).
4. Discussion
In the heart, concerted Ca2+ release through RyR2 is the essential step for initiating a robust myocardial contraction. Consequently, defects in RyR2 regulation cause contractile dysfunction in a variety of cardiac pathologies [7,8]. RyR2 has been characterized as highly sensitive to oxidative stress, with multiple sites that undergo redox modifications, including disulfide intersubunit cross-linking [38]. We have previously shown that the level of RyR2 cross-linking correlates with the degree of SR Ca2+ leak in rabbit ventricular myocytes [29]. Moreover, myocytes isolated from a rabbit HF model had a significant fraction of the cross-linked RyR2 [18]. However, the functional significance of this redox modification for human RyR2 remains largely unknown. We developed a simple expression system to study the redox regulation of hRyR2 in a well-controlled live-cell system that mimics aspects of cardiac Ca2+ handling. HEK293 cells can be easily transfected with large cDNA plasmids. Also, an advantage of the HEK cell line is that it lacks endogenous RyR, thereby permitting the study of only recombinant RyR2. This system, however, has its own limitations. Although we reconstructed cardiac-like Ca2+ handling by adding SERCA2a and CaM to the system, some mechanisms of RyR2 regulation are missing (e.g. phosphorylation). In addition, the dyadic organization of Ca2+ handling cannot be recapitulated in the expressing cell system. Despite these limitations, studies of recombinant human RyR2 provide important insights into the channel structure-function relationships that can uniquely inform therapeutic development targeting a range of serious clinical indications promoted by dysregulated Ca2+ [39].
In this study, we showed that hRyR2 recombinantly expressed in HEK293 cells undergoes intersubunit cross-linking during diamide exposure (Fig. 1A). The reducing agent DTT restored the monomeric pattern of hRyR2, confirming the reversibility of this redox modification. We compared the sensitivity of hRyR2 cross-linking to cysteine oxidation within the roGFP sensor. By fusing roGFP to the N-terminus of ER transmembrane protein PLB, we were able to measure the level of oxidative stress at the cytosolic side of ER next to hRyR2 (Fig. 1B). In permeabilized cells expressing hRyR2WT, 25 μM diamide caused 100% hRyR2 cross-linking that corresponded only to 70% oxidation of roGFP-PLB cysteines (Fig. 1D). It appears that cysteines involved in the RyR2 intersubunit cross-linking are highly sensitive to oxidative stress. While a structural basis for such high sensitivity of the cross-linked cysteines is unclear, it is possible that these RyR2 cysteines are more exposed to the cytosolic milieu, and thus more accessible to the oxidizing agent, than the cysteines in the roGFP-PLB biosensor.
To characterize the link between hRyR2 cross-linking and the channel's function, we studied the effect of diamide on hRyR2-mediated Ca2+ release (Fig. 2). Expression of both hRyR2 and SERCA2a in HEK293 cells creates a Ca2+ release/uptake machinery that generates “cardiac-like” Ca2+ waves [31]. Diamide decreased ER Ca2+ load followed by the cessation of Ca2+ waves. As a certain [Ca2+]ER is required to trigger spontaneous CICR, the disappearance of Ca2+ waves was a result of ER Ca2+ load depletion. Since SERCA Ca2+ uptake is not affected by the diamide concentrations used in this study [31], the decline in ER Ca2+ load is likely to be mediated by an increase in ER Ca2+ leak due to hRyR2 oxidation. The maximal effect of diamide on ER Ca2+ load and Ca2+ leak correlates with the maximal level of hRyR2 cross-linking, suggesting the functional importance of this redox modification. Since intersubunit dynamics within the RyR tetramer determines the channel's gating [[40], [41], [42], [43], [44]], it is not surprising that the intersubunit cross-linking has a significant impact on hRyR2 function.
