

Summary
NANOG is an essential transcriptional factor for the maintenance of embryonic stem cells (ESCs) and cancer stem-like cells (CSCs) in prostate cancer (PCa). However, the regulation mechanism of NANOG protein stability in cancer progression is still elusive. Here, we report that NANOG is degraded by SPOP, a frequently mutated tumor suppressor of PCa. Cancer-associated mutations of SPOP or the mutation of NANOG at S68Y abrogated the SPOP-mediated NANOG degradation, leading to elevated PCa cancer stemness and poor prognosis. In addition, SPOP-mediated NANOG degradation is controlled by AMPK-BRAF signal axis through the phosphorylation of NANOG at Ser68, which blocked the interaction between SPOP and NANOG. Thus, our study provides a regulation mechanism of PCa stemness controlled by phosphorylation-mediated NANOG stability, which helps to identify novel drug targets and improve therapeutic strategy for PCa.
Graphical Abstract
In Brief:
Wang et al. uncover a regulation mechanism by which NANOG stability, which is involved in the regulation of the stem-like characteristics of prostate cancer cells, is dictated by SPOP-mediated degradation and is controlled by AMPK-BRAF mediated phosphorylation of NANOG at Ser68.
Introduction
Most cancers have a small population of cells with stem cell-like properties, termed as cancer stem-like cells (CSCs) that are capable of self-renewal and tumor-initiation (O'Brien et al., 2007, Patrawala et al., 2006, Singh et al., 2003). Moreover, CSCs express pluripotency-related transcription factors of embryonic stem cells (ESCs), such as NANOG, OCT3/4 and SOX2 (Pece et al., 2010, Wong et al., 2008, Chambers and Tomlinson, 2009). The expression levels of these pluripotency transcriptional factors in cancers are tightly related to the development and progression of various cancers (Po et al., 2010, Suva et al., 2013, Boumahdi et al., 2014). Prostate cancer (PCa) is one of the most common cancers in the worldwide and the second leading cause of cancer-related deaths in men (Siegel et al., 2013). Accumulating evidence indicates that presence of CSCs is responsible for prostate cancer initiation, progression and chemotherapy resistance (Jeter et al., 2011, Liu et al., 2010b). Therefore, in-depth understanding of the molecular underpinnings in PCa stem cell-like traits may shed light on the development of CSC-specific targeted therapy for PCa.
NANOG, an embryonic stem cell transcription factor, maintains the self-renewal and pluripotency of ES cells (Chambers et al., 2003, Mitsui et al., 2003). NANOG is highly expressed in various cancers and plays a pleiotropic role in the tumorigenesis cascade, such as chemotherapy and radiation resistance, metabolism reprogramming and CSCs population modulation (Iv Santaliz-Ruiz et al., 2014, Chen et al., 2016). Additionally, NANOG is also a critical regulator for PCa progression (Ugolkov et al., 2011), its overexpression promotes the tumorigenesis in both androgen-sensitive and -insensitive PCa cells (Zhang et al., 2014a), indicative of poor prognosis (Mathieu et al., 2011, Miyazawa et al., 2014). It has been reported that PCa cells are endowed with the stem-like properties by NANOG expression, especially under the stabilized and accumulated condition (Jeter et al., 2011, Kawamura et al., 2015). However, the regulatory mechanism of NANOG stability in cancers remains unclear.
The ubiquitin-proteasome system (UPS) is one of the key pathways that regulate stem cells differentiation and function (Buckley et al., 2012). Speckle-type POZ protein (SPOP) is a protein possessing bric-a-brac-tramtrack-broad/poxvirus and zinc finger (BTB/POZ) domain, an adaptor for the E3 ubiquitin ligase Cullin3 (Zhuang et al., 2009). SPOP gene mutation is highly relevant to human prostate cancers, with a mutation rate of 10% to 15% (Cancer Genome Atlas Research, 2015). SPOP can flexibly degrade various protein substrates such as androgen receptor (AR) (An et al., 2014, Geng et al., 2014), steroid receptor coactivator 3 (SRC-3) (Li et al., 2011), DEK, TRIM24 (Theurillat et al., 2014), BRD4 (Janouskova et al., 2017, Dai et al., 2017, Zhang et al., 2017), PD-L1 (Zhang et al., 2018) and ERG (Gan et al., 2015, An et al., 2015), and thus regulate the proliferation and invasion of prostate cancers. However, the role of SPOP in mediating the stemness and pluripotency of PCa stem cells and ESCs, remains largely unknown.
In this study, we report that SPOP inhibits the self-renewal and stem-like characteristics of PCa via the ubiquitin-dependent degradation of NANOG. The cancer-associated NANOG S68Y mutant is refractory for SPOP-mediated degradation since NANOG Ser68 is required for the direct interaction between SPOP and NANOG. In parallel, AMPK activation promotes the NANOG degradation through blocking the binding of NANOG to BRAF that phosphorylates NANOG at Ser68. Thus, our study uncovers a de novo regulation mechanism of NANOG stability dictated by SPOP-induced degradation, which is abrogated by the phosphorylation of NANOG at Ser68 and thereby acts synergistically with AMPK-BRAF signaling axis.
Results
NANOG is degraded by SPOP
NANOG is often expressed in prostate cancer stem cells (CSCs) (Miyazawa et al., 2014, Jeter et al., 2009) and its expression is intimately correlated to poor prognosis in human prostate cancers. Our study indicated that NANOG is a short-lived protein. Treatment with the proteasome inhibitor MG132 dramatically increased the protein level and prolonged the half-life of NANOG (Figures S1A-B). Similar results were obtained with the treatment of MLN4924 (Figures S1A-B), a potent inhibitor of Cullin-RING ligases by depressing the NEDD8-activating enzyme (NAE) (Ohh et al., 2002), suggesting that NANOG is targeted for degradation through a Cullin E3 ubiquitin-ligase-mediated UPS pathway. To search for the Cullin-dependent E3 ligase that targets NANOG for degradation, we screened various dominant negative forms of Cullins (dnCuls) together with the co-expression of NANOG. Our data showed that the dominant negative form of Cullin3 (dnCul3), but not other dnCuls, significantly stabilized NANOG (Figures 1A and S1C). We further demonstrated that the depletion of Cullin3, but not other Cullins, markedly increased protein level of NANOG in DU145 PCa cells (Figures 1B and 1C). Together, these data indicate that Cullin3 E3 ligase involves in the control of NANOG degradation in PCa cells.
Figure 1. Cullin3/SPOP E3 Ubiquitin Ligase Targets NANOG for Ubiquitination and Degradation.

(A) HA-NANOG and Flag-dnCullins were coexpressed in HEK293T cells. Protein levels of NANOG and Cullins were analyzed by western blotting (WB).
(B) DU145 cells were transfected with control siRNA or Cullins siRNA. The endogenous NANOG and Cullins were analyzed by WB.
(C) DU145 cells were transfected with control siRNA or three independent Cul3-specific siRNAs. The endogenous NANOG were analyzed by WB.
(D) Comparison of putative SPOP binding site in NANOG with the SPOP-binding consensus (SBC) motif defined in the known SPOP substrates.
(E) PC3 cells were transfected with the indicated siRNA. The endogenous NANOG were analyzed by WB.
(F) DU145 SPOP WT or KO cell lines were constructed by lenti-CRISPR system, cells were treated with cycloheximide (CHX, 10 μg/ml) for indicated times. Protein levels of NANOG and SPOP were analyzed by WB.
(G) Flag-NANOG, HA-SPOP and His-ubiquitin were coexpressed in HEK293T cells. After treatment with MG132 (10 μM) for 6 hrs, the Ni-NTA ubiquitination assay was performed and analyzed by WB.
(H) DU145 cells were transfected with the indicated siRNA and constructs for 3 days, the cells were treated with MG132 (5 μM) for 12 hrs, cell lysate was immunoprecipitated (IP) with anti-NANOG antibody and the ubiquitination of NANOG was detected by WB using the anti-His antibody.
(I) Flag-SPOP and indicated HA-tag plasmids were coexpressed in HEK293T cells. Cell lysates were prepared for Co-IP and WB. Cells were treated with MG132 (10 μM) for 6 hrs before harvesting.
(J) DU145 cell lysates were prepared for Co-IP with SPOP antibody and WB. Cells were treated with MG132 (10 μM) for 6 hrs before harvesting.
(K) E14 mESCs cell lysates were prepared for Co-IP with SPOP antibody and WB. Cells were treated with MG132 (10 μM) for 6 hrs before harvesting.
(L) Representative sphere images from each condition of DU145 cells. Scale bar, 200 μm.
(M) Indicated retrovirus infected DU145 tumor spheres were dissociated and equal numbers of cells were passaged for three generations. Spheres counts are normalized to the first generation scrambled shRNA spheres. Data are means ± SEM (n=3). **P<0.01 vs shCon (Student’s t-test).
(N-O) Growth of DU145 cells (N) and 22RV1 cells (O) derived tumors in nude mice infected with retrovirus expressing indicated shRNAs of three different cell densities. Frequency of CSCs is estimated as per ELDA calculating website and P value represents overall test for differences in stem cell frequencies between the two groups, 1/1 in SPOP shRNA tumors means stem cell frequency (100%) *P<0.05, ***P<0.001 vs shNC.
To identify the adaptor of Cullin3 which determines NANOG degradation, we analyzed the protein sequence of NANOG and revealed that NANOG contains an evolutionarily conserved SPOP-binding consensus motif (Φ-π-S-S/T-S/T; Φ, nonpolar residues; π, polar residues) (Zhuang et al., 2009) using online software (http://elm.eu.org/) (Figure 1D). We therefore examined whether NANOG is a potential substrate of SPOP. To this end, we co-expressed NANOG with SPOP in HEK293T cells and examined the protein level of NANOG. We found that co-expression of SPOP significantly shortened the half-life of NANOG (Figures S1D-E). Moreover, both the protein level and the half-life of endogenous NANOG protein were significantly increased upon SPOP depletion in PCa cells and mESCs (Figures 1E and S1F-G). To further confirm this result, we generated two SPOP-deficient DU145 cell lines. Our data showed that the half-life of NANOG is significantly prolonged in SPOP-deficient DU145 cells (Figure 1F), indicating that NANOG is targeted for degradation by SPOP.
Next, we tested whether SPOP targets NANOG for ubiquitination. Our data showed that co-expression of SPOP significantly promoted NANOG ubiquitination in HEK293T cells (Figure 1G). In addition, depletion of SPOP markedly decreased NANOG ubiquitination in DU145 cells (Figure 1H). Moreover, Cul3-SPOP complex promoted the ubiquitination of NANOG in vitro (Figure S1H). Deletion of the BTB domain in SPOP, which is required for its interaction with Cul3, failed to target NANOG for ubiquitination (Figure S1H), supporting an important role of Cullin3 enzyme activity in SPOP-mediated ubiquitination of NANOG.
Furthermore, we examined whether NANOG is a binding partner of SPOP. Our data indicated that SPOP specifically interacts with NANOG, but not other stem cell transcriptional factors, including OCT4, SOX2, and KLF4 (Figure 1I). Notably, the interaction between endogenous SPOP and NANOG could be readily detected in PCa cells (Figures 1J and S1I) and E14 mESCs (Figure 1K). Moreover, our data showed that NANOG specifically interacts with SPOP, but not other Cullin3-based E3 ligase adaptor proteins (Figure S1J). These results together indicate that SPOP is the adaptor responsible for interacting with NANOG and targeting it for ubiquitination by Cullin3 E3 ubiquitin ligase complex.
SPOP represses stem cell-like properties via its regulation of NANOG stability
NANOG expression in PCa is closely correlated to cancer stem cell characteristics, we thus examined whether SPOP can regulate cancer stem cell-like properties by targeting NANOG for degradation. To this end, we examined the effect of SPOP on the oncosphere-forming capacity of PCa cells, which is a standard for the measurement of cancer stem cell functions (Zhang et al., 2010). The number of oncospheres formed over multiple passages represents the self-renewal activity, while the size of sphere indicates cell proliferation capability (Dontu et al., 2004). Our data showed that compared to control cells infected with empty vector, both AR-negative DU145 and AR−positive 22RV1 cells lacking of SPOP had a progressive increase in self-renewal capacity and the ability to form daughter spheres. Notably, simultaneous depletion of NANOG abolished the phenotype induced by SPOP depletion (Figures 1L-M and S1L-M). Moreover, SPOP-KO DU145 cells also showed an increased capability of forming oncospheres (Figure S1K).
