

Abstract
The purpose of this study was to develop solid dispersions of fenretinide(4HPR), incorporate them into poly (lactic-co-glycolic)(PLGA) millicylindrical implants, and evaluate the resulting implants in vitro and in vivo for future applications in oral cancer chemoprevention. Due to the extreme hydrophobicity of 4HPR, 4HPR-polyvinylpyrrolidone (PVP) amorphous solid dispersions(ASDs) were prepared for solubility enhancement. The optimal PVP-4HPR ratio of 9/1 (w/w) provided a 50-fold solubility enhancement in aqueous media, which was sustained over 1 week. PVP-4HPR ASD particles were loaded into PLGA millicylinders and drug release was evaluated in vitro in PBST and in vivo by recovery from subcutaneous injection in rats. While initial formulations of PLGA PVP-4HPR millicylinders only released 10% 4HPR in vitro after 28 days, addition of the plasticizer triethyl-o-acetyl-citrate(TEAC) into PVP-4HPR ASDs resulted in a 5.6-fold total increase in drug release. Remarkably, the TEAC-PVP-4HPR PLGA implants demonstrated slow, continuous, and nearly complete release over 1 month in vivo compared to a 25% release for our previously reported formulation incorporating solubilizers and pore-forming agents. Hence, a combination of PLGA plasticizer and ASD formation provides an avenue for long-term controlled release in vivo for the exceptionally difficult drug to formulate, 4HPR, and a suitable formulation for future evaluation in rodent models of oral cancer.
Keywords: Fenretinide (4HPR), polyvinylpyrrolidone (PVP), amorphous solid dispersion (ASD), Solubility enhancement, Poly(lactic-co-glycolic) (PLGA), In vivo release, long-acting release (LAR)
1. Introduction
The synthetic analogue of all-trans retinoic acid, fenretinide (4HPR) is an excellent chemopreventive drug that induces both differentiation and apoptosis in a variety of human precancerous cells in vitro (Berni and Formelli, 1992). Despite success in cell culture, clinical trials that employed systemic administration of 4HPR for oral squamous cell carcinoma (OSCC) prevention were ineffective (Garaventa et al., 2003; Peng et al., 1989; Formelli et al., 1989). These negative data likely reflect poor 4HPR bioavailability at the target site due to high clearance during first pass metabolism resulting in sub-therapeutic sera levels (Formelli et al., 1989). Attempts to increase systemic dosing induced deleterious side effects that included nyctalopia and elevated sera lipids without enhancing therapeutic effects (Formelli et al., 1993; Radu et al., 2005). Therefore, benefits of sustained release local 4HPR implants are readily apparent, i.e. maintenance of therapeutic levels while eliminating issues with compliance, bioavailability and deleterious drug-related side effects.
Due to the water insolubility of 4HPR, owing to its high hydrophobicity with a 6.31 log [octanol–water partition coefficient] (Wishart et al., 2018), there is an extreme challenge for submucosal delivery to interstitial fluid from controlled release implants. Many techniques have been employed in an attempt to increase aqueous solubility of poorly soluble drug molecules like 4HPR, such as micellar solubilization (Orienti et al., 2012; Li et al., 1996), reduction in particle size via milling, conjugation with amphiphilic moieties (Orienti et al., 2007; Orienti et al., 2016), complexation to dextrans (Orienti et al., 2009), PEGylation (Orienti et al., 2012), encapsulation into PLGA microparticles (Ying Zhang et al., 2016; Wischke et al., 2010; Graves et al., 2015), liposomal encapsulation (Trapasso et al., 2009; Parchment et al., 2014), formation of drug-salt (Orienti et al., 2016), formation of cocrystals (Almarsson et al., 2011), and preparation of amorphous solid dispersions (ASDs). Although solubilizers and surfactants can solubilize a highly lipophilic drug, the large amount of excipient needed to achieve the required drug loading is often not feasible to incorporate into the delivery vehicle. For example, we have determined that the bile salt, sodium deoxycholate (NaDC) greatly enhances 4HPR water solubility 50-fold at 1 mg/mL, although, 200-fold higher solubilizer to drug was required on a weight basis (Nieto et al., 2018; Desai et al., 2011; Wu et al., 2012). Our lab has also screened many 4HPR solubility and permeation enhancing excipients including: polysorbates, polyvinyl alcohol, Soluplus, ASDs with polyethylene glycol (PEG), and organic cosolvent systems among others (Wischke et al., 2010; Desai et al., 2011; Wu et al., 2012). Although these additives effectively solubilized 4HPR, the high water solubility of the additive resulted in immediate drug release-which is not amenable to the design of a long-acting release local delivery systems. These considerations prompted us to explore other solubilization options for this poorly water-soluble crystalline drug.
The solubility enhancement of poorly soluble drugs in amorphous dispersions of polyvinylpyrrolidone (PVP) is well known (Bhardwaj et al., 2014). The amorphous drug dissolves much more rapidly than the crystalline form in water due to the combined effects of a lack of crystalline lattice and the elevated water solubility of the polymer, leading to a supersaturated drug concentration (Chen et al., 2016). ASDs enable formation of super-saturated aqueous drug concentrations as the ASD inhibit drug crystallization. Due to liquid–liquid phase separation and eventual drug precipitation, the drug solubilization will decrease over time in non-optimized ASDs. The stability of the ASD can be measured by determining the molecular mobility of the polymer by means such as dielectric relaxation, where a slower relaxation time would indicate a more stable system (Bhardwaj et al., 2014). Preservation of super-saturation over time reflects the robustness of the ASD system, and can be achieved by optimization of the polymer type, polymer molecular weight and chain length, and mass ratio of polymer to drug (Chen et al., 2016).
PVP, which was discovered in 1938, has been widely used in pharmaceutical, cosmetic, food, and industrial applications, due to its ability to readily dissolve in a variety of solvents including water, alcohols, amides, chlorinated hydrocarbons. It is odorless, colorless, nonirritating to the skin, non-ionic, absorbs large amounts of water, and has adhesive and thickening properties (BASF. PVP and More. In: BASF, editor., 2009). PVP has been used as a blood plasma substitute and to stabilize beer and alcohol polyphenols via carbonyl and hydroxyl group interactions (Wu et al., 2012; Bhardwaj et al., 2014; Chen et al., 2016). Conceptually, PVP could exhibit similar interactions between the redox-reactive 4′hydroxyl phenol group of 4HPR. In addition, PVP interacts with the hydroxyl surface groups of oxides via an acid-base interaction with the carbonyl bond in the PVP backbone (Pattanaik and Bhaumik, 2000), which acts as a Lewis base in aqueous solutions (Cohen Stuart and Fleer, 1982). In the pharmaceutical industry, PVP is used to form films for tablet coatings, as a disintegrant in tablets, and a crystallization inhibitor. Notably, PVP, is generally regarded as safe (GRAS) by the US FDA (GRAS Substances, 2016). For example, the FDA approved topical tretinoin (a retinoid derivative) gel formulations incorporated 0.06% PVP as a crystallization inhibitor (Farng et al., 1998). Recently, Laurent Pharmaceuticals developed PVP-4HPR solid dispersion tablets for oral dosing to enhance 4HPR bioavailability (Laurent, 2016). Another group prepared PVP-4HPR nanoparticles and determined that a 4:1 PVP-4HPR ratio provided greatest cell uptake in Caco2 cells (Ledet et al., 2015).
The aims of this work were two-fold. First, we sought to enhance 4HPR solubility by PVP-4HPR ASD formation. Secondly, we evaluated the possibility to exploit the resulting 4HPR-ASD in poly(lactic-co-glycolic acid) (PLGA) millicylinders for controlled release of 4HPR in vitro and in vivo. The PLGA implants were specifically designed for their capacity to provide local long-acting release during a 1- to 2-month dosing period and thereby circumvent issues with systemic delivery, e.g., inability to achieve therapeutic levels at the target site. We have reported 4HPR release from PLGA millicylindrical implants in vivo is undesirably slow and has a similar release profile relative to the solid drug depot (Nieto et al., 2018). These data reflect rate-controlling slow dissolution of the crystalline drug within the polymeric implant. Optimized 4HPR solubilization should, therefore, facilitate its release to a greater extent into the aqueous tissue environment and prevent crystallization. Below we describe our efforts to optimize 4HPR ASD formulations, incorporate them in PLGA implants and successfully demonstrate their desired delivery properties in vitro and in vivo.
2. Materials and methods
2.1. Materials
PLGAs (50:50) were purchased from Evonik, including 503H and 503 (24–38 kDa, acid and ester end-capped, respectively). 4HPR was generously supplied by Merck Co. Due to its photo-instability, 4HPR light exposure was minimized by protecting samples with foil. Excipients utilized included: polyvinylpyrrolidone (PVP K30, 40 kDa, BASF) and triethyl-o-acetyl-citrate (TEAC, Sigma-Aldrich., St. Louis, MO) (See Fig. 1 for chemical structures). All other materials were reagent or pharmaceutical grade including: magnesium carbonate (MgCO3), acetone, dichloromethane (DCM), ethanol (EtOH), tetra-hydrofuran (THF), and Tween 80 (polysorbate 80). Solvents for UPLC-UV and LC-MS analysis were HPLC or MS grade including acetonitrile (ACN), methanol (MeOH), and phosphoric acid (H3PO4). Silicone tubing (0.8 mm i.d.) for extrusion of implants was from BioRad Laboratories). Additional media for the solubility study included fetal bovine serum (FBS) and Dulbecco’s Modified Eagle Medium (DMEM Cat# 11995065, Life Technologies, Thermo Fisher Scientific, Waltham, MA).
Fig. 1.
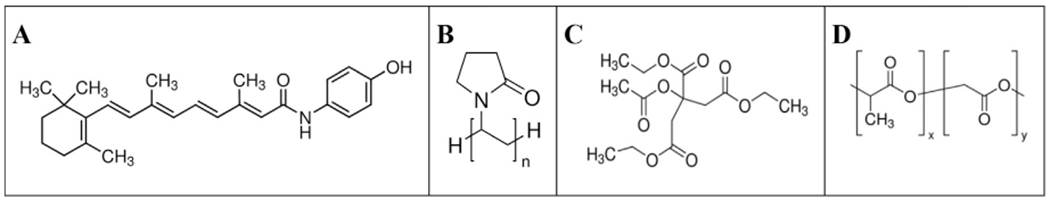
Chemical structures of a) 4HPR, b) PVP, c) TEAC, and d) PLGA.
2.2. Solubilization and crystallization inhibition of 4HPR by PVP
4HPR solubility in PVP aqueous solutions (1, 2, 5, 10, and 20% w/v in ddH2O) was evaluated by adding 2 mg of 4HPR to 1 mL of PVP solution and incubating gently at 37° C on a rotator. Prior to sampling at day 1, the suspensions were centrifuged and a 10 μL aliquot of the supernatant was assayed by UPLC-UV as described below. The suspensions were re-dispersed by vortexing and sampled again for analysis at day 7.
