

Abstract
Contrary to frequent reports in the literature, hydroxyl radical is not a key species participating in endogenous oxidative DNA damage. Instead, carbonate radical anion is formed from the Fenton reaction under cellular conditions and from decomposition of nitrosoperoxycarbonate generated during inflammation. Carbonate radical anion is a potent one-electron oxidant capable of generating base radical cations that can migrate over long distances in duplex DNA, ultimately generating 8-oxo-7,8-dihydroguanine at a redox-sensitive sequence such as GGG. Such a mechanism enables G-quadruplex-forming sequences to act as long-range sensors of oxidative stress, impacting gene expression via the DNA repair mechanism that reads and ultimately erases the oxidized base. With a writing, reading and erasing mechanism in place, oxidative ‘damage’ to DNA might be relabeled as ‘epigenetic’ modifications.
Oxidative stress is a cellular imbalance in oxidizing vs. reducing species. Mammalian cells typically have dioxygen concentrations near 200 μM, and while O2 itself is not very reactive, except with transition metals, oxidative phosphorylation in mitochondria reduces O2 to superoxide (O2∙ ─) that is converted to hydrogen peroxide (H2O2) and other reactive species. These along with others are collectively known as “reactive oxygen species” or ROS and are counterbalanced by cellular reductants, the most abundant of which is the thiol glutathione, present in low millimolar concentration.1 Along with ROS, cells can produce reactive nitrogen species (RNS) such as nitric oxide (NO) that is trapped by superoxide to generate peroxynitrite (ONOO─), which further reacts with dissolved CO2, ultimately producing carbonate radical anion, CO3∙ ─.2 Together, ROS and RNS react covalently with biomolecules such as DNA to modify or “damage” the carrier of genetic information.3 The fact that cells generate these reactive species naturally during metabolism, hypoxia, and also the inflammatory response to ward off invading microorganisms sets up another tug-of-war, that between DNA damage and DNA repair, whose imbalance can result in disease.1,3
Among ROS, hydroxyl radical (HO∙) is often cited as a key player, and bad actor, in the endogenous DNA damage pathway, and in particular its generation from the iron- or copper-catalyzed Fenton reaction of O2∙ ─ or H2O2 is blamed for mutagenic DNA base oxidation and ribose hydrogen abstractions that lead to DNA strand breaks and abasic sites.4 In contrast, recent work by Meyerstein and coworkers, who thoroughly characterized the kinetics of intermediate and product formation, demonstrated that hydroxyl radical is not the product of the Fenton reaction when physiological bicarbonate is present.5 Instead, the inner-sphere reaction of metal-bound carbonate and hydroperoxide generates carbonate radical anion, CO3∙ ─, as the sole ROS, according to Eqn. 1:
| (1) |
The production of carbonate radical anion by Eqn. 1 predominates over hydroxyl radical formation when the bicarbonate concentration exceeds about 100 μM. Given that intracellular [HCO3─] is typically 10–40 mM, essentially no hydroxyl radical should be formed by the Fenton reaction in mammalian cells. A similar conclusion was reached when chelated FeII was studied with the ligand being citrate in physiologically relevant concentrations.6
Both hydroxyl radical and carbonate radical anion are potent one-electron oxidants with reduction potentials of 2.4 and 1.6 V vs. NHE, respectively. The most sensitive base toward oxidation in DNA is guanine (1.3 V vs. NHE), and not surprisingly, oxidation of 2’-deoxyguanosine is a common outcome for both of these free radical oxidants. However, the chemistry of HO∙ with DNA is messy—it forms adducts to all four bases with low selectivity and abstracts hydrogen atoms from any solvent accessible site in 2’-deoxyribose leading to base release (from H1’ abstraction), 5’,8-cyclopurines (from H5’ abstraction) and a cascade of reactions leading to strand scission from hydrogen atom abstraction at these and other positions.4 Basically, hydroxyl radical is so reactive that it reacts covalently with any organic compound or biomolecule that it encounters. On the other hand, CO3∙ ─ serves principally as a one-electron oxidant with DNA, forming guanine radical cation (G∙+), and ultimately leading to C8 or C5 oxidation products (Fig. 1).7
Figure 1.