In skeletal RyR1, C3635 has been identified as a key residue involved in redox regulation [25,27]. This cysteine is located within the CaM binding domain and can participate in intersubunit cross-linking. To evaluate a role of the corresponding C3602 in hRyR2 cross-linking, we analyzed the sensitivity of the hRyR2C3602A mutant to diamide oxidation. We found that the C3602A mutation does not protect from hRyR2 cross-linking nor hRyR2 activation by diamide (Fig. 4). In agreement with these findings, recent structural studies suggest that C3602 is too far from a neighboring RyR2 subunit to form a disulfide bond within the RyR2 tetramer [37]. It appears that other sites within hRyR2 are responsible for the formation of intersubunit disulfide bonds. The cross-linking sites might include cysteines in the N-terminal and the central domains. For example, the N-terminus of RyR2 has been reported to be involved in the redox-sensitive tetramer formation [45]. In addition to C3635 in RyR1, 11 other cysteines have been shown to undergo endogenous and exogenous redox modifications [25]. Among these cysteines, C36, C2326 and C2363 can be oxidized to disulfide bonds [25]. While there is no homologue of C2326 in hRyR2, our pilot studies revealed that C36 and C2363 (corresponding C36 and C2330 in hRyR2) are not involved in intersubunit cross-linking (Fig. 3 Sup). Therefore, despite 65% homology, RyR1 and RyR2 display fundamental differences in mechanisms of redox regulation.
The CaM-RyR2 interaction plays an important role in regulation of CICR. CaM binds tightly to each RyR2 subunit and inhibits RyR2 activity via a Ca2+-dependent mechanism [46]. It has been shown that oxidative stress can increase SR Ca2+ leak in ventricular myocytes in part by displacing CaM from the RyR2 complex [22,23]. In the present study, we found that the diamide concentration producing maximal hRyR2 cross-linking resulted in the maximal CaM dissociation from hRyR2 (Fig. 3). Since diamide interacts mainly with cysteine thiol groups (which are absent in CaM), the observed effect was probably mediated by hRyR2 oxidation. The C3602A mutation prevented the diamide-induced CaM dissociation only by ~24% (Fig. 5), indicating that C3602 oxidation has a limited role in this effect. These results suggest that conformational changes within the hRyR2 tetramer caused by the cross-linking (outside of the CaM binding site) play an important role in CaM dissociation from hRyR2. We found that, under control conditions, CaM decreased hRyR2-Ca2+ release by promoting early CICR termination and by delaying CICR activation (Fig. 6). As a result of hRyR2 inhibition, CaM increased ER Ca2+ load. The diamide-induced hRyR2 oxidation increased ER Ca2+ leak and depleted ER Ca load in part by reducing the CaM inhibitory action. As expected based on the CaM binding experiments (Fig. 5), C3602 played only a limited role in the diamide-induced hRyR2 activation.
Collectively, these studies reveal a previously unknown mechanism of human RyR2 regulation. hRyR2 oxidation increases ER Ca2+ release/leak via at least two main mechanisms: (1) by promoting CaM dissociation due to intersubunit cross-linking and C3602 oxidation and (2) by direct activation of hRyR2 due to intersubunit cross-linking. This is in addition to CaM methionine oxidation, which can also contribute to ER Ca2+ leak by reducing the CaM binding to RyR [23,47,48].
Author contributions
R.N. E.B., D.D.T., R.L.C and A.V.Z. contributed to the conception and design of the study. R.N., E.B., D.K. and R.T.R performed the experimental work and analysis of results. R.N., E.B., D.D.T., R.L.C and A.V.Z. contributed to writing of the manuscript. All authors have approved the version to be published.
Declaration of competing interest
The authors declare that there is no conflict of interest regarding the publication of this article.