The 4HPR crystallization inhibition by PVP was evaluated by spiking 4HPR dissolved in acetone (0.5 mL of 10 mg 4HPR/mL) into 2 mL of 0%, 1% or 10% w/v PVP in ddH2O, in an open glass vial, and stirred at room temperature at 350 rpm for 24 h to allow for acetone evaporation. An aliquot of each suspension was filtered, and the solubilized 4HPR concentration assayed by UPLC. The 3 mixtures were then capped, and were held at ambient temperature and exposed to room light for 4 months. After 4 months, the 4HPR precipitate was harvested from the three suspensions (water, PVP/water, and acetone/water) by centrifuging, decanting the supernatant, followed by drying of the solid material. The resulting solids were examined for crystalline character by scanning electron microscopy (SEM) and compared to the crystal structures of a) 4HPR without any treatment and, b) 4HPR recrystallized from acetone after solvent evaporation.
2.3. Formation of PVP-4HPR and PVP-4HPR-TEAC amorphous solid dispersions
PVP (50% w/w) was dissolved in MeOH, and then 4HPR was added at mass ratios of 9/1, 8/2, or 7/3 PVP-4HPR. Later studies incorporated TEAC at a mass ratio of 9/1/1 PVP-4HPR-TEAC. The suspensions were vigorously mixed, shaken for 2 h to maximize polymer-drug interactions, poured into a Teflon-lined petri dish to form a thin film (solvent casting method), slowly dried at room temperature for 2 days, and then further dried in vacuum oven at 40° C for 2 additional days. The resulting solvent cast film was cryomilled (Retsch swing mill cryomill, PN 20.749.001) and at a frequency of 1/30 sec for 30 min, and particles were sieved to < 90 μm. The film morphology was inspected for drug crystallization via light microscopy, and the cryomilled particles were imaged via SEM. The 4HPR loading in PVP-4HPR ASD particles was determined by dissolving 2 mg of particles in 1 mL MeOH, diluting into mobile phase, and assaying by UPLC-UV as described in Section 2.9.
2.4. 4HPR-PVP solubilization enhancement
Dissolution studies were conducted by addition of 5 mg of PVP-4HPR ASD particles (9/1, 8/2, 7/3, all < 90 μm) into 1 mL of media (ddH2O, PBST 0.02%, or PBST 2% pH 7.4) and placing on a shaking platform at 37 °C, and compared to the dissolution of crystalline 4HPR. Media were sampled after 1, 6, and 12 h, and then daily for up to 7 days and assayed for 4HPR by UPLC-UV. The solubility enhancement was calculated by dividing the effective solubility of the 4HPR from PVP-4HPR ASD (CS, PVP-4HPR) by 4HPR’s crystalline solubility (CS, 4HPR). To assess the impact of serum, the effective solubility of 9/1 PVP-4HPR particles was also determined in DMEM cell culture medium supplemented with 5% FBS) after 24 h.
Statistical significance of the solubility enhancement of the ASD formulations compared to 4HPR solubility in each media, was determined by one way ANOVA, Bartlett’s test for equal variance, followed by Dunnett’s multiple comparison test. Statistically significant results were determined by a p-value < 0.05.
2.5. Formulation of controlled release PLGA PVP-4HPR ASD millicylindrical implants
Due to solubility differences of hydrophilic PVP and hydrophobic PLGA in organic solvents used in millicylinder fabrication processes, different approaches were investigated for incorporating PVP-4HPR ASDs into PLGA millicylinders. All implants contained 5% w/w 4HPR theoretical loading, and the formulation strategies are outlined in Table 1.
Table 1.
Formulation Strategies for Long-Acting Release PLGA + 4HPR-PVP ASD Millicylinders.
| Formulation | Preparation Methods | Rationale |
|---|---|---|
| 1–5 | Load varying ratios of PVP-4HPR particles into PLGA dissolved in acetone | Maximize 4HPR loading anddetermine optimal PLGA type |
| 6 | Co-dissolve PVP, 4HPR, PLGA in DCM (dichloromethane)Coat with PLGAs | All components soluble in DCMReduce release rate of PVP |
| 7,8 | Coat PVP-4HPR and PVP-4HPR-TEAC core implants with PLGAs | ASD implant encapsulated in PLGA |
| 9–11 | PVP-4HPR-TEAC (9/1/1) particles loaded into PLGACompared to control PLGA-TEAC-4HPR | TEAC may preserve integrity of PVP-4HPR ASD |
The initial formulations (#1–5) investigated effects of the ratio of PVP-4HPR particles (9/1, 8/2 or 7/3) and PLGA polymer type (acid or ester end-capped) on drug release profiles. Briefly, 60% w/w PLGA 503H or 503 were dissolved in acetone, and the required amount of PVP-4HPR particles to obtain 5% w/w 4HPR loading was added to the PLGA solution and vigorously stirred. It is important to note that different PVP-4HPR ratios resulted in different levels of PLGA and PVP in the implants. The resulting PVP-4HPR/PLGA mixture was loaded into a 3 mL syringe equipped with an 18G blunt end needle attached to silicone rubber tubing (BioRad, 0.8 mm i.d.), slowly extruded, and then dried at room temperature for 48 h, and then under vacuum oven at 40 °C for another 48 h. The tubing was then removed and implants were cut to the desired length of 1 cm.
Formulation #6 was prepared in similar fashion to #1–5, with the exception of use of DCM as the carrier solvent instead of acetone to dissolve PLGA, PVP and 4HPR. Millicylinders were coated with PLGA coatings to delay the release of the water soluble PVP: PLGA 503H, PLGA 503H + 3% MgCO3 (a basic pore forming salt, < 90 μm sized particles), and PLGA 503 + 3% MgCO3. PLGA coatings were prepared by dissolving 50% w/w PLGA in acetone and adding MgCO3 (to augment pore formation) to the mixture before extrusion around the core PLGA-PVP-4HPR implants. The samples were then dried under vacuum (40 °C x 48 h), and the tubing was removed and cut to 1 cm.
In the next set of formulations (#7, 8), PVP-4HPR (9/1) or PVP-4HPR-TEAC (9/1/1) core implants were coated with PLGAs as described in the above coating procedure. To prepare PVP-4HPR (and TEAC) ASD millicylinders, PVP was dissolved at 60% w/v in MeOH, and then 4HPR and TEAC were added and vigorously stirred, followed by the aforementioned millicylinder extrusion and drying procedure.
The final set of formulations (#9–11) incorporated PVP-4HPR-TEAC particles into PLGA, and were compared to PLGA-4HPR-TEAC implants (without PVP) to determine whether TEAC and/or PVP caused the accelerated 4HPR release. PVP-4HPR-TEAC 9/1/1 particles were added to 50% PLGA 503H dissolved in acetone with a total implant composition of PLGA/PVP/4HPR/TEAC 40/50/5/5 on a weight basis. A control formulation (#10) was prepared by adding 7.5% TEAC and 5% 4HPR to PLGA 503H + 4HPR, which is representative of the PLGA-TEAC ratio (11:1) in formulation #9. Additional 4% TEAC (#11) was added to the PLGA solution prior to adding the PVP-4HPR-TEAC 9/1/1 particles to assess its release effects.
2.6. 4HPR loading in PLGA millicylinders
To determine the amount to 4HPR loaded into PLGA millicylinders, a millicylinder was weighed into a 15 mL centrifuge tube, PLGA and 4HPR were co-dissolved by addition of 0.5 mL THF, followed by precipitation of PLGA upon addition of 9.5 mL EtOH. The sample was then centrifuged, and supernatant was assayed by UPLC/UV. 4HPR loading in millicylinders was well-controlled and encapsulation efficiency ranged from 97 to 103%.
2.7. In vitro release of 4HPR and PVP from PLGA-PVP millicylinders
4HPR and PVP in vitro release from millicylinders was performed in triplicate by incubating 1 millicylinder (5–7 mg) in a non-solubilizing buffer of 4 mL PBS pH 7.4 + 0.02% polysorbate 80 at 37 °C on a shaking platform (200 rpm). The solutions were sampled by complete media replacement over 6 weeks.
2.8. In vivo release of 4HPR from PLGA-PVP millicylinders
All experiments were conducted in accordance to University of Michigan’s AALAC protocols. The animals were retained under standard 12 h light/12 h dark vivarium conditions and had constant access to water and standard rat chow ad lib. Prior to millicylinder implantation, male Sprague-Dawley rats were anesthetized with 5% isofluorane via inhalation, and pre-weighed millicylinders were implanted subcutaneously (s.c.) in the dorsal region of the rat via a 12G trocar. For every formulation, 3 implants were implanted in each rat, and 1 rat per time point (days 1, 14, 28) was sacrificed, and millicylinders were harvested. The amount of 4HPR released was determined by assaying the amount of 4HPR remaining in the recovered millicylinder using the loading assay described in Section 2.6. The results of the in vivo release profiles of the optimized formulations were compared for statistical significance by a paired t-test, with significance indicated by p-value < 0.05.
2.9. 4-HPR UPLC-UV assay
4HPR levels in PVP-4HPR particles, millicylinder loading, and in vitro release media were determined by UPLC/UV. The reverse phase UPLC/UV analysis utilized a Waters Acquity UPLC system and Empower software under the following conditions: Acquity BEH C18 2.1x100 mm column, mobile phase 80:20 ACN: ddH2O + 0.1% H3PO4, isocratic flow rate of 0.65 mL/min, UV detection at 365 nm, and total analysis time of 2 min. 4HPR calibration standards were prepared in mobile phase (0.5–100 μg/mL) from a 0.5 mg/mL 4HPR stock solution in ACN, and reflects the large linear dynamic calibration range and a LLOQ of 5 ng/mL.
2.10. PVP UPLC-UV SEC assay
PVP levels in in vitro release media were determined by UPLC/UV size exclusion chromatography (SEC) following a previously validated PVP detection method (Tavlarakis et al., 2011). The analyses were carried out on a Waters Acquity UPLC system and Empower software under the following conditions: Acquity BEH H125 1.7 μm 4.6x150 mm column (SEC column), mobile phase 80:20 0.1 M sodium acetate buffer in ddH2O: MeOH, isocratic flow rate of 0.7 mL/min, UV detection at 220 nm, and total analysis time of 4 min.
2.11. Particle and millicylinder morphology via scanning electron microscopy (SEM)
The morphology of 4HPR, 4HPR-PVP particles, and millicylinder cross sections were examined by scanning electron microscopy (SEM) using a Phillips XL FEG SEM. All samples were completely dried, sputter coated with gold for 90 sec, and images obtained with a 3 kV electron beam.
2.12. Differential scanning calorimetry (DSC) and Thermogravimetric analysis (TGA)
Glass transition temperature (Tg) of the PVP-4HPR particles and millicylinders were determined via DSC (Discovery, TA instruments, New Castle, DE). The DSC and TGA instruments were calibrated by heating an indium standard under nitrogen, performed routinely during the instrument’s preventative maintenance schedule. Prior to analysis, millicylinders were stored in a well capped container and placed into a sealed bag to protect from light that contained desiccant to prevent uptake of atmospheric contents including water. For the DSC analysis, 2–3 mg of the PVP-4HPR particles or millicylinders were added to a hermetic pan, and samples were heated with a modulating temperature program from −10 °C to 180 °C. The amount of residual solvent trapped in the cryomilled films and millicylinders after drying was estimated by Thermogravimetric Analysis (TGA, Discovery, TA instruments). The following temperature ramping profile was used for the TGA analyses: 10 °C/min until 300° C, then 20 °C/min until 600° C.