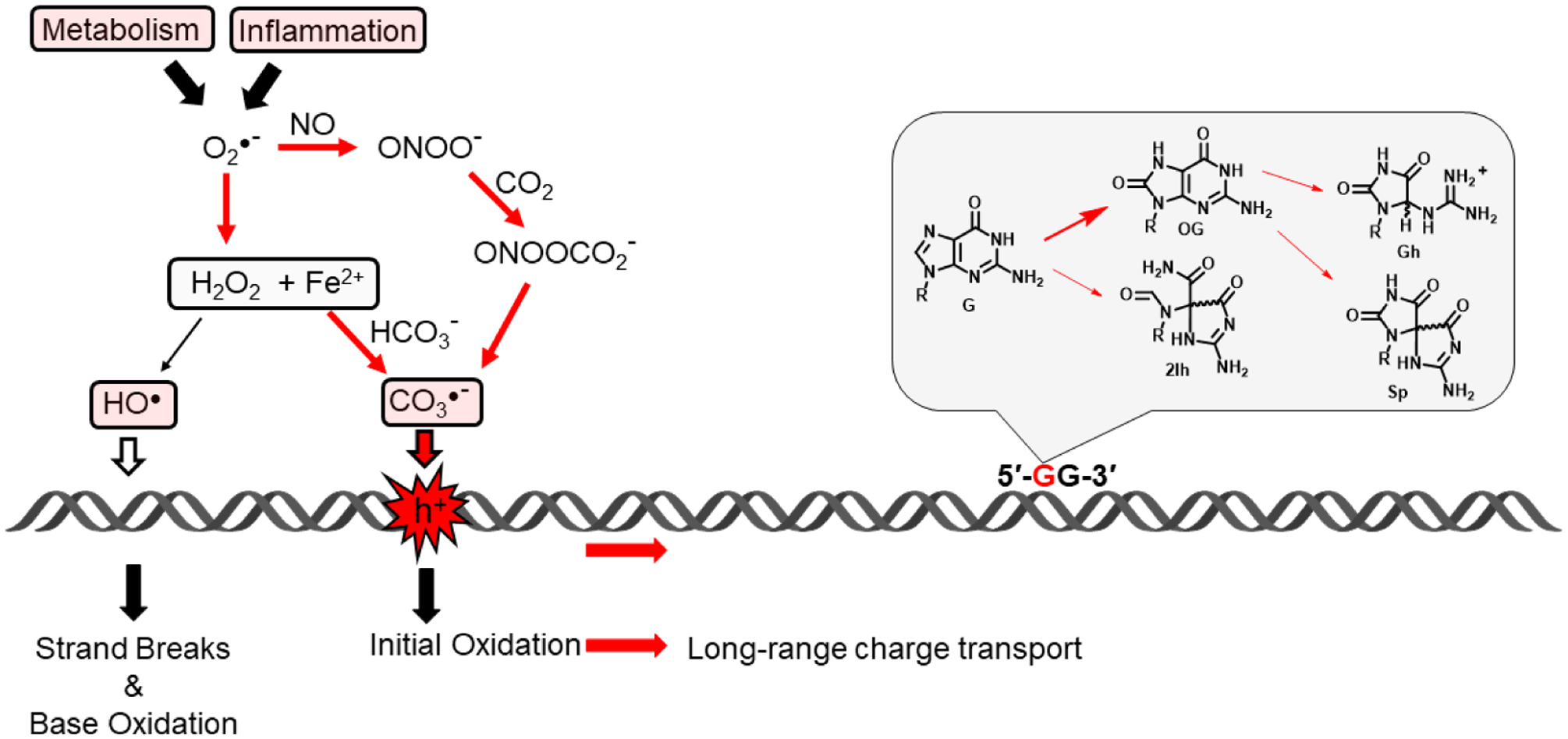
Cellular metabolism and the inflammatory response generate reactive oxygen and nitrogen species including superoxide, nitric oxide and hydrogen peroxide. Ultimate production of hydroxyl radical would lead to indiscriminate DNA base damage and strand breaks, whereas production of carbonate radical anion leads to electron hole (h+) formation that can migrate over long distances before finding a low energy site where further chemistry generates 8-oxoguanine (OG) and the hydantoins 2Ih, Gh, and Sp.
