

Abstract
We report highly β-selective bis-thiourea-catalyzed 1,2-cis-O-pyranosylations employing easily accessible acetonide-protected donors. A wide variety of alcohol nucleophiles, including complex natural products, glycosides, and amino acids were β-mannosylated and rhamnosylated successfully using an operationally simple protocol under mild and neutral conditions. Less nucleophilic acceptors such as phenols were also glycosylated efficiently in excellent yields and with high β-selectivities.
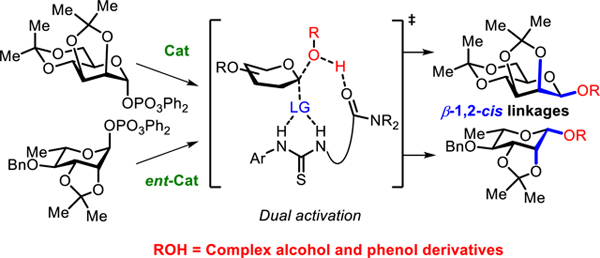
TOC graphic
INTRODUCTION
Pyranosides bearing β−1,2-cis-O-glycosidic linkages are found in many biologically important oligosaccharides that play crucial roles ranging from structural support to cellular recognition.1 For example, β-mannosides are key constituents of plant primary cell wall2 and are found at the branch-points of asparagine-linked glycopeptides, the structure of which is known to define unique antibody-effector function for a given isotype.3 β-Rhamnosides, although largely xenobiotic to humans, are the antigenic components of many pathogenic bacterial polysaccharides,4 and improved understanding of their immunological effects may be of value in the development of therapeutics.4a The construction of well-defined β−1,2-cis-linkages is therefore an important objective in synthetic chemistry, but one that presents fundamental challenges. β−1,2-cis-O-Glycosides are intrinsically less stable than their α-diastereomers as a result of diminished anomeric stabilization5 and increased steric congestion. These effects are also manifested in glycosylation pathways, resulting in a strong kinetic preference for α-products in typical mannosylation and rhamnosylation reactions (Figure 1A).
Figure 1.

(A) Challenges associated with β−1,2-cis-pyranosyl glycosidic bond construction. (B) Traditional approaches to achieving selectivity in glycosylation. (C) Catalyst-controlled approach to β−1,2-cis linkages with a bis-hydrogen-bond donor.
Significant efforts have been devoted to overcoming the inherent α-selectivity and developing β−1,2-cis glycosylation methods.6 Successful approaches include intramolecular aglycone delivery,7 hydrogen-bond-mediated aglycone delivery,8 the use of 1,2-anhydropyranose donors with borinic and boronic acid catalysts,9 protecting-group control such as with 4,6-benzylidene acetals10 and 2,6-lactones,11 and anomeric O-alkylation12 (Figure 1B). Each of these strategies employs donors with specific protecting group schemes requiring multi-step syntheses. Although the strongly Lewis acidic or oxidizing reaction conditions required for these couplings are generally compatible with carbohydrate substrates and can therefore be applied to oligosaccharide synthesis,13 they display poor tolerance toward a wide variety of common functional groups and therefore have limited utility for the glycosylation of complex acceptors.14,15 Finally, no general methods for 1,2-cis glycosylation of less reactive nucleophiles such as neutral phenols have been identified to date. In particular, electron-rich O-aryl glycosides are difficult to access as they are prone to Fries-type rearrangement pathways to afford C-glycosides under Lewis acidic conditions.16
The use of hydrogen-bond-donor and chiral phosphoric acid organocatalysts in glycosylation reactions has been reported by several groups.14,17 With the goal of developing a mild, functional-group-tolerant, and broadly applicable method for β−1,2-cis glycosylations employing easily accessible donors, we hypothesized that our recently reported stereospecific approach catalyzed by precisely designed bis-thiourea derivatives could be applied in this most challenging context (Figure 1C).18 While construction of β−1,2-trans linkages18a,b and β−1,2-cis-O-furanosyl18c linkages has been demonstrated using the appropriate α-glycosylphosphate donors with linked bis-thiourea catalysts such as 1, very limited success has been achieved as yet in the application of this strategy to β−1,2-cis-O-pyranosyl linkages. Herein, we describe the discovery of remarkable protecting group effects in catalytic glycosylations of mannosyl and rhamnosyl phosphate derivatives, leading to the development of highly selective, operationally simple, and general protocols for β−1,2-cis glycosylations of amino acids, carbohydrates, complex natural products, and pharmaceuticals.