3. Results
3.1. 4HPR solubility in PVP solutions and crystallization inhibition
As shown in Fig. 2A, the solubility of 4HPR only rose marginally in PVP aqueous solutions. Even at 20% PVP, only 0.7 μg/mL 4HPR was soluble after 1 week at 37 °C. PVP, however, diminished 4HPR precipitation for 1 day at room temperature as observed after the spiking of solubilized 4HPR in acetone into PVP aqueous solutions. While the control ddH2O solution showed wall-adhering 4HPR aggregates, the presence of PVP resulted in cloudy suspensions. PVP also appears to have suppressed 4HPR photo-degradation, whereby the retention of the 4HPR’s native yellow color was maintained over 4 months, compared to the orange-colored drug 4HPR observed in water mixtures without PVP (Fig. 2B). Additional photolytic degradation studies, are necessary to elucidate PVP’s apparent inhibition of 4HPR photodegradation.
Fig. 2.
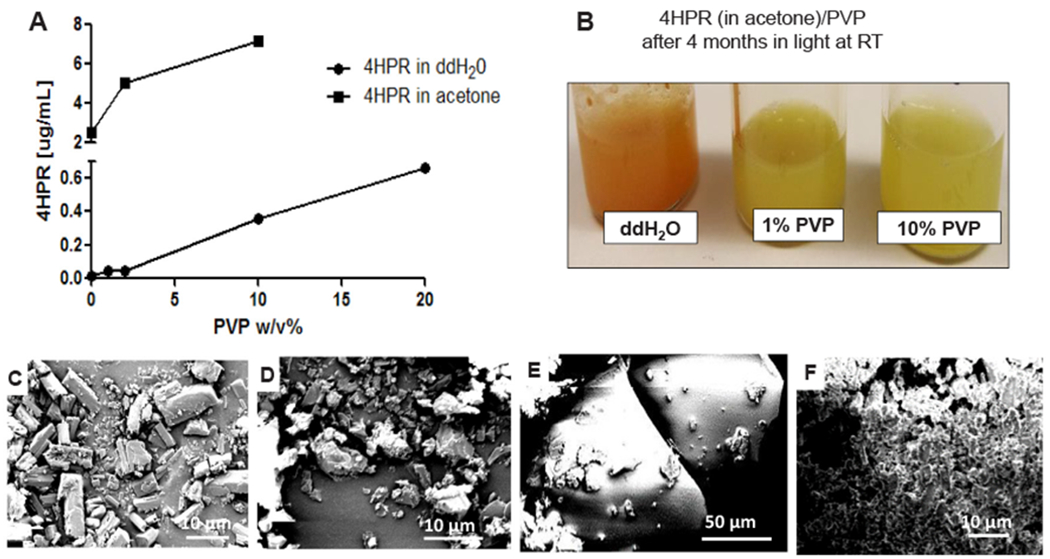
4HPR solubility, stability, and morphology in solid state and in PVP and PVP-acetone solutions. A) Dependence of 4HPR apparent solubility in solutions of PVP in ddH20 after 7 days at 37 °C (solid squares) and 4HPR crystallization inhibition dissolving 4HPR first in acetone (0.5 mL of 10 mg 4HPR/mL in 2 mL 1–10% PVP aqueous solutions) after 1 day at room temperature (solid circles). B) Effect of PVP on 4HPR apparent photostability during crystallization inhibition experiment (4HPR in acetone) after 4 months of constant light exposure at room temperature. SEM images of C) 4HPR solid, D) 4HPR recrystallized in acetone, E) 4HPR dissolved in acetone then precipitated in 10% PVP aqueous solution (appreciably smaller particles present), and F) 4HPR dissolved in acetone, then precipitated in ddH2O.
The solubility of 4HPR/acetone in PVP suspensions increased 10-fold compared to PVP acetone-free suspensions, findings that may reflect enhanced solubility from the residual, non-evaporated acetone (1:4 v/v acetone:ddH2O level). 4HPR crystal morphology reflected its respective solvent (Fig. 2C–F). 4HPR recrystallization in acetone (Fig. 2D) led to a more disordered, irregularly-shaped crystal structure relative to the original cuboidal-shaped 4HPR crystals (Fig. 2C). Further, during precipitation from PVP solutions (Fig. 2E), 4HPR appeared to interact with PVP, resulting in a markedly smaller particle size with distinct morphology relative to the aggregate, syncytium-like 4HPR precipitation in ddH2O (Fig. 2F).
3.2. Formation of PVP-4HPR ASDs and solubility enhancement
In Fig. 3A and 3B, the light microscopy of 4HPR amorphous dispersions (ASDs), formed by solvent casting after co-dissolution in PVP and MeOH are displayed. A translucent appearance was observed with solubilization of 4HPR at the high PVP/drug ratios of 8/2 and 7/3 PVP/4HPR, while the cracks present in the films demonstrate the brittle nature of PVP. The corresponding PVP-4HPR particle SEM images (Fig. 3C–F) depict the physical interactions between PVP and 4HPR, and demonstrate the effects of varied PVP: 4HPR ratios on particle morphology. Future additional analyses (FITR or Raman Spectroscopy) are planned to elucidate the nature of these PVP-4HPR chemical interactions. In the presence of the PVP polymer, 4HPR’s regular cuboidal crystal structure (Fig. 2C) was no longer present. The 4HPR loading in the PVP-4HPR particles is listed in Table 2. While the morphology of the 9/1 and 8/2 PVP-4HPR particles was similar, the 7/3 formulation was notably different. 4HPR loading was naturally the highest in the 7/3 formulation, because of the highest PVP/4HPR ratio used in the ASD. The addition of the TEAC (9/1/1 PVP-4HPR-TEAC particles) resulted in particles with a distinct morphology compared to the 9/1 PVP-4HPR particles. Introduction of PVP-TEAC generated finer particles (likely PVP) that were nestled on larger particles (likely 4HPR).
Fig. 3.

Morphology of PVP-4HPR (varying ratios) and PVP-4HPR-TEAC ASDs. Light microscopic images of the solvent cast films with A) 8/2 and B) 7/3 wt ratios of PVP to 4HPR. SEM images of PVP-4HPR ASD particles with C) 7/3, D) 8/3 and E) 9/1 wt ratios of PVP to 4HPR, and particles of F) 9/1/1 wt ratio of PVP to 4HPR to TEAC. All particles (< 90 μm) were prepared from cryomilling films.
Table 2.
4HPR loading in PVP-4HPR ASD particles. Mean ± SE, n = 3.
| ASD Particle Composition | % 4HPR |
|---|---|
| 9/1 PVP-4HPR | 9.7 ± 0.0 |
| 8/2 PVP-4HPR | 18.3 ± 0.3 |
| 7/3 PVP-4HPR | 26.1 ± 0.4 |
| 9/1/1 PVP-4HPR-TEAC | 9.2 ± 0.2 |
The effect of different PVP-4HPR particle ratios (< 90 μm) on 4HPR solubilization was evaluated in water and PBST with low and high surfactant levels (0.02% and 2% polysorbate 80) (Fig. 4). Notably, the net final PVP concentration in all studies was < 0.5% w/v, which alone only yielded a modest solubility increase (Fig. 2, < 0.7 ug 4HPR/ml). The 9/1 PVP-4HPR ASDs provided maximal solubilization, where > 300 μg/mL 4HPR was maintained in ddH2O for 7 days, corresponding to a 50 to 1000-fold solubility enhancement and considered a significant (p < 0.001) compared to 4HPR solubility alone in water (Fig. 4A, D). Higher drug loaded ASD’s (8/2 and 7/3 PVP-4HPR) showed little solubility enhancement (2.4 and 1.3-fold increase, respectively) after 7 days in ddH2O. Introduction of PBS buffer with 0.2 or 2.0% polysorbate 80 reduced 4HPR solubility in the PVP-4HPR ASDs. This finding likely reflects the recognized ability of salts to disrupt polymer-drug interactions in ASDs (Pattanaik and Bhaumik, 2000) (Fig. 4). In PBST 0.02%, the 8/2 and 9/1 PVP-4HPR ASD’s significantly (p < 0.001) enhance 4HPR solubility compared to the drug alone, resulting in a sustained a 50 to 170-fold solubility enhancement over 7 days (Fig. 4B, E). However, the 7/3 PVP-4HPR ASDs showed appreciably lower solubility enhancement in PBST 0.02% (11-fold after 7 days), and was insignificant compared to 4HPR. (See Fig. 4B,E). All ASD’s displayed a limited solubility in 2% PBST (between a 1 to 6-fold solubility advantage), findings that likely reflect competitive solubilization by the surfactant polysorbate 80. The 4HPR solubilization trend was reversed in 2% PBST: 7/3 > 8/2 > 9/1 PVP-4HPR up to 24 h, which again suggests a competition between surfactant-mediated micellar solubilization and PVP-4HPR ASD. The solubility advantage provided by 7/3 PVP-4HPR was considered significant in the PBST 2% compared to the 4HPR alone (p = 0.013). After 24 h in PBST 2%, 4HPR solubility steeply declined until day 4, where equilibrium was reached and all groups show comparable solubility to the drug alone. The time to reach max solubility varied: in ddH2O PVP-4HPR 9/1 took 6 h, 8/2 and 7/3 took 48 h, and in PBST all took 6 h. Lower levels of PVP and saline (PBST) are accompanied by decreased solubility over time in every formulation. Further, introduction of competing micellar solubilization enhancement with 2% polysorbate 80 abolished PVP’s solubility enhancement after 48 h
Fig. 4.