Despite at least four products being formed from the G∙+ reaction channel, oxidation of DNA by CO3∙ ─ is considerably less messy than that by HO∙. No ribose oxidation occurs (therefore, no direct strand breaks), and only the guanine base is oxidized to G∙+ when the Fenton reaction is conducted in the presence of bicarbonate.8 Also, only one DNA repair pathway is needed to reverse the oxidative modifications imposed by CO3∙ ─ and return to canonical base pairs.9 Non-bulky base modifications formed from oxidative stress are recognized and removed from DNA by a specialized set of DNA glycosylases in the base excision repair (BER) pathway.10 The major product of DNA oxidation in the cell is 8-oxo-7,8-dihydro-2’-deoxyguanosine (OG, Fig. 1), which is recognized and removed from an OG:C base pair in duplex DNA by the enzyme OGG1 through cleavage of the glycosidic bond between the ribose and the base, releasing the oxidized base and generating an apurinic (AP) site in DNA.10 The AP site is cleaved with the aid of AP-endonuclease-1 (APE1), and re-synthesis of undamaged DNA can then occur using the cytosine base opposite as a template for installation of an undamaged G nucleotide (Fig. 2).
Figure 2.
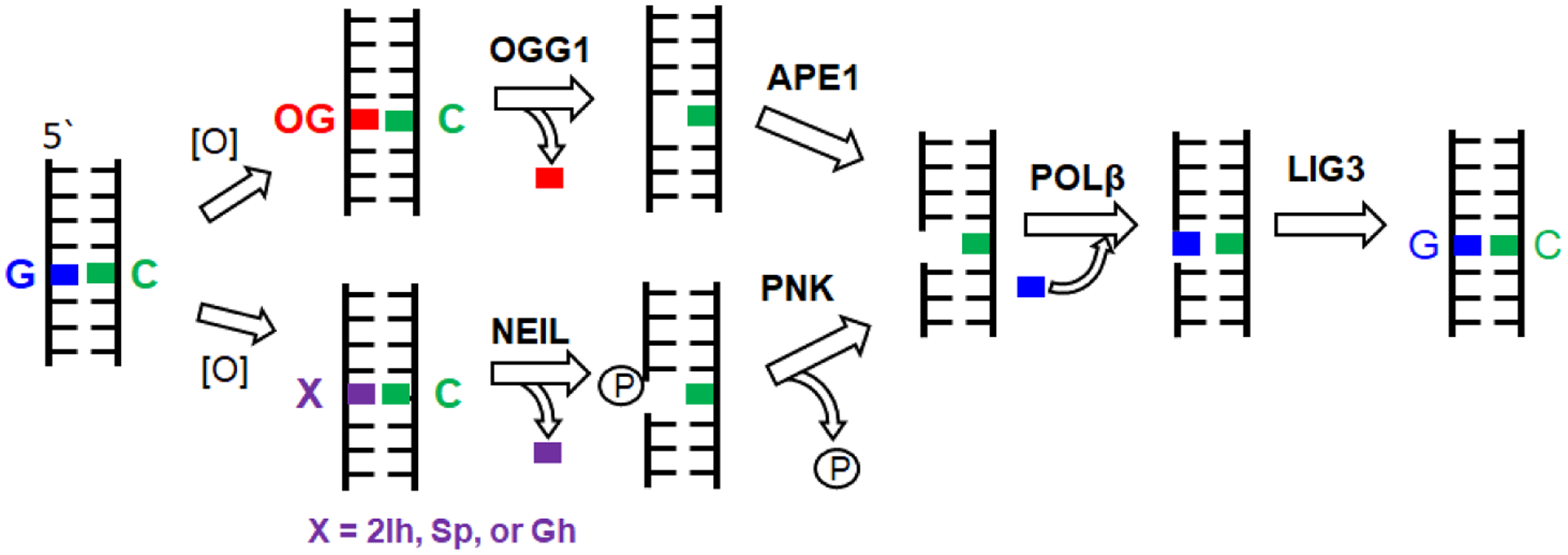
The BER pathway of oxidized G lesions.
