

Abstract
Lysyl oxidase like-2 (LOXL2) is an amine oxidase with both intracellular and extracellular functions. Extracellularly, LOXL2 promotes collagen and elastin crosslinking, whereas intracellularly, LOXL2 has been reported to modify histone H3, stabilize SNAIL, and reduce cell polarity. LOXL2 promotes liver and lung fibrosis, but little is known regarding its role in renal fibrosis. This study explored whether LOXL2 influences kidney disease in COL4A3 (−/−) Alport mice. Alport mice were treated with a small molecule inhibitor selective for LOXL2 (LOXL2i) or vehicle and assessed for glomerular sclerosis and fibrosis, albuminuria, BUN, lifespan, pro-fibrotic gene expression and GBM ultrastructure. GBM laminin α2 deposition and mesangial filopodial invasion of the glomerular capillaries was also assessed. LOXL2 inhibition significantly reduced interstitial fibrosis and mRNA expression of MMP-2, MMP-9, TGF-β1, and TNF-α. LOXL2i treatment also reduced glomerulosclerosis, expression of MMP-10, MMP-12, and MCP-1 mRNA in glomeruli, and decreased albuminuria and BUN. Mesangial filopodial invasion of the capillary tufts was blunted, as was laminin α2 deposition in the GBM, and GBM ultrastructure was normalized. There was no effect on lifespan. We conclude that LOXL2 plays an important role in promoting both glomerular and interstitial pathogenesis associated with Alport syndrome in mice. Other etiologies of CKD are implicated with our observations.
Keywords: glomerulonephritis, fibrosis, Alport syndrome, Lysyl oxidase
Introduction:
LOXL2 is a member of the lysyl oxidase (LOX) family of amine oxidases which share a conserved C-terminus containing a LTQ-cofactor-dependent, copper-dependent catalytic domain. The secreted, extracellular LOX enzymes catalyze the oxidative deamination of peptidyl lysines and hydroxylysines in collagen and elastin molecules, thus promoting the formation of covalent interchain crosslinks.1 Collagen cross-linking increases the stiffness of the extracellular matrix (ECM) which promotes fibrosis progression and limits the reversibility of established fibrosis. The amino terminal ends of the LOX family proteins are more variable and confer more specific functional roles, including regulation of catalytic activity and potential protein-protein interactions.2,3 For example, LOXL2 contains four amino-terminal Scavenger Receptor Cysteine Rich (SRCR) domains and is synthesized as an active ~100 kDa full-length protein that can be cleaved by an unknown serine protease to a ~65 kDa form which maintains catalytic activity. In contrast, LOX and LOXL1 lack SRCR domains in their amino termini and are produced as catalytically inactive proteins that require cleavage of the basic propeptide in order to generate the active protein.
Intracellular LOXL2 has been functionally linked to transcriptional regulation 4,5 as it relates to epithelial plasticity and metastasis.6-8 Specifically, LOXL2 has been shown to induce epithelial to mesenchymal transition (EMT) 9 as well as neovascularization,10 both important processes that contribute significantly to the aggressiveness of tumors. Elevated expression levels of LOXL2 in tumor biopsy specimen have been linked to poor survival in patients with colon cancer. Thus, LOXL2 has been investigated as a possible target for limiting tumor growth and metastasis.11
Given its activities in ECM crosslinking and EMT, it was obvious to explore the role(s) of LOXL2 in fibrosis progression. Elevated levels of LOXL2 have been detected in cardiac, liver, and pulmonary fibrotic diseases.12-17 Both small molecule inhibitors and therapeutic antibodies have been recently developed to specifically target LOXL2.18-20 Targeting LOXL2 with the therapeutic antibody approach was shown to improve heart function and inhibit fibrogenesis in a mouse model of cardiac interstitial fibrosis.13 Liver fibrosis was also successfully treated using the therapeutic antibody (AB0023), where the reversal of fibrosis was observed.21 Similarly, AB0023 treatment improved fibrosis in the mouse bleomycin model of lung fibrosis.17 Simtuzumab (the humanized version of AB0023) is no longer in clinical development due to disappointing results in idiopathic pulmonary fibrosis (IPF) and nonalcoholic steatohepatitis (NASH) patient trials.21 It should be noted that the antibody does not completely inhibit catalytic activity22 and it is unclear whether it was able to penetrate the dense extracellular matrix where pathologic LOXL2 is located. Techniques to measure target engagement in the Simtuzumab studies were not available; therefore, it is currently unclear if the lack of efficacy was due to a limited role of LOXL2 in those diseases or limitations in the antibody approach.
The role for LOXL2 in renal fibrosis has not yet been explored, however, its expression is upregulated in a number of human nephropathies. 23 Recent publications have demonstrated a role for the ~65 kDa proteolytically processed form of LOXL2 to cross-link the 7S domain of collagen IV, a structural domain that stabilizes collagen IV in the basement membrane.24,25 Here we show that the levels of LOXL2 mRNA and protein are elevated in renal cortex and glomeruli of the COL4A3 (−/−) Alport mice, a model for chronic kidney disease. Blocking LOXL2 activity using a selective small molecule inhibitor markedly reduced fibrosis and glomerulosclerosis and improved renal function, thus suggesting that inhibition of LOXL2 using a small molecule inhibitor is a useful approach in slowing the progression of fibrosis in Alport syndrome, as well as other forms of CKD.
Results:
LOXL2 protein and mRNA is upregulated in both the glomerulus and renal cortex of Alport mice.
Seven week-old 129 Sv wild type and Alport mice were used to assess LOXL2 mRNA and protein expression in both renal cortex and isolated glomeruli. Figure 1 shows that LOXL2 mRNA (upper panels) and protein (middle panels) are significantly induced in both cortex and glomeruli in Alport mice relative to wild-type mice. In the cortex, the majority of the LOXL2 protein is the ~100 kDa full-length form. Based on recent findings, the 150 kDa band observed in glomerular extracts is likely a 65kDa isoform of LOXL2 covalently bound to the 7S domain of type IV collagen.25 In the glomeruli of Alport mice, LOXL2 protein is primarily increased in the cytoplasm and close proximity to podocytes (labeled with anti-WT1 or anti-synaptopodin antibodies, Figure 2) consistent with an earlier report.26 In the tubulointerstitium, LOXL2 protein is expressed by both proximal (identified as CD13-positive) and distal (identified as NCC-positive) tubule cells (Figure 2 M-X), with possible extracellular staining. It is clear that LOXL2 protein is higher in Alport glomeruli relative to wild type, while differences in the tubulointerstitium are less apparent, in contrast to the western blot results (Figure 1).
Figure 1.