RESULTS AND DISCUSSION
Bis-thiourea 1, the enantiomer of which had been identified previously as an effective catalyst for stereospecific β−1,2-trans-pyranosylation reactions,18b was evaluated in the mannosylation of the model acceptor 3a (Figure 2A).19 Variation of the protecting groups on the mannosyl diphenylphospate donor resulted in pronounced effects on anomeric selectivity.20 Simple alkyl protecting schemes such as perbenzyl (2a) or permethyl (2b) resulted in largely unselective coupling reactions (1:1 to 1:2 α:β). Incorporation of the 4,6-benzylidene acetal protecting group (as in 2c) pioneered by Crich for β-selective mannosylations under strong Lewis acid conditions again led to an unselective reaction in the presence of catalyst 1. However, a dramatic increase in β-selectivity was observed under catalytic conditions when the C2 and C3 substituents were bridged by an acetonide protecting group. Thus, coupling reactions with donors 2d-f all afforded high β-selectivity (1:16–32 α:β) with the relatively unhindered acceptor 3a.21 Mannosylation of the more sterically congested secondary nucleophile 3b was found to be substantially more β-selective with 2f than with 2d (1:24 vs 1:10 α:β, Figure 2B). The bis-acetonide-protected mannose derivative 2f also offers important practical advantages, being prepared in a simple two-step sequence from d-mannose, and affording glycosylation products that may be selectively or fully deprotected under mild conditions (vide infra).22 As observed previously in related bis-thiourea-catalyzed glycosylations of glycosyl phosphates, the inclusion of 4 Å molecular sieves is crucial for minimizing undesired hydrolysis of donors and sequestering the phosphoric acid by-product, which has been shown to promote glycosyations unselectively.18b,c Effects of varying the stoichiometry of donor 2f and acceptor 3c were also investigated (Figure 2C). While slightly higher conversions were obtained when the stoichiometry of either 2f or 3c was increased, those improvements were offset by observable decreases in β-selectivity. Accordingly, subsequent scope studies (Figures 4 and 5) were carried out with equimolar ratios of donor and acceptor.
Figure 2.

(A) Protecting-group effects on bis-thiourea-catalyzed and TMSOTf-promoted mannosylations of primary alcohol 3a. (B) Protecting-group effect on mannosylation reactions with secondary alcohol nucleophile 3b catalyzed by 1. (C) Stoichiometry optimization. Conversions were determined by 1H NMR analysis of crude product mixtures with mesitylene as an internal standard. Selectivities were determined by 1H NMR analysis of crude product mixtures. aConversions after 3 h. Selectivies remained constant over time. bReaction run at –40 °C.
Figure 4.

Acceptor scope of β-mannosylations. Yields of reactions catalyzed by 1 reflect isolated yields of pure β-products. Yields of TMSOTf reactions reflect combined yield of the α- and β-products and were determined by 1H NMR analysis of crude product mixtures with mesitylene as an internal standard. Selectivities were determined by 1H NMR analysis of crude product mixtures. a2.0 equiv. nucleophilic coupling partners were used. bReaction was run at 40 °C. c48 h reaction time. d63 h reaction time. Additional substrates are provided in Figure S1.
Figure 5.

Acceptor scope of β-rhamnosylations. Yields of reactions catalyzed by ent-1 reflect isolated yields of pure β-products. Yields of TMSOTf reactions reflect combined yield of the α- and β-products and were determined by 1H NMR analysis of crude product mixtures with mesitylene as an internal standard. Selectivities were determined by 1H NMR analysis of crude product mixtures. aReaction was run at 50 °C for 48 h. b48 h reaction time. cReaction was run at 40 °C. Additional substrates are provided in Figure S2.
We sought to elucidate the origin of the remarkably beneficial effect of the 2,3-acetonide protecting group on β-selective mannosylations with catalyst 1 through density functional theory computations (Figure 3).23 B3LYP/6-311+G(d,p)//B3LYP/6-31G(d) were selected because they have been previously applied to compute the transition states of glycosylation reactions and geometries and energies of oxocarbenium ion intermediates.24 Oxocarbenium ion intermediates were computed in isolation due to the observation of undesired ion-pair collapse into glycosyl phosphates when computations were performed in the presence of diphenyl phosphate anions. Crich has proposed that 4,6-benzylidene acetal protection of mannose serves to destabilize the corresponding oxocarbenium ion species relative to acyclic protection schemes (e.g. 5b vs 5a), thereby favoring stereospecific glycosylation pathways via transient α-mannosyl triflate intermediates.10d–e,25 Computational modeling of the oxocarbenium ions 5a and 5b fully supports that hypothesis, with the 4,6-benzylidene-protected derivative 5b calculated to be destabilized by 4.4 and 3.2 kcal/mol (4H3 and B2,5 conformation respectively relative to the unbridged analog 5a. The oxocarbenium ion 5c derived from 4.6-acetonide-protected donor is conformationally similar to 5b, and displays a similar destabilizing effect. However, a similar analysis of 2,3-acetonide-protected oxocarbenium ion 5d revealed that it was instead stabilized by 1.3 kcal/mol relative to the unbridged analog 5a. Thus, the dramatic increases in β-selectivity resulting from 2,3-acetonide protection cannot be ascribed to destabilization of an α-selective SN1-type pathway.