Other products that form from G∙+ are the C5 oxidation product of G, 2Ih, and the further oxidation products of OG (i.e., those oxidized at both C8 and C5), namely Sp and Gh (Fig. 1).7 Their mechanistic pathways have been reviewed.9 These products arise because OG has a significantly lower reduction potential, by at least 600 mV, and it is therefore very sensitive to further oxidation. These three hydantoin structures, 2Ih, Gh and Sp, are the best known substrates for the NEIL glycosylases, of which there are three that specialize in removal of hydantoins from various DNA contexts.7 Thus, the overall picture of DNA oxidation is greatly simplified for the case in which carbonate radical anion acts as the predominant cellular ROS; a single family of guanine oxidation products is formed, and these in turn are recognized and repaired by either OGG1 or NEIL1–3 of the base excision repair pathway.9,10
What is the biological relevance of simplifying the chemical pathway to DNA oxidation? By funneling ROS-induced DNA modifications to a narrower set of products, chiefly derived from G and recognized by a small set of BER glycosylases, the genome can use oxidative modifications as a signaling pathway to respond to oxidative stress.11,12 In an ideal scenario, oxidative damage to guanine in DNA would be used as a signal to upregulate DNA repair genes or other genes that need to respond to an imbalance in the cellular redox state. Upregulating repair genes would have the advantage of aiding in the repair of the damage that caused the signal in the first place. For such a scenario to function, the oxidative damage would need to occur in a regulatory region of the genome such as a promoter/enhancer sequence or the 5’ or 3’-untranslated regions (UTRs). At a glance, this seems to ask the impossible because normal cellular levels of OG are only about 1 in 106 guanines.9 The amount of OG can go up nearly an order of magnitude during oxidative stress; nevertheless, one would consider OG a rare modification in the genome.
In order for a one-in-a-million phenomenon like oxidation of G to be responsible for a signaling event in DNA, the chemical reaction should not only be focused on a single type of nucleotide, G, it should also be localized to specific sequences in gene elements such as promoters where the oxidative modification can impact initiation of transcription. Two features of the DNA duplex assist this process.1 First, the redox potential of G is sequence dependent in π-stacked double-stranded DNA; runs of consecutive Gs, whether GG, GGG or GGGG, are measurably easier to oxidize than a 5’-HGH-3’ (H=A, C, T) sequence, and the site of oxidation is any G that has a 3’ adjacent G.13 Thus, in the sequence 5’-GGGG-3’, the first 3 Gs would be low energy sites for a G∙+ to reside, and the 3’-most G would be rarely oxidized. Migration of the electron hole formed by DNA’s reaction with carbonate radical anion is possible due to charge transport between adjacent π-stacked bases.
The second key feature of duplex DNA is that a well-stacked, intact duplex is capable of long-range charge transport as has been well described by the Barton lab over the past two decades.14 Most recently, electron hole migration has been observed in vitro up to a few thousand base pairs distant from the original site of oxidation.14 This means that the one-in-a-million oxidative damage in DNA can migrate to specific sites in the genome that are optimally reactive toward one-electron oxidation, namely oligo-G tracks. As long as the initial oxidation event is somewhere near a promoter region, OG will be formed in a sensitive site, and the initiation of repair can then impact gene expression. Given that G-rich sequences show lower nucleosome occupancy and that euchromatin appears to be more sensitive to DNA damage, it seems just plausible that oxidative stress could lead to enhanced DNA oxidation near G-rich promoters leading to a final focusing event on the oligo-G tracks via long-range charge transport.
Enter G-quadruplexes. These sequences comprise four or more tracks of oligo-G, interspersed with one or more loop nucleotides, generally following the formula 5’-(Gx≥3 Ny≤7 Gx≥3 Ny≤7 Gx≥3 Ny≤7 Gx≥3)-3’.15 The GGG tracks in one strand of the duplex can refold via Hoogsteen hydrogen bonding to form layers of G-tetrads connected by the intervening loops and binding K+ ions between layers. These GGG tracks are ideal sites for G oxidation by a one-electron mechanism, and thus we have proposed that these G-rich sequences marry these two features of oligo-G—their sensitivity to oxidation and their propensity to refold as G-quadruplexes (G4s)—to render them sensors of oxidative stress in the genome.