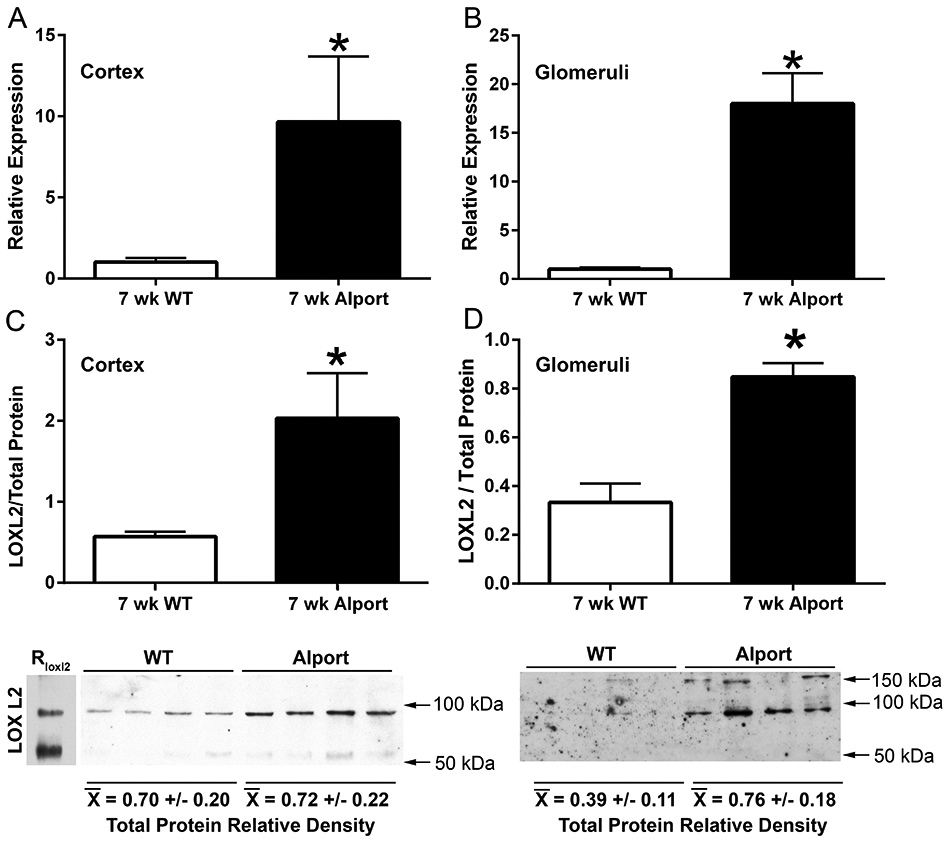
LOXL2 mRNA and protein is induced in both glomeruli and cortex of Alport mice relative to wild type mice. Real time PCR analysis of RNA from cortex (A) or isolated glomeruli (B) from 7-week old wild type and Alport mice was analyzed by real time RT-PCR for expression of LOXL2 mRNA. LOXL2 protein was also measured by western blot, the bands normalized to total protein (C and D). LOXL2 protein levels were significantly elevated in both glomerular and cortical extracts from Alport mice compared to wild type. Mean ± SEM *p<0.05.
Figure 2.
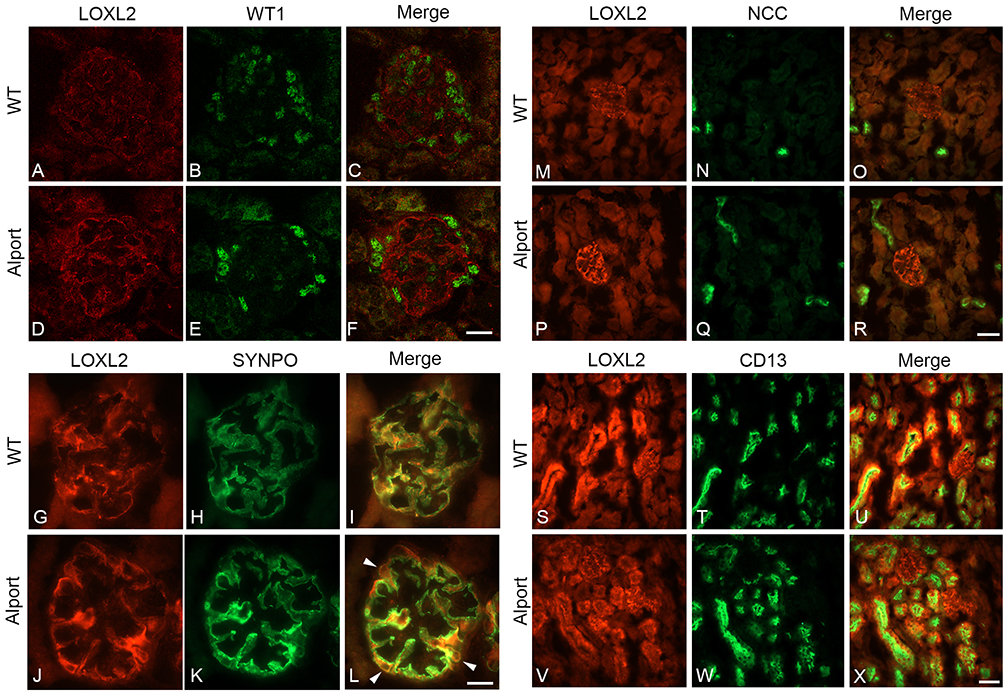
LOXL2 protein expression is elevated in glomerular podocytes. Panels A-F: Dual immunofluorescence with antibodies specific for LOXL2 (red) and podocyte-specific nuclear marker WT-1 (green); Panels G-L LOXL2 and podocyte marker synaptopodin demonstrate elevated expression of LOXL2 emanates primarily from the cytoplasm of the podocyte compartment. LOXL2 is expressed in both distal tubule cells (distil tubule marker NCC, panels M-R), and proximal tubule cells (proximal tubule marker CD13, panels S-X). Bar Panels F and L = 25 μm; Panels R and X = 75 μM.
LOXL2 inhibition ameliorates glomerular and interstitial fibrosis and significantly reduces both albuminuria and blood urea nitrogen levels in Alport mice.
To test the role of LOXL2 in renal fibrosis, we used PAT-1251, a small molecule inhibitor selective for LOXL2 over LOX and LOXL3.18, 19 The inhibitor (referred to as LOXL2i) was administered once daily at 30 mg/kg and achieved concentrations in the Alport mice that were approximately equal to the IC90 in a mouse whole blood assay (data not shown). Therefore, a dose of 30 mg/kg LOXL2i was selected for the study and Alport mice and wild type littermates were dosed orally once daily from 2 to 7 weeks of age. Tissue sections were stained with Mason’s trichrome or by immunofluorescence with anti-collagen I and anti-CD45 (pan leukocyte) antibodies, or with anti-smooth muscle actin for myofibroblasts (Figure 3). Inhibition of LOXL2 was associated with near absent leukocytic (CD45-positive) infiltration in the treated Alport mice (Figure 3A-F), as well as reduced accumulation of myofibroblasts as shown by the decrease in α-smooth muscle actin-positive cells in LOXL2i treated Alport mice (Figure 3D, E, and F). Masson’s trichrome histochemical staining showed clear cellular infiltration and tubule loss in vehicle-treated Alport mice, but not in LOXL2i-treated Alport mice (Figure 3G, H and I).
Figure 3.