Figure 3.

Computed energies of oxocarbenium ion intermediates relative to the corresponding α-mannosyl diphenyl phosphates. Calculations were performed at B3LYP/6–311+G(d,p)//B3LYP/6–31G(d) with D3BJ dispersion corrections and PCM (H2O). Free energy corrections were determined at the B3LYP/6–31G(d) level of theory at 298.15 K using Grimme’s quasi-harmonic free energy correction. Similar trends were observed in ether solvation model and other levels of theory (see SI for full details).
Recently, high β-selectivity has been demonstrated in mannosylations utilizing 4,6-benzylidene acetal-protected donors in systems in which the formation of transient α-mannosyl triflates is not possible.26 In those cases, the stereoselectivity has been ascribed to preferential axial attack of nucleophiles on the energetically accessible B2,5 conformation of the oxocarbenium ion intermediates,26b,27 a similar conformation to that calculated for 5d and 5e. However, if this mode of stereoselectivity were operative in reactions catalyzed by bis-thiourea 1, comparable levels of β-selectivity might be expected with 2c and 2f because of the similarity in the conformations of 5b_B2,5 and 5e. In fact, dramatically higher β-selectivity is observed with donor 2f (α:β =1:32 vs 1:1 for 2c, Figure 2A). Furthermore, the divergent selectivities observed with 2d–f in 1-catalyzed and trimethylsilyl triflate (TMSOTf)-promoted reactions (1:16–32 vs 3–8:1 α:β, Figure 2A) appear to be inconsistent with stereoselective SN1 addition to B2,5 oxocarbenium ion intermediates in both pathways. We propose instead that the enhancement in β-selectivity is achieved by acceleration of a stereospecific SN2-type pathway promoted by the bis-thiourea catalyst 1, consistent with the observation that increased reactivity is observed with 2,3-acetonide protection 2d relative to unbridged analogs 2a–b (39% vs 11%, 15% conversion respectively). However, the basis for the acceleration effect resulting from the interplay between protecting group and catalyst remains to be determined, and the possibility that reactions proceed through a catalyst-promoted β-selective SN1 pathway cannot be strictly ruled out. Further examination of the mechanism of substitutions catalyzed by 1 and related bis-thioureas is the focus of continuing investigation.28
The scope of catalytic, β-selective mannosylation reactions with the bis-acetonide donor 2f was explored, with particular attention to cases where the mild and neutral conditions could provide a unique advantage over traditional Lewis acid-promoted glycosylation protocols (Figures 4 and S1). Sugar derivatives were found to be excellent substrates for the methodology, with both primary (6-OH-galactose 3c and 6-OH-glucose 3d) and secondary glycosyl acceptors (2-OH-galactose 3e and 4-OH-rhamnose 3f) undergoing mannosylation with high β-selectivity.29 The β-mannoside of C4-linked N-acetylglucosamine derivative 3g is of particular interest as it is a conserved branch-point on all N-glycans.3a A wide variety of other alcohols containing Lewis basic functional groups including esters, carbamates, and tertiary amines (3a, 3h–i, and S3f) were also found to undergo β-mannosylations selectively and cleanly, with no decomposition of nucleophiles observed.
Substrates bearing multiple hydroxyl groups such as deoxycholic acid octyl ester (S3g), pleuromutilin (3j) and FK-506 (3k) were found to undergo glycosylation at the least hindered position with high site-selectivity.30,31 Highly sterically congested secondary alcohols and tertiary alcohols underwent mannosyation with reduced level of selectivity and reactivity (Figure S1).
Phenolic acceptors were found to display excellent reactivity toward 2f under catalysis by bis-thiourea 1 to afford β-mannosylation products (Figures 4 and S1). A wide assortment of phenol derivatives, including sterically hindered substrates such as clofoctol (3l) and α-tocopherol (3m) were mannosylated with high β-selectivity. Excellent functional group tolerance was again demonstrated, such as in the glycosylation of the β-lactam-containing amoxicillin derivative 3n. Fries-type rearrangement of O-aryl glycosides to C-glycosides, which is common with electron-rich phenol derivatives (e.g. 3o and 3q) under Lewis acidic conditions,16 was not observed in any reactions promoted by 1.
Mannosylations with donor 2f using catalyst 1 were compared to reactions promoted by the Lewis acid TMSOTf.32 In all the examples illustrated in Figure 4, the reactions catalyzed by 1 afforded the mannosylation products cleanly and with high β-selectivity. In contrast, TMSOTf-promoted reactions yielded α-anomers as the major glycosylation products in almost all cases, consistent with the expected intrinsic preference for α-product in mannosylations proceeding via oxocarbenium ion intermediates. Functionally complex acceptors such as FK-506 (3k) were unstable to the Lewis acid activation conditions, while substrates possessing Lewis basic functionality such as quetiapine (3h) afforded none of the desired glycosylation products. Other sensitive substrates such as amoxicillin (3n) and serotonin (3o) underwent mannosylation in low yield and anomeric selectivity under TMSOTf conditions, with formation of multiple side products.