We further investigated whether SPOP can affect the tumor progression via its mediation of NANOG degradation using a nude mouse xenograft model. Our data showed that depletion of SPOP in AR-negative DU145 and AR-positive 22RV1 PCa cells significantly promoted tumor growth, whereas simultaneous depletion of NANOG abolished such phenotype induced by SPOP depletion (Figure S1N-O). Furthermore, SPOP-depleted oncospheres developed more tumors while simultaneous knockdown of NANOG abolished the phenotype derived from SPOP depletion (Figure 1N-O). Moreover, our data showed that knockdown of NANOG greatly reduced the ability of tumor formation when examined by the injection of as few as 1000 oncospheres into nude mice (Figure 1N-O and S1N-O)
To further examine the role of SPOP in prostate cancer stem cell function, we performed a clonal serial in vivo repopulation assay, which is a widely-accepted functional assay to examine the self-renewal capacity of maintaining the long-term clonal growth of cancer stem cells (Kreso and Dick, 2014). Our data from triplicate experiments of serial transplantation showed that depletion of SPOP in both androgen (AR) negative DU145 and AR positive 22RV1 PCa cells elevated the in vivo cell renewal efficacy in a NANOG-dependent manner (Figure 1N-O and S1N-O). These data together indicated that SPOP negatively regulates PCa stem cell maintenance in a NANOG-dependent manner and confirmed that regulation of PCa stem cells properties by SPOP-mediated NANOG degradation is independent of AR.
PCa-associated SPOP mutants cannot target NANOG for degradation
SPOP contains a MATH domain that is responsible for the substrate binding and a BTB domain that is required for its interaction with Cul3 (Zhuang et al., 2009) (Figure 2A). Next, we determined the molecular basis of the interaction between NANOG and SPOP. Our data showed that wild-type SPOP directly interacted with NANOG whereas the absence of the MATH domain completely abrogated the binding in vitro and in vivo (Figures 2B and S2A). While deletion of BTB domain in SPOP also reduced the binding to its substrate, the plausible explanation is that the dimerization of SPOP through its BTB domain is required for sufficient Interaction with substrates (Marzahn et al., 2016, Zhang et al., 2014b). Furthermore, our data showed that deletion of either the MATH domain or BTB domain abolished the SPOP-mediated NANOG degradation and ubiquitination (Figures 2C-D), while overexpression of SPOP, but not its deletion mutants ΔBTB or ΔMATH) in E14 mESCs, led to a loss of mESCs morphology and reduced AP staining (Figures S2B-D).
Figure 2. Prostate Cancer-Associated SPOP Mutants are Defective in Promoting NANOG Ubiquitination and Degradation.

(A) Schematic diagram of SPOP domains and prostate cancer-associated mutations.
(B) HA-NANOG and indicated Flag-tag SPOP plasmids were coexpressed in HEK293T cells. Cell lysates were prepared for Co-IP and WB. Cells were treated with MG132 (10 μM) for 6 hrs before harvesting.
(C) Flag-NANOG and HA-SPOP or mutants were coexpressed in HEK293T cells, protein levels of NANOG and SPOP were analyzed by WB.
(D) Flag-NANOG, HA-SPOP or mutants and His-ubiquitin were coexpressed in HEK293T cells. After treatment with MG132 (10 μM) for 6 hrs, the Ni-NTA ubiquitination assay was performed and analyzed by WB.
(E) Flag-NANOG and indicated HA-tag SPOP plasmids were coexpressed in HEK293T cells. Cell lysates were prepared for Co-IP and WB. Cells were treated with MG132 (10 μM) for 6 hrs before harvesting.
(F) Flag-NANOG was coexpressed in HEK293T cells with indicated HA-tag SPOP plasmids. After treating cells with CHX (10 μg/ml) for indicated time intervals, protein levels of NANOG and SPOP were analyzed by WB.
(G) The NANOG protein abundance in (F) was quantified by ImageJ and plotted as indicated.
(H) Flag-NANOG, HA-SPOP or mutants and His-ubiquitin were coexpressed in HEK293T cells. After treatment with MG132 (10 μM) for 6 hrs, the Ni-NTA ubiquitination assay was performed and analyzed by WB.
(I) DU145 cells were infected with lentivirus expressed SPOP or F133L mutants, then the cells were treated with MG132 (10 μM) for 6 hrs, cell lysate was IP with anti-NANOG antibody and the ubiquitination of NANOG was detected by WB using the anti-Ub antibody.
(J) Representative sphere images from each condition of DU145 cells. Scale bar, 200 μm.
(K) Frequency of tumor spheres formed from DU145 cells in (J). Sphere counts are normalized to mock infected spheres. Data are means ± SEM (n=3). **P<0.01 vs EV; ##P < 0.01 vs WT (Student’s t-test).
(L) Representative sphere images from each condition of DU145 cells. Scale bar, 200 μm.
(M) Indicated lentivirus infected DU145 tumor spheres were dissociated and equal numbers of cells were passaged for three generations. Spheres counts are normalized to the first generation scrambled shRNA spheres. Data are means ± SEM (n=3). *P<0.05,**P<0.01 vs Vector (Student’s t-test).
(N) AP staining after 48 hrs SPOP or SPOP mutants overexpression in E14 mESCs cells (left) and the expression of SPOP or its mutants and NANOG protein are shown (right).
(O) Percentage of undifferentiated colonies counted in (N). Data are means ± SEM (n=3). ##P< 0.01 vs EV, **P < 0.01 vs WT (Student’s t-test).
(P) Immunofluorescent staining analysis for lentivirus expressed SPOP or mutants (green) and endogenous NANOG (red) in E14 mESCs cells. Scale bar, 100 μm.
Importantly, various somatic mutations have been identified in the MATH domain of SPOP in prostate cancer tissues. Notably, these PCa-associated SPOP mutations, such as Y87C/N, F102C and F133L/V, mainly occur on the amino acids residues located on the surface of the substrate-binding pocket of the MATH domain and thus lost the ability to bind and degrade its substrates (Barbieri et al., 2012, Blattner et al., 2014, Zhuang et al., 2009). Our data indicate that these PCa-associated SPOP mutants are unable to interact with NANOG (Figure 2E). Consistently, ectopic expression of these PCa-associated SPOP mutants failed to degrade NANOG (Figures 2F-G). Moreover, these mutants were incapable of promoting NANOG ubiquitination (Figure 2H-I). Functionally, the mutants (Y87C and F133L) failed to inhibit the sphere formation of PCa cells (Figures 2J-K). Besides, our data showed that cells ectopically expressing NANOG exhibited a great potential to form more and larger spheres than control samples, while such effect was inhibited by WT-SPOP, but not its F133L mutant in both DU145 and 22RV1 cells for three consecutive generations (Figures 2L-M and S2E-F). Furthermore, expression of WT-SPOP, but not its mutants including Y87C and F133L impaired the stemness of mESCs (Figures 2N-P).
To examine whether the mutation of SPOP is correlated to aberrant NANOG-mediated gene expression in prostate cancers, we analyzed the gene-expression-based signatures from human clinical specimens with high expression level of NANOG versus SPOP mutation. By two-class unpaired significance analysis of microarray data obtained from tumor and normal samples with or without SPOP mutation, as well as the patient cases with high expression of NANOG from TCGA database, we observed an intimate correlation between the enhanced gene expression of mutant SPOP and high level of NANOG in tumors (fold change >2, q< 0.05) (Figure S2G). More importantly, our analysis demonstrated that 1815 genes including 756 upregulated genes and 1059 downregulated genes, are affected by SPOP mutations and the high expression level of NANOG, emphasizing the crucial roles of both SPOP and NANOG in cancer progression (Figures S2H-I). Indeed, the selected ones from forementioned 1815 genes were influenced by the expression of NANOG (Figure S2J-K). Together, these data indicate that SPOP mutation affects the NANOG-regulated gene expression in PCa cells.
Identification of the SPOP binding consensus (SBC) motif in NANOG
NANOG contains a conserved putative SBC motif 66PDSST70 at its N-terminus (Figure 3A). We then examined whether the putative SBC motif is involved in the interaction between SPOP and NANOG. Our data showed that deletion of the putative SBC motif (ΔPDSST) completely abolished the binding between NANOG and SPOP in vivo and in vitro (Figures 3B-C). Moreover, deletion of the NANOG SBC motif dramatically prolonged its half-life (Figures S3A-B) and increased its transcriptional activity (Figure S3C). Importantly, SPOP was unable to promote the ubiquitination and degradation of the SBC-deleted NANOG mutant (Figures 3D-3F). These data indicate that the SBC motif 66PDSST70 of NANOG is essential for its interaction with SPOP and resultant SPOP-mediated NANOG degradation.
Figure 3. The 66PDSST70 Motif in NANOG Is the Degron Recognized by SPOP.

(A) Sequence alignment of NANOG with the SPOP binding motif (SBC) among different species.
(B) Flag-SPOP and indicated HA-tag NANOG plasmids were coexpressed in HEK293T cells (3A: S68A, S69A, T70A). Cell lysates were prepared for Co-IP and WB. Cells were treated with MG132 (10 μM) for 6 hrs before harvesting.
(C) GST and GST-SPOP proteins were purified from E.coli and incubated with NANOG or PDSST deletion protein IP from HEK293T cells. The products of pull-down assay were analyzed by WB.
(D) Flag-NANOG, HA-SPOP and His-ubiquitin were coexpressed in HEK293T cells. After treatment with MG132 (10 μM) for 6 hrs, the Ni-NTA ubiquitination assay was performed and analyzed by WB.
(E) Flag-NANOG or PDSST deletion mutant was coexpressed in HEK293T cells with HA-SPOP. After treating cells with CHX (10 μg/ml) for indicated time intervals, protein levels of NANOG and SPOP were analyzed by WB.
(F) The NANOG protein abundance in (E) was quantified by ImageJ and plotted as indicated.
(G) HA-SPOP and indicated Flag-tag NANOG plasmids were coexpressed in HEK293T cells. Cell lysates were prepared for Co-IP and WB. Cells were treated with MG132 (10 μM) for 6 hrs before harvesting.
(H) Ectopically expressed SPOP promotes protein degradation of exogenous WT-NANOG but not D67A or S68A mutants. 293T cells were transfected with indicated constructs for 24 hrs followed by WB.
(I) Flag-NANOG or mutants were coexpressed in HEK293T cells with HA-SPOP. After treating cells with CHX (10 μg/ml) for indicated time intervals, protein levels of NANOG and SPOP were analyzed by WB.
(J) The NANOG protein abundance in (I) was quantified by ImageJ and plotted as indicated.
(K) Binding was examined using biotinylated peptide pull-down assay and analyzed using WB.
We next explored whether the residues located in the SBC motif involve in the interaction between NANOG and SPOP. The simultaneous replacement of Ser68, Ser69 or Thr70 with Ala (3A) in NANOG completely abolished its binding to SPOP (Figure 3B). We also generated a series of NANOG point mutations, including S65A, P66A, D67A, S68A, S69A, T70A, S71A and P72A and detected their interaction with SPOP. Co-IP assays showed that D67A and S68A, but not other mutants, failed to interact with SPOP (Figure 3G). Consistent with these results, SPOP did not promote the degradation of NANOG-D67A and S68A mutants (Figures 3H-J). Moreover, data from the biotin-labeled peptide pull-down assay showed that SPOP bound to the peptide containing 66PDSST70, but not the mutated 66PDAST70 motif of NANOG (Figure 3K). These data together indicate that the SBC motif of NANOG, especially D67 and S68 residues, are essential for SPOP-mediated NANOG degradation.