Acknowledgment
This work was supported by the National Institutes of Health Grants HL130231 (to A.V.Z.), HL092097 and HL138539 (to R.L.C), and AG026160 (to D.D.T). This study was also supported by a James DePauw pilot grant, Loyola University Chicago (to A.V.Z.). The authors would like to thank Dr. Christopher George (University of Cardiff, UK) for providing the vector encoding the GFP-hRyR2 and Dr. Ino for donating the R-CEPIA1er vector.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101729.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
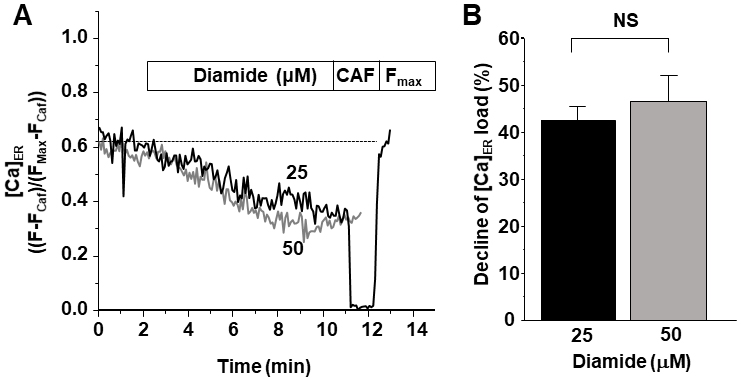
Effects of diamide on ER Ca load at 100 nM cytosolic [Ca2+]. A, representative traces of [Ca2+]ER decline in two HEK293 cells expressing hRyR2WT during the application of 25 and 50 μM diamide. At the end of the experiment, caffeine was applied to completely deplete [Ca]ER. B, effect of diamide on ER Ca load. On average, the decline in ER Ca2+ load induced by 25 μM and 50 μM diamide were not significantly different (p = 0.57; n = 11 and 10 cells).
{kind=link}
A. Sequence alignment of CaM binding site for human, porcine, rabbit and mouse RyR1 and RyR2. Conserved residues are shown in bold. The position of a cysteine residue corresponding to hRyR2 C3602 is boxed with yellow background. B. A superimposition of Ca2+-CaM (white) bound to RyR1 CaM binding peptide (cyan, PDB ID: 2BCX) and Ca2+-CaM (gray) bound to porcine RyR2 CaM binding site (green, PDB ID: 6JIY). Cysteines in CaM binding site that correspond to C3603 in porcine RyR2 (or C3602 in hRyR2) and C3635 in RyR1 are shown as yellow spheres. Images are obtained using Python 1.7.
{kind=link}
Western blot images of hRyR2 cross-linking induced by diamide in intact HEK 293 cells expressing hRyRC36A or hRyR2C2330A mutants.
{kind=link}
References
- 1.Coronado R., Morrissette J., Sukhareva M., Vaughan D.M. Structure and function of ryanodine receptors. Am. J. Physiol. Cell Physiol. 1994 doi: 10.1152/ajpcell.1994.266.6.c1485. [DOI] [PubMed] [Google Scholar]
- 2.Meissner G. Ryanodine receptor/Ca2+ release channels and their regulation by endogenous effectors. Annu. Rev. Physiol. 1994 doi: 10.1146/annurev.ph.56.030194.002413. [DOI] [PubMed] [Google Scholar]
- 3.Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 1983;245:C1–14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- 4.Bers D.M. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 5.Shannon T.R., Ginsburg K.S., Bers D.M. Quantitative assessment of the SR Ca 2+ leak-load relationship. Circ. Res. 2002;91:594–600. doi: 10.1161/01.RES.0000036914.12686.28. [DOI] [PubMed] [Google Scholar]
- 6.Zima A.V., Bovo E., Bers D.M., Blatter L.A. Ca 2+ spark-dependent and -independent sarcoplasmic reticulum Ca 2+ leak in normal and failing rabbit ventricular myocytes. J. Physiol. 2010;588:4743–4757. doi: 10.1113/jphysiol.2010.197913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yano M. Ryanodine receptor as a new therapeutic target of heart failure and lethal arrhythmia. Circ. J. 2008;72:509–514. doi: 10.1253/circj.72.509. JST.JSTAGE/circj/72.509 [pii] [DOI] [PubMed] [Google Scholar]
- 8.Blayney L.M., Lai F.A. Ryanodine receptor-mediated arrhythmias and sudden cardiac death. Pharmacol. Ther. 2009;123:151–177. doi: 10.1016/j.pharmthera.2009.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Priori S.G., Chen S.R.W. Inherited dysfunction of sarcoplasmic reticulum Ca2+ handling and arrhythmogenesis. Circ. Res. 2011;108:871–883. doi: 10.1161/CIRCRESAHA.110.226845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suzuki Y.J., Ford G.D. Redox regulation of signal transduction in cardiac and smooth muscle. J. Mol. Cell. Cardiol. 1999;31:345–353. doi: 10.1006/jmcc.1998.0872. [DOI] [PubMed] [Google Scholar]