We sought to explore whether the 2,3-acetonide protection strategy applied successfully in β−1,2-cis mannosylations catalyzed by 1 might be extended to other interesting glycosyl donors. Rhamnose is the C6-deoxy analogue of mannose, and it presents another long-standing challenge as a coupling partner in β−1,2-cis glycosylation reactions. Because the C6 hydroxyl of mannose is lacking, the 4,6-benzylidene acetal protecting group strategy pioneered by Crich for β-mannosylations is not applicable. Alternative strategies for β-rhamnoside synthesis have been identified, but the reported successful examples have been limited to simple alcohols as nucleophilic coupling partners.7e,8b,9c,23f,33 The 2,3-acetonide-protected l-rhamnose phosphate donor 6 was evaluated with 3a as a model acceptor (Figure S14). l-Rhamnose is pseudoenantiomeric with d-mannose, and we observed that a large improvement in β-selectivity was achieved by using the enantiomer of catalyst 1 (1:7 α:β with catalyst 1 vs 1:32 α:β with ent-catalyst 1, Figure S14). Such stereochemical matching points to precise catalyst-substrate interactions in the catalytic glycosylation event and is consistent with observations we have made previously in less challenging glycosylation reactions.18b,c The nucleophile scope in β-rhamnosylations catalyzed by ent-1 was examined and proved to be similarly broad to that documented above in β-mannosylations (Figures 5 and S2). Thus, excellent functional group compatibility was demonstrated with substrates bearing Lewis basic groups such as tertiary amines (3h), imide (3i), and carbamates (3o–p), as well as with complex natural products (e.g. 3k and 3t).34 Excellent β-selectivity and reactivity were observed in the rhamnosylation of phenols as well. Reactions promoted by TMSOTf displayed limited functional group tolerance and generally favored the α-rhamnosylation products in cases where glycosylation product was obtained.
The acetonide-protected glycosylation products are readily unmasked under mild conditions.22c Thus, both acetonides on disaccharide 4d were hydrolyzed in a 4:1 acetic acid:water mixture at room temperature, with the β-mannoside product isolated in 81% yield with no erosion of anomeric purity (Figure 6A). Similarly, mannosylated FK-506 (4k) was also deprotected in 86% yield with no detectable anomerization, demonstrating how even highly sensitive functional groups are tolerant to the mildly acidic hydrolytic conditions. The potential practicality of the mannosylation procedure was demonstrated in a multi-millimole-scale synthesis of methyl β-d-mannopyranoside 8c. The shortest prior reported stereoselective synthesis of 8c required 7 steps from d-mannose.35 Using the approach outline herein and in Figure 6B, 8c was prepared in 2 steps from 2f and isolated in >95% purity without column chromatography (4 steps from d-mannose). The glycosylation step was performed on 2.5 mmol scale using 1 mol% catalyst 1 to provide the acetonide protected-mannoside quantitatively,36 and that material was subjected to the deprotection protocol directly to afford the fully deprotected β-mannoside 8c in 1:99 α:β selectivity.
Figure 6.

(A) Protocol for bis-acetonide deprotection under mildly acidic hydrolytic conditions. (B) Multi-millimole-scale synthesis of methyl β-d-mannopyranoside. Yields reflect isolated yields of pure β-products. Selectivities were determined by 1H NMR analysis of crude product mixtures.
CONCLUSION
We have demonstrated that β-mannosides and β-rhamnosides can be mildly and selectively accessed under catalyst control using readily accessible 2,3-acetonide-protected glycosyl phosphate donors. The catalytic protocol is compatible with extensive functional group complexity on the nucleophilic coupling partner, so we anticipate this method may enable exploration of the effect of specific O-glycosylation on a wide range chemical matter, although the application of the current methodology to highly sterically congested nucleophiles is still limited. Work is currently underway to elucidate the origins of selectivity imparted by the 2,3-acetonide protecting group in connection with the bis-thiourea catalyst, and to further illuminate the scope of potential coupling partners in these catalytic glycosylation reactions.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the NIH through GM132571 and the Common Fund Glycoscience Program (U01 GM116249), and by an NSF pre-doctoral fellowship to S.M.L. We thank Dr. Shao-Liang Zheng (Harvard University) for X-ray data collection and structure determination.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental and characterization data of catalyst and substrate syntheses, details of computational studies (PDF). Crystallographic data for 4p (CIF). Crystallographic data for S8 (CIF).
This material is available free of charge via the Internet at http://pubs.acs.org.
Notes
The authors declare no competing financial interests.