Cancer-associated NANOG mutation within the SBC motif is resistant to SPOP-mediated degradation
Our data showed that SBC motif is essential for NANOG stability, we thus analyzed whether there is any mutation at the SBC motif of NANOG in cancers. Interestingly, a Ser68Tyr (S68Y) mutation in the SBC motif of NANOG was identified in endometrium carcinoma tissues by analyzing the TCGA and COSMIC database (https://cancer.sanger.ac.uk/cosmic/) (Figure 4A). Through computational modeling analyses, we found that the amino acid sequence 66PDSST70 of NANOG binds to an extended groove in the MATH domain of SPOP (Figure S4A). However, the cancer-associated mutation 66PDYST70 in the NANOG mutant increased steric hindrance and impaired its interaction with SPOP (Figures S4B), suggesting that the cancer-associated mutant S68Y may affect the interaction between NANOG and SPOP.
Figure 4. Oncogenic NANOG Mutations within SBC Are Resistant to SPOP-Mediated Degradation.

(A) Sequence alignment of NANOG and cancer associated mutation within the SPOP binding motif (SBC).
(B) HA-SPOP and indicated Flag-tag NANOG plasmids were coexpressed in HEK293T cells. Cell lysates were prepared for Co-IP and WB. Cells were treated with MG132 (10 μM) for 6 hrs before harvesting.
(C) Binding was examined using biotinylated peptide pull-down assay and analyzed using WB.
(D) Flag-NANOG or S68Y were expressed in HEK293T cells. After treating cells with CHX (10 μg/ml) for indicated time intervals, protein levels of NANOG were analyzed by WB.
(E) Flag-NANOG or mutants were coexpressed in HEK293T cells with HA-SPOP. After treating cells with CHX (10 μg/ml) for indicated time intervals, protein levels of NANOG and SPOP were analyzed by WB.
(F) The NANOG protein abundance in (E) was quantified by ImageJ and plotted as indicated.
(G) Flag-NANOG or S68Y, HA-SPOP and His-ubiquitin were coexpressed in HEK293T cells. After treatment with MG132 (10 μM) for 6 hrs, the Ni-NTA ubiquitination assay was performed and analyzed by WB.
(H) Frequency of tumor spheres formed from DU145 cells. Sphere counts are normalized to mock infected spheres. Data are means ± SEM (n=3). *P<0.05, **P < 0.01 vs EV (Student's t-test); ##P < 0.01 vs WT (Student's t-test).
(I) Representative sphere images from each condition of DU145 cells. Scale bar, 200 μm.
(J) Growth of DU145 cells derived tumors in nude mice infected with lentivirus expressing indicated constructs.
(K) Tumor growth curves (Related to J) are shown. Data are means ± SEM (n=6). ***P < 0.001 vs SPOP (−) (Student's t-test).
To confirm this result, we examined the interaction between NANOG S68Y and SPOP. Our data showed that NANOG S68Y mutant almost completely lost the ability to interact with SPOP (Figure 4B). Moreover, SPOP bound to the peptide motif of WT-NANOG containing 66PDSST70, but not the point-mutated one within the sequence of 66PDYST70 (Figure 4C). Furthermore, SPOP promoted the degradation of WT-NANOG but not NANOG-S68Y (Figures 4D-F). Consistently, SPOP promoted the ubiquitination of NANOG-WT but not NANOG-S68Y (Figure 4G). Also, NANOG S68Y mutant exhibited an increased transcriptional activity compared with that of WT NANOG (Figure S4C).
Next, we examined whether NANOG-S68Y mutant regulates the function of cancer stem cell. Sphere formation assays indicated that overexpression of NANOG-WT or NANOG S68Y significantly increased the efficacy of sphere formation (Figures 4H-I). In contrast, co-expression of SPOP dramatically reduced the sphere-formation activity of NANOG-WT, but not that of the NANOG S68Y mutant (Figures 4H-I). In addition, SPOP inhibited the effect of WT NANOG but not the NANOG SBC-deleted or S68Y mutants on migration and proliferation of PCa cells (Figures S4D-F). Subcutaneous injection of DU145 cells expressing NANOG S68Y mutant into nude mice promoted tumor growth even under the condition of SPOP overexpression (Figure 4J-K). Together, these data indicate that NANOG bearing mutations within the SBC motif is oncogenic and resistant to SPOP-mediated degradation.
Phosphorylation of NANOG at Ser68 blocks its degradation by SPOP
Although our data identified that cancer-associated Ser68 mutation of NANOG abolished SPOP-mediated degradation, the mutation frequency of NANOG Ser68 is very low by our analysis from TCGA database. This triggered us to explore other possible mechanism in mediating the stability regulation of NANOG. A pervious study showed that NANOG could be phosphorylated at Ser68 using high-resolution mass spectrometry (Brumbaugh et al., 2014). To investigate whether the phosphorylation of Ser68 affects its interaction with SPOP, we constructed a phosphorylation-mimic mutant of NANOG, S68D (Figure 5A). Our data indicated that S68D mutant lost the ability to interact with SPOP (Figure 5B). Moreover, SPOP could not promote the degradation of NANOG S68D (Figures S5A-C). Our data from a peptide pull-down assay showed that SPOP bound to the peptide of 66PDSST70 in WT-NANOG, but not the phosphorylated peptide of 66PDS(P)ST70 at Ser68 (Figure S5D), suggesting that the phosphorylation of NANOG at Ser68 blocked its interaction with SPOP.
Figure 5. AMPK promotes SPOP-mediated destruction of NANOG through down-regulated Ser68 phosphorylation of NANOG.

(A) Sequence alignment of phosphorylation of NANOG within the SPOP binding motif (SBC).
(B) HA-SPOP was coexpressed with Flag-NANOG or mutations in HEK293T cells. Cell lysates were prepared for Co-IP and WB. Cells were treated with MG132 (10 μM) for 6 hrs before harvesting.
(C) SPOP WT or KO DU145 cells were treated with Compound C (6.6 μM ) for 4 hrs before performing the CHX (10 μg/ml) chase analysis. Expression levels of NANOG and SPOP were analyzed by WB.
(D) The NANOG protein abundance in (C) was quantified by ImageJ and plotted as indicated.
(E) AMPK WT and KO MEF cells were infected with lentivirus expressed NANOG, then treated with Compound C (6.6 μM) for 4 hrs before performing the CHX (10 μg/ml) chase analysis.
(F) The NANOG protein abundance in (E) was quantified by ImageJ and plotted as indicated.
(G) DU145 cells were treated with Compound C (6.6 μM) for 4 hrs, cell lysates were prepared for Co-IP with NANOG antibody and WB.
(H) DU145 cells were treated with AICAR (2 mM) for 4 hrs, cell lysates were prepared for Co-IP with NANOG antibody and WB.
(I) DU145 cells were treated with DMSO or Compound C (6.6 μM) for 4 hrs, cell lysates were prepared for Co-IP with SPOP antibody, and the associated NANOG was analyzed by WB.
(J-K) SPOP WT or KO DU145 cells were infected with lentivirus expressed His-Ub. before treated with Compound C (6.6 μM) or 2-DG (25 mM) for 4 hrs. cell lysates were IP by NANOG antibody and the ubiquitinated NANOG was analyzed by WB.
(L) Representative sphere images from each condition of DU145 cells. The SPOP WT or KO DU145 cells were maintained in DMEM supplemented with 5 mM Glucose and treated with DMSO or Compound C (3.3 μM), Scale bar, 200 μm.
(M) Frequency of tumor spheres formed from DU145 cells in (L). Sphere counts are normalized to mock infected spheres. Data are means ± SEM (n=3). **P<0.01 vs EV (Student's t-test).
(N) Representative sphere images from each condition of DU145 cells. The SPOP WT or KO DU145 cells were treated with 2-DG (25 mM), AICAR (2 mM) or Metformin (2 mM) Scale bar, 100 μm.
(O) Frequency of tumor spheres formed from DU145 cells in (N). Sphere counts are normalized to mock infected spheres. Data are means ± SEM (n=3). *P<0.05, **P<0.01 vs DMSO (Student's t-test).
(P) Representative sphere images from each condition of DU145 cells. The DU145 cells were treated with Metformin (2 mM). Scale bar, 100 μm.
(Q) Frequency of tumor spheres formed from DU145 cells in (P). Sphere counts are normalized to mock infected spheres. Data are means ± SEM (n=3). *P<0.05, **P < 0.01 vs EV (Student's t-test).
To further investigate the mechanism underlying NANOG Ser68 phosphorylation, we generated an antibody that could specifically recognize the Ser68-phosphorylated NANOG (p-S68 NANOG). Our data clearly showed that the antibody specifically targeted to Ser68-phosphorylated NANOG since it did not recognize the NANOG peptide mutated at Ser68 (Figure S5E).
AMPK attenuates the phosphorylation of NANOG at Ser68
Next, we aimed to identify the protein kinase that involves in the phosphorylation of NANOG at Ser68. Since the phosphorylation of Ser68 may affect the protein stability of NANOG, it is a reasonable assumption that the kinases involved in the phosphorylation of NANOG should affect NANOG protein stability. To this end, we generated a cell line that is stably expressing NanoLuc-NANOG fusion protein, and screened 244 kinase inhibitors by monitoring the NanoLuc activity. Our data showed that treatment of MG132 or MLN4924 efficiently increased the NanoLuc activity, indicating that this assay could identify the unique compound that affects NANOG stability (Figure S5F). Surprisingly, we found that the NanoLuc activity was significantly increased with the treatment of Compound C (Dorsomorphin), a specific inhibitor of AMPK (Figure S5F), suggesting that Compound C may have an important effect on the stability of NANOG. Furthermore, the effect of Compound C on NANOG stability was confirmed by western-blotting (Figures 5C-D and S5G-H). Importantly, we found that the regulatory role of Compound C on NANOG stability is dependent on the presence of SPOP (Figures 5C-D). To further confirm that AMPK is involved in the regulation of NANOG stability, we expressed NANOG in AMPKα1/α2 −/− MEF cells and evaluated the half-life of NANOG. Our data clearly demonstrated that the half-life of NANOG was significantly prolonged in AMPKα1/α2 −/− MEF cells compared to that of WT MEFs. However, Compound C failed to stabilize NANOG in AMPKα1/α2 −/− MEF cells (Figures 5E-F).
We therefore examined whether the activation of AMPK could promote NANOG degradation. To this end, 2-DG, a glucose molecule that can activate AMPK by increasing the cellular concentration of AMP/ATP was used (Wang et al., 2015). Defects in glucose metabolism create energy stress in cells and thereby activate AMPK kinase (Jones and Thompson, 2009). Our data showed that the half-life of NANOG was significantly reduced upon the treatment of 2-DG or withdrawal of glucose (Figures S5I-J). These data together indicate that AMPK is a negative regulator of NANOG stability.
Then we examined whether AMPK affects the phosphorylation of NANOG at Ser68. Our data showed that inhibition of AMPK with Compound C significantly increased the phosphorylation of NANOG at Ser68 (Figures 5G and S5K). On the other hand, AMPK activator AICAR reduced the phosphorylation of NANOG at Ser68 (Figures 5H and S5L). Moreover, inhibition of AMPK abrogated the interaction between SPOP and NANOG, while activation of AMPK strengthened the association between SPOP and NANOG (Figures 5I and S5M).
Next, we examined whether AMPK affects NANOG stability dependent on the presence of SPOP. Our data indicated that inhibition of AMPK by Compound C prolonged the half-life of NANOG in SPOP+/+, but not SPOP−/− DU145 cells (Figures 5C-D). Consistently, AMPK affected the ubiquitination of NANOG in SPOP+/+, but not SPOP−/− cells (Figures 5J-K and S5N). These data indicate that AMPK affects NANOG degradation and ubiquitination in a SPOP-dependent manner.
Functionally, we found that inhibition of AMPK dramatically increased the sphere-forming potential and cell proliferation in SPOP+/+, but not SPOP−/− DU145 cells (Figures 5L-M). On the other hand, the self-renewal capacity and proliferation of DU145 cells decreased upon the activation of AMPK (Figures 5N-O). Meanwhile, Metformin (AMPK activator) reduced the sphere formation of the DU145 cells expressing WT-NANOG, but not those overexpressing cancer-related NANOG S68Y mutant (Figures 5P-Q), which established the concept that AMPK regulates PCa stem cell traits in a SPOP-dependent manner. These results together suggest that AMPK affects the stemness of PCa cells via regulating NANOG phosphorylation at Ser68.