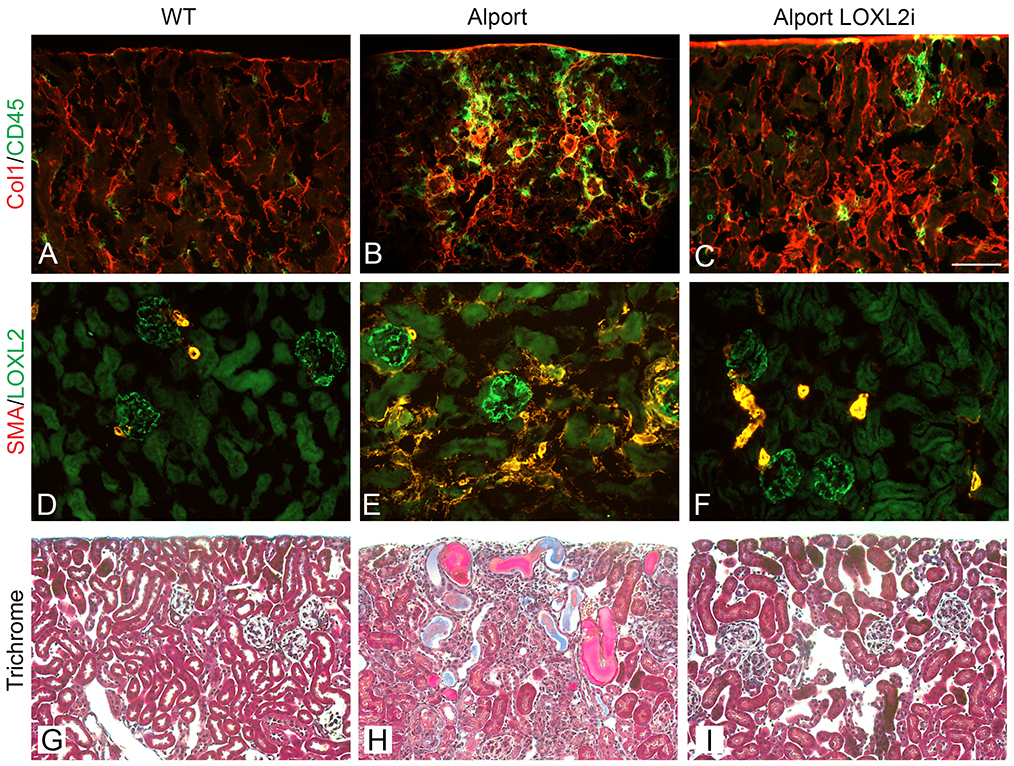
Renal fibrosis is ameliorated in Alport mice treated with LOXL2i. Panels A-C show dual immunofluorescence staining for collagen I and CD45 (pan leukocyte antigen). Panels D-F show that LOXL2i treatment blocks the genesis of interstitial and glomerular myofibroblasts, as assessed by anti-smooth muscle actin immunostaining. Panels G-I show Masson’s trichrome histochemical staining with clear cellular infiltration and tubule loss in vehicle-treated Alport mice (Panel H), but not in LOXL2i-treated Alport mice (Panel I). (Bar = 100 μm). All IF images are the same magnification.
The ability of 30 mg/kg LOXL2i to inhibit LOXL2 in vivo was confirmed by demonstrating an 80% reduction in LOXL2 activity using plasma from LOXL2i-dosed mice relative to the vehicle treated mice (Figure 4A). the data represented by Figure 3 was scored blindly for % fibrosis and glomerulosclerosis. Treatment with LOXL2i significantly reduced % fibrosis (Figure 4B) and glomerular sclerosis (Figure 4C) in the Alport mice relative to the vehicle-treated Alport mice and kidney function was also improved as indicated by significantly reduced blood urea nitrogen (BUN, Figure 4D) and albuminuria (Figure 4E) in LOXL2i treated Alport mice relative to the vehicle-treated Alport mice. BUN levels in the treated Alport mice were not significantly different from that of wild type mice. The renoprotective effects of LOXL2i observed at week 7did not translate to increased lifespan (> 10 weeks). Rather the treated animals trended towards a shorter lifespan (Figure 4F; p=0.08) with BUN levels reaching that of untreated Alport mice by 10 weeks of age (Figure 4G). We attribute this to an increase in podocyte loss since podocyte numbers were significantly reduced in both wild type and Alport mice treated with LOXL2i (Figure 4H). It is important to note that podocyte loss was not observed in Sprague-Dawley rats treated with high doses of LOXL2i in dose-range finding toxicology studies nor adult C57Bl/6 mice dosed at 30 mg/kg/day for 5 weeks as described herein (data not shown). This suggests that the observation in Alport mice is either strain or species-specific.
Figure 4.
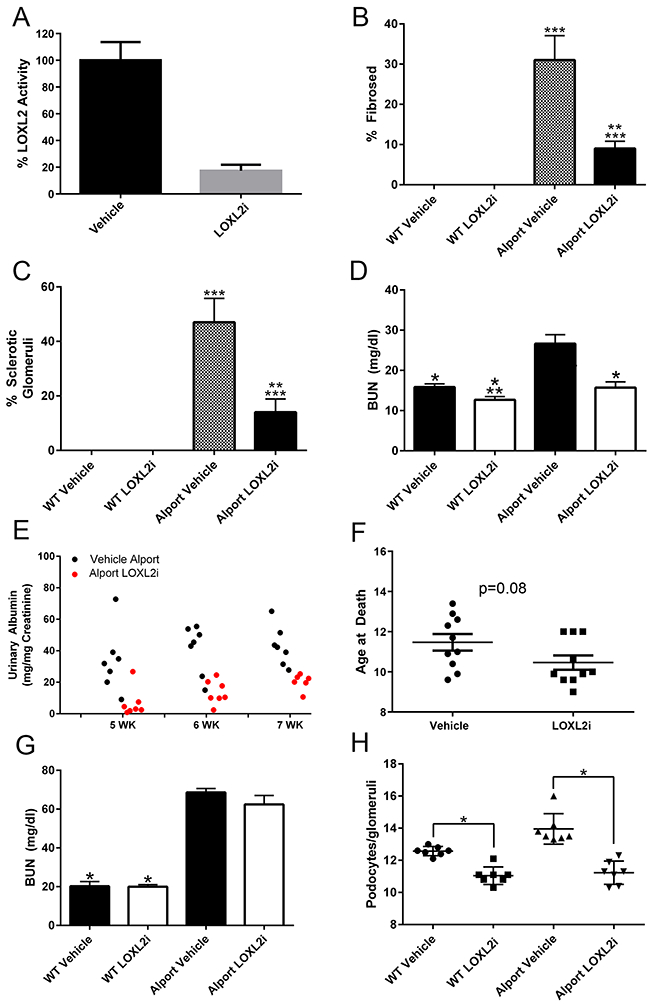
Treatment of Alport mice with LOXL2i normalizes renal morphology and function, but does not improve lifespan. (Panel A) Plasma LOXL2 activity in mice orally dosed with PAT-1251 relative to vehicle control mice (Ave % ± SEM) (Panels B-E) Wild type and Alport mice were treated with LOXL2i from 2-7 weeks of age and scored for % fibrosis (panel B), % glomerulosclerosis (panel C), blood urea nitrogen levels (BUN) (panel D) and albuminuria normalized to urinary creatinine as measured by ELISA (panel E). Panel F shows that lifespan of LOXL2i treated Alport mice is shorter than vehicle-treated Alport mice. Panel G shows BUN levels at 10 weeks (PAT-1251 dosing from weeks 2-10) and Panel H shows a significant reduction in podocyte numbers in LOXL2i-treated mice (weeks 2-7) relative to vehicle-treated mice. Mean ± SEM *p<0.05
LOXL2 inhibition significantly reduces expression of pro-fibrotic/pro-inflammatory genes in both glomeruli and renal cortex in Alport mice, prevents accumulation of laminin α2 in the GBM, reduces mesangial filopodial invasion of the glomerular capillaries and ameliorates GBM damage in Alport mice.
RNA from magnetic bead-isolated glomeruli or renal cortex isolated from either LOXL2i-treated or vehicle treated Alport mice and wild type littermates was analyzed by real time PCR for the transcripts indicated in Figure 5. Transcripts encoding MMP-2, MMP-9, TGFβ−1, and TNF-α, previously shown to be induced in fibrotic renal cortex of Alport mice,27were significantly reduced in Alport mice treated with LOXL2i. Likewise, in glomeruli from vehicle-treated Alport mice, transcripts for MMP-10, MMP-12, IL-6 and MCP-1 were all markedly elevated relative to wild type mice as shown previously.27,28 With the exception of IL-6, which is typically more variable within cohorts, all transcripts were significantly reduced in glomeruli from LOXL2i-treated Alport mice compared to vehicle-treated Alport mice.
Figure 5.