REFERENCES
- (1).(a) Varki A; Cummings RD; Esko JD; Stanley P; Hart GW; Aebi M; Darvill AG; Kinoshita T; Packer NH; Prestegard JH; Schnaar RL; Seeberger PH Essentials of Glycobiology, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, 2009. [PubMed] [Google Scholar]; (b) Faille C; Michalski JC; Strecker G; Mackenzie DWR; Camus D; Poulain D. Immunoreactivity of Neoglycolipids Constructed from Oligomannosidic Residues of the Candida albicans Cell Wall. Infect. Immun 1990, 58, 3537–3544. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fengel D; Wegener G Wood: Chemistry, Ultrastructure, Reactions; Walter de Gruyter: Berlin, 1984. [Google Scholar]; (d) Shibata N; Fukusawa S; Kobayashi H; Tojo M; Ambo A; Ohkubo Y; Suzuki S. Structural Analysis of Phospho-d-Mannan-Protein Complexes Isolated from Yeast and Mold form Cells of Candida albicans NIH A-207 Serotype A Strain. Carbohydr. Res 1989, 187, 239–253. [DOI] [PubMed] [Google Scholar]
- (2).(a) Aspinall GO The Polysaccharides; Academic Press: London, 1982. [Google Scholar]; (b) Schröder R; Atkinson RG; Redgwell RJ Re-interpreting the Role of Endo-β-Mannanases as Mannan Endotransglycosylase/hydrolases in the Plant Cell Wall. Ann. Bot 2009, 104, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Sjöström E Wood Chemistry: Fundamentals and Applications, 2nd ed.; Academic Press: New York, 1993. [Google Scholar]
- (3).(a) Moremen KW; Tiemeyer M; Nairn AV Vertebrate Protein Glycosylation: Diversity, Synthesis and Function. Nat. Rev. Mol. Cell Biol 2012, 13, 448–462. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wada R; Matsui M; Kawasaki N. Influence of N-Glycosylation on Effector Functions and Thermal Stability of Glycoengineered IgG1 Monoclonal Antibody with Homogeneous Glycoforms. mAbs 2019, 11, 350–372. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Larkin M; Childs RA; Matthews TJ; Thiel S; Mizuochi T; Lawson AM; Savill JS; Haslett C; Diaz R; Feizi T. Oligosaccharide-Mediated Interactions of the Envelope Glycoprotein gp120 of HIV-1 that are Independent of CD4 Recognition. AIDS 1989, 3, 793–798. [DOI] [PubMed] [Google Scholar]
- (4).(a) Emmadi M; Khan N; Lykke L; Reppe K; S GP; Lisboa MP; Wienhold SM; Witzenrath M; Pereira CL; Seeberger PH A Streptococcus pneumoniae Type 2 Oligosaccharide Glycoconjugate Elicits Opsonic Antibodies and Is Protective in an Animal Model of Invasive Pneumococcal Disease. J. Am. Chem. Soc 2017, 139, 14783–14791. [DOI] [PubMed] [Google Scholar]; (b) Chernyak AY; Weintraub A; Kochetkov NK; Lindberg AA The β-Configuration of the Rhamnosidic Linkage in Salmonella serogroups C2 and C3, Lipopolysaccharide is Important for the Immunochemistry of the O-Antigen 8. Mol. Immunol 1993, 30, 887–893. [DOI] [PubMed] [Google Scholar]; (c) Feng L; Senchenkova SN; Wang W; Shashkov AS; Liu B; Shevelev SD; Liu D; Knirel YA; Wang L. Structural and Genetic Characterization of the Shigella boydii Type 18 O-Antigen. Gene 2005, 355, 79–86. [DOI] [PubMed] [Google Scholar]
- (5).(a) Kirby AJ The Anomeric Effect and Related Stereoelectronic Effects at Oxygen; Springer-Verlag: Berlin, 1983. [Google Scholar]; (b) Deslongchamps P Stereoelectronic Effects in Organic Chemistry; Pergamon: Oxford, 1983. [Google Scholar]
- (6). For reviews on β−1,2-cis glycosylation methods, see. [Google Scholar]; (a) El Ashry ESH; Rashed N; Ibrahim ESI Strategies of Synthetic Methodologies for Constructing β-Mannosidic Linkage. Curr. Org. Synth 2005, 2, 175–213. [Google Scholar]; (b) Sasaki K; Tohda K. Recent Topics in β-Stereoselective Mannosylation. Tetrahedron Lett. 2018, 59, 496–503. [Google Scholar]; (c) Crich D. Chemistry of Glycosyl Triflates: Synthesis of β-Mannopyranosides, J. Carbohydr. Chem 2002, 21, 663–686. [Google Scholar]