Phosphorylation of NANOG at Ser68 by BRAF
As AMPK activation reduced the phosphorylation of NANOG at Ser68, it is unlikely that AMPK is the protein kinase that directly phosphorylates NANOG. To identify the protein kinase that is directly involved in the phosphorylation of NANOG at Ser68, we searched the online website (www.phosphonet.ca/kinasepredictor.) and found that multiple kinases were potential candidates, including Casein Kinase, Raf Kinase, GSK3β and AKT. To determine which kinase is the right one, we coexpressed NANOG with these kinases, respectively, and the cells were treated with MG132 to avoid the influence from protein degradation. The phosphorylation of NANOG was examined using p-S68 antibody. Our data showed that BRAF kinase, but not other kinases listed above, specially increased the phosphorylation of NANOG at Ser68 (Figures 6A and B). In parallel, treatment with BRAF kinase inhibitor AZ628 or LY03009120 inhibited the phosphorylation of NANOG Ser68 (Figure S6A). Kinase dead mutant K483M (BRAF) failed to promote the phosphorylation of NANOG at Ser68 (Figure S6B), suggesting that the kinase activity of BRAF is required for NANOG phosphorylation. Furthermore, we found that BRAF can bind to NANOG in cells (Figure S6C). Together, these data suggest that BRAF is a protein kinase that accounts for the phosphorylation of NANOG at Ser68.
Figure 6. Phosphorylation of NANOG at Ser68 within SBC Motif by BRAF blocks SPOP-mediated destruction of NANOG.

(A) Flag-NANOG was coexpressed with the indicated kinases in HEK293T cells. The protein level of pS68-NANOG was analyzed by WB.
(B) Flag-NANOG or S68A was coexpressed with BRAF in HEK293T cells. The protein level of pS68-NANOG was analyzed by WB.
(C) Flag-NANOG was coexpressed with Flag-BRAF or K483M in HEK293T cells. After treating cells with CHX (10 μg/ml) for indicated time intervals, protein levels of NANOG and SPOP were analyzed by WB.
(D) The NANOG protein abundance in (C) was quantified by ImageJ and plotted as indicated.
(E) SPOP WT or KO DU145 cells were treated with SB590885 (10 μM) for 4 hrs before performing the CHX (10 μg/ml) chase analysis. Expression levels of NANOG and SPOP were analyzed by WB.
(F) The NANOG protein abundance in (E) was quantified by ImageJ and plotted as indicated.
(G) DU145 cells were treated with DMSO or SB590885 (10 μM) for 4 hrs, cell lysates were prepared for Co-IP with SPOP antibody, the associated NANOG was analyzed by WB.
(H) DU145 cells were transfected with control siRNA or BRAF siRNA. After 72 hrs, cells were treated with DMSO or Compound C (6.6 μM) for 4 hrs. Expression levels of NANOG were analyzed by WB.
(I) Flag-BRAFs and HA-NANOG were coexpressed in HEK293T cells. After 24 hrs, cells were treated with Metformin (2 mM) for 4 hrs. Cell lysates were prepared for Co-IP and WB. Cells were treated with MG132 (10 μM) for 6 hrs before harvesting.
(J) Flag-NANOG was coexpressed in HEK293T cells with indicated BRAF plasmids. After treating cells with CHX (10 μg/ml) for indicated time intervals, protein levels of NANOG were analyzed by WB.
(K) The NANOG protein abundance in (J) was quantified by ImageJ and plotted as indicated.
(L) Representative sphere images from each condition of DU145 cells. SPOP WT or KO DU145 cells were treated with AZ628 (2 μM) or SB590885 (2 μM). Scale bar, 200 μm.
(M) Frequency of tumor spheres formed from DU145 cells in (L). Sphere counts are normalized to mock infected spheres. Data are means ± SEM (n=3). *P<0.05,**P<0.01 vs DMSO (Student's t-test).
(N) Representative sphere images from each condition of DU145 cells. The DU145 cells were treated with SB590885 (2 μM). Scale bar, 200 μm.
(O) Frequency of tumor spheres formed from DU145 cells in (N). Sphere counts are normalized to mock infected spheres. Data are means ± SEM (n=3). *P<0.05, **P < 0.01 vs EV (Student's t-test).
Next, we examined whether BRAF affects the protein stability of NANOG. Our data showed that the expression of BRAF, but not kinase dead mutant K483M, strongly increased the protein level and prolonged the half-life of NANOG (Figures 6C-D). On the other hand, knockdown of BRAF reduced the NANOG protein level (Figure S6D). The half-life of NANOG was also shortened with the treatment of BRAF inhibitor AZ628 and SB590885 (Figures S6F-G).
Since BRAF can phosphorylate NANOG at Ser68, we examined whether BRAF affects the interaction between NANOG and SPOP. Our data showed that inhibition of BRAF increased the interaction between SPOP and NANOG (Figures 6G) and thereby promoted NANOG ubiquitination in DU145 cells (Figure S6E). Consistently, BRAF inhibitor had minor effect on the half-life of NANOG in the SPOP−/− DU145 cells (Figures 6E-F), suggesting that BRAF affects NANOG stability in a SPOP-dependent manner.
Furthermore, in vitro phosphorylation assay demonstrated that BRAF could phosphorylate NANOG at Ser68 and the phosphorylation of NANOG at Ser68 prevented the interaction between SPOP and NANOG in vitro (Figures S6H-I).
Phosphorylation of BRAF at Ser729 by AMPK blocks its interaction with NANOG
We next examined whether the effect of AMPK on NANOG phosphorylation is dependent on BRAF. Our data showed that knockdown of BRAF abolished the inhibitory effect on the protein level of NANOG exerted by AMPK signaling (Figure 6H), suggesting that AMPK may affect the NANOG stability via BRAF.
Previous report showed that AMPK phosphorylates BRAF at Ser729, disrupted the heterodimerization of BRAF with KSR1 and then prevented the paradoxical activation of MEK-ERK signaling (Shen et al., 2013). We thus examined whether AMPK affects the function of NANOG by phosphorylating BRAF. To this end, we generated a phosphorylation-mimic mutant, BRAF Ser729D, and detected its interaction with NANOG. Our data showed that mutation of Ser729 to Asp (S729D) completely blocked the interaction between NANOG and BRAF (Figure 6I), suggesting that Ser729 is essential for its interaction with NANOG. Importantly, our data showed that mutation of BRAF at Ser729 lost the ability to stabilize NANOG in cells (Figures 6J-K).
Functionally, BRAF inhibitor SB590885 impaired the sphere formation and cell proliferation of DU145 cells (Figures 6L-M and S6J). Moreover, SB590885 led to a marked decrease in the sphere formation of DU145 cells expressing WT-NANOG, but not those expressing NANOG S68Y, indicating that BRAF largely governs PCa stem cell traits via the regulation of SPOP (Figures 6N-O). Taken together, these results demonstrate that BRAF directly phosphorylates NANOG at Ser68 and increases the self-renewal capacity and cell proliferation of PCa cells by impairing the interaction between SPOP and NANOG.
Regulation of NANOG by SPOP in clinical applications
We next examined whether the regulation of NANOG by SPOP is correlated to prostate cancers clinically. To this end, we analyzed the mutations of the MATH domain of SPOP in 33 PCa samples by Sanger sequencing and found that 7 of the 33 cases harbored SPOP mutations (Figure S7A). Interestingly, in addition to the well-characterized F133V, W131G and R139K/T mutations, we identified a novel SPOP mutation, F104C, located in the MATH domain. Our data showed that the SPOP F104C mutant was also deficient in promoting NANOG degradation (Figure S7B).
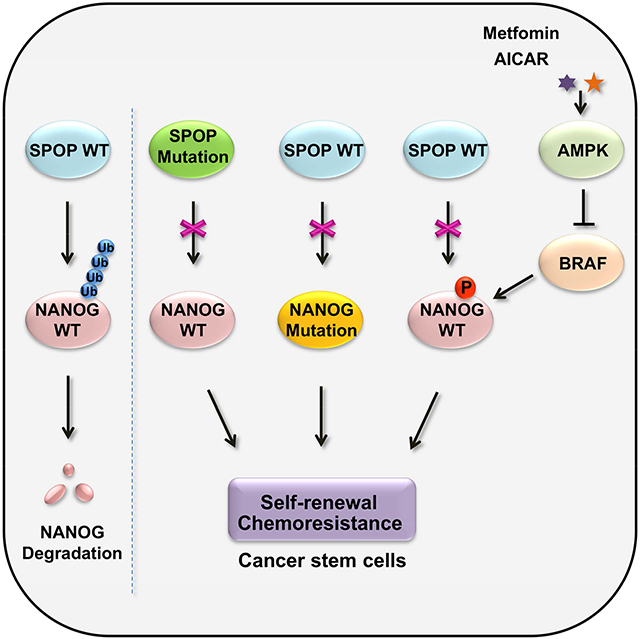
The expression of NANOG in above-mentioned samples was also analyzed by IHC. The specificity of NANOG antibody was examined by IHC assay in NANOG ectopically expressed HEK293T cells (Figure S7C). The staining of NANOG was scored as negative (0), weak (1), intermediate (2), or strong (3). We found that the protein expression of NANOG was higher in SPOP-mutated tumors than that of the SPOP-WT tumors (Figure 7A-B). Moreover, we found that the percentage of cells with high expression of NANOG in SPOP mutated tissues was significantly more than that of wild-type tissues (Figure 7C). Surprisingly, we also found that NANOG is highly expressed in 34.6% of SPOP wild-type tissues (Figure 7B). As we found that SBC is essential for SPOP-mediated NANOG degradation, we examined whether NANOG is mutated in SBC in those tissues. However, we did not find mutations of NANOG at Ser68 or other sites via Sanger sequencing analysis (Data not show). We therefore examined whether the phosphorylation of NANOG at Ser68 is correlated to the expression level of NANOG in prostate cancers. To this end, we analyzed the phosphorylation of S68 in WT-SPOP PCa tissues. The specificity of NANOG Ser68 phosphorylation antibody was examined by IHC assay in HEK293T cells expressing NANOG ectopically (Figure S7D). Interestingly, we found that the phosphorylated protein level of NANOG at Ser68 was closely correlated to the total protein level of NANOG in PCa specimens (Figures 7D-F). These data support the notion that the stability of NANOG in prostate cancers is controlled at multiple levels (Figures 7G).
Figure 7. SPOP mutation and phosphorylation of NANOG-Ser68 contributed to elevated NANOG expression in human prostate tumor specimens.

(A) Representative images of NANOG immunohistochemical staining in SPOP mutated or wild-type prostate tumor specimens, scale bar (up), 50 mm; scale bar (down), 12.5 mm.
(B) Quantification of NANOG expression levels in 26 cases of SPOP wild-type and 7 cases of SPOP-mutated prostate tumor specimens. NANOG staining was scored as negative (0), weak (1), intermediate (2) or strong (3).
(C) Percentage of high NANOG expression cells in SPOP wild-type and mutated prostate tumor specimens by counting the NANOG-high cell numbers using ImageJ.
(D) Representative images of NANOG and pS68-NANOG immunohistochemical staining in SPOP wild-type prostate tumor specimens, scale bar, 12.5 mm.
(E) Quantification of SPOP mutation and pS68-NANOG expression cases in NANOG high tumors.
(F) The correlation analysis of NANOG and pS68-NANOG protein level in prostate tumor specimens. R indicates the spearman correlation coefficient.
(G) Model for SPOP negatively regulates NANOG.