A G-quadruplex-forming sequence in a promoter can act as a redox sensor by switching from the standard DNA duplex to a G4 fold after one of the Gs is oxidized.1 Upon initial inspection, it seems that oxidation of a G would do the opposite and disrupt hydrogen bonding in a G-tetrad layer, thus favoring the duplex state in which an OG modification is well accommodated as an OG:C base pair rather than in the quadruplex. However, most promoter G4s have extra Gs in their G-tracks, often 4 or 5 instead of only 3, and about half of them have a 5th G run, which we propose can function as a “spare tire,” swapping in to replace a damaged G track (Fig. 3).16 Nevertheless, the presence of OG may not be enough to shift the equilibrium from duplex to quadruplex. However, once the OG base is excised by OGG1 in the base excision repair process, the AP site so formed is very destabilizing to the duplex, encouraging refolding to the quadruplex if the AP can be placed into a loop or a bulge while still forming a stable G4 (Fig. 3). Thermal stability measurements on DNA oligomers containing OG vs. AP support this structure-switching concept when extra Gs in the sequence permit the lesion to be looped out from a stable G4 core.12
Figure 3.
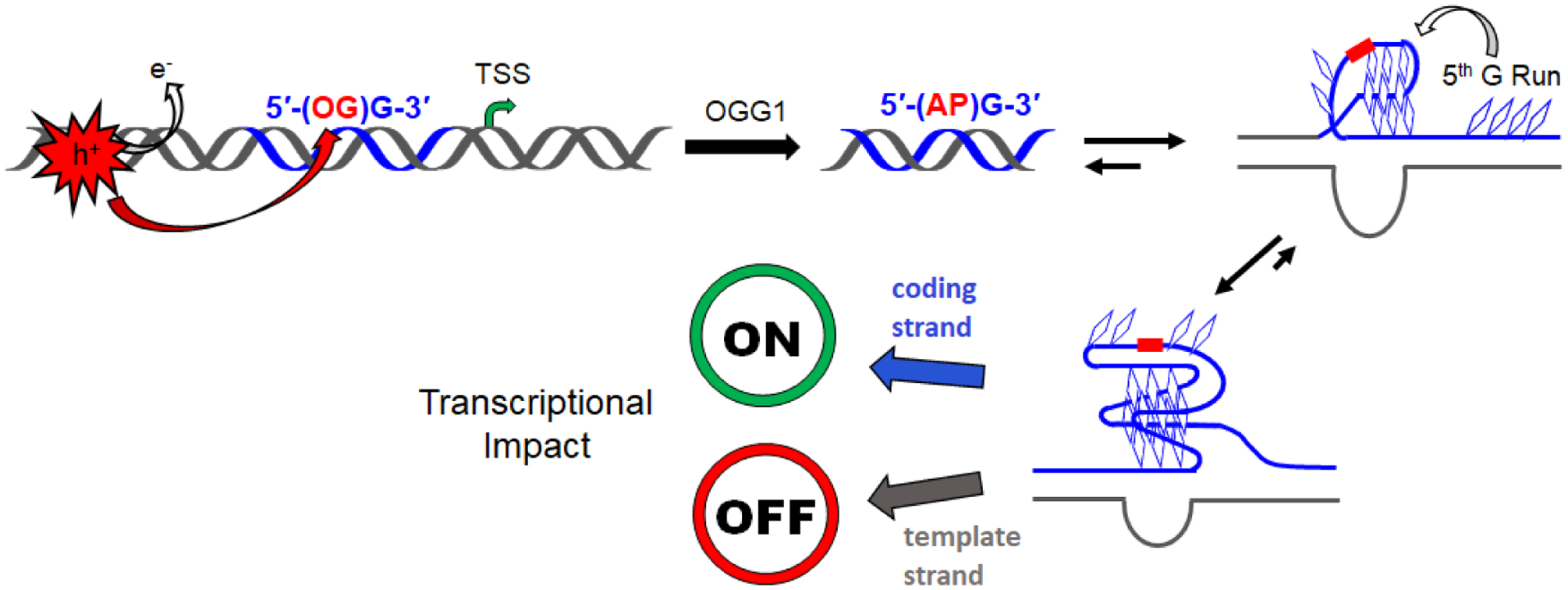
Scheme for refolding of an OG-modified G-rich sequence in a promoter to a G4 during base excision repair, ultimately impacting gene expression. The AP intermediate is duplex destabilizing leading to G4 folding that loops out the damage site. In the coding (non-template) strand, APE1 is involved in gene upregulation; in the template strand, CSB is involved in gene downregulation.