- (7).(a) Barresi F; Hindsgaul O. Synthesis of β-Mannopyranosides by Intramolecular Aglycon Delivery. J. Am. Chem. Soc 1991, 113, 9376–9377. [Google Scholar]; (b) Ito Y; Ogawa T. A Novel Approach to the Stereoselective Synthesis of β‐Mannosides. Angew. Chem., Int. Ed 1994, 33, 1765–1767. [Google Scholar]; (c) Stork G; La Clair JJ Stereoselective Synthesis of β-Mannopyranosides via the Temporary Silicon Connection Method. J. Am. Chem. Soc 1996, 118, 247–248. [Google Scholar]; (d) Ito Y; Ohnishi Y; Ogawa T; Nakahara Y. Highly Optimized β-Mannosylation via p-Methoxybenzyl Assisted Intramolecular Aglycon Delivery. Synlett 1998, 1102–1104. [Google Scholar]; (e) Lee YJ; Ishiwata A; Ito Y. Stereoselective Synthesis of β-l-Rhamnopyranosides. J. Am. Chem. Soc 2008, 130, 6330–6331. For a review, see: [DOI] [PubMed] [Google Scholar]; (f) Fairbanks AJ Intramolecular Aglycon Delivery (IAD): The Solution to 1,2-cis Stereocontrol for Oligosaccharide Synthesis? Synlett 2003, 1945–1958. [Google Scholar]
- (8).(a) Pistorio SG; Yasomanee JP; Demchenko AV Hydrogen-Bond-Mediated Aglycone Delivery: Focus on β-Mannosylation. Org. Lett 2014, 16, 716–719. [DOI] [PubMed] [Google Scholar]; (b) Lei J-C; Ruan Y-X; Luo S; Yang J-S Stereodirecting Effect of C3-Ester Groups on the Glycosylation Stereochemistry of l-Rhamnopyranose Thioglycoside Donors: Stereoselective Synthesis of α- and β-l-Rhamnopyranosides. Eur. J. Org. Chem 2019, 6377–6382. [Google Scholar]
- (9).(a) Nishi N; Nashida J; Kaji E; Takahashi D; Toshima K. Regio- and Stereoselective β-Mannosylation using a Boronic Acid Catalyst and Its Application in the Synthesis of a Tetrasaccharide Repeating Unit of Lipopolysaccharide Derived from E. coli O75. Chem. Commun 2017, 53, 3018–3021. [DOI] [PubMed] [Google Scholar]; (b) Tanaka M; Nashida J; Takahashi D; Toshima K. Glycosyl-Acceptor-Derived Borinic Ester-Promoted Direct and β-Stereoselective Mannosylation with a 1,2-Anhydromannose Donor. Org Lett. 2016, 18, 2288–2291. [DOI] [PubMed] [Google Scholar]; (c) Nishi N; Sueoka K; Iijima K; Sawa R; Takahashi D; Toshima K. Stereospecific β-l-Rhamnopyranosylation through an SNi-Type Mechanism by Using Organoboron Reagents. Angew. Chem. Int. Ed 2018, 57, 13858–13862. [DOI] [PubMed] [Google Scholar]
- (10).(a) Crich D; Sun S. Direct Synthesis of β-Mannopyranosides by the Sulfoxide Method. J. Org. Chem 1997, 62, 1198–1199. [Google Scholar]; (b) Crich D; Sun S. Direct Formation of β-Mannopyranosides and Other Hindered Glycosides from Thioglycosides. J. Am. Chem. Soc 1998, 120, 435–436. [Google Scholar]; (c) Crich D; Sun S. Formation of β-Mannopyranosides of Primary Alcohols Using the Sulfoxide Method. J. Org. Chem 1996, 61, 4506–4507. [DOI] [PubMed] [Google Scholar]; (d) Kim KS; Kim JH; Lee YJ; Lee YJ; Park J. 2-(Hydroxycarbonyl)benzyl Glycosides: A Novel Type of Glycosyl Donors for Highly Efficient β-Mannopyranosylation andOligosaccharide Synthesis by Latent-Active Glycosylation. J. Am. Chem. Soc 2001, 123, 8477–8481. For reviews, see [DOI] [PubMed] [Google Scholar]; (e) Aubry S; Sasaki K; Sharma I; Crich D Influence of Protecting Groups on the Reactivity and Selectivity of Glycosylation: Chemistry of the 4,6-O-Benzylidene Protected Mannopyranosyl Donors and Related Species In Reactivity Tuning in Oligosaccharide Assembly; Fraser-Reid B, Cristóbal López J, Eds.; Top. Curr. Chem 2011, 301, 141–188. [DOI] [PubMed] [Google Scholar]; (f) Crich D. Mechanism of a Chemical Glycosylation Reaction. Acc. Chem. Res 2010, 43, 1144–1153. [DOI] [PubMed] [Google Scholar]
- (11).(a) Hashimoto Y; Tanikawa S; Saito R; Sasaki K. β-Stereoselective Mannosylation Using 2,6-Lactones. J. Am. Chem. Soc 2016, 138, 14840–14843. [DOI] [PubMed] [Google Scholar]; (b) Sasaki K; Hashimoto Y. 2,6-Lactones as a New Entry in Stereoselective Glycosylations. Synlett 2017, 28, 1121–1126. [Google Scholar]