Discussion
In current study, we demonstrated that the E3 ubiquitin ligase SPOP governs prostate cancer stem cell traits by modulating NANOG stability. Clinically, cancer-associated mutations in the MATH domain of SPOP disrupt the interaction between SPOP and NANOG, thereby preventing SPOP-mediated destruction of NANOG, stabilizing NANOG from AMPK-BRAF-signaling-axis-induced phosphorylation, and consequently promoting the maintenance of PCa stem-like cells. Our results offer fresh insight into CSC formation and show that SPOP, as a tumor suppressor, promotes NANOG ubiquitination and degradation in PCa cells and thus regulates prostate cancer progression.
AMPK, a master metabolic regulator, has emerged as a potential therapeutic target for various cancer treatments, such as breast cancer and prostate cancers. AMPK activators such as AICAR and Metformin have been shown to inhibit prostate cancer cell proliferation (Huang et al., 2008, Zakikhani et al., 2008). A direct activator of AMPK inhibits prostate cancer cell growth in the androgen-sensitive and castration-resistant PCa (CRPC) models (Zadra et al., 2014). Metformin works together with chemotherapy to block tumor growth and prolong remission via targeting cancer stem cells in breast cancer cell lines (Hirsch et al., 2009). However, the mechanism by which Metformin targets cancer stem cells remains unknown. In this study, we found that activation of AMPK attenuated NANOG phosphorylation at Ser68 and thus destabilized NANOG. Thus, our study suggests that AMPK activation may function as a novel therapeutic strategy for PCa treatments since it specifically targets at CSCs and thereby offering a very promising way to eliminate the tumor from root.
BRAF is an oncogenic protein and its gain-of-function mutation has been demonstrated to be tightly correlated with cancer progression in various cancers, such as colon cancer and melanoma (Makrodouli et al., 2011, Lu et al., 2016). Gao’s study showed that activation of B-Raf/Erk pathway can act synergistically to promote androgen independence in the context of the prostate microenvironment in vivo (Gao et al., 2006). However, Raf signaling in prostate cancer stem cells is not well understood yet. In our study, we found that BRAF can phosphorylate and stabilize NANOG to promote PCa stem cell-like traits. Although low frequency of BRAF mutation was reported in prostate cancers, our data showed that regulation of NANOG by BRAF is controlled by AMPK-mediated BRAF phosphorylation at Ser729. To further examine the role of Raf in prostate cancers, we analyzed human epidemiological data using the PrognoScan database and found that higher BRAF expression predicted worse patient survival in human prostate cancer (data not show). Together, our study unveiled a novel mechanism by which BRAF regulates cancer stem cell activity via controlling NANOG stability.
As an essential E3 ubiquitin ligase, the role of SPOP in the maintenance of stem cell properties remains unknown. Our study presents the evidence showing that SPOP controls PCa stemness by degrading NANOG. We also found that overexpression of SPOP in mESCs results in the differentiation of mESCs. Our study discovered an unexplored area in regulating CSC characteristics, which not only contributes to a better understanding of tumorigenesis, but also shed light on a novel strategy of anti-tumor therapeutics targeting at a thorough elimination of the “seeds”, CSCs.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHAREING
Further information and requests for reagents may be directed to, and will be fulfilled by the Lead Contact, Dr. Ping Wang (wangp@tongji.edu.cn).
EXPERIMETNTAL MODEL AND SUBJECT DETAILS
Cell culture
PC3 (male), 22RV1 (male), LNCaP (male) cells were cultured in RPMI 1640 medium supplemented with 10% FBS. DU145 (male), SPOP WT and KO DU145 (male), AMPKα1/α2 WT and KO MEFs (a kind gift from Dr.Jia Li, National Center for Drug Screening, China) and human embryonic kidney 293T (from Cell Bank, Chinese Academy of Sciences) cells were cultured in DMEM medium supplemented with 10% heat-inactivated fetal bovine serum. E14 mESCs (a kind gift from Dr.Xin Xie, National Center for Drug Screening, China) were maintained on feeder-free gelatin-coated plastics cultured by mES medium (DMEM with 15% FBS (Gibco 10099), 2mM GlutaMAX, 0.1 mM non-essential amino acids (NEAA), 0.1 mM β-mercaptoethanol, 100 U/ml penicillin and 100 μg/ml streptomycin) supplemented with 1000 U/ml LIF and passaged every 3 days. Cells were cultured at 37°C supplied with 5% CO2. Transfection was performed by using calcium phosphate-DNA coprecipitation for HEK293T cells and using Lipofectamine 2000 for DU145, LNCaP, PC3 and E14 cells.
In vivo xenograft assay
All treatments were administered according to the guidelines of Institution Animal Care and Use Committee and all the protocols were approved by East China Normal University and Tongji University. Mice were caged in the groups of five in a laminar airflow cabinet under specific pathogen-free conditions, fed with sterilized food and water, and kept on a 12 hr light/dark cycle. Serial limiting dilution DU145 (AR−) or 22RV1 (AR+) cells were admixed with Matrigel and injected into the backside of male 6-week-old BALB/cA nude mice. The animals were checked two to three times per week. Mean of primary, secondary and tertiary tumor volumes at 5 weeks.
Human prostate tumor tissue and IHC staining
This study was approved by the ethical committee of Fudan University Shanghai Cancer Center (FUSCC), and each patient signed the consent form before participation (No.050432-4-1212B). Human prostate cancer tumor tissue arrays were provided by FUSCC. The arrays were stained by using the Histostain-plus IHC Kit (Miao Tong Biological Science and Technology Co.Ltd, Shanghai, China) as described previously (Liao et al., 2014). Briefly, the tissue sections were deparaffinized and incubated in citrate buffer at 95°C for 40 mins for antigen retrieval and then incubated overnight at 4°C with the primary antibodies. After three times washing, tissue sections were incubated with biotinylated anti-rabbit IgG for 1 hr at room temperature and then washed three times, after which streptavidin–horseradish peroxidase conjugates were added and the slides were incubated for 45 mins. After three washes with PBS, DAB solution was added and the slides were counterstained with haematoxylin. The stained slides were examined under a microscope, and images were acquired. Staining intensity was scored in a blinded manner.
METHOD DETAILS
Alkaline Phosphatase (AP) staining
Alkaline Phosphatase staining was performed using a leukocyte AP kit (Sigma, catalog No 85L3R) according to the manufacturer's protocol. Briefly, the cultured cells were fixed with 4% paraformaldehyde (PFA) in PBS for 30 s and rinsed three times with PBS, then the cells were incubated at room temperature in the solution containing naphthol AS-BI phosphate and fresh fast red violet LB salt for 15 mins.
In Vitro phosphorylation assay
Flag-BRAF was expressed in HEK293T cells and purified using M2 beads for immunoprecipitation and eluted using Flag peptide, purified Flag-BRAF was incubated with 0.5 μg His-NANOG in 50 μl of kinase buffer (25mM Tris-HCl (pH 7.5), 5 mM glycerophosphate, 10 mM MgCl2, 1mM DTT and 0.1 mM Na3VO4) containing 100 μM ATP at 30°C for 1 hr. The reaction was terminated by the addition of 17 μl of 4XLaemmli SDS sample buffer. The reaction mixtures were boiled and separated by SDS–PAGE and phosphorylation NANOG was immunobloted by anti-pSer68-NANOG antibody.
Sphere formation assay
Oncosphere assays were performed as previously described (Liu et al., 2007). Oncospheres were enriched from DU145 cells. Single-cell suspension of DU145 and PC3 cells (200 cells per well, repeat 10 holes in each group) were plated on 96-well ultra-low Attachment Plates (Corning Incorporated, catalog number: 3474) and cultured in Dulbecco’s Modified Eagle’s Medium/F12 (Gibco) supplemented with 5 μg/ml insulin (Sigma), 20 ng/ml EGF (Sigma), 1:50 B27 (Gibco), 10 ng/ml bFGF, and 0.4% BSA for 10 days alone. Floating spheres that grew in 2 weeks were counted. Tumor spheres were visualized under phase contrast microscope, photographed and counted and represented graphically. Tumor spheres larger than 100 μm were counted, and the “relative % of sphere” represent the relative increase number of sphere per dish. Spheres were digested with trypsin 0.05% EDTA and filtered through a 40 mm filter.
RNA interference
Non-specific control siRNA and siRNAs for SPOP, Cullins and BRAF were purchased from GenePharma. siRNA transfection of cells was performed according to the manufacturer’s instructions. Briefly, siRNA and Lipofectamine® 2000 reagent were mixed and incubated in serum-free 1640 medium for 15 mins at room temperature, respectively, and then added to cells with 70% confluence. Virus was produced by transfected 293T cells, by harvesting the viral supernatant 48-72 hrs after transfection, passed through 0.45 μm filter, diluted 2:3 with fresh medium containing 2 μg/ml polybrene and used to infect the target cells at 70% confluence. shRNA target sequences:
SPOP shRNA1: CCAAGGGAGAAGAAACCAA,
SPOP shRNA2: CAACUAUCAUGCUUCGGAU,
SPOP shRNA (3-UTR): CUCCGUUUAAUUUCCAGAATT,
NANOG shRNA: UGAUUGUUCCAGGAUUGGGUG,
Scramble shRNA: TTCTCCGAACGTGTCACGTTT.
Generation of the SPOP KO cell lines
SPOP knockout cell lines were generated using lentiCRISPR methods (Shalem et al., 2014). Briefly, guide RNA (sgRNA) was constructed into the lentiviral expression vector for Cas9 and sgRNA (lentiCRISPR). The lentiCRISPR vector was linearized using BsmBI. The sequence of sgRNA is:
sgRNA SPOP-1# : CACCGGGTTCCAAGTCCTCCACCTC,
sgRNA SPOP-2# : CACCGGGGAGGATTTGGTGGACGAA.