Presentation of DNA damage in the loop of a G4 can then be recognized by a reader protein, APE1 in the case of an AP lesion or NEIL3 in the case of a hydantoin lesion. Indeed, NEIL3 prefers to bind to lesions in the G4 context.17 If OG is still present after G4 folding, it is possible that OGG1 might bind for signaling purposes, but this repair enzyme has a strict requirement of the duplex OG:C context for excision of OG.10 Studies in our laboratory and others have shown that APE1 plays a significant role in its capacity as a redox effector factor to bind to non-canonical structures and recruit transcription factors such as hypoxia-inducible factor 1α (HIF-1α).12,18–20 The complete mechanism for gene regulation would then be as shown in Fig. 3.
Conveniently for this hypothesis, the majority of human gene promoters are G-rich, and nearly half contain potential G4 sequences, typically just upstream of the transcription start site—in other words, in an ideal location to impact initiation of gene induction.1,15 We examined 191 genes of the DNA repair pathway and found more than half of them have a potential G4 sequence in their promoter region, and many of these contain a 5th track or spare tire.1 Additionally, many oncogenes and cancer-associated genes (VEGF, c-MYC, KRAS, etc.) have active G4s in their promoters, and both misregulation of gene expression and increased oxidative stress are hallmarks of cancer cells. However, not all genes are responsive to oxidative stress, not all genes that are responsive to oxidative stress are immediately upregulated, and not all genes have G-quadruplexes in their promoters. Much is yet to be learned about the cellular choreography of gene expression following oxidative stress.
We tested the hypothesis that the interplay of DNA repair with oxidative damage in a promoter G4 would impact gene expression by constructing a reporter plasmid in which a luciferase gene was driven by a G4-containing promoter in its coding (non-template) strand, just ahead of the transcription start site.1,12 We inserted a synthetic oligonucleotide containing either OG or AP in specific sites of the VEGF, NTHL1, NEIL3, PCNA, or other promoter G4s into the plasmid, and then transfected the plasmid into various normal and cancer cells. Consistently, we observed about a threefold enhancement of gene expression when OG was present in the non-template strand, and a threefold decrease in gene expression when the lesion-containing G4 was in the template strand.1 In the latter case, the effect was dependent on the Cockayne Syndrome B (CSB) protein that is involved in transcription-coupled repair. In the now many cases of increased transcription from oxidative damage located in the non-template (coding) strand, the increase depended on the presence of BER enzymes, particularly APE1.
Nature’s logic in placing redox-sensitive G-rich sequences in regulatory regions of the genome only makes sense if the ROS generated during endogenous oxidative stress are able to target these sequences specifically. Does long-range charge transport provide enough focusing power to actually target promoter G4 sequences effectively? A factor in favor of oxidation happening near the promoter is the fact that G-rich promoter sequences have lower histone occupancy,21 and therefore they are most exposed to diffusible ROS. Another interesting proposal is that there may actually be a protein ‘writer’ of OG; Avvedimento and coworkers found evidence for the chromatin remodeler LSD1, which oxidatively demethylates histone lysines, to also produce OG and recruit OGG1 in the same vicinity.22 Both of these mechanisms, introduction by carbonate radical anion plus long-range CT or an oxidation wave from chromatin remodeling near a promoter, could cooperate to place OG in a promoter G4 to impact gene expression. Further experiments will help paint this overall landscape in more detail.
Endogenous oxidative damage to DNA should be mainly attributed to carbonate radical anion, the product of both the Fenton reaction in the intracellular medium and the decomposition of peroxynitrite in the presence of CO2.1–3,5–7 Hydroxyl radical chemistry with DNA may still be important in radiation damage during tumor treatment with ionizing radiation or from environmental exposure to x-rays or intense UV light.4 In addition, formation of ROS in highly localized concentrations may permit HO∙ to exact its non-specific damage on DNA when oxidative stress is rampant. Fortunately, multiple DNA repair mechanisms exist, and repair glycosylases often act on a wide range of substrates beyond the endogenously produced lesions they have evolved to recognize. For example, the NEIL glycosylases excise a broad family of oxidized pyrimidines including thymine glycol, 5-hydroxyC, and 5-hydroxyU, that could form from HO∙ attack at the 5,6-double bond of pyrimidines, in addition to the hydantoin lesions derived from G.9 Ionizing radiation also causes direct single- and double-strand breaks, and this damage recruits a suite of proteins for repair, signaling and gene expression via more complex pathways.