- 11.Zima A.V., Blatter L.A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc. Res. 2006;71:310–321. doi: 10.1016/j.cardiores.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 12.Hool L.C., Corry B. Redox control of calcium channels: from mechanisms to therapeutic opportunities. Antioxidants Redox Signal. 2007;9:409–435. doi: 10.1089/ars.2006.1446. [DOI] [PubMed] [Google Scholar]
- 13.Xu L. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]
- 14.Donoso P., Sánchez G., Bull R., Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front. Biosci. 2011;16:553–567. doi: 10.2741/3705. [DOI] [PubMed] [Google Scholar]
- 15.Terentyev D., Györke I., Belevych A.E., Terentyeva R., Sridhar A., Nishijima Y. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ. Res. 2008;103:1466–1472. doi: 10.1161/CIRCRESAHA.108.184457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belevych A.E., Terentyev D., Viatchenko-Karpinski S., Terentyeva R., Sridhar A., Nishijima Y. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc. Res. 2009;84:387–395. doi: 10.1093/cvr/cvp246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santos C.X.C., Anilkumar N., Zhang M., Brewer A.C., Shah A.M. Redox signaling in cardiac myocytes. Free Radic. Biol. Med. 2011;50:777–793. doi: 10.1016/j.freeradbiomed.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bovo E., Mazurek S.R., Zima A.V. The role of RyR2 oxidation in the blunted frequency-dependent facilitation of Ca2+transient amplitude in rabbit failing myocytes. Pflueg. Arch. Eur. J. Physiol. 2018;470:959–968. doi: 10.1007/s00424-018-2122-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan Y., Liu J., Wei C., Li K., Xie W., Wang Y. Bidirectional regulation of Ca2+ sparks by mitochondria-derived reactive oxygen species in cardiac myocytes. Cardiovasc. Res. 2008;77:432–441. doi: 10.1093/cvr/cvm047. [DOI] [PubMed] [Google Scholar]
- 20.Prosser B.L., Ward C.W., Lederer W.J. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011:80. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- 21.Bovo E., Lipsius S.L., Zima A.V. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J. Physiol. 2012;590:3291–3304. doi: 10.1113/jphysiol.2012.230748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ono M., Yano M., Hino A., Suetomi T., Xu X., Susa T. Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca2+ release in heart failure. Cardiovasc. Res. 2010;87:609–617. doi: 10.1093/cvr/cvq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oda T., Yang Y., Uchinoumi H., Thomas D.D., Chen-Izu Y., Kato T. Oxidation of ryanodine receptor (RyR) and calmodulin enhance Ca release and pathologically alter, RyR structure and calmodulin affinity. J. Mol. Cell. Cardiol. 2015;85:240–248. doi: 10.1016/j.yjmcc.2015.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Voss A.A., Lango J., Ernst-Russell M., Morin D., Pessah I.N. Identification of hyperreactive cysteines within ryanodine receptor type 1 by mass spectrometry. J. Biol. Chem. 2004;279:34514–34520. doi: 10.1074/jbc.M404290200. [DOI] [PubMed] [Google Scholar]
- 25.Aracena-Parks P., Goonasekera S.A., Gilman C.P., Dirksen R.T., Hidalgo C., Hamilton S.L. Identification of cysteines involved in S-nitrosylation, S-glutathionylation, and oxidation to disulfides in ryanodine receptor type 1. J. Biol. Chem. 2006;281:40354–40368. doi: 10.1074/jbc.M600876200. [DOI] [PubMed] [Google Scholar]