Real-Time Quantitative PCR
Total RNAs were extracted with TriZol (sigma) from various cells by RNA preparation kit as indicated. After their quantification using a Nanodrop 1000 spectrophotometer (Thermo Scientific), one microgram of total RNA was reverse-transcribed into complementary DNA using the ReverTra Ace qPCR RT Kit (TOYOBO). Quantitative PCR was performed with SYBR Green Realtime PCR Master Mix (TOYOBO) and quantified by the Step One Real-Time PCR System (Applied Biosystems). Primers for real-time PCR are shown in KEY RESOURCES TABLE.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Flag | Sigma | Cat#F3165 RRID:AB_259529 |
| Anti-HA | Santa Cruz | Cat#sc-7392 RRID:AB_627809 |
| Anti-GFP | Santa Cruz | Cat#sc-81045 RRID:AB_1123764 |
| Anti-KLF4 | Santa Cruz | Cat#sc-20691 RRID:AB_669567 |
| Anti-Tubulin | Proteintech | Cat#11224-1-AP RRID:AB_2210206 |
| Anti-Actin | Proteintech | Cat#60008-1-Ig RRID:AB_2289225 |
| Anti-SPOP | Proteintech | Cat#16750-1-AP |
| Anti-OCT4 | Abcam | Cat#ab18976 RRID:AB_444714 |
| Anti-GST | Abcam | Cat#ab19256 RRID:AB_444809 |
| Anti-phospho-BRAF (Ser729) | Abcam | Cat#ab124794 RRID:AB_10976055 |
| Anti-Cul1 | Epitomics | Cat#2436-1 RRID:AB_1267087 |
| Anti-Cul3 | Epitomics | Cat#2506-1 RRID:AB_1267090 |
| Anti-Cul4A/B | Epitomics | Cat#2527-1 RRID:AB_1267096 |
| Anti-SOX2 | Epitomics | Cat#2683-1 RRID:AB_2302581 |
| Anti-NANOG | Millipore | Cat#AB5731 RRID:AB_2267042 |
| Anti-ERK | Cell Signaling Technology | Cat#4695 RRID:AB_390779 |
| Anti-p-ERK | Cell Signaling Technology | Cat#4370 RRID:AB_2315112 |
| Anti-AMPKα | Cell Signaling Technology | Cat#5831 RRID:AB_10622186 |
| Anti-phospho-ACC (Ser79) | Cell Signaling Technology | Cat#3661 RRID:AB_330337 |
| anti-BRAF | Cell Signaling Technology | Cat#9433 RRID:AB_2259354 |
| Rabbit polyclonal antibody against phospho-Ser68-NANOG | In our lab | N/A |
| Bacterial and Virus Strains | ||
| DH5α | TIANGEN | Cat#CB101 |
| BL21 | TIANGEN | Cat#CB105 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MG132 | Sigma | Cat#C2211 |
| MLN4924 | Synthesized by GL Biochem (Shanghai) Ltd | N/A |
| NI-NTA agarose | QIAGEN | Cat#30310 |
| 2-DG | Sigma | Cat#D8375 |
| cycloheximide | Sigma | Cat#C7698 |
| Compound C | Selleck | Cat#S7306 |
| AZ628 | Selleck | Cat#S2746 |
| LY03009120 | Selleck | Cat#S7842 |
| LIF | Millipore | Cat#LIF1010 |
| AICAR | Dr.Jia Li, National Center for Drug Screening, China | N/A |
| SB590885 | Dr.Jia Li, National Center for Drug Screening, China | N/A |
| Metformin | Dr.Jia Li, National Center for Drug Screening, China | N/A |
| Protein Kinase Inhibitor Library | Dr.Jia Li, National Center for Drug Screening, China | N/A |
| GlutaMAX | Gibco | Cat#35050061 |
| NEAA (non-essential amino acids) | Gibco | Cat#11140050 |
| Penicillin-streptomycin | Gibco | Cat#15070063 |
| β-mercaptoethanol | Gibco | Cat#21985023 |
| GST recombinant proteins | In our lab | N/A |
| GST-SPOP | In our lab | N/A |
| GST-SPOPΔBTB | In our lab | N/A |
| GST-SPOPΔMATH | In our lab | N/A |
| His-NANOG recombinant proteins | In our lab | N/A |
| His-NANOGΔPDSST | In our lab | N/A |
| Experimental Models: Cell Lines | ||
| HEK293T | Cell Bank, Chinese Academy of Sciences | N/A |
| DU145 | Cell Bank, Chinese Academy of Sciences | N/A |
| 22RV1 | Cell Bank, Chinese Academy of Sciences | N/A |
| LNCaP | Cell Bank, Chinese Academy of Sciences | N/A |
| PC3 | Stem Cell Bank, Chinese Academy of Sciences. | N/A |
| E14 mESCs | Dr.Xin Xie, National Center for Drug Screening, China | N/A |
| AMPKα1/α2 WT and KO MEFs | Dr.Jia Li, National Center for Drug Screening, China | N/A |
| SPOP WT and KO cell lines | In our lab | N/A |
| Experimental Models: Organisms/Strains | ||
| BALB/cA nude mice | ShanghaiSippr-BK laboratory animal Co. Ltd. | N/A |
| Oligonucleotides | ||
| Spop | F:ACCCTCTGCAGTAACCTGTC; R:GTCTCCAAGACATCCGAAGC |
N/A |
| Yap1 | F:AGATGGAGAAGGAGAGGCTG; R:AGTGTTGGTAACTGGCTACG |
N/A |
| Adcy10 | F:TCTACCACCTGATGGCTTAC; R:TTACCCTGCCTGCTACAA |
N/A |
| Igfbp2 | F:TCTGGAGCACCTCTACTCCCTG; R:GTCTACTGCATCCGCTGGGT |
N/A |
| Acad8 | F:CTCCAGGGTCCACCAGAT; R:GAATAGAGCGGCCCACAT |
N/A |
| Trpm8 | F:AAAGCCAACGACACCTCA; R:AAACAGGCACCATTCTGG |
N/A |
| Birc5 | F:AGCATTCGTCCGGTTGCGCT; R: TCGATGGCACGGCGCACTTT |
N/A |
| Pfkfb1 | F:TGAATGTGGAGGCCGTGAA; R:AAGGCAGAGTAGGAGAAGAGCA |
N/A |
| Arx | F:CCGACACCCAGCTTTCATC; R:TGGAGGGCAGCCTTTAGC |
N/A |
| siCul1 | UAGACAUUGGGUUCGCCGUTT | N/A |
| siCul2 | CCCUUGGAGAAAGACUUUAUA | N/A |
| siCul4B | AAGGTGCGAGAAGATGTA | N/A |
| siCul3–1 | AAGGUGCGAGAAGAUGUATT | N/A |
| siCul3–2 | AACAACUUUCUUCAAACGCUA | N/A |
| siCul3–3 | AACAACACUUGGCAAGGAGAC | N/A |
| siSPOP-1 | CAACUAUCAUGCUUCGGAU | N/A |
| siSPOP-2 | GGUAAAGGUUCCUGAGUGC | N/A |
| siSPOP-3 | AAAUGGUGUUUGCGAGUAA | N/A |
| siSPOP-4 | UAGAACUUUUAAUGACUUCAC | N/A |
| siBRAF-1 | GGUGUGGAAUAUCAAACAATT | N/A |
| siBRAF-2 | GGCUCACUAACUAACGUGATT | N/A |
| siBRAF-3 | AAACGUUUUUCGUACAAAGUU | N/A |
| siBRAF-4 | AUUCAUACAGAACAAUUCCAA | N/A |
| Recombinant DNA | ||
| pcDNA3.1-HA-NANOG | In our lab | N/A |
| pcDNA3.1-Flag-dnCul1 | In our lab | N/A |
| pcDNA3.1-Flag-dnCul2 | In our lab | N/A |
| pcDNA3.1-Flag-dnCul3 | In our lab | N/A |
| pcDNA3.1-Flag-dnCul4A | In our lab | N/A |
| pcDNA3.1-Flag-dnCul4B | In our lab | N/A |
| pcDNA3.1-Flag-GFP | In our lab | N/A |
| pcDNA3.1-HA-SPOP | In our lab | N/A |
| pcDNA3.1-Flag-NANOG | In our lab | N/A |
| pcDNA3.1-His-ubiquitin | In our lab | N/A |
| pcDNA3.1-HA-OCT4 | In our lab | N/A |
| pcDNA3.1-HA-SOX2 | In our lab | N/A |
| pcDNA3.1-HA-KLF4 | In our lab | N/A |
| pcDNA3.1-Flag-SPOP | In our lab | N/A |
| pcDNA3.1-Flag-SPOPΔBTB | In our lab | N/A |
| pcDNA3.1-Flag-SPOPΔMATH | In our lab | N/A |
| pcDNA3.1-HA-SPOPΔBTB | In our lab | N/A |
| pcDNA3.1-HA-SPOPΔMATH | In our lab | N/A |
| pcDNA3.1-HA-SPOP-Y87C | In our lab | N/A |
| pcDNA3.1-HA-SPOP-Y87N | In our lab | N/A |
| pcDNA3.1-HA-SPOP-F102C | In our lab | N/A |
| pcDNA3.1-HA-SPOP-F133V | In our lab | N/A |
| pcDNA3.1-HA-SPOP-F133L | In our lab | N/A |
| Lenti-pWPI-Flag-SPOP | In our lab | N/A |
| Lenti-pWPI-Flag-SPOP-F133L | In our lab | N/A |
| Lenti-pWPI-Flag-SPOP-Y87C | In our lab | N/A |
| Lenti-pWPI-Flag-NANOG | In our lab | N/A |
| pcDNA3.1-HA-NANOG-ΔPDSST | In our lab | N/A |
| pcDNA3.1-HA-NANOG-3A | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-ΔPDSST | In our lab | N/A |
| pGEX-4T-2-GST-SPOP | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-S65A | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-P66A | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-D67A | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-S68A | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-S69A | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-T70A | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-S71A | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-P72A | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-S68Y | In our lab | N/A |
| Lenti-pWPI-Flag-NANOG-S68Y | In our lab | N/A |
| pcDNA3.1-Flag-NANOG-S68D | In our lab | N/A |
| NANOG reporter plasmid | Dr. Duanqing Pei, Guangzhou Institutes of Biomedicine and Health | N/A |
| pcDNA3.1-HA-CK1α | In our lab | N/A |
| pcDNA3.1-HA-CK1ε | In our lab | N/A |
| pcDNA3.1-HA-GSK3β | In our lab | N/A |
| pcDNA3.1-HA-AKT | In our lab | N/A |
| pcDNA3.1-HA-ARAF | In our lab | N/A |
| pcDNA3.1-HA-BRAF | In our lab | N/A |
| pcDNA3.1-HA-CRAF | In our lab | N/A |
| pcDNA3.1-Flag-BRAF | In our lab | N/A |
| pcDNA3.1-Flag-BRAF-K483M | In our lab | N/A |
| pcDNA3.1-Flag-BRAF-S729D | In our lab | N/A |
| pcDNA3.1-Flag-BRAF-V600E | In our lab | N/A |
| pcDNA3.1-HA-SPOP-F104C | In our lab | N/A |
| pET28a-His-NANOG | In our lab | N/A |
| Retro-Migr-Venus-Mir30-shSPOP | In our lab | N/A |
| Retro-Migr-Venus-Mir30-shNANOG | In our lab | N/A |
| pGEX-4T-2-GST-SPOP-ΔBTB | In our lab | N/A |
| pGEX-4T-2-GST-SPOP-ΔMATH | In our lab | N/A |
| pcDNA3.1-Flag-KCTD2 | In our lab | N/A |
| pcDNA3.1-Flag-KCTD5 | In our lab | N/A |
| pcDNA3.1-Flag-BACURD2 | In our lab | N/A |
| pcDNA3.1-Flag-KLHL9 | In our lab | N/A |
| pcDNA3.1-Flag-KLHL13 | In our lab | N/A |
| pcDNA3.1-Flag-KLHL15 | In our lab | N/A |
| pcDNA3.1-Flag-KLHL17 | In our lab | N/A |
| pcDNA3.1-Flag-KLHL22 | In our lab | N/A |
| pcDNA3.1-Flag-KLHL24 | In our lab | N/A |
| Lenti-pWPI-Flag-NANOG-ΔPDSST | In our lab | N/A |
Immunoprecipitation (IP) and Western blot
IP and Western blot were conducted as previously described (Liu et al., 2010a). Transfected HEK293T cells were lysed in CoIP lysis buffer (50 mM Tris-cl, pH 7.4, 0.5% NP-40, 150 mM NaCl, 1 mM EDTA, 10% glycerophosphate, and a cocktail of proteinase inhibitors). After lysis for 30 mins, the soluble fraction of the cell lysates was isolated via centrifugation at 12,000 rpm in a microcentrifuge for 15 mins at 4°C. For IP, the cell lysates were centrifuged to remove the cell debris and were incubated in HA-conjugated beads (Abmart) or M2 beads (Sigma) for 2-3 hrs. Endogenous SPOP was immunoprecipitated using an anti-SPOP polyclonal antibody. The beads were boiled after extensive washing; The proteins were boiled, resolved via SDS-PAGE gel electrophoreses, and analyzed via immunoblotting. The proteins were detected using the Odyssey system (LI-COR Biosciences).
Immunofluorescence
Cells were seeded on fibronectin-coated glass coverslips in 24-well tissue culture plates. After 24 hrs, the cells were rinsed once with PBS and fixed in 4% paraformaldehyde for 15 mins at room temperature. The fixed cells were permeabilized using 0.1% Triton X-100 and rinsed twice with PBS. The coverslips were blocked with blocking buffer for 1 hr (0.3% BSA in PBS) and incubated in a primary antibody in blocking buffer overnight at 4°C. Next, the coverslips were rinsed twice with blocking buffer and incubated in secondary antibodies for 1 hr at room temperature in the dark, followed by tyramide signal amplification. The glass coverslips were mounted using Mowiol and were examined using a Zeiss LSM 510 Meta confocal system.
Biotinylated peptides pull down assay
The biotinylated peptides were synthesized by GL Biochem (Shanghai) Ltd and dissolved in PBS solution. Briefly, 2 μg of peptides was preloaded onto 10 μl of Neutravidin agarose (Thermo). Then, 293T cells were transfected with HA-SPOP. 24 hrs post-transfection, cells were collected, lysed, and the lysates were incubated with the peptide-preloaded agarose for 3 hrs at 4°C in binding buffer (20 mM HEPES (pH7.9), 150 mM KCl, 1 mM DTT, 1 mM PMSF, 10% glycerol, 0.1% NP-40 and proteinase inhibitors). After five washes with washing buffer (20 mM HEPES (pH7.9), 150 mM KCl, 1 mM DTT, 1 mM PMSF, 0.1% NP-40, and proteinase inhibitors), the bound proteins were eluted by boiling in 2XLaemmli SDS loading buffer, resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and subjected to nitrocellulose membranes (Millipore), followed by western blotting analysis.