The predominance of CO3∙ ─ over HO∙ as the intracellular reactive oxygen species during endogenous oxidative stress reduces the chemical pathway to a single set of products derived from G∙+, and it permits the location of the damage to be focused on G-rich sites in gene regulatory elements such as G4s. Experiments to date suggest that this chemistry enables a feedback mechanism to up- or downregulate gene expression for a large set of redox-responsive genes, thus imbuing oxidative damage lesions with epigenetic qualities. Importantly, carbonate radical anion chemistry reveals how G-quadruplex-forming sequences can act as sensors of oxidative stress via DNA repair.1
Acknowledgements:
The authors thank coworkers in the Burrows lab for helpful discussions and experimental results described in the papers referenced. The research was supported by the U.S. National Cancer Institute grant no. R01 CA090689.
References
- 1.Fleming AM and Burrows CJ, J. Am. Chem. Soc, 2020, 142, 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koppenol WH, Serrano-Luginbuehl S, Nauser T and Kissner R, Chem. Res. Toxicol, 2020, 33, 1516. [DOI] [PubMed] [Google Scholar]
- 3.Lonkar P and Dedon PC, Int. J. Cancer, 2011, 128, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cadet J, Davies KJA, Medeiros MH, Di Mascio P, et al. , Free Radic. Biol. Med, 2017, 107, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Illes E, Mizrahi A, Marks V and Meyerstein D, Free Radic. Biol. Med, 2019, 131, 1. [DOI] [PubMed] [Google Scholar]
- 6.Illés E, Patra SG, Marks V, Mizrahi A, et al. , J. Inorg. Biochem, 2020, 206, 111018. [DOI] [PubMed] [Google Scholar]
- 7.Rokhlenko Y, Geacintov NE and Shafirovich V, J. Am. Chem. Soc, 2012, 134, 4955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fleming AM and Burrows CJ, Chem. Commun, 2020, 10.1039/D0CC04138F. [DOI] [Google Scholar]
- 9.Fleming AM and Burrows CJ, Free Radic. Biol. Med, 2017, 107, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.David SS, O’Shea VL and Kundu S, Nature, 2007, 447, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hao W, Wang J, Zhang Y, Wang C, et al. , FASEB J, 2020, doi: 10.1096/fj.201902243R. [DOI] [Google Scholar]
- 12.Fleming AM, Ding Y and Burrows CJ, Proc. Natl. Acad. Sci. U.S.A, 2017, 114, 2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saito I, Takayama M, Sugiyama H, Nakatani K, et al. , J. Am. Chem. Soc, 1995, 117, 6406. [Google Scholar]
- 14.Tse ECM, Zwang TJ, Bedoya S and Barton JK, ACS Cent. Sci, 2019, 5, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varshney D, Spiegel J, Zyner K, Tannahill D, et al. , Nat. Rev. Mol. Cell Biol, 2020, doi: 10.1038/s41580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fleming AM, Zhou J, Wallace SS and Burrows CJ, ACS Cent. Sci, 2015, 1, 226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou J, Fleming AM, Averill AM, Burrows CJ, et al. , Nucleic Acids Res, 2015, 43, 4039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broxson C, Hayner JN, Beckett J, Bloom LB, et al. , Nucleic Acids Res, 2014, 42, 7708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Antoniali G, Lirussi L, D’Ambrosio C, Dal Piaz F, et al. , Mol. Biol. Cell, 2014, 25, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pastukh V, Roberts JT, Clark DW, Bardwell GC, et al. , Am. J. Physiol. Lung Cell Mol. Physiol, 2015, 309, L1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fenouil R, Cauchy P, Koch F, Descostes N, et al. , Genome Res, 2012, 22, 2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Perillo B, Ombra MN, Bertoni A, Cuozzo C, et al. , Science, 2008, 319, 202. [DOI] [PubMed] [Google Scholar]