- 26.Moore C.P., Zhang J.Z., Hamilton S.L. A role for cysteine 3635 of RYR1 in redox modulation and calmodulin binding. J. Biol. Chem. 1999;274:36831–36834. doi: 10.1074/jbc.274.52.36831. [DOI] [PubMed] [Google Scholar]
- 27.Sun J., Xin C., Eu J.P., Stamler J.S., Meissner G. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc. Natl. Acad. Sci. U. S. A. 2001;98:11158–11162. doi: 10.1073/pnas.201289098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sun J., Xu L., Eu J.P., Stamler J.S., Meissner G. Nitric oxide, NOC-12, and S-nitrosoglutathione modulate the skeletal muscle calcium release channel/ryanodine receptor by different mechanisms: an allosteric function for O2 in S-nitrosylation of the channel. J. Biol. Chem. 2003;278:8184–8189. doi: 10.1074/jbc.M211940200. [DOI] [PubMed] [Google Scholar]
- 29.Mazurek S.R., Bovo E., Zima A.V.( Regulation of sarcoplasmic reticulum Ca(2+) release by cytosolic glutathione in rabbit ventricular myocytes. Free Radic. Biol. Med. 2014;68:159–167. doi: 10.1016/j.freeradbiomed.2013.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Mi T., Xiao Z., Guo W., Tang Y., Hiess F., Xiao J. Role of Cys 3602 in the function and regulation of the cardiac ryanodine receptor. Biochem. J. 2015;467:177–190. doi: 10.1042/BJ20141263. [DOI] [PubMed] [Google Scholar]
- 31.Bovo E., Nikolaienko R., Bhayani S., Kahn D., Cao Q., Martin J.L. Novel approach for quantification of endoplasmic reticulum Ca2+ transport. Am. J. Physiol. Heart Circ. Physiol. 2019 doi: 10.1152/ajpheart.00031.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cornea R.L., Nitu F., Gruber S., Kohler K., Satzer M., Thomas D.D. FRET-based mapping of calmodulin bound to the RyR1 Ca2+ release channel. Proc. Natl. Acad. Sci. U. S. A. 2009;106:6128–6133. doi: 10.1073/pnas.0813010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morgan B., Sobotta M.C., Dick T.P. Free radical biology & medicine measuring E GSH and H 2 O 2 with roGFP2-based redox probes. Free Radic. Biol. Med. 2011;51:1943–1951. doi: 10.1016/j.freeradbiomed.2011.08.035. [DOI] [PubMed] [Google Scholar]
- 34.Wu X., Bers D.M. Free and bound intracellular calmodulin measurements in cardiac myocytes. Cell Calcium. 2007;41:353–364. doi: 10.1016/j.ceca.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hakamata Y., Nakai J., Takeshima H., Imoto K. Primary structure and distribution of a novel ryanodine receptor/calcium release channel from rabbit brain. FEBS Lett. 1992;312:229–235. doi: 10.1016/0014-5793(92)80941-9. [DOI] [PubMed] [Google Scholar]
- 36.Maximciuc A.A., Putkey J.A., Shamoo Y., MacKenzie K.R. Complex of calmodulin with a ryanodine receptor target reveals a novel, flexible binding mode. Structure. 2006;14:1547–1556. doi: 10.1016/j.str.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 37.Gong D., Chi X., Wei J., Zhou G., Huang G., Zhang L. Modulation of cardiac ryanodine receptor 2 by calmodulin. Nature. 2019 doi: 10.1038/s41586-019-1377-y. [DOI] [PubMed] [Google Scholar]
- 38.Zima A.V., Mazurek S.R. Functional impact of ryanodine receptor oxidation on intracellular calcium regulation in the heart. Rev. Physiol. Biochem. Pharmacol. 2016;159:39–62. doi: 10.1007/112_2016_2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kushnir A., Wajsberg B., Marks A.R. Ryanodine receptor dysfunction in human disorders. Biochim. Biophys. Acta Mol. Cell Res. 2018;1865:1687–1697. doi: 10.1016/j.bbamcr.2018.07.011. [DOI] [PubMed] [Google Scholar]