GST pull-down assay
The His-NANOG protein was purified from E.coli and incubated in 10 μg of purified GST or GST-SPOP protein. The GST proteins were purified using glutathione sepharose 4B, and the bound His-NANOG was detected via Western blotting.
Ubiquitination assay
For the in vivo ubiquitination assay using Ni-NTA beads, the cells were transfected with His-ubiquitin. Then, the transfected cells were lysed using denaturing Buffer A (6 M guanidine-HCl, 0.1 M Na2HPO4/NaH2PO4, 10 mM imidazole, pH 8.0), and the ubiquitinated proteins were purified using Ni-NTA beads. The beads were then washed sequentially with Buffer A, Buffer B (8 M urea, 0.1 M, Na2HPO4/NaH2PO4, pH 8.0, 0.01 M Tris-HCl, pH 8.0, 10 mM β-mercaptoethanol), Buffer C (8 M urea, 0.1 M Na2HPO4/NaH2PO4, pH 6.3, 0.01 M Tris-HCl, pH 6.3, 10 mM β-mercaptoethanol) containing 0.2% Triton X-100 and Buffer C. The washed beads were incubated in 40 μl of elution buffer (200 mM imidazole, 0.15 M Tris-HCl, pH 6.7, 30% glycerol, 5% SDS, 0.72 M β-mercaptoethanol) at room temperature for 30 mins. The input fractions and eluates were analyzed via western blotting. For the in vitro ubiquitination assay, Flag-Cul3, HA-Rbx1 and HA-SPOP-WT or ΔBTB were co-transfected in 293T cells and purified by anti-HA beads. The precipitated protein complexes were incubated with 250 ng E1, 200 ng E2 (UbcH5a and UbcH3), 5 μg ubiquitin (Ub), His-NANOG, 0.2 μl 100 mM ATP, 1 μl 10 x ubiquitin reaction buffer (500 mM Tris-HCl (pH7.5), 50 mM KCl, 50 mM NaF, 50 mM MgCl2 and 5 mM DTT) at 37°C for 2 hrs, followed by NANOG immunoprecipitated for 2 hrs at 4°C. The beads were washed four times with the lysis buffer and then boiled in 30 μl 2XLaemmli SDS loading buffer for western blotting.
ELISA assay
To test the specificity of pS68-NANOG antibody, the peptides (10 ng/μl) were coated to ELISA plate overnight at 4°C, Plates were washed followed by block at room temperature for 1 hr. After wash and incubated with the pS68-NANOG antibody and then Avidin-HRP*, the TMB solution was added to each well after wash, read plate at 450nm after Stop solution was added.
NANOG Peptide WT: DLLIQDSPDSSTSPKGKQP,
NANOG Peptide Ser68(P): DLLIQDSPD(p)SSTSPKGKQP,
NANOG Peptide S68A: DLLIQDSPDASTSPKGKQP.
Genome DNA extraction and SPOP sequencing
Genomic DNA from prostate tumor specimens was isolated using a QIAamp DNA Mini Kit (50) (Qiagen) and subjected to an initial pre-PCR amplification step to enrich the SPOP exons 6, 7 and 8. Then SPOP mutation status was determined by Sanger sequencing. SPOP primers (Exons 6, 7 and 8) were used for amplification and sequencing:
SPOP-6F 5’-CCTATTTAATTGCTTCCTG-3’,
SPOP-6R 5’-ACAGTTAGACGTATTCTTCC-3’,
SPOP-7F 5’-GTCTGATTTCCAGTTCTATC-3’,
SPOP-7R 5’-ATGAACTTCTGGATGTGAAACTT-3’,
SPOP-8F 5’-CCAGAAGTTTCATACACTGACAA-3’,
SPOP-8R 5’-CTACCAATACTCATCAGATCT-3’.
TCGA data analysis
Level 3 data for mRNA expression from TCGA were downloaded and processed using standard methods. Gene expression was analyzed using two-class unpaired significance analysis of microarrays (SAM) (http://statweb.stanford.edu/~tibs/SAM/) for the indicated tumors versus normal samples. Differences in expression were considered to be statistically significant when the fold change>2 and q<0.05.
Structure modeling
Homology modeling was used to build a structural model of the MATH domain of SPOP with the NANOG peptide (aa 66–70; PDSST). The structure of a SBC peptide from phosphatase Puc (PucSBC1) in a complex with the MATH domain of SPOP (PDB entry 3IVV) (Zhuang et al., 2009) was used as a template. The homology modeling was performed in PyMOL (The PyMOL Molecular Graphics System; Version 1.5.0.3).
Statistical analysis
Statistical analyses were performed with a two-tailed unpaired Student’s t-test. The data are presented as the means ± SEM. The mean was calculated from truly independent experiments. P values < 0.05 were considered statistically significant.
Supplementary Material
Highlights:
SPOP targets NANOG for degradation
Cancer-associated SPOP mutants lost the ability to degrade NANOG
NANOG stability is regulated by AMPK-BRAF-mediated phosphorylation
SPOP represses prostate cancer stemness via targeting NANOG
Acknowledgements
We thank Dr. Duanqing Pei and Chengji Wang for kindly providing reagents. We thank Dr. Dianqing Wu and Filippo G. Giancotti for critical reading of the manuscript. This work was supported by grants from the National Natural Science Foundation of China to P.W. (81625019), G.X. (81502559 & 81874198), X.J. (31771573), F.L. (31501051), DW.Y (81472377), FN.W (81502192) and Shanghai Sailing Program to J.J.L. (18YF1419300).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no conflict of interests.
References
- AN J, REN S, MURPHY SJ, DALANGOOD S, CHANG C, PANG X, CUI Y, WANG L, PAN Y, ZHANG X, ZHU Y, WANG C, HALLING GC, CHENG L, SUKOV WR, KARNES RJ, VASMATZIS G, ZHANG Q, ZHANG J, CHEVILLE JC, YAN J, SUN Y & HUANG H 2015. Truncated ERG Oncoproteins from TMPRSS2-ERG Fusions Are Resistant to SPOP-Mediated Proteasome Degradation. Mol Cell, 59, 904–16. [DOI] [PubMed] [Google Scholar]
- AN J, WANG C, DENG Y, YU L & HUANG H 2014. Destruction of full-length androgen receptor by wild-type SPOP, but not prostate-cancer-associated mutants. Cell Rep, 6, 657–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARBIERI CE, BACA SC, LAWRENCE MS, DEMICHELIS F, BLATTNER M, THEURILLAT JP, WHITE TA, STOJANOV P, VAN ALLEN E, STRANSKY N, NICKERSON E, CHAE SS, BOYSEN G, AUCLAIR D, ONOFRIO RC, PARK K, KITABAYASHI N, MACDONALD TY, SHEIKH K, VUONG T, GUIDUCCI C, CIBULSKIS K, SIVACHENKO A, CARTER SL, SAKSENA G, VOET D, HUSSAIN WM, RAMOS AH, WINCKLER W, REDMAN MC, ARDLIE K, TEWARI AK, MOSQUERA JM, RUPP N, WILD PJ, MOCH H, MORRISSEY C, NELSON PS, KANTOFF PW, GABRIEL SB, GOLUB TR, MEYERSON M, LANDER ES, GETZ G, RUBIN MA & GARRAWAY LA 2012. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat Genet, 44, 685–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLATTNER M, LEE DJ, O'REILLY C, PARK K, MACDONALD TY, KHANI F, TURNER KR, CHIU YL, WILD PJ, DOLGALEV I, HEGUY A, SBONER A, RAMAZANGOLU S, HIERONYMUS H, SAWYERS C, TEWARI AK, MOCH H, YOON GS, KNOWN YC, ANDREN O, FALL K, DEMICHELIS F, MOSQUERA JM, ROBINSON BD, BARBIERI CE & RUBIN MA 2014. SPOP mutations in prostate cancer across demographically diverse patient cohorts. Neoplasia, 16, 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOUMAHDI S, DRIESSENS G, LAPOUGE G, RORIVE S, NASSAR D, LE MERCIER M, DELATTE B, CAAUWE A, LENGLEZ S, NKUSI E, BROHEE S, SALMON I, DUBOIS C, DEL MARMOL V, FUKS F, BECK B & BLANPAIN C 2014. SOX2 controls tumour initiation and cancer stem-cell functions in squamous-cell carcinoma. Nature, 511, 246–50. [DOI] [PubMed] [Google Scholar]
- BRUMBAUGH J, RUSSELL JD, YU P, WESTPHALL MS, COON JJ & THOMSON JA 2014. NANOG is multiply phosphorylated and directly modified by ERK2 and CDK1 in vitro. Stem Cell Reports, 2, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUCKLEY SM, ARANDA-ORGILLES B, STRIKOUDIS A, APOSTOLOU E, LOIZOU E, MORAN-CRUSIO K, FARNSWORTH CL, KOLLER AA, DASGUPTA R, SILVA JC, STADTFELD M, HOCHEDLINGER K, CHEN EI & AIFANTIS I 2012. Regulation of pluripotency and cellular reprogramming by the ubiquitin-proteasome system. Cell Stem Cell, 11, 783–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CANCER GENOME ATLAS RESEARCH, N. 2015. The Molecular Taxonomy of Primary Prostate Cancer. Cell, 163, 1011–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHAMBERS I, COLBY D, ROBERTSON M, NICHOLS J, LEE S, TWEEDIE S & SMITH A 2003. Functional expression cloning of Nanog, a pluripotency sustaining factor in embryonic stem cells. Cell, 113, 643–55. [DOI] [PubMed] [Google Scholar]
- CHAMBERS I & TOMLINSON SR 2009. The transcriptional foundation of pluripotency. Development, 136, 2311–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN CL, UTHAYA KUMAR DB, PUNJ V, XU J, SHER L, TAHARA SM, HESS S & MACHIDA K 2016. NANOG Metabolically Reprograms Tumor-Initiating Stem-like Cells through Tumorigenic Changes in Oxidative Phosphorylation and Fatty Acid Metabolism. Cell Metab, 23, 206–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAI X, GAN W, LI X, WANG S, ZHANG W, HUANG L, LIU S & ZHONG Q 2017. Prostate cancer-associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. 23, 1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DONTU G, JACKSON KW, MCNICHOLAS E, KAWAMURA MJ, ABDALLAH WM & WICHA MS 2004. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. Breast Cancer Res, 6, R605–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAN W, DAI X, LUNARDI A, LI Z, INUZUKA H, LIU P, VARMEH S, ZHANG J, CHENG L, SUN Y, ASARA JM, BECK AH, HUANG J, PANDOLFI PP & WEI W 2015. SPOP Promotes Ubiquitination and Degradation of the ERG Oncoprotein to Suppress Prostate Cancer Progression. Mol Cell, 59, 917–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAO H, OUYANG X, BANACH-PETROSKY WA, GERALD WL, SHEN MM & ABATE-SHEN C 2006. Combinatorial activities of Akt and B-Raf/Erk signaling in a mouse model of androgen-independent prostate cancer. Proc Natl Acad Sci U S A, 103, 14477–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GENG C, RAJAPAKSHE K, SHAH SS, SHOU J, EEDUNURI VK, FOLEY C, FISKUS W, RAJENDRAN M, CHEW SA, ZIMMERMANN M, BOND R, HE B, COARFA C & MITSIADES N 2014. Androgen receptor is the key transcriptional mediator of the tumor suppressor SPOP in prostate cancer. Cancer Res, 74, 5631–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRSCH HA, ILIOPOULOS D, TSICHLIS PN & STRUHL K 2009. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res, 69, 7507–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG X, WULLSCHLEGER S, SHPIRO N, MCGUIRE VA, SAKAMOTO K, WOODS YL, MCBURNIE W, FLEMING S & ALESSI DR 2008. Important role of the LKB1-AMPK pathway in suppressing tumorigenesis in PTEN-deficient mice. Biochem J, 412, 211–21. [DOI] [PubMed] [Google Scholar]