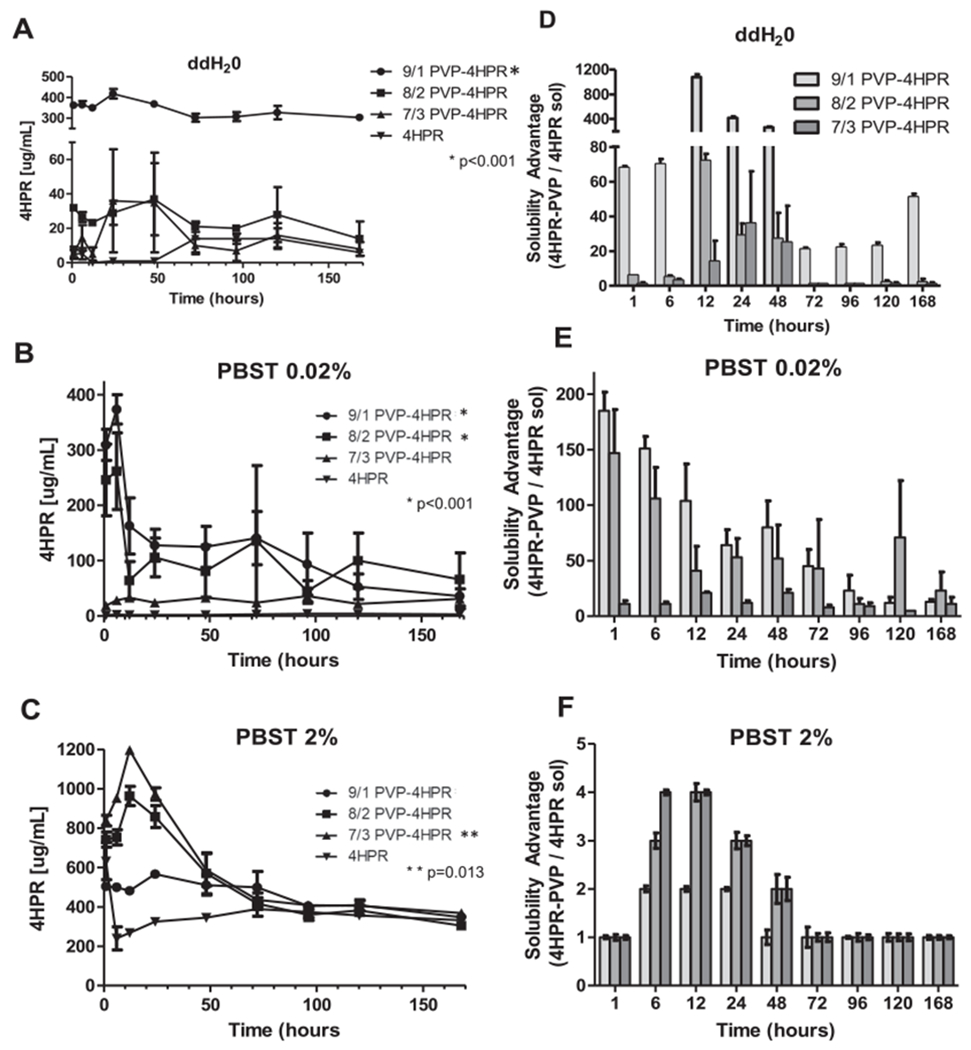
Effects of varying ratios of 4HPR-PVP in the ASDs on kinetics of saturated 4HPR solubility (A, B, and C) and corresponding 4HPR solubility advantage (D, E, and F) in micellar solubilizing media (PBST with 2% polysorbate 80 (C and F)) and non-solubilizing water (A and D) or PBST with 0.02% polysorbate 80 (B and E) media over time. Data represents mean ± SE, n = 3, *denotes statistical significance of the ASD compared to 4HPR, *p < 0.001 and **p = 0.013.
In contrast to a reduced solubilization effect in saline, the presence of serum (DMEM medium + 5% FBS) increased PVP-4HPR (9/1) solubilization. The 4HPR concentration was solubilized to 337 ± 11 μg/mL 4HPR equating to 87 ± 3% solubilized (24 h, data not shown). PVP-4HPR 9/1 solubility enhancement was sustained for > 7 days. These results are favorable for the proposed local drug delivery chemopreventive applications and imply the potential to augment 4HPR tissue dispersion.
3.3. Characterization of PVP-4HPR ASDs by Microcalorimetry: DSC and TGA
The presence of one Tg indicates formation of an ASD (Sun et al., 2010), which occurred with all compositions of PVP-4HPR (9/1, 8/2, 7/3) (Fig. 5A). Interestingly, the Tg decreased with increased 4HPR content. The 9/1 PVP-4HPR ASD particles had the greatest Tg (155 °C) relative to the 8/2 and 7/3 particles (131, 133 °C, respectively). All Tg values for 4HPR-PVP ASDs were lower than the Tm of 4HPR (174 °C) and Tg of PVP (164 °C), and could likely reflect either a reduction in crystalline solids in the more amorphous structure, or presence of residual solvents used in the manufacturing of the ASD (Fig. 5). The Tg dramatically decreased to 91 °C with addition of the plasticizer, TEAC, as expected. The rationale for including both Tm and Tg is to clearly depict the physical and chemical differences between 4HPR and PVP. These data demonstrate that 4HPR exists as a crystalline solid (as indicated by a Tm lacking a Tg) whereas PVP is a glassy polymer. Notably, when PVP-4HPR form an ASD, the Tm disappears, and the Tg for the ASD is less than that of the PVP Tg, indicating that a true ASD has formed. A Thermogram of 4HPR can be referenced in Supplemental Fig S2, and thermograms of the PVP-4HPR particles and the PVP-4HPR PLGA millicylinders can be referenced in Supplemental Fig S3. Residual solvent levels of 1–6% based on mass loss reading at 100 °C (representative TGA spectra in Supplemental Information, Fig. S4) confirmed that the solvent contribution to Tg lowering was minimal.
Fig. 5.
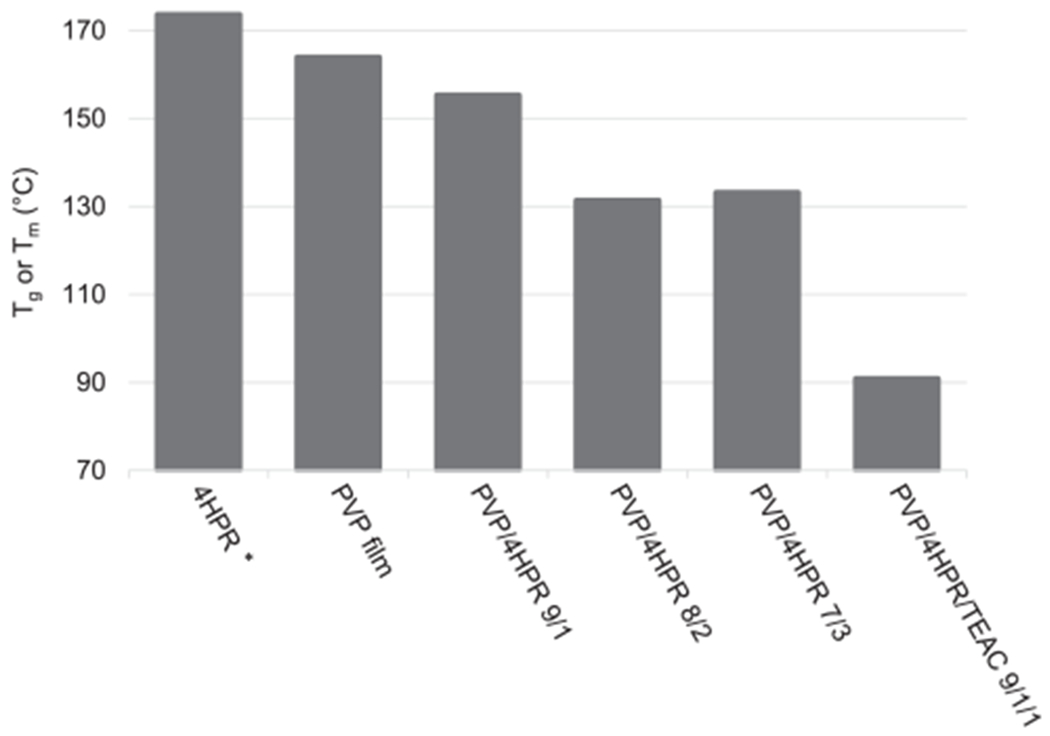
DSC of PVP-4HPR ASDs. *Indicates a Tm value. All other values are expressed as Tg.
3.4. In vitro release of PVP-4HPR particles from PLGA millicylinders
3.4.1. Effect of polymer type and PVP/4HPR ratios
The first set of formulations (#1–5) were developed to investigate the different ratios of PVP-4HPR particles loaded into either PLGA 503H or 503, with a goal of using the lower ratio of PVP-4HPR (i.e. 8/2 or 7/3) to achieve a greater drug loading in the PLGA implant. As seen in Fig. 6, the 9/1 PVP-4HPR in PLGA (503H, acid end-capped) implants (#1) showed a 10-fold more 4HPR released after 28 days (7.7% vs. 0.7–4.2%). These results are consistent with the dissolution studies, where 9/1 PVP-4HPR greatly enhanced solubility. These data reinforce that the 9/1 PVP-4HPR ratio should be retained in PLGA millicylinders, and validate the benefits of utilizing the acid end-capped PLGA (503H) polymer to provide slow and continuous release of this ASD. This formulation showed a large PVP burst release (> 80% after 3 days), likely due to greater percent solids or relatively less PLGA in the implant. The ester-capped PLGA 503 (#4, 5) released more 4HPR than the 503H with similar PVP-4HPR composition (Fig. 6). Overall, more PVP resulted in higher 4HPR release in both the 503 and 503H PLGA implants. None of the implant formulations, however, showed an optimal release profile, i.e., slow, continuous and complete drug release. Although the PLGA 503H provided the greatest 4HPR release, it was only < 7% total over one month. In Table 3 a summary of implant compositions and release profiles is displayed. PVP level also affected implant erosion, with increasing erosion accompanying lower PVP content. (See Supplemental Fig. S1 for complete implant erosion data.)
Fig. 6.
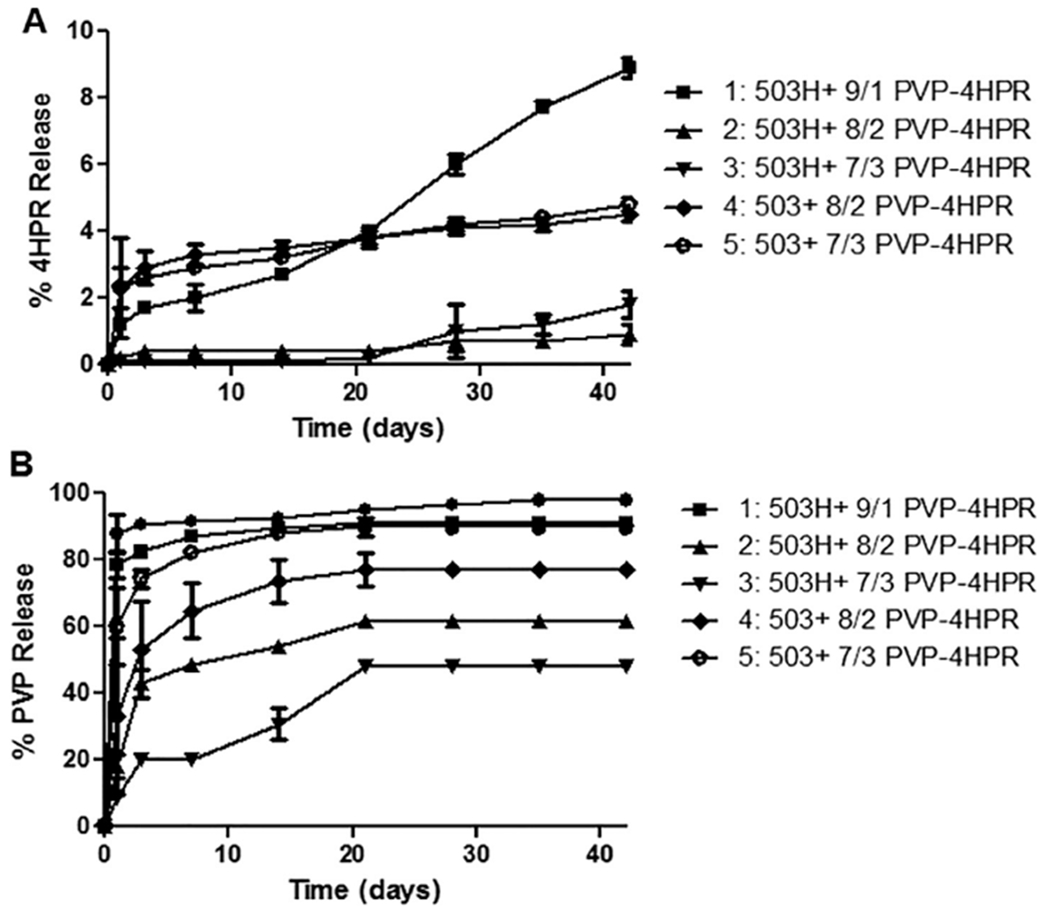
Effects of ASD PVP-4HPR ratio and polymer type on in vitro release of A) 4HPR and B) PVP from PLGA implants (mean ± SE, n = 3) in PBST with 0.02% polysorbate 80 at 37 °C. PLGA implants were prepared by loading either PLGA 503 or 503H with PVP-4HPR ASD particles (with 9/1, 8/2, 7/3 wt ratios) to yield implants containing ~ 5% 4HPR.