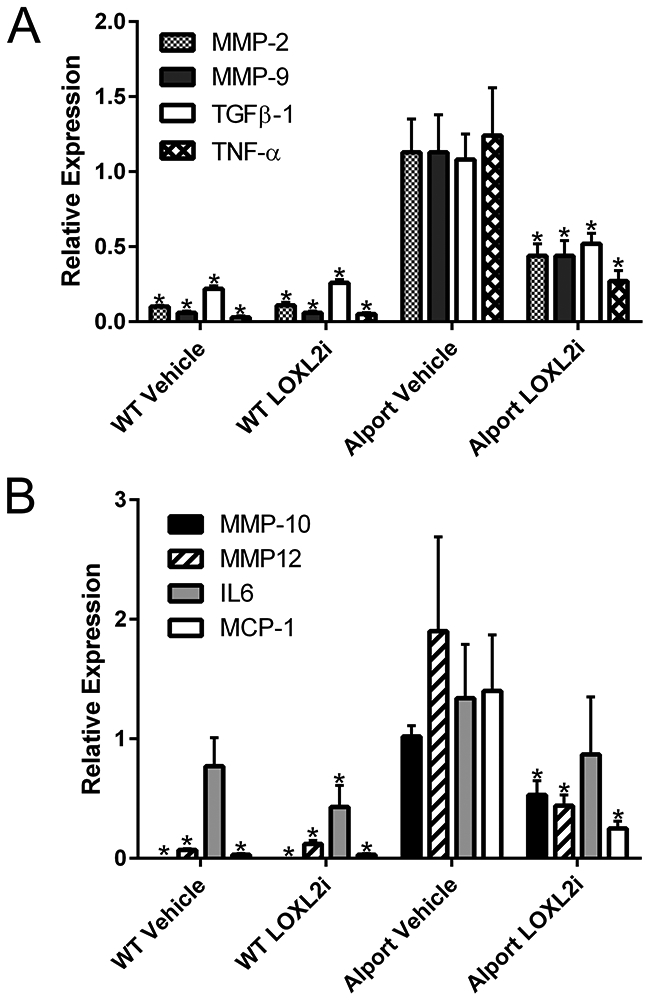
Expression of genes known to promote glomerular and tubulointerstitial damage are significantly reduced in RNA from LOXL2i-treated Alport mice relative to vehicle-treated Alport mice. RNA from either cortex (panel A) or isolated glomeruli (panel B) were analyzed by real time RT-PCR for the indicated transcripts. Statistically significant differences when comparing Alport-vehicle samples with the other cohorts are indicated by asterisks. Mean ± SEM * p<0.05.
Two events are endemic to Alport glomerular pathogenesis, including the progressive invasion of glomerular capillaries by mesangial processes and the progressive accumulation of laminin α2 in the GBM.27-30 Activation of these events are thought to underlie the mechanism of Alport disease initiation.31,32 Dual immunofluorescence staining was performed on 7-week old wild-type and vehicle-or LOXL2i-treated Alport mice. As shown in Figures 6 and 7, both GBM laminin α2 accumulation (Figure 6) and mesangial cell filopodial invasion of the GBM (Figure 7) were markedly attenuated in glomeruli from LOXL2i-treated Alport mice relative to vehicle treated Alport mice, thus suggesting attenuation of the molecular events that underlie glomerular disease initiation and progression.27-30
Figure 6.
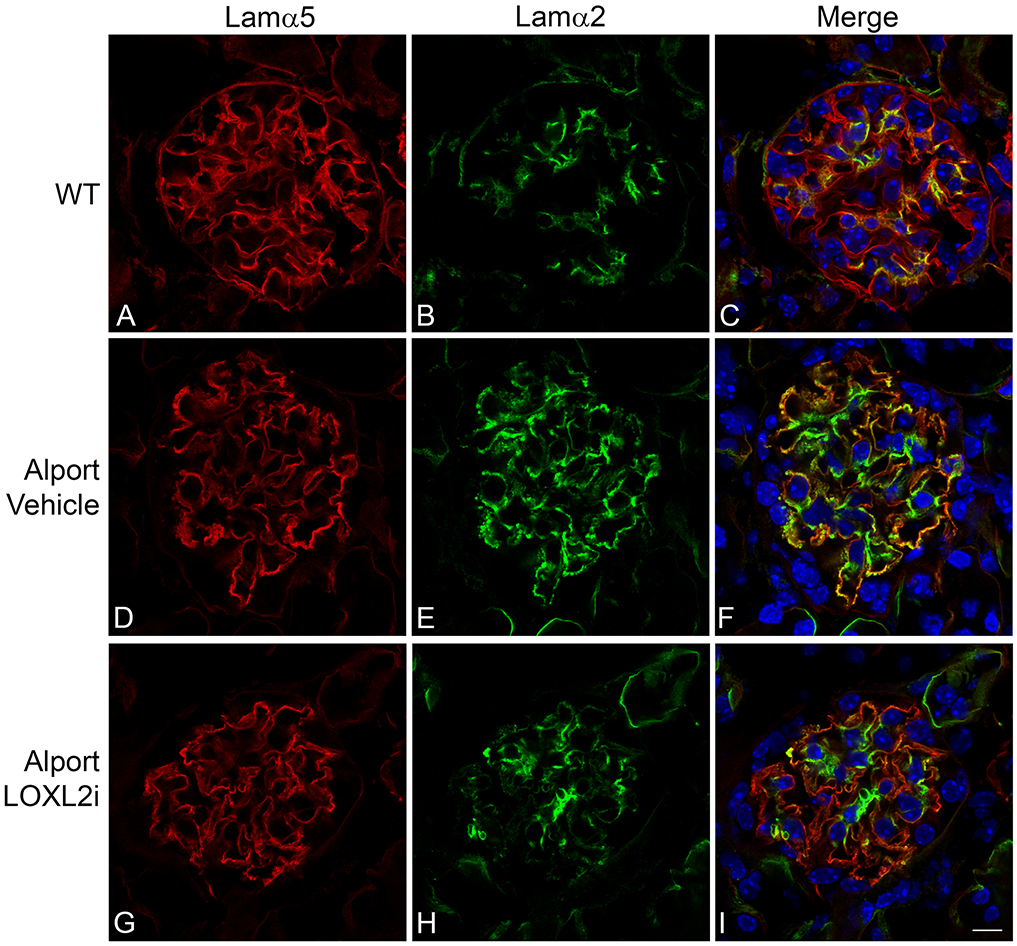
LOXL2i attenuates the accumulation of laminin 211 in the GBM. Dual immunofluorescence immunostaining using antibodies specific for GBM marker laminin α5 (red) and the mesangial cell-specific laminin α2 (green). Punctate yellow staining in the GBM is indicative of laminin α2 deposits (panel F). These punctate laminin deposits are nearly absent from the glomeruli of mice treated with LOXL2i (panel I). Bar Panel I = 10 μm.
Figure 7.