- 40.Orlova E.V., Serysheva I.I., Van Heel M., Hamilton S.L., Chiu W. Two structural configurations of the skeletal muscle calcium release channel. Nat. Struct. Biol. 1996 doi: 10.1038/nsb0696-547. [DOI] [PubMed] [Google Scholar]
- 41.Tung C.-C., Lobo P. a, Kimlicka L., Van Petegem F. The amino-terminal disease hotspot of ryanodine receptors forms a cytoplasmic vestibule. Nature. 2010;468:585–588. doi: 10.1038/nature09471. [DOI] [PubMed] [Google Scholar]
- 42.Zalk R., Clarke O.B., des Georges A., Grassucci R.A., Reiken S., Mancia F. Structure of a mammalian ryanodine receptor. Nature. 2015;517:44–49. doi: 10.1038/nature13950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.des Georges A., Clarke O.B., Zalk R., Yuan Q., Condon K.J., Grassucci R.A. Structural basis for gating and activation of RyR1. Cell. 2016 doi: 10.1016/j.cell.2016.08.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santulli G., Lewis D., des Georges A., Marks A.R., Frank J. Ryanodine receptor structure and function in health and disease. Subcell. Biochem. 2018 doi: 10.1007/978-981-10-7757-9_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zissimopoulos S., Viero C., Seidel M., Cumbes B., White J., Cheung I. N-terminus oligomerization regulates the function of cardiac ryanodine receptors. J. Cell Sci. 2013;126:5042–5051. doi: 10.1242/jcs.133538. [DOI] [PubMed] [Google Scholar]
- 46.Yang Y., Guo T., Oda T., Chakraborty A., Chen L., Uchinoumi H. Cardiac myocyte Z-line calmodulin is mainly RyR2-bound, and reduction is arrhythmogenic and occurs in heart failure. Circ. Res. 2014;114:295–306. doi: 10.1161/CIRCRESAHA.114.302857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Balog E.M., Norton L.E., Bloomquist R.A., Cornea R.L., Black D.J., Louis C.F. Calmodulin oxidation and methionine to glutamine substitutions reveal methionine residues critical for functional interaction with ryanodine receptor-1. J. Biol. Chem. 2003;278:15615–15621. doi: 10.1074/jbc.M209180200. [DOI] [PubMed] [Google Scholar]
- 48.Balog E.M., Norton L.E., Thomas D.D., Fruen B.R. Role of calmodulin methionine residues in mediating productive association with cardiac ryanodine receptors. Am. J. Physiol. Heart Circ. Physiol. 2006;290:794–799. doi: 10.1152/ajpheart.00706.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effects of diamide on ER Ca load at 100 nM cytosolic [Ca2+]. A, representative traces of [Ca2+]ER decline in two HEK293 cells expressing hRyR2WT during the application of 25 and 50 μM diamide. At the end of the experiment, caffeine was applied to completely deplete [Ca]ER. B, effect of diamide on ER Ca load. On average, the decline in ER Ca2+ load induced by 25 μM and 50 μM diamide were not significantly different (p = 0.57; n = 11 and 10 cells).
A. Sequence alignment of CaM binding site for human, porcine, rabbit and mouse RyR1 and RyR2. Conserved residues are shown in bold. The position of a cysteine residue corresponding to hRyR2 C3602 is boxed with yellow background. B. A superimposition of Ca2+-CaM (white) bound to RyR1 CaM binding peptide (cyan, PDB ID: 2BCX) and Ca2+-CaM (gray) bound to porcine RyR2 CaM binding site (green, PDB ID: 6JIY). Cysteines in CaM binding site that correspond to C3603 in porcine RyR2 (or C3602 in hRyR2) and C3635 in RyR1 are shown as yellow spheres. Images are obtained using Python 1.7.
Western blot images of hRyR2 cross-linking induced by diamide in intact HEK 293 cells expressing hRyRC36A or hRyR2C2330A mutants.