Table 3.
PLGA + PVP-4HPR Millicylinder Implant Formulations: Composition and In Vitro Performance.
| # | Preparation Method | PLGA type | Theoretical Loadings | In vitro Release Results | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| % PLGA | % 4HPR | %PVP | % TEAC | %4HPR | % PLGA coating | % 4HPR released, Day 28 | Time (days) to 80% PVP release | |||
| 1 | PLGA + 9/1 PVP-4HPR ASD in acetone | 503H | 50 | 5 | 45 | – | 4.1 ± 0.5 | – | 7.7 ± 0.2 | 3 |
| 2 | PLGA + 8/2 PVP-4HPR ASD in acetone | 503H | 75 | 5 | 20 | – | 3.5 ± 0 | – | 0.7 ± 0.3 | > 35 |
| 3 | PLGA + 7/3 PVP-4HPR ASD in acetone | 503H | 83 | 5 | 12 | – | 3.1 ± 0.3 | – | 1.0 ± 0.8 | > 35 |
| 4 | PLGA + 8/2 PVP-4HPR ASD in acetone | 503 | 75 | 5 | 20 | – | 2.3 ± 0.6 | – | 4.1 ± 0.2 | 28 |
| 5 | PLGA + 7/3 PVP-4HPR ASD in acetone | 503 | 83 | 5 | 12 | – | 3.1 ± 0.1 | – | 4.2 ± 0.2 | 7 |
| 6a | PLGA + 9/1 PVP-4HPR ASD in DCM + PLGA/DCM coating | 503H/503H coat | 45 | 5 | 50 | – | 3.4 | < 0.1 | 4.2 ± 0.5 | > 42 |
| 6b | PLGA + 9/1 PVP-4HPR ASD in DCM +3% MgCO3/PLGA/DCM coating | 503H/503H coat | 2.9 | 0.3 | 5.8 ± 0.8 | 28 | ||||
| 6c | PLGA + 9/1PVP-4HPR ASD in DCM +3% MgCO3/PLGA/DCM coating | 503H/503 coat | 3.1 | 0.1 | 3.1 ± 0.4 | > 42 | ||||
| 6d | PLGA + 9/1 PVP-4HPR ASD in DCM | 503H, no coat | 3.2 | – | 9.5 ± 0.7 | 35 | ||||
| 7a | PVP-4HPR-TEAC (9/1/1) core in MeOH + PLGA/acetone coating | Coat 503H | – | 9 | 82 | 9 | 5.9 | 1.7 | 9.4 ± 1.6 | 7 |
| 7b | PVP-4HPR-TEAC (9/1/1) core in MeOH + 3% MgCO3/PLGA/acetone coating | Coat 503H | 5.8 | 1.8 | 6.3 ± 0.2 | 28 | ||||
| 7c | PVP-4HPR-TEAC (9/1/1) core in MeOH + 3% MgCO3/PLGA/DCM coating | Coat 503 | 5.8 | 1.9 | 9.5 ± 0.0 | 1 | ||||
| 7d | PVP-4HPR-TEAC (9/1/1) core in MeOH | no PLGA | 7.6 | – | – | – | ||||
| 8a | PVP-4HPR (9/1) core in MeOH + PLGA/acetone coating | Coat 503H | – | 10 | 90 | – | 7.0 | – | 10.9 ± 0.2 | 28 |
| 8b | PVP-4HPR (9/1) core in MeOH +3% MgCO3/PLGA/acetone coating | Coat 503H | 7.2 | 2.1 | 12.7 ± 0.2 | 14 | ||||
| 8c | PVP-4HPR (9/1) core in MeOH +3% MgCO3/PLGA/DCM coating | Coat 503 | 7.1 | 2.0 | 7.2 ± 0.2 | 3 | ||||
| 8d | PVP-4HPR (9/1) core in MeOH | no PLGA | 9.1 | 2.1 | – | – | ||||
| 9 | PLGA + PVP-4HPR-TEAC (9/1/1) ASDin acetone | 503H | – | 5 | 45 | 5 | 5.5 | – | 33.5 ± 1.3 | 3 |
| 10 | PLGA + TEAC + 4HPR ASDin acetone | 503H | 87.5 | 5 | – | 7.5 | 5.9 ± 0.4 | – | 0.7 ± 0.2 | – |
| 11 | PLGA + TEAC + PVP-4HPR-TEAC(9/1/1) ASD in acetone | 503H | 41 | 5 | 45 | 9 | 2.7 ± 0 | – | 22.8 ± 0 | 7 |
All percentages are expressed as w/w. Optimal performance was achieved with highlighted Formulation #9.
3.4.2. PLGA-PVP-4HPR millicylinders co-dissolved in DCM + PLGA coatings
The solvent DCM was introduced to allow for the PVP-4HPR ASD to be encapsulated in PLGA by solubilizing all components, while PLGA coatings were added to assess their effects on PVP release. As seen in Fig. 7, the uncoated implant (#6D) displayed the fastest release of 4HPR without excessive PVP release. Only 10% 4HPR was released after 42 days. As anticipated, the coatings slowed 4HPR release, with the slowest release of 4HPR with coating 503H and 503 + 3% MgCO3. PVP release was slowest from uncoated implants, while the fastest release occurred with the 503H + 3% MgCO3 coating (opposite of 4HPR). These data suggest that the hydrophobic PLGA coatings repel the hydrophilic PVP, and promote faster PVP release.
Fig. 7.
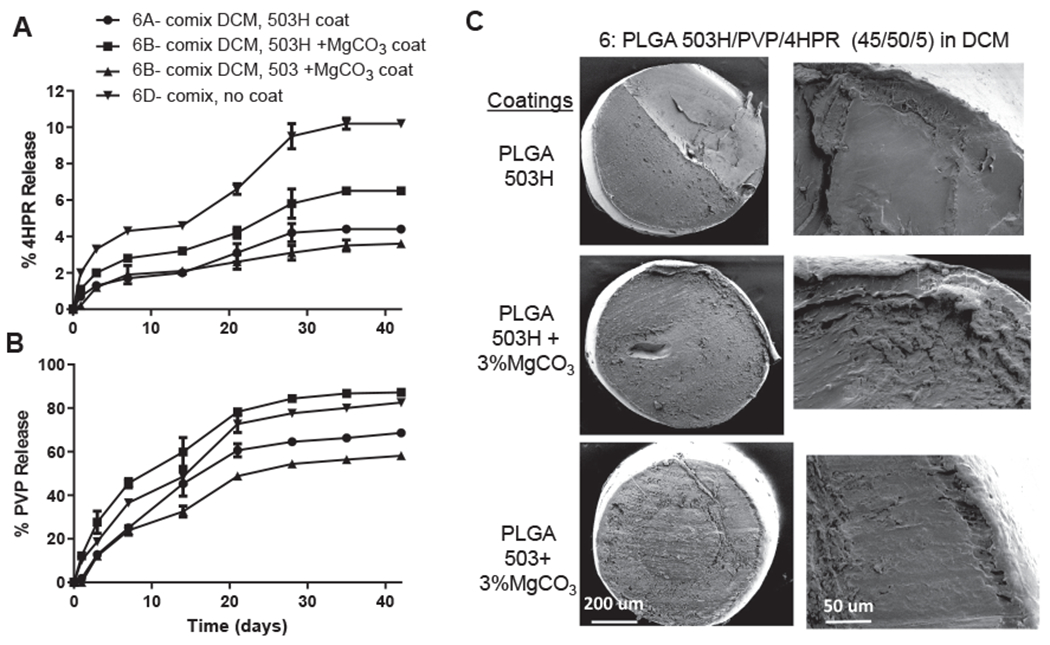
In vitro release kinetics of A) 4HPR and B) PVP in PBST with 0.02% polysorbate 80 at 37 °C from PLGA coated millicylinder #6 (PLGA 503H/PVP/4HPR 45/50/5, prepared by co-mixing in DCM) (mean ± SE, n = 3). C) SEMs of cross-sectional morphology of #6 PLGA coated implants, including a close up of the coating-core edge.
3.4.3. PLGA coated PVP-4HPR and PVP-4HPR-TEAC core implants
In the next set of millicylinder formulations, we sought to create a reservoir of the soluble drug by preparing core implants with PVP-4HPR (#7) and PVP-4HPR-TEAC (#8), and coating with PLGAs to slowly release the solubilized drug. These millicylinders were open-ended to facilitate drug release. TEAC was added to the PVP implant (#8) in an effort to preserve the PVP-4HPR ASD interactions, reduce PVP release, and provide controlled release of solubilized 4HPR. While TEAC is not soluble in PLGA, it is soluble in PVP. Addition of TEAC to PVP-4HPR had no effect on release from these PLGA coated implants, as both implants released ~ 10% 4HPR after 28 days (See Fig. 8A–D). The PVP-4HPR core implant dissolved in a short time of a few hours, resulting in rapid PVP release, with a remnant of the PLGA shell containing crystallized 4HPR. As shown in Fig. 8, the PLGA coating suppressive effect on 4HPR release followed the following trend (in decreasing suppression of release): 503 + 3% MgCO3 > 503H > 503H + 3% MgCO3.
Fig. 8.