- IV SANTALIZ-RUIZ LE, XIE X, OLD M, TEKNOS TN & PAN Q 2014. Emerging role of nanog in tumorigenesis and cancer stem cells. Int J Cancer, 135, 2741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JANOUSKOVA H, EL TEKLE G, BELLINI E, UDESHI ND, RINALDI A, ULBRICHT A, BERNASOCCHI T, CIVENNI G, LOSA M, SVINKINA T, BIELSKI CM, KRYUKOV GV & CASCIONE L 2017. Opposing effects of cancer-type-specific SPOP mutants on BET protein degradation and sensitivity to BET inhibitors. 23, 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JETER CR, BADEAUX M, CHOY G, CHANDRA D, PATRAWALA L, LIU C, CALHOUN-DAVIS T, ZAEHRES H, DALEY GQ & TANG DG 2009. Functional evidence that the self-renewal gene NANOG regulates human tumor development. Stem Cells, 27, 993–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JETER CR, LIU B, LIU X, CHEN X, LIU C, CALHOUN-DAVIS T, REPASS J, ZAEHRES H, SHEN JJ & TANG DG 2011. NANOG promotes cancer stem cell characteristics and prostate cancer resistance to androgen deprivation. Oncogene, 30, 3833–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONES RG & THOMPSON CB 2009. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev, 23, 537–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAWAMURA N, NIMURA K, NAGANO H, YAMAGUCHI S, NONOMURA N & KANEDA Y 2015. CRISPR/Cas9-mediated gene knockout of NANOG and NANOGP8 decreases the malignant potential of prostate cancer cells. Oncotarget, 6, 22361–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KRESO A & DICK JE 2014. Evolution of the cancer stem cell model. Cell Stem Cell, 14, 275–91. [DOI] [PubMed] [Google Scholar]
- LI C, AO J, FU J, LEE DF, XU J, LONARD D & O'MALLEY BW 2011. Tumor-suppressor role for the SPOP ubiquitin ligase in signal-dependent proteolysis of the oncogenic co-activator SRC-3/AIB1. Oncogene, 30, 4350–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIAO P, WANG W, SHEN M, PAN W, ZHANG K, WANG R, CHEN T, CHEN Y, CHEN H & WANG P 2014. A positive feedback loop between EBP2 and c-Myc regulates rDNA transcription, cell proliferation, and tumorigenesis. Cell Death Dis, 5, e1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU JC, DENG T, LEHAL RS, KIM J & ZACKSENHAUS E 2007. Identification of tumorsphere- and tumor-initiating cells in HER2/Neu-induced mammary tumors. Cancer Res, 67, 8671–81. [DOI] [PubMed] [Google Scholar]
- LIU N, LI H, LI S, SHEN M, XIAO N, CHEN Y, WANG Y, WANG W, WANG R, WANG Q, SUN J & WANG P 2010a. The Fbw7/human CDC4 tumor suppressor targets proproliferative factor KLF5 for ubiquitination and degradation through multiple phosphodegron motifs. J Biol Chem, 285, 18858–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU T, XU F, DU X, LAI D, LIU T, ZHAO Y, HUANG Q, JIANG L, HUANG W, CHENG W & LIU Z 2010b. Establishment and characterization of multi-drug resistant, prostate carcinoma-initiating stem-like cells from human prostate cancer cell lines 22RV1. Mol Cell Biochem, 340, 265–73. [DOI] [PubMed] [Google Scholar]
- LU H, LIU S, ZHANG G, KWONG LN, ZHU Y, MILLER JP, HU Y, ZHONG W, ZENG J, WU L, KREPLER C, SPROESSER K, XIAO M, XU W, KARAKOUSIS GC, SCHUCHTER LM, FIELD J, ZHANG PJ, HERLYN M, XU X & GUO W 2016. Oncogenic BRAF-Mediated Melanoma Cell Invasion. Cell Rep, 15, 2012–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAKRODOULI E, OIKONOMOU E, KOC M, ANDERA L, SASAZUKI T, SHIRASAWA S & PINTZAS A 2011. BRAF and RAS oncogenes regulate Rho GTPase pathways to mediate migration and invasion properties in human colon cancer cells: a comparative study. Mol Cancer, 10, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARZAHN MR, MARADA S, LEE J, NOURSE A, KENRICK S, ZHAO H, BEN-NISSAN G, KOLAITIS RM, PETERS JL, POUNDS S, ERRINGTON WJ, PRIVE GG, TAYLOR JP, SHARON M, SCHUCK P, OGDEN SK & MITTAG T 2016. Higher-order oligomerization promotes localization of SPOP to liquid nuclear speckles. EMBO J, 35, 1254–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATHIEU J, ZHANG Z, ZHOU W, WANG AJ, HEDDLESTON JM, PINNA CM, HUBAUD A, STADLER B, CHOI M, BAR M, TEWARI M, LIU A, VESSELLA R, ROSTOMILY R, BORN D, HORWITZ M, WARE C, BLAU CA, CLEARY MA, RICH JN & RUOHOLA-BAKER H 2011. HIF induces human embryonic stem cell markers in cancer cells. Cancer Res, 71, 4640–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITSUI K, TOKUZAWA Y, ITOH H, SEGAWA K, MURAKAMI M, TAKAHASHI K, MARUYAMA M, MAEDA M & YAMANAKA S 2003. The homeoprotein Nanog is required for maintenance of pluripotency in mouse epiblast and ES cells. Cell, 113, 631–42. [DOI] [PubMed] [Google Scholar]
- MIYAZAWA K, TANAKA T, NAKAI D, MORITA N & SUZUKI K 2014. Immunohistochemical expression of four different stem cell markers in prostate cancer: High expression of NANOG in conjunction with hypoxia-inducible factor-1alpha expression is involved in prostate epithelial malignancy. Oncol Lett, 8, 985–992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'BRIEN CA, POLLETT A, GALLINGER S & DICK JE 2007. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature, 445, 106–10. [DOI] [PubMed] [Google Scholar]
- PATRAWALA L, CALHOUN T, SCHNEIDER-BROUSSARD R, LI H, BHATIA B, TANG S, REILLY JG, CHANDRA D, ZHOU J, CLAYPOOL K, COGHLAN L & TANG DG 2006. Highly purified CD44+ prostate cancer cells from xenograft human tumors are enriched in tumorigenic and metastatic progenitor cells. Oncogene, 25, 1696–708. [DOI] [PubMed] [Google Scholar]
- PECE S, TOSONI D, CONFALONIERI S, MAZZAROL G, VECCHI M, RONZONI S, BERNARD L, VIALE G, PELICCI PG & DI FIORE PP 2010. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell, 140, 62–73. [DOI] [PubMed] [Google Scholar]
- PO A, FERRETTI E, MIELE E, DE SMAELE E, PAGANELLI A, CANETTIERI G, CONI S, DI MARCOTULLIO L, BIFFONI M, MASSIMI L, DI ROCCO C, SCREPANTI I & GULINO A 2010. Hedgehog controls neural stem cells through p53-independent regulation of Nanog. EMBO J, 29, 2646–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHALEM O, SANJANA NE, HARTENIAN E, SHI X, SCOTT DA, MIKKELSON T, HECKL D, EBERT BL, ROOT DE, DOENCH JG & ZHANG F 2014. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science, 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHEN CH, YUAN P, PEREZ-LORENZO R, ZHANG Y, LEE SX, OU Y, ASARA JM, CANTLEY LC & ZHENG B 2013. Phosphorylation of BRAF by AMPK impairs BRAF-KSR1 association and cell proliferation. Mol Cell, 52, 161–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIEGEL R, NAISHADHAM D & JEMAL A 2013. Cancer statistics, 2013. CA Cancer J Clin, 63, 11–30. [DOI] [PubMed] [Google Scholar]
- SINGH SK, CLARKE ID, TERASAKI M, BONN VE, HAWKINS C, SQUIRE J & DIRKS PB 2003. Identification of a cancer stem cell in human brain tumors. Cancer Res, 63, 5821–8. [PubMed] [Google Scholar]
- SUVA ML, RIGGI N & BERNSTEIN BE 2013. Epigenetic reprogramming in cancer. Science, 339, 1567–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THEURILLAT JP, UDESHI ND, ERRINGTON WJ, SVINKINA T, BACA SC, POP M, WILD PJ, BLATTNER M, GRONER AC, RUBIN MA, MOCH H, PRIVE GG, CARR SA & GARRAWAY LA 2014. Prostate cancer. Ubiquitylome analysis identifies dysregulation of effector substrates in SPOP-mutant prostate cancer. Science, 346, 85–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UGOLKOV AV, EISENGART LJ, LUAN C & YANG XJ 2011. Expression analysis of putative stem cell markers in human benign and malignant prostate. Prostate, 71, 18–25. [DOI] [PubMed] [Google Scholar]
- WANG W, XIAO ZD, LI X, AZIZ KE, GAN B, JOHNSON RL & CHEN J 2015. AMPK modulates Hippo pathway activity to regulate energy homeostasis. Nat Cell Biol, 17, 490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WONG DJ, LIU H, RIDKY TW, CASSARINO D, SEGAL E & CHANG HY 2008. Module map of stem cell genes guides creation of epithelial cancer stem cells. Cell Stem Cell, 2, 333–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZADRA G, PHOTOPOULOS C, TYEKUCHEVA S, HEIDARI P, WENG QP, FEDELE G, LIU H, SCAGLIA N, PRIOLO C, SICINSKA E, MAHMOOD U, SIGNORETTI S, BIRNBERG N & LODA M 2014. A novel direct activator of AMPK inhibits prostate cancer growth by blocking lipogenesis. EMBO Mol Med, 6, 519–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZAKIKHANI M, DOWLING RJ, SONENBERG N & POLLAK MN 2008. The effects of adiponectin and metformin on prostate and colon neoplasia involve activation of AMP-activated protein kinase. Cancer Prev Res (Phila), 1, 369–75. [DOI] [PubMed] [Google Scholar]
- ZHANG J, BU X, WANG H, ZHU Y, GENG Y, NIHIRA NT, TAN Y, CI Y, WU F, DAI X, GUO J, HUANG YH, FAN C, REN S, SUN Y, FREEMAN GJ, SICINSKI P & WEI W 2018. Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature, 553, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG J, WANG X, CHEN B, XIAO Z, LI W, LU Y & DAI J 2010. The human pluripotency gene NANOG/NANOGP8 is expressed in gastric cancer and associated with tumor development. Oncol Lett, 1, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG K, FOWLER M, GLASS J & YIN H 2014a. Activated 5'flanking region of NANOGP8 in a self-renewal environment is associated with increased sphere formation and tumor growth of prostate cancer cells. Prostate, 74, 381–94. [DOI] [PubMed] [Google Scholar]
- ZHANG P, GAO K, TANG Y, JIN X, AN J, YU H, WANG H, ZHANG Y, WANG D, HUANG H, YU L & WANG C 2014b. Destruction of DDIT3/CHOP protein by wild-type SPOP but not prostate cancer-associated mutants. Hum Mutat, 35, 1142–51. [DOI] [PubMed] [Google Scholar]
- ZHANG P, WANG D, ZHAO Y, REN S, GAO K, YE Z, WANG S, PAN CW, ZHU Y, YAN Y, YANG Y, WU D, HE Y, ZHANG J, LU D, LIU X, YU L, ZHAO S, LI Y, LIN D, WANG Y & WANG L 2017. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT-mTORC1 activation. 23, 1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHUANG M, CALABRESE MF, LIU J, WADDELL MB, NOURSE A, HAMMEL M, MILLER DJ, WALDEN H, DUDA DM, SEYEDIN SN, HOGGARD T, HARPER JW, WHITE KP & SCHULMAN BA 2009. Structures of SPOP-substrate complexes: insights into molecular architectures of BTB-Cul3 ubiquitin ligases. Mol Cell, 36, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.