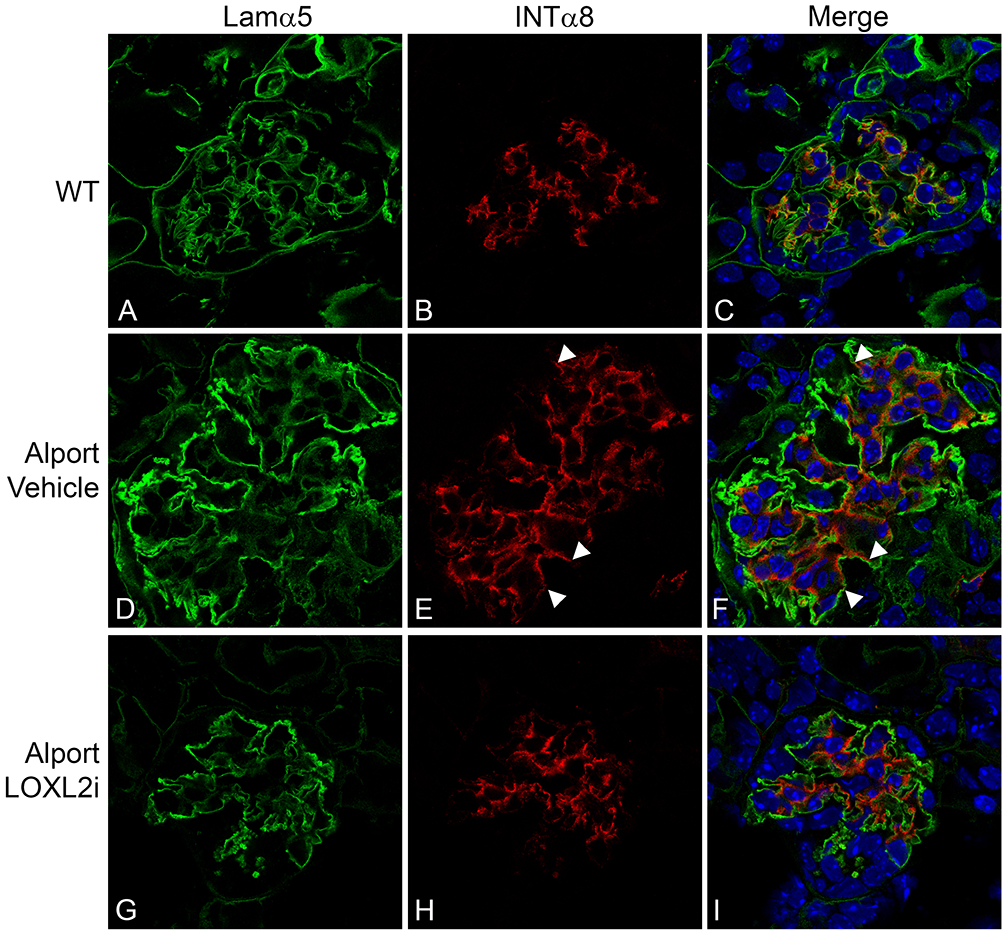
Treatment of Alport mice with LOXL2i reduces the degree of mesangial filopodial invasion into the endothelial aspect of the GBM. Dual immunofluorescence staining for GBM marker laminin α5 (green) and mesangial cell marker integrin α8 (red) demonstrates that invasion of the capillary loops by mesangial cell processes (panel F arrowheads) is markedly reduced in Alport mice treated with LOXL2i (Panel I). Bar panel I = 10 μm.
The hallmark of Alport GBM disease is the multi-laminar GBM ultrastructure punctuated with irregular thickening and thinning of the GBM.32,33 We examined the GBM architecture of 7-week old vehicle-treated Alport mice compared to Alport mice treated with LOXL2i and wild-type littermates. As shown in Figure 8, Alport mice treated with LOXL2i are dramatically normalized in terms of the GBM architecture with reduced GBM irregularities. Alport GBM shows both increased thickening and focal thinning of the glomerular basement membrane. This can be seen in the cumulative distribution of glomerular thickness (Figure 8G) with increased frequency of both thinner and thicker regions of GBM (WT vs. Alport p<0.0001 by KS test). Treatment with LOXL2i normalizes the distribution reducing both the areas of GBM thickening and premature thinning (LOXL2i vs. Alport p<0.0001; LOXL2i vs. WT p=0.45 by KS test).
Figure 8.
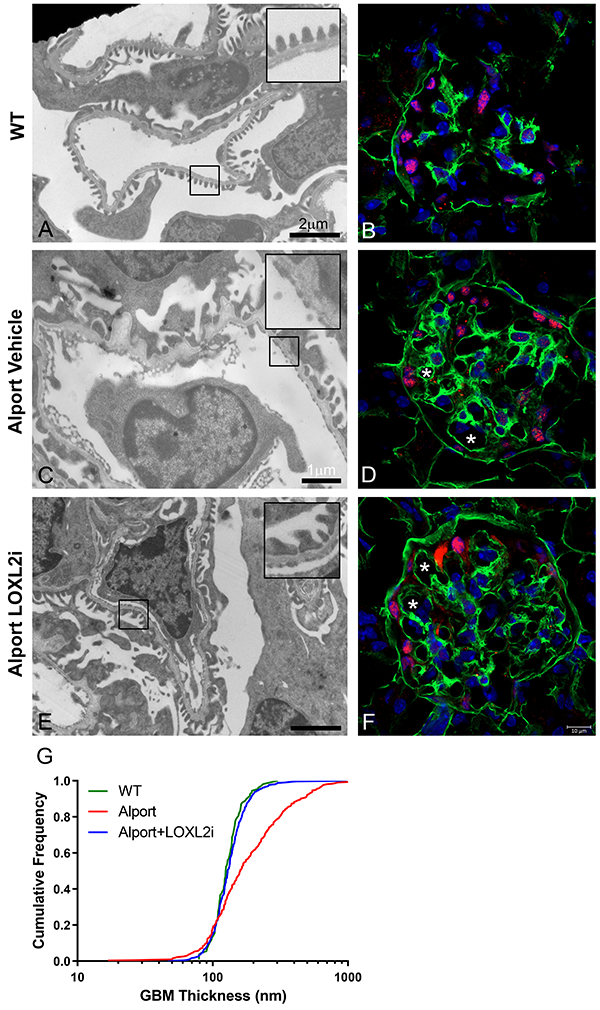
Treatment of Alport mice with LOXL2i greatly improves GBM architecture. Renal cortex from wild type mice (panel A), vehicle-treated Alport mice (panel C) or LOXL2i –treated Alport mice (panel E) were subjected to transmission electron microscopic analysis as described in the methods. The irregular thickening and thinning of the GBM characteristic of Alport mice (C) is ameliorated in Alport mice treated with LOXL2i (E). Panels B, D, and F are wild type (B), vehicle-treated Alport (D) and LOXL2i-treated Alport (F) stained with anti α1(IV) collagen antibodies provided as a frame of reference. Glomerular capillaries are labeled with asterisks. Quantitative morphometric analysis for GBM thickness (Panel G) shows near complete normalization of GBM thickness in the LOXL2i-treated Alport mice. Mean ± SEM * p<0.05.
This is consistent with our demonstration of reduced proteinuria and BUN measures (at 7 weeks), as well as reduced glomerular and interstitial pro-inflammatory cytokine and MMP expression, and likely accounts for the improved renal function.
Discussion:
Collectively, this work demonstrates that LOXL2 plays an important role in driving renal disease progression in COL4A3/Alport mice. LOXL2 expression is upregulated in a number of human nephropathies , and a recent study showed that LOXL2 expression in the kidney was up-regulated in a folic acid-induced model for tubulointerstitial fibrosis; however, a causative connection between LOXL2 and renal pathology has not previously been established.26 LOXL2 has been shown to drive fibrosis progression in models of cardiac and hepatic fibrosis using both a gene knockout and a therapeutic antibody.13,14 Inhibitors to LOXL2 have also demonstrated activity in models of lung fibrosis and cancer.17,34 Taken together it was logical to test whether LOXL2 plays an important role in fibrosis progression and renal failure in the Alport mouse model.
Like all LOX proteins (LOX, LOXL1, LOXL2, LOXL3, and LOXL4), LOXL2 contains a conserved carboxy-terminal domain that comprises a copper and quinone-dependent amine oxidase. This family of enzymes can oxidize the amino group at the epsilon-position of lysine and hydroxylysine residues within collagen and elastin molecules, this converting them to aldehydes. The aldehydes spontaneously condense to form intra- and intermolecular crosslinks. Collagen cross-linking increases the stiffness of the extracellular matrix (ECM) which promotes fibrosis progression through the activation of resident fibroblasts and limits the reversibility of established fibrosis.35,36 In addition, this stiffening is thought to activate the processes that underlie EMT, further resulting in the accumulation of myofibroblasts which drive fibrosis. Inhibition of LOXL2 prevented the accumulation of both interstitial and glomerular α-smooth muscle actin-positive myofibroblasts (Figure 3F) and prevented both interstitial fibrosis and glomerulosclerosis (Figure 4 A,B), consistent with this notion.