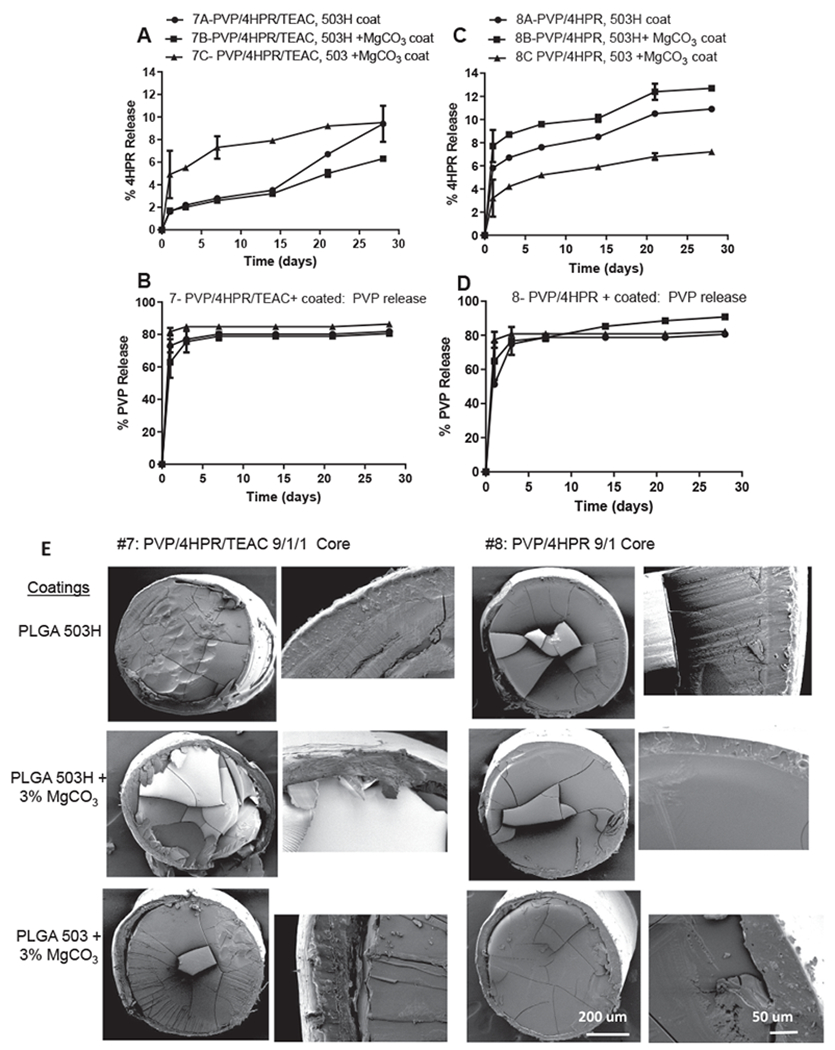
In vitro release kinetics of 4HPR (A, C) and PVP (B, D) from PVP-4HPR-TEAC 9/1/1 (A, B) or PVP-4HPR 9/1 (C, D) core implants coated with PLGA (503H, 503H + 3% MgCO3 or 503 + 3% MgCO3) (mean ± SE, n = 3) in PBST with 0.02% polysorbate 80 at 37 °C. E) SEMs depict cross-sectional morphology of #7 and #8 PLGA coated implant, including a close up of the coating-core edge for each implant.
3.4.4. PLGA + PVP-4HPR-TEAC ASDs millicylinders enhance 4HPR release
The aim of the next formulations (#9–11) was to incorporate TEAC into the PVP-4HPR particles to preserve the ASD structure. As shown in Fig. 9, the PLGA millicylinder loaded with TEAC-PVP-4HPR particles 9/1/1 (#9), resulted in a 5.6-fold increase in 4HPR release after 28 days (34% vs. 6% 4HPR respectively) compared to formulation #1 without TEAC. To determine if the plasticizing effects of TEAC on PLGA modulated 4HPR release from #9, a control PVP-free implant was evaluated (#10: PLGA 503H + 4% TEAC + 5% 4HPR). Implant #10 only released 0.5% 4HPR after 28 days. In the last formulation (#11), TEAC was added to PLGA prior to PVP-4HPR-TEAC ASD to determine if this approach enhanced 4HPR release. Formulation #11 released slightly less 4HPR than #9, (23% after day 28). Our results imply that TEAC exerts its effects by stabilizing the PVP-4HPR complex as opposed to PLGA plasticization. The solubility of each of these components in PLGA can be seen in SEM cross-sectional images (Fig. 9C). Fig. 9 depicts the importance of constituents and the timing of their addition on implant structure.
Fig. 9.
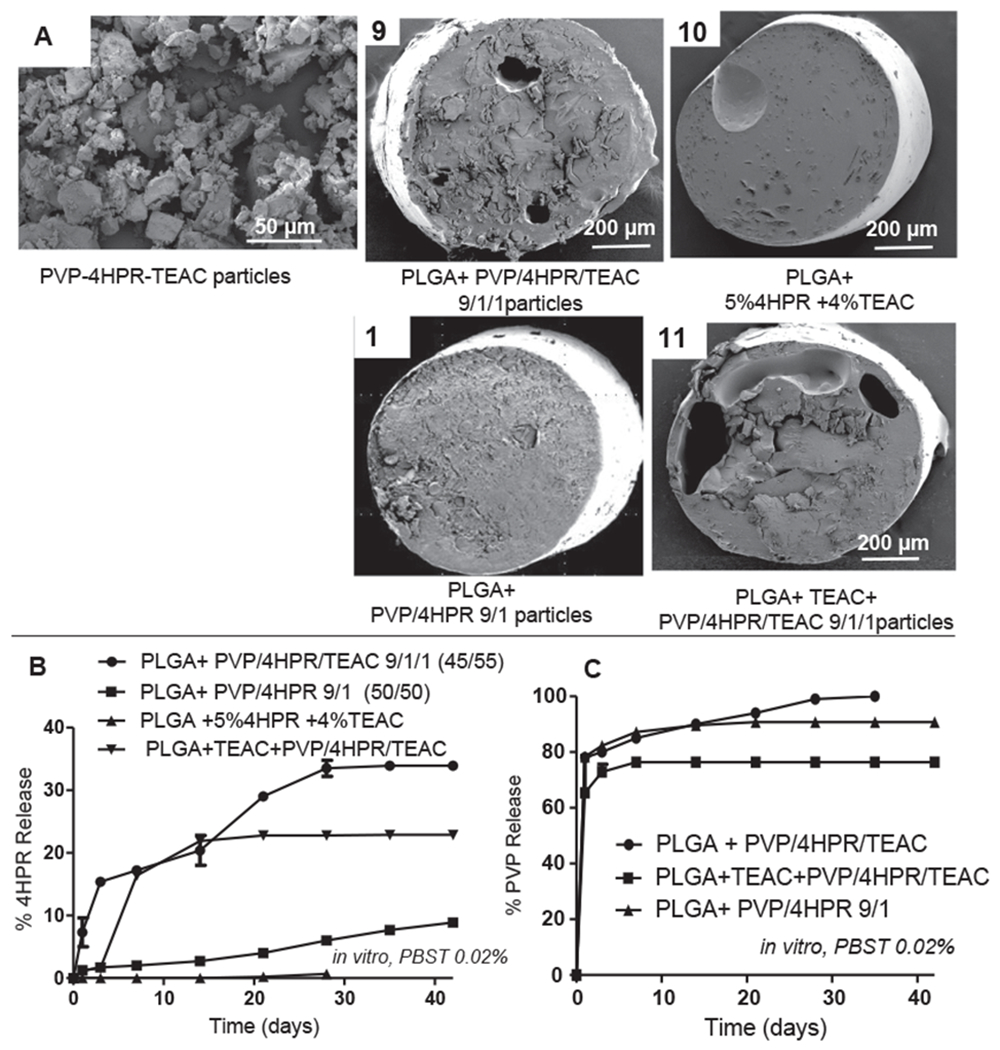
Effects of ASD particles and plasticizer on PLGA implant structure and 4HPR release rates in vitro in PBS + 0.02% Tween 80 at 37 °C. A) SEM images of PVP-4HPR-TEAC particles and four PLGA-4HPR implant cross sections. B) Effect of TEAC incorporation in the PVP-4HPR ASD particles on in vitro 4HPR release. C) Effect of TEAC incorporation on in vitro PVP release from PLGA implants. Data represents mean ± s.e., n = 3.
3.5. In vivo release from the PLGA + PVP-4HPR-TEAC implant
The optimized formulation, as defined by highest sustained 4HPR release, [#9, i.e. PLGA + PVP-4HPR-TEAC (9/1/1)], was then evaluated for release profiles after s.c. implantation relative to an immediately dissolving PVP-4HPR (9/1) implant (Fig. 10). While both formulations provided slow and continuous release in vivo over 30 days, the PLGA formulation provided more complete drug release (approximately ~ 90% relative to ~ 65% release), and was determined to be a significant improvement over the ASD without PLGA, with a p-value of 0.0478. Both formulations showed appreciable improvement relative to an earlier formulation (containing solubilizing agents and pore forming agents) that provided only 25% 4HPR release over the same period (Nieto et al., 2018). Additionally, these ASDs are capable of delivering the drug from non-crystallized 4HPR with inhibition of crystallization, which should be more bioavailable to the local tissues.
Fig. 10.
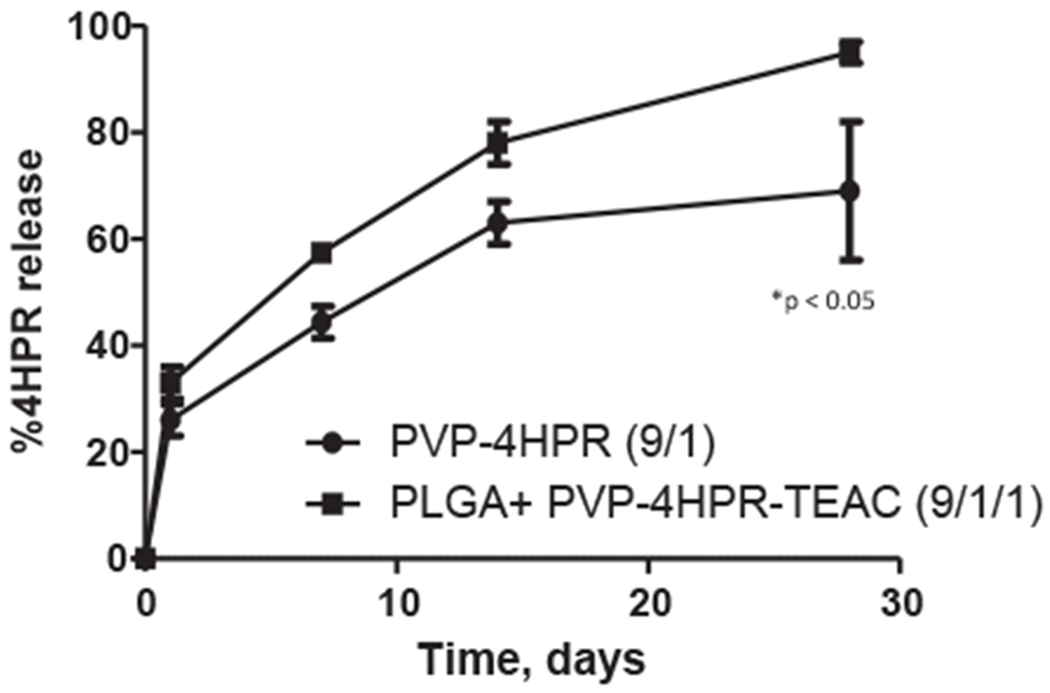
In vivo release of 4HPR from PLGA + PVP-4HPR implants and PVP-4HPR-TEAC ASD implants over 1 month after s.c. implantation into rats. Data represents mean ± s.e., n = 3, p < 0.05.