- (12).Nguyen H; Zhu D; Li X; Zhu J. Stereoselective Construction of β-Mannopyranosides by Anomeric O-Alkylation: Synthesis of the Trisaccharide Core of N-linked Glycans. Angew. Chem. Int. Ed 2016, 55, 4767–4771. [DOI] [PubMed] [Google Scholar]
- (13).(a) Crich D; Li W; Li H. Direct Chemical Synthesis of the β-Mannans: Linear and Block Syntheses of the Alternating β-(1→3)-β-(1→4)-Mannan Common to Rhodotorula glutinis, Rhodotorula mucilaginosa, and Leptospira biflexa. J. Am. Chem. Soc 2004, 126, 15081–15086. [DOI] [PubMed] [Google Scholar]; (b) Wang Z; Chinoy ZS; Ambre SG; Peng W; McBride R; de Vries RP; Glushka J; Paulson JC; Boons GJ Science 2013, 341, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Levi SM; Jacobsen EN Catalyst-Controlled Glycosylations. Org. React 2019, 100, 801–852. [Google Scholar]
- (15). Glycosylation of small molecule, peptide and protein pharmaceuticals has shown significant potential for enhancing biodistribution by affecting molecular structure, hydrophilicity, stability, and bioavailability. For a review, see: [Google Scholar]; (a) Sola RJ; Griebenow K. Effects of Glycosylation on the Stability of Protein Pharmaceuticals J. Pharm. Sci 2009, 98, 1223–1245. For selected, specific examples, see: [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baudyš M; Uchio T; Mix D; Kim SW; Wilson DJ Pharm. Sci 1995, 84, 28–33. [DOI] [PubMed] [Google Scholar]; (c) Wang C; Eufemi M; Turano C; Giartosio A. Influence of the Carbohydrate Moiety on the Stability of Glycoproteins. Biochemistry 1996, 35, 7299–7307. [DOI] [PubMed] [Google Scholar]; (d) Albert R; Marbach P; Bauer W; Briner U; Fricker G; Bruns C; Pless J. SDZ CO 611: a Highly Potent Glycated Analog of Somatostatin with Improved Oral Activity. Life Sci. 1993, 53, 517–525. [DOI] [PubMed] [Google Scholar]; (e) Negri L; Lattanzi R; Tabacco F; Scolaro B; Rocchi R. Glycodermorphins: Opioid Peptides with Potent and Prolonged Analgesic Activity and Enhanced Blood-Brain Barrier Penetration. Br. J. Pharmacol 1998, 124, 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Fu Z-H; Wen C; Ye Q-M; Huang W; Liu X-M; Jiang R-W An Efficient Strategy for the Glycosylation of Total Bufadienolides in Venenum Bufonis. ACS Omega 2019, 4, 6819–6825. [Google Scholar]
- (16).(a) Mahling J-A; Schmidt RR Aryl C-Glycosides from O-Glycosyl Trichloroacetimidates and Phenol Derivatives with Trimethylsilyl Trifluoromethanesulfonate (TMSOTf) as the Catalyst. Synthesis 1993, 325–328. [Google Scholar]; (b) Palmacci ER; Seeberger PH Synthesis of C-Aryl and C-Alkyl Glycosides Using Glycosyl Phosphates. Org. Lett 2001, 3, 1547–1550. For reviews, see [DOI] [PubMed] [Google Scholar]; (c) Suzuki K. Lessons from Total Synthesis of Hybrid Natural Products. Chem. Rec 2010, 10, 291–307. [DOI] [PubMed] [Google Scholar]; (d) dos Santos RG; Jesus AR; Caio JM; Rauter AP Fries-type Reactions for the C-Glycosylation of Phenols. Curr. Org. Chem 2011, 15, 128–148. [Google Scholar]
- (17). For reviews of organocatalytic glycosylations, see ref. 14 and: [Google Scholar]; (a) Williams R; Galan MC Recent Advances in Organocatalytic Glycosylations. Eur. J. Org. Chem 2017, 6247–6264. [Google Scholar]; (b) Wang HY; Blaszczyk SA; Xiao G; Tang W. Chiral Reagents in Glycosylation and Modification of Carbohydrates. Chem. Soc. Rev 2018, 47, 681–701. For examples of chiral phosphoric acid-catalyzed glycosylations, see: [DOI] [PubMed] [Google Scholar]; (c) Cox DJ; Smith MD; Fairbanks AJ Glycosylation Catalyzed by a Chiral Brønsted Acid. Org. Lett 2010, 12, 1452–1455. [DOI] [PubMed] [Google Scholar]; (d) Kimura T; Sekine M; Takahashi D; Toshima K. Chiral Brønsted Acid Mediated Glycosylation with Recognition of Alcohol Chirality. Angew. Chem. Int. Ed 2013, 52, 12131–12134. [DOI] [PubMed] [Google Scholar]; (e) Tay J-H; Argüelles AJ; DeMars II MD; Zimmerman PM; Sherman DH; Nagorny P. Regiodivergent Glycosylations of 6-Deoxy-erythronolide B and Oleandomycin-Derived Macrolactones Enabled by Chiral Acid Catalysis. J. Am. Chem. Soc 2017, 139, 8570–8578. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Palo-Nieto C; Sau A; Williams R; Galan MC Cooperative Brønsted Acid-Type Organocatalysis for the Stereoselective Synthesis of Deoxyglycosides. J. Org. Chem 2017, 82, 407–414. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Liu D; Sarrafpour S; Guo W; Goulart B; Bennett CS Matched/Mismatched Interactions in Chiral Brønsted Acid-Catalyzed Glycosylation Reactions with 2-Deoxy-Sugar Trichloroacetimidate Donors. J. Carbohydr. Chem 2014, 33, 423–434. For examples of hydrogen-bond-donor-catalyzed glycosylations, see: [Google Scholar]; (h) Peng P; Geng Y; Göttker-Schnetmann I; Schmidt RR 2-Nitro-thioglycosides: α-and β-Selective Generation and Their Potential as β-Selective Glycosyl Donors. Org. Lett 2015, 17, 1421–1424. [DOI] [PubMed] [Google Scholar]; (i) Geng Y; Kumar A; Faidallah HM; Albar HA; Mhkalid IA; Schmidt RR Cooperative Catalysis in Glycosidation Reactions with O-Glycosyl Trichloroacetimidates as Glycosyl Donors. Angew. Chem. Int. Ed 2013, 52, 10089–10092. [DOI] [PubMed] [Google Scholar]; (j) Sun L; Wu X; Xiong DC; Ye XS Stereoselective Koenigs-Knorr Glycosylation Catalyzed by Urea. Angew. Chem. Int. Ed 2016, 55, 8041–8044. [DOI] [PubMed] [Google Scholar]; (k) Medina S; Harper MJ; Balmond EI; Miranda S; Crisenza GE; Coe DM; McGarrigle EM; Galan MC Stereoselective Glycosylation of 2-Nitrogalactals Catalyzed by a Bifunctional Organocatalyst. Org. Lett 2016, 18, 4222–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Kimura T; Eto T; Takahashi D; Toshima K. Stereocontrolled Photoinduced Glycosylation Using an Aryl Thiourea as an Organophotoacid. Org. Lett 2016, 18, 3190–3193. [DOI] [PubMed] [Google Scholar]; (m) Xu C; Loh CCJ An Ultra-low Thiourea Catalyzed Strain-release Glycosylation and a Multicatalytic Diversification Strategy. Nat. Commun 2018, 9, 4057. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Li S; Kobayashi Y; Takemoto Y. Organocatalytic Direct α-Selective N-Glycosylation of Amide with Glycosyl Trichloroacetimidate. Chem. Pharm. Bull 2018, 66, 768–770. [DOI] [PubMed] [Google Scholar]
- (18).(a) Park Y; Harper KC; Kuhl N; Kwan EE; Liu RY; Jacobsen EN Macrocyclic Bis-thioureas Catalyze Stereospecific Glycosylation Reactions. Science 2017, 355, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Levi SM; Li Q; Rötheli AR; Jacobsen EN Catalytic Activation of Glycosyl Phosphates for Stereoselective Coupling Reactions. Proc. Natl. Acad. Sci. U. S. A. 2019, 116, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mayfield AB; Metternich JB; Trotta AH; Jacobsen EN Stereospecific Furanosylations Catalyzed by Bis-thiourea Hydrogen-Bond Donors. J. Am. Chem. Soc 2020, 142, 4061–4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).A systematic evaluation of dimeric hydrogen-bond donors led to the identification of 1 as the optimal catalyst for the β-mannosylation reactions. Ent-1 and catalysts with less rigid structures or different arylpyrrolidine components were found to be less β-selective and less reactive (see Figures S3–S5 for details).
- (20).(a) A screen of phosphate leaving groups revealed the diphenyl derivatives as optimal. The more electron-withdrawing bis-(4-cholorophenyl) phosphates were found to be more reactive but less β-selective in the presence of 1 (Figure S12). [Google Scholar]; (b) Comparable conversions and selectivities were obtained in di-n-butyl ether (see Figures S6–S7). Diisopropyl ether was selected because of its greater ability to solubilize acceptors.
- (21).Higher reactivity of 2f was achieved by increasing reaction temperature to 50 °C, albeit with slightly lower β