More recently, it has been demonstrated that LOXL2 functions in the nucleus as a transcriptional regulator. LOXL2 interacts with the transcription factor Snail1, which enhances the ability of Snail1 to repress expression of the CDH1 gene, reducing cadherin-1 expression which directly promotes EMT.5 In addition, LOXL2 has been shown to deaminate lysyine residues on histone H3, resulting in further repression of CDH1 expression.37 More generally, this modification of histone H3 is thought to facilitate the Snail1/LOXL2 dependent transient release of heterochomatin protein 1-alpha (HP1α), resulting in “loosened” heterochromatin that facilitates expression of other genes necessary for EMT to occur.38
Recently published work demonstrates that LOXL2 crosslinks the 7S domains (amino terminal ends) of type IV collagen and thus contributes to the assembly of the collagen networks in basement membranes.25,34 The authors suggested that LOXL2 might be a critical component for basement membrane stabilization (including GBM), however our 5 week treatment with LOXL2i in wild-type mice resulted in no observable GBM abnormalities either ultrastructurally or functionally, suggesting that redundant pathways for 7S domain crosslinking exist (likely other members of the LOX family).
The combined effect of LOXL2 on ECM stiffening and potentially EMT, along with the fact that LOXL2 inhibition has proven to exert antifibrotic effects in heart, liver, and lung fibrosis models, provides support for the use of this molecule as a means to influence the progression of fibrosis in a broader array of chronic kidney diseases.
Methods:
Mice:
For all experiments, the Col4a3 null mouse, an autosomal model for Alport syndrome on the 129 Sv background was used.39 All experiments were done in accordance with an approved IACUC protocol with full attention and compliance with USDA recommendations for the care and use of animals in research. Every effort was made to minimize the number of animals employed, and to minimize pain and distress.
Treatment of mice with LOXL2i:
Four groups of eight 129sv mice were treated: Wild type with 0.5% methylcellulose (SIGMA) as vehicle, Wild type with PAT-1251 (provided by PharmAkea, Inc.) at 30 mg/kg in 0.5% methocel, Alport with vehicle, and Alport with PAT-1251 at 30 mg/kg in 0.5% methylcellulose. The treatments were given once daily by gavage needle from 2-7 weeks of age.
Urine was collected weekly from 4-7 weeks. Albumin was measured using Molecular Innovations (Novi, MI) Mouse Albumin Antigen ELISA Kit Cat# MSAKT. The albumin levels were normalized to creatinine measured using BioAssay Systems (Hayward, CA) Creatinine Assay Kit (DICT-500) according to the manufacturer’s instructions. BUN measurements were determined with BioAssay Systems Urea Assay Kit (DIUR-100) according to the manufacturer’s instructions.
Assaying plasma LOXL2 activity in PAT-1251 dosed mice:
Two groups of 5 mice were dosed with 30 mpk PAT-1251 or vehicle and blood collected 1 hour post-dosing into heparin vacutainer tubes. Plasma was isolated and stored at −80°C until analysis. Because the endogenous plasma LOXL2 concentrations are very low, measurable LOXL2 activity can only be observed after addition of mouse recombinant LOXL2 (Sino Biologicals (Beijing, China)) to the plasma. To assay for inhibition of plasma LOXL2 activity, 25 μL plasma from each animal was added in triplicate to a black-walled clear bottom 96-well optical plate. Mouse recombinant LOXL2 was added to two of the triplicate wells to a final concentration of 50 ng/μL and the pan-LOX family inhibitor, βAPN, was added to one of the two LOXL2 spiked wells at a final concentration of 300 μM to serve as the maximum (100%) inhibition controls. After incubation of the plate at 37°C for 2 hours, 25 μL of 40 mM 1,5 diaminopentane (Sigma) and 50 μL of Amplex Red Mix (48.5 μL of 50 mM sodium borate buffer pH 8 + 0.5 μL 10 mM Amplex Red (Invitrogen) + 1 μL 500 U/mL Horse Radish Peroxidase) was added to each well and the plate immediately loaded into a FlexStation3 to perform a kinetic read of the fluorescence at excitation = 544 nm and emission = 590 nm. LOXL2 activity or Vmax was determined from the slope of the linear portion of the curve (from 2 to 20 minutes). Maximum LOXL2 activity was determined from wells containing plasma from vehicle-dosed mice spiked with mouse recombinant LOXL2 and 100% inhibition of LOXL2 activity was determined from wells containing plasma from vehicle-dosed mice spiked with mouse recombinant LOXL2 and βAPN. LOXL2 activity from the PAT-1251-dosed animals was calculated relative to the vehicle-dosed mice.
Antibodies:
Antibodies used for immunofluorescence or western blot: Rat anti-CD13 (Bio-Rad, Hercules, CA, USA, Cat #: MCA2183), rat anti-CD45 (Thermo Fisher Scientific, Waltham, MA, USA, Cat #: 14-0451-85), rabbit anti-Collagen type 1 (CederLane Labs, Burlington, NC, USA, Cat #: CL50151AP), rabbit-anti Collagen α1 IV (Biodesign, Saco, ME, USA, Cat #: T40261R), goat anti-Integrin α8 (R&D Systems, Inc., Minneapolis, MN, USA, Cat #: AF4076), rat anti-Laminin α2 (MilliporeSigma, St.Louis, MO, USA, Cat #: L-0663), rabbit anti-Laminin α5 (Dr. Jeff Miner, Washington University, St. Louis, MO, USA), goat anti-Lysyl Oxidase Homolog 2/LOXL2 (R&D Systems, Inc., Cat #: AF2639), rabbit anti-LOXL2 (Abcam, San Francisco, CA, USA, Cat #: ab96233), rabbit anti-NCC (StressMarq Biosciences, Victoria, British Columbia, Canada, Cat #: SPC-402), anti-Actin, α-Smooth Muscle-cy3 (MilliporeSigma, Cat#: C6198), rabbit anti-Synaptopodin (MilliporeSigma, Cat #: S9442), rabbit anti-WT1 (Santa Cruz, Dallas, TX, USA, Cat #: sc-192), and anti-rabbit IgG-peroxidase (MilliporeSigma, A9169).
Immunofluorescence microscopy:
Fresh frozen OCT-embedded kidneys were sectioned at 8-μm and fixed in acetone for 10 minutes at −20°C. Slides were incubated overnight at 4°C in the appropriate primary antibody and blocking solution. The dual stains of collagen IV at 1:500, integrin α8 at 1:1000, laminin α2 at 1:200, laminin α5 at 1:2000, Synaptopodin at 1:2000, and WT1 at 1:50 were incubated in 0.3% PBST (Triton X-100) + 5% FBS. CD45 at 1:100 and collagen type 1 at 1:200 were incubated in 7% milk blocking solution. Slides were rinsed with 1X PBS and incubated for 1 hour at room temperature in Alexa Fluor secondary antibodies and blocking solutions at 1:500. For the LOXL2 duals and α-SMA-cy3 stain, kidneys were sectioned at 8-μm and fixed in 4% paraformaldehyde for 10 minutes at room temperature, permeabilized in 0.3% PBST for 10 minutes and then rinsed. Slides were incubated in CD13 at 1:200, LOXL2 (R&D Systems) at 1:100, NCC at 1:200 and Synaptopodin at 1:2000 in 7% milk blocking solution overnight at 4°C. Slides were rinsed with 1X PBS and incubated for 1 hour at room temperature in the appropriate secondary antibodies or α-SMA-cy3 at 1:200 in 7% milk blocking solution. All slides were rinsed in 1X PBS following the secondary antibodies. Slides stained with α-SMA-cy3 were fixed in 4% paraformaldehyde for 10 minutes at room temperature before mounting. All slides were mounted using Prolong Gold Antifade Mountant with DAPI (Thermo Fisher, Cat #: P36931). Images captured using a Zeiss Axio Imager A.1 with a 20x/0.8 objective, 40x/1.3 oil, or 100x/1.4 oil. Confocal images captured using a Zeiss LSM 800 with a 63x NA: 1.4 oil objective. Final figures compiled using Adobe Photoshop and Illustrator software (Adobe Systems, San Jose, CA, USA).