4. Discussion
Water-insoluble 4HPR has well established chemopreventive properties, including for our desired application of secondary and tertiary OSCC chemoprevention (Holpuch et al., 2011). This retinoid, however, is a very difficult drug to deliver to the body. Previous OSCC chemoprevention studies, which employed orally administered fenretinide capsules, were ineffective (Holpuch et al., 2012). Furthermore, at the highest oral capsule dosing (900 mg/m2 b.i.d., 3 week cycles, 1 week on drug, 2 without), fenretinide remained ineffective and the lack of efficacy combined with drug-induced toxicity resulted in early trial discontinuation (Holpuch et al., 2011). Considering fenretinide’s significant first pass metabolism to the inactive methylated derivative 4-methoxy fenretinide (Illingworth et al., 2011) and the lack of vessels in the target surface epithelial tissue, these negative data are logical and reflect issues with drug delivery but not fenretinide. Notably, human oral epithelium does not possess the imine N-methyltransferases responsible for 4HPR inactivation (Holpuch et al., 2011). To address this shortcoming for 4HPR systemic treatment of intraoral cancer, we have investigated multiple site-specific delivery approaches such as buccal mucoadhesive patches (Desai et al., 2011; Wu et al., 2012; Holpuch et al., 2012) and PLGA microspheres (Zhang et al., 2016) as well as PLGA in-situ forming (Wischke et al., 2010) and solid millicylindrical implants (Nieto et al., 2018) for submucosal injection. PLGA delivery of 4HPR suffers from the inability of the polymer to control the release of drug as opposed to the slow dissolution control from the encapsulated drug crystals (Nieto et al., 2018). Long-acting injectable drug products release rates are often controlled by the dissolution of the solid drug, or partitioning into the surrounding media, making pharmacokinetic properties difficult to predict (Owen and Rannard, 2016). Therefore, the goal this investigation was to adjust the formulation to solubilize and control the release in vivo beyond what has been possible when encapsulating 4HPR drug crystals with solubilizers and other additives in PLGA. Our lab has previously conducted and published time-based pharmacokinetic analyses in rats dosed with long acting PLGA formulations (Zhang et al., 2016). While these data were promising, our data indicated that bioavailability of released 4HPR was negatively impacted by drug crystallization. The focus of this current study was to enhance drug bioavailability following PLGA release. Our data, which depict formation of bioavailable PVP-4HPR ASDs, confirm this goal was achieved. Previous studies from our lab have confirmed the pharmacologic advantage i.e. therapeutic local levels in the absence of systemic blood levels, obtained by local 4HPR delivery (Holpuch et al., 2012). During application of multiple permeation-enhanced 4HPR patches to rabbit oral mucosa (approximately 80% of oral mucosa covered by patches), 4HPR levels were not detectable rabbit sera by LC/MS analyses [LLQO = 1 ng/ml (0.0025 μmol/L) (Wu et al., 2012). For these reasons, PK studies were restricted to local tissue levels for these current analyses.
PVP aqueous solutions provide limited solubilization of crystalline 4HPR (Fig. 2A). PVP appears to function as a wetting agent and formed a cloudy colloidal drug suspension, which strongly inhibited 4HPR crystal formation, reduced its photosensitization, and preserved its native yellow color. Formulation of a 4HPR solubility enhancing ASD by co-dissolving 4HPR and the stabilization polymer PVP in MeOH (9/1 PVP-4HPR), resulted in a 50-fold increase in 4HPR solubility (ddH2O) which was sustained for a week at physiological temperature (Fig. 4).
Advantages of amorphous dispersions of poorly soluble drugs include faster dissolution and greater apparent solubility for extended periods relative to the crystalline drug. Corresponding disadvantages include decreased physical stability such as the potential formation of different crystal structures (polymorphs) during storage and after dissolution. The presence of multiple polymorphic states was confirmed by our 4HPR acetone recrystallization studies (Fig. 2C and 2F). Two polymorphs of 4HPR are known, with melting points of 173–175 °C and 178–180 °C (Graves et al., 2015). In addition, solid-state stability of amorphous drug systems is influenced by the Tg and intermolecular interactions (Alhnan and Basit, 2011; Yao et al., 2013). The high Tg of PVP (164 °C) improves the stability of the PVP-4HPR amorphous solid dispersion due to inhibition of drug diffusion and precipitation in the glassy matrix. The presence of a single glass transition temperature can be used to validate a completely amorphous and highly homogenous dispersed drug (Laurent, 2016). The amorphous character of our PVP-4HPR ASDs was confirmed by preservation of a homogenous system with a sustained Tg (Fig. 5). These results agreed favorably with the PVP-4HPR ratios (20%) for ASDs prepared by Laurent Pharmaceuticals (Farng et al., 1998) and Ledet et al. (Laurent, 2016). Our data, which showed improved 4HPR release with the higher PVP content (9/1 PVP/drug), likely reflects prevention of PVP-4HPR dissociation and/or increased hydrogen bonding. Using PVP-nifedipine glassy dispersions, Mehta et al. determined that increased hydrogen bonding resulting from higher PVP levels alleviated the ASD dissociation (Mehta et al., 2016). Hydrogen bonding between the carbonyl and hydroxyl group of 4HPR is the most probable stability-enhancing mechanism.
Our results indicate that PVP-4HPR ASD implants are capable of providing continuous release of solubilized 4HPR over 1 month in vitro and in vivo. Interestingly, the PVP in vitro release data shows that the majority of PVP releases within the first couple of days, while the 4HPR release rate is apparently still enhanced by the presence a small amount of PVP remaining. The PVP HPLC-SEC assay appeared to have a high degree of accuracy, specificity, good level of quantitation (10 μg/ml PVP), and the actual loading of PVP into the implants was invariably the same as the theoretical loading. As we were aware that many substances demonstrate absorption at 220 nm, we utilized a validated size exclusion chromatography method capable of detecting PVP in aqueous solutions. Specifically, PVP’s molecular weight of 40 kDa is appreciably greater than other water soluble compounds that would be released in the PBST release media, and therefore, we do not expect substantial interference from other millicylinder excipients in this assay. Several implants exhibited 80–90% PVP release after the first week, and no additional PVP detected in in vitro release media thereafter. When optimal implants were evaluated in vivo, the target 4HPR release profile was met. Because the in vivo release data is obtained by assaying the amount of 4HPR remaining in the millicylinder by a digestion assay specific to 4HPR, the PVP in vivo release data was not obtained due to a different loading assay requirement. Obtaining in vivo PVP release data along with the drug release data will be of interest in future work to more fully understand the critical role of PVP in this formulation.
When considering PLGA encapsulation of PVP-4HPR ASDs, the three key constituents for the formulation, i.e., 4HPR, PVP and PLGA, all possess different chemical and solubility properties. A total of 11 different implant formulations were prepared, which varied in the solvents for dissolving the PLGA, PVP:drug ratios in the ASD, presence of polymer coatings, and inclusion of the plasticizer. By virtue of its in vitro sustained release profile over 1 month and total 4HPR released (36%), implant #9 - PLGA + PVP-4HPR-TEAC (9/1/1) was identified as the lead formulation. Surprisingly, this formulation performed similarly well in vivo as the in vitro release did, whereby it released 4HPR slowly and continuously nearly completely over 1 month (Fig. 10). While this result is very promising from a practical perspective, several important questions remain. First the in vitro release assays that are reasonable to use for this drug have proven inadequate to predict in vivo release behavior. However, relative differences in release rates have been born out in the few formulations that have underwent in vivo evaluation. Due to fenretinide’s very poor aqueous solubility (which is encountered in vivo), it was very challenging to generate in vitro/in vivo correlation data. While the use of a solubilizing buffer (PBST 2%) for the in vitro release media, as done in previous studies (Nieto et al., 2018), provides sink conditions for the in vitro analyses, these data did not extrapolate well to in vivo physiologic conditions. Previous studies from our lab have demonstrated 4HPR solubility differences reflect presence or absence of solubilizing agents (i.e. 4HPR solubility in PBST 2% Tween80 = 300 μg/ml and 2 μg/ml in PBST 0.02% Tween80 at physiological temperature) (Wu et al., 2012). These studies presented here utilized a non-solubilizing buffer (PBST 0.02%) for the in vitro release media to develop the implant formulations. While sink conditions were not maintained in this non-solubilizing release media, it was a better predictor for in vivo performance of 4HPR release rates from implants compared to the solubilizing media, and had greater correlation to the in vivo physiological conditions. A second question relates to the release kinetics of the multiple excipients in vivo, as noted for PVP above. For example, if the PVP released slower in vivo than in vitro this could explain the more complete drug release in vivo (Fig. 10) than in vitro (Fig. 9B). A final important question is whether this lead formulation will release in an equivalent manner in the submucosal region where blood flow is greater than the subcutaneous space. These questions will be important to evaluate in the future using the lead formulation described here as a starting point.
5. Conclusions
A significant enhancement of 4HPR solubility can be achieved by formation of PVP-4HPR ASDs at the optimum PVP-drug weight ratio of 9 to 1. This solubilized, amorphous form of 4HPR can be incorporated into long-acting release PLGA millicylinders for sustained release local delivery. Introduction of the plasticizer, TEAC, into the PVP-4HPR particles, provides a 5.6-fold increase in 4HPR in vitro release relative to TEAC-free implants. Furthermore, in vivo release from the PLGA PVP-4HPR-TEAC implants demonstrates marked improvement by complete and continuous 4HPR release, 90% 4HPR over 1 month, relative to the release of 25% 4HPR from an earlier PLGA-4HPR millicylinder prototype based on surfactants and pore-forming agents. In the future, local delivery of PVP-4HPR from ASD loaded PLGA implants will be examined for chemoprevention activity in animal models of oral cancer.
Supplementary Material
Acknowledgment
This study was funded in part by NIH R01 CA211611 and R01 CA227273.
Abbreviations:
- ASD
Amorphous solid dispersion
- OSCC
Oral squamous cell carcinoma
- PVP
Polyvinylpyrrolidone
- PLGA
Poly(lactic-co-glycolic acid)
- 4HPR
Fenretinide
- TEAC
Triethyl-o-acetyl-citrate
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ijpharm.2020.119475.
References
- Alhnan MA, Basit AW, 2011. In-process crystallization of acidic drugs in acrylic microparticle systems: influence of physical factors and drug-polymer interactions. J. Pharm. Sci 100 (8), 3284–3293 Epub 2011/04/19. [DOI] [PubMed] [Google Scholar]
- Almarsson Ö, Hickey MB, Peterson ML, Zaworotko MJ, Moulton B, Rodriguez-Hornedo N. Pharmaceutical co-crystal compositions. Google Patents; 2011. https://patents.google.com/patent/EP1608339A1/en [Accessed 02/2017]. [Google Scholar]
- BASF. PVP and More. In: BASF, editor. 2009. https://www.google.com/url?sa=t&rct=j&q=&esrc=s&source=web&cd=2&ved=2ahUKEwjHkIXY-9boAhVXJ80KHe8wBy4QFjABegQIARAB&url=https%3A%2F%2Fpharmaceutical.basf.com%2Fglobal%2Fimages%2Fb_03_110921e_solubility_enhance_compendium.pdf&usg=AOvVaw0w9X9XsNIv-xCALjtZi8S_ [Accessed 06/2016]. [Google Scholar]
- Berni R, Formelli F, 1992. In vitro interaction of fenretinide with plasma retinolbinding protein and its functional consequences. FEBS Lett. 308 (1), 43–45. [DOI] [PubMed] [Google Scholar]
- Bhardwaj SP, Arora KK, Kwong E, Templeton A, Clas SD, Suryanarayanan R, 2014. Mechanism of amorphous itraconazole stabilization in polymer solid dispersions: role of molecular mobility. Mol. Pharm 11 (11), 4228–4237 Epub 2014/10/18. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wang S, Liu C, Su C, Hageman M, Hussain M, et al. , 2016. Initial Drug Dissolution from Amorphous Solid Dispersions Controlled by Polymer Dissolution and Drug-Polymer Interaction. Pharm. Res 33 (10), 2445–2458 Epub 2016/06/11. [DOI] [PubMed] [Google Scholar]
- Cohen Stuart MA, Fleer GJ, 1982. Bijsterbosch BH. Adsorption of poly(vinyl pyrrolidone) on silica. II. The fraction of bound segments, measured by a variety of techniques. J. Colloid Interface Sci 90 (2), 321–334. [Google Scholar]
- Desai KG, Mallery SR, Holpuch AS, Schwendeman SP, 2011. Development and in vitro-in vivo evaluation of fenretinide-loaded oral mucoadhesive patches for site-specific chemoprevention of oral cancer. Pharm. Res 28 (10), 2599–2609 Epub 2011/06/16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farng RK, Gendimenico GJ, Mezick JA, Ng SM, Wrobel SB. Aqueous gel vehicles for retinoids. Google Patents; 1998. https://patents.google.com/patent/US5643584A/en [Accessed 09/2015]. [Google Scholar]
- Formelli F, Carsana R, Costa A, Buranelli F, Campa T, Dossena G, et al. , 1989. Plasma Retinol Level Reduction by the Synthetic Retinoid Fenretinide: A One Year Follow-up Study of Breast Cancer Patients. Cancer Res. 49 (21), 6149–6152. [PubMed] [Google Scholar]
- Formelli F, Clerici M, Campa T, Di Mauro MG, Magni A, Mascotti G, et al. , 1993. Five-year administration of fenretinide: pharmacokinetics and effects on plasma retinol concentrations. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol 11 (10), 2036–2042 Epub 1993/10/01. [DOI] [PubMed] [Google Scholar]
- Garaventa A, Luksch R, Lo Piccolo MS, Cavadini E, Montaldo PG, Pizzitola MR, et al. , 2003. Phase I trial and pharmacokinetics of fenretinide in children with neuroblastoma. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res 9 (6), 2032–2039 Epub 2003/06/11. [PubMed] [Google Scholar]
- GRAS Substances, (SCOGS Database) [database on the Internet]. 01/July/2016. [accessed 09/20/2016]. Available from: http://www.accessdata.fda.gov/scripts/fdcc/?set=SCOGS.
- Graves RA, Ledet GA, Glotser EY, Mitchner DM, Bostanian LA, Mandal TK, 2015. Formulation and evaluation of biodegradable nanoparticles for the oral delivery of fenretinide. Eur. J. Pharm. Sci 76, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holpuch AS, Desai KG, Schwendeman SP, Mallery SR, 2011. Optimizing therapeutic efficacy of chemopreventive agents: A critical review of delivery strategies in oral cancer chemoprevention clinical trials. J. Carcinogen 10, 23 Epub 2011/10/21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holpuch AS, Phelps MP, Desai KG, Chen W, Koutras GM, Han BB, et al. , 2012. Evaluation of a mucoadhesive fenretinide patch for local intraoral delivery: a strategy to reintroduce fenretinide for oral cancer chemoprevention. Carcinogenesis 33 (5), 1098–1105 Epub 2012/03/20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illingworth NA, Boddy AV, Daly AK, Veal GJ, 2011. Characterization of the metabolism of fenretinide by human liver microsomes, cytochrome P450 enzymes and UDP-glucuronosyltransferases. Br. J. Pharmacol 162 (4), 989–999 Epub 2010/11/09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent P, BETANCOURT A, Lemieux M, Thibert R, inventors; Google Patents, assignee. Solid oral formulations of fenretinide, 2016. https://patents.google.com/patent/WO2016011535A1/en [Accessed 09/2016].
- Ledet GA, Graves RA, Glotser EY, Mandal TK, Bostanian LA, 2015. Preparation and in vitro evaluation of hydrophilic fenretinide nanoparticles. Int J Pharm. 479 (2), 329–337 Epub 2014/12/30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C-Y, Zimmerman C, Wiedmann T, 1996. Solubilization of Retinoids by Bile Salt/Phospholipid Aggregates. Pharm. Res. 13 (6), 907–913. [DOI] [PubMed] [Google Scholar]
- Mehta M, McKenna GB, Suryanarayanan R. 2016. Molecular mobility in glassy dispersions. J. Chem. Phys 144(20). [DOI] [PubMed] [Google Scholar]
- Nieto K, Pei P, Wang D, Mallery SR, Schwendeman SP, 2018. In vivo controlled release of fenretinide from long-acting release depots for chemoprevention of oral squamous cell carcinoma recurrence. Int. J. Pharm 538 (1-2), 48–56 Epub 2017/11/25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orienti I, De MMR, Zeuner AP. Fenretinide complexes. Google Patents; 2016. https://patents.google.com/patent/WO2016038534A1/no {Accessed 03/2017]. [Google Scholar]
- Orienti I, Zuccari G, Bergamante V, Carosio R, Gotti R, Cilli M, et al. , 2007. Fenretinide-polyvinylalcohol Conjugates: New Systems Allowing Fenretinide Intravenous Administration. Biomacromolecules 8 (10), 3258–3262. [DOI] [PubMed] [Google Scholar]
- Orienti I, Zuccari G, Carosio RG, Montaldo P, 2009. Improvement of aqueous solubility of fenretinide and other hydrophobic anti-tumor drugs by complexation with amphiphilic dextrins. Drug Delivery 16 (7), 389–398. [DOI] [PubMed] [Google Scholar]
- Orienti I, Zuccari G, Falconi M, Teti G, Illingworth NA, Veal GJ, 2012. Novel micelles based on amphiphilic branched PEG as carriers for fenretinide. Nanomed. Nanotechnol. Biol. Med 8 (6), 880–890. [DOI] [PubMed] [Google Scholar]
- Owen A, Rannard S, 2016. Strengths, weaknesses, opportunities and challenges for long acting injectable therapies: Insights for applications in HIV therapy. Adv. Drug Deliv. Rev 103, 144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parchment RE, Jasti BR, Boinpally RR, Rose SE, Holsapple ET. Liposomal nanoparticles and other formulations of fenretinide for use in therapy and drug delivery. Google Patents; 2014. https://patentscope.wipo.int/search/en/detail.jsf?docId=US73540944&docAn=12301587 [Accessed 08/2016]. [Google Scholar]
- Pattanaik M, Bhaumik SK, 2000. Adsorption behaviour of polyvinyl pyrrolidone on oxide surfaces. Mater. Lett 44 (6), 352–360. [Google Scholar]
- Peng YM, Dalton WS, Alberts DS, Xu MJ, Lim H, Meyskens FL Jr., 1989. Pharmacokinetics of N-4-hydroxyphenyl-retinamide and the effect of its oral administration on plasma retinol concentrations in cancer patients. Int. J. Cancer. 43 (1), 22–26 Epub 1989/01/15. [DOI] [PubMed] [Google Scholar]
- Radu RA, Han Y, Bui TV, Nusinowitz S, Bok D, Lichter J, et al. , 2005. Reductions in Serum Vitamin A Arrest Accumulation of Toxic Retinal Fluorophores: A Potential Therapy for Treatment of Lipofuscin-Based Retinal Diseases. Invest. Ophthalmol. Vis. Sci 46 (12), 4393–4401. [DOI] [PubMed] [Google Scholar]
- Sun Y, Tao J, Zhang GGZ, Yu L, 2010. Solubilities of Crystalline Drugs in Polymers: An Improved Analytical Method and Comparison of Solubilities of Indomethacin and Nifedipine in PVP, PVP/VA, and PVAc. J. Pharm. Sci 99 (9), 4023–4031. [DOI] [PubMed] [Google Scholar]
- Tavlarakis P, Urban JJ, Snow N, 2011. Determination of total polyvinylpyrrolidone (PVP) in ophthalmic solutions by size exclusion chromatography with ultraviolet-visible detection. J. Chromatogr. Sci 49 (6), 457–462 Epub 2011/06/21. [DOI] [PubMed] [Google Scholar]
- Trapasso E, Cosco D, Celia C, Fresta M, Paolino D, 2009. Retinoids: new use by innovative drug-delivery systems. Expert Opin Drug Deliv. 6 (5), 465–483 Epub 2009/05/06. [DOI] [PubMed] [Google Scholar]
- Wischke C, Zhang Y, Mittal S, Schwendeman SP, 2010. Development of PLGA-based injectable delivery systems for hydrophobic fenretinide. Pharm. Res 27 (10), 2063–2074 Epub 2010/07/30. [DOI] [PubMed] [Google Scholar]
- Wishart DS FY, Guo AC, Lo EJ, Marcu A, Grant JR, Sajed T, Johnson D, Li C, Sayeeda Z, Assempour N, Iynkkaran I, Liu Y, Maciejewski A, Gale N, Wilson A, Chin L, Cummings R, Le D, Pon A, Knox C, Wilson M. 2017. DrugBank 5.0: a major update to the DrugBank database for 2018. Nucl. Acids Res November 8. doi: 10.1093/nar/gkx1037 [Accessed 03/2017]. [DOI] [PMC free article] [PubMed]
- Wu X, Desai KG, Mallery SR, Holpuch AS, Phelps MP, Schwendeman SP, 2012. Mucoadhesive fenretinide patches for site-specific chemoprevention of oral cancer: enhancement of oral mucosal permeation of fenretinide by coincorporation of propylene glycol and menthol. Mol. Pharm 9 (4), 937–945 Epub 2012/01/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao J, Shi NQ, Wang XL. 2013. [The development of co-amorphous drug systems]. Yao xue xue bao = Acta Pharmaceut. Sin 48(5):648–54. Epub 2013/07/31. [PubMed] [Google Scholar]
- Ying Zhang, C W, Mittal Sachin, Mitra Amitava, Schwendeman Steven P., 2016. Design of controlled release PLGA microspheres for hydrophobic fenretinide. Mol. Pharm 13 (8), 2622–2630. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Wischke C, Mittal S, Mitra A, Schwendeman SP, 2016. Design of Controlled Release PLGA Microspheres for Hydrophobic Fenretinide. Mol Pharm. 13 (8), 2622–2630 Epub 2016/05/05. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.