Immunohistochemistry microscopy:
Kidneys were prepared by immersion fixation in 4% paraformaldehyde. Tissue was processed for paraffin embedding, cut, and stained for Masson’s Trichrome by UNMC Tissue Sciences Facility (Omaha, NE, USA). Images were captured using a Zeiss Axio Imager A.1 with a 20x/0.8 objective.
Fibrosis and glomerulosclerosis scoring:
Total number of sclerotic glomeruli and total number of glomeruli were recorded for each tissue. Fibrosis scoring was performed blinded by visually estimating the % of interstitial fibrosis within the kidney sample. Podocytes per glomeruli were determined by blindly imaging kidneys stained with WT-1, a podocyte-specific nuclear marker. 15 glomeruli were imaged. Statistical analysis was performed using two-tailed student’s t-test.
Western blot:
Renal cortex and glomerular lysates were collected in T-PER™ Tissue Reagent (Thermo 78510) containing Protease Inhibitor Cocktail (Sigma P 8340) and quantified using Coomassie Protein Assay Reagent™ (Thermo 1856209). 20 μg of cortex and 10 μg of glomerular lysates were boiled for 3 minutes in 6X SDS loading buffer containing mercapoethanol and fractionated on duplicate 7.5 % SDS-PAGE gels. One gel was stained with 0.1 % G-250 Coomassie Blue (Sigma) and the other electro transferred onto PVDF membrane (Thermo 88518) in Tris-glycine buffer for one hour at 100 V. Coomassie stained gels were destained, scanned and total protein quantified for loading normalization.40 PVDF membranes were stained with MemCode™ Reversible Stain Kit (Thermo 24585) and evaluated for consistency of transfer. Membranes were blocked in 5 % milk PBST (0.1% Triton X-100) and incubated with a 1:5000 dilution of anti-LOXL2 antibody (abcam) overnight at 4°. Following 3X wash in PBST, blots were incubated with a 1:100,000 dilution of α rabbit whole molecule HRP antibody (Sigma) then washed and developed using SuperSignal® West Femto Maximum Sensitivity Substrate (Thermo 34095). Appropriate bands were quantified and normalized using ImageJ 1.47v (NIH).
Real Time qRT-PCR:
Glomeruli, isolated as previously described,41 and a 0.2 cm2 piece of renal cortex were placed in 200 μl of TRIZOL™ (Invitrogen, Walthan, MA). Cortex were homogenized and RNA isolated utilizing PureLink® (Invitrogen) from both tissues. cDNA was generated with SuperScript® VILO™ (Invitrogen). Real time PCR was performed using TaqMan™ Gene Expression Master Mix (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA, USA) and a StepOnePlus™ Real-Time PCR System (Applied Biosystems). The following TaqMan™ Gene Expression Assay Probes (Applied Biosystems) were used to analyze the samples: IL-6 (Cat# 4331182, ID# Mm00446190_m1), LOXL2 (Cat# 4331182, ID# Mm00804740_m1), MCP-1 (Cat# 4331182, ID# Mm00441242_m1), MMP-2 (Cat# 4331182, ID# Mm00439508_m1), MMP-9 (Cat# 4331182, ID# Mm00442991_m1), MMP-10 (Cat# 4331182, ID# Mm00444630_m1), MMP-12 (Cat# 4331182, ID# Mm00500554_m1), TGFβ−1 (Cat# 4331182, ID# Mm01178820_m1), and TNF-α (Cat# 4331182, ID# Mm00443258_m1). All transcripts were normalized to mouse GAPDH Endogenous Control VIC® Probe (Applied Biosystems, Cat# 4352339E). Samples ran in triplicate with a final volume of 20 μL using the following cycling parameters: 50°C for 2 minutes, 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. The difference in gene expression was calculated using the comparative CT method. Statistical analysis performed using the one sample student’s t-test (hypothetical mean value equal to 1).
Transmission electron microscopy:
Quantitative analysis of GBM thickness:
Thin sections (70 nm) mounted on copper grids, counterstained with uranyl acetate and lead citrate were examined in a Hitachi H-7500 transmission electron microscope. Digitized images were obtained and archived with an ORCA camera with IC-PCI framegrabber and AMT 12-HR software. Point to point measurements of GBM width were made from images taken at 12,000x. GBM measurements were taken from at least five different glomeruli from 2 wild type (>100 measurements), 2 Alport-Vehicle (>300 measurements) and 5 LOLX2i treated Alport mice (>700 measurements). Given the increased variability inherent in GBM thickness in the Alport mice, data are graphed using a cumulative distribution plot and analyzed using pairwise Komolgorov-Smirnov (KS) tests (GraphPad Prism 6.0).
Lifespan studies.
Ten animals per group either treated with LOXL2i or vehicle were dosed until they lost 15% of peak body weight, which occurs within a few days of death. This precaution was used to avoid unnecessary stress and suffering from uremia.
Acknowledgements:
We thank John (Skip) Kennedy for graphic art work. Confocal microscopy was performed at the Advanced Microscopy Core Facility, UNMC, Omaha, NE (NIH P30GM106397). Transmission electron microscopy was conducted at the Microscopy and Digital Imaging Core of the Research Center for Auditory and Vestibular Studies at Washington University (P30 DC004665). We also thank Gina Ma, Kristen Shannon and Katherine Nguyen for execution of some of the in vivo studies.
Footnotes
Disclosures: This work was conducted as a collaborative effort between PharmAkea, Inc., and Dr. Cosgrove’s laboratory. While the work in Dr. Cosgrove’s lab was conducted under a Sponsored Research Agreement with PharmAkea, which funded the work, Dr. Cosgrove maintains no financial interest in the Company as of completion of the project, which preceded this publication.
References:
- 1.Molnar J, Fong KS, He QP, et al. Structural and functional diversity of lysyl oxidase and the LOX-like proteins. Biochim Biophys Acta. 2003;1647:220–224. [DOI] [PubMed] [Google Scholar]
- 2.Kagan HM, Li W. Lysyl oxidase: properties, specificity, and biological roles inside and outside of the cell. J Cell Biochem. 2003;88:660–672. [DOI] [PubMed] [Google Scholar]
- 3.Finney J, Moon HJ, Ronnenbaum T, et al. Human copper-dependent amine oxidases. Arch. Biochem. Biophys. 2014;546:19–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peinado H, Del Carmen Iglesias-de la Cruz M, Olmeda D, et al. A molecular role for lysyl oxidase-like 2 enzyme in snail regulation and tumor progression. EMBO J. 2005;24:3446–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iturbide A, García de Herreros A, Peiró S. A new role for LOX and LOXL2 proteins in transcription regulation. FEBS J.2015;282:1768–1773. [DOI] [PubMed] [Google Scholar]
- 6.Rodríguez D, Rodríguez-Sinovas A, Martínez-González J. Lysyl oxidase as a potential therapeutic target. Drug News Perspect. 2008;21:218–224. [DOI] [PubMed] [Google Scholar]
- 7.Barker HE, Cox TR, Erier JT. The rationale for targeting the LOX family in cancer. Nat Rev Cancer. 2012;12:540–552. [DOI] [PubMed] [Google Scholar]
- 8.Xiao Q, Ge G. Lysyl oxidase, extracellular matrix remodeling and cancer metastasis. Cancer Microenviron. 2012;5:261–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cuevas EP, Eraso P, Mazón MJ. LOXL2 drives epithelial-mesenchymal transition via activation of IRE1-XBP1 signaling pathway. Sci Rep. 2017;7:44988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Bergen T, Spangler R, Marshall D, et al. The role of LOX and LOXL2 in pathogenesis of an experimental model of choroidal neovascularization. Invest Ophthalmol Vis Sci. 2015;56:5280–5209. [DOI] [PubMed] [Google Scholar]
- 11.Wu L, Zhu Y. The function and mechanisms of action of LOXL2 in cancer (Review). Int J Mol Med. 2015;36:1200–1204. [DOI] [PubMed] [Google Scholar]
- 12.Chien JW, Richards TJ, Gibson KF, et al. Serum lysyl oxidase-like 2 levels and idiopathic pulmonary fibrosis disease progression. Eur Respir J. 2014;43:1430–1438. [DOI] [PubMed] [Google Scholar]
- 13.Yang J, Savvatis K, Kang JS, et al. Targeting LOXL2 for cardiac interstitial fibrosis and heart failure treatment. Nat Commun. 2016;7:13710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vadasz Z, Kessler O, Akiri G, et al. Abnormal deposition of collagen around hepatocytes in Wilson’s disease is associated with hepatocyte specific expression of lysyl oxidase and lysyl oxidase like protein-2. J Hepatol. 2005;43:499–507. [DOI] [PubMed] [Google Scholar]
- 15.Aumiller V, Strobel B, Romeike M, et al. Comparative analysis of lysyl oxidase (like) family members in pulmonary fibrosis. Sci Rep. 2017;7:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dongiovanni P, Meroni M, Baselli GA, et al. Insulin resistance promotes lysyl oxidase like 2 induction and fibrosis accumulation in non-alcoholic fatty liver disease. Clin Sci (Lond). 2017;131:1301–1315. [DOI] [PubMed] [Google Scholar]
- 17.Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase-like-2 impedes the development of a pathologic microenvironment. Nature Med. 2010;9:1009–1017. [DOI] [PubMed] [Google Scholar]
- 18.Meissner EG, McLaughlin M, Matthews L, et al. Simtuzumab treatment of advanced liver fibrosis in HIV and HCV-infected adults: results of a 6-month open-label safety trial. Liver Int. 2016;36:1783–1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hutchinson JH, Rowbottom MW, Lonergan D, et al. Small molecule lysyl oxidase-like 2 (LOXL2) inhibitors: The identification of an inhibitor selective for LOXL2 over LOX. ACS Med Chem Lett. 2017;8:423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rowbottom MW, Bain G, Calderon I, et al. Identification of 4-(Aminomethyl)-6-(trifluoromethyl)-2-(phenoxy) pyridine derivatives as potent, selective, and orally efficacious inhibitors of the copper-dependent amine oxidase, Lysyl oxidase-like 2 (LOXL2). J Med Chem. 2017;60:4403–4423. [DOI] [PubMed] [Google Scholar]
- 21.Ikenaga N, Peng ZW, Vaid KA, et al. Selective targeting of lysyl oxidase-like 2 (LOXL2) suppresses hepatic fibrosis progression and accelerates its reversal. Gut. 2017;66:1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rodriguez HM, Vaysberg M, Mikels A, et al. Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. J. Biol. Chem 2010;285:20964–20974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haase VH. Hypoxia-inducible factor signaling in the development of kidney fibrosis. Fibrogenesis and Tissue Repair 2012;5:S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anazco C, Lopez-Jimenez AJ, Rafi M et al. Lysyl oxidase-like-2 cross-links collagen IV of glomerular basement membrane. J. Biol. Chem 2016;291:25999–26012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lopez-Jimenez AJ, Basak T, Vanacore RM. Proteolytic processing of lysyl oxidase like-2 in the extracellular matrix is required for crosslinking of basement membrane collagen IV. J. Biol. Chem 2017;117:798603. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi SE, Jeon N, Choi HY, et al. Lysyl oxidase-like 2 is expressed in kidney tissue and is associated with the progression of tubulointerstitial fibrosis. Mol Med Rep. 2017;16:2477–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hinz B, Phan SH, Thannickal VJ, et al. Recent developments in myofibroblast biology: paradigms for connective tissue remodeling. Am J Pathol. 2012;180:1340–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dufek B, Meehan DT, Delimont D, et al. Endothelin A receptor activation on mesangial cells initiates Alport glomerular disease. Kidney Int. 2016;90:300–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Delimont D, Dufek BM, Meehan DT, et al. Laminin α2-mediated focal adhesion kinase activation triggers Alport glomerular pathogenesis. PLoS One. 2014;9:e99083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zallocchi M, Johnson BM, Meehan DT, et al. α1β1 integrin/Rac1-dependent mesangial invasion of glomerular capillaries in Alport syndrome. Am J Pathol. 2016;183:1269–1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark SD, Nabity MB, Cianciolo RE, et al. X-lined Alport dogs demonstrate mesangial filopodial invasion of the capillary tuft as an early event in glomerular damage. PLoS One. 2016:11:e0168343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cosgrove D, Liu S. Collagen IV diseases: A focus on the glomerular basement membrane in Alport syndrome. Matrix Biol. 2017;57–58:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kashtan CD. Diagnosis of Alport syndrome. Kidney Int. 2004;66:1290–1291. [DOI] [PubMed] [Google Scholar]
- 34.Chang J, Lucas MC, Leonte LE, et al. Pre-clinical evaluation of small molecule LOXL2 inhibitors in breast cancer. Oncotarget 2017;8:26066–26078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williamson PR, Kagan HM. Reaction pathway of bovine aortic lysyl oxidase. J Biol Chem. 1986;261:9477–9482. [PubMed] [Google Scholar]
- 36.Smith-Mungo LI, Kagan HM. Lysyl oxidase: properties, regulation and multiple functions in biology. Matrix Biol. 1998;16:387–398. [DOI] [PubMed] [Google Scholar]
- 37.Herranz N, Dave N, Millanes-Romero A, et al. Lysyl oxidase-like 2 deaminates lysine 4 in histone H3. Mol Cell. 2012;46:369–376. [DOI] [PubMed] [Google Scholar]
- 38.Millanes-Romero A, Herranz N, Perrera V, et al. Regulation of heterochromatin transcription by Snail1/LOXL2 during epithelial-to-mesenchymal transition. Mol Cell. 2013;52:746–757. [DOI] [PubMed] [Google Scholar]
- 39.Cosgrove D, Meehan DT, Grunkemeyer JA, et al. Collagen COL4A3 knockout: a mouse model for autosomal Alport syndrome. Genes Dev. 1996;121:84–98. [DOI] [PubMed] [Google Scholar]
- 40.Eaton SL,Roche SL, Hurtado ML, et al. Total Protein Analysis as a Reliable Loading Control for Quantitative Fluorescent Western Blotting. PLOS ONE 2013;8: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rao VH, Meehan D, Delimont D, Nakajima M, Wada T, Gratton MA, and Cosgrove D Role for MMP-12 in GBM damage associated with Alport syndrome. Am. J. Pathol 2006;169:32–46. [DOI] [PMC free article] [PubMed] [Google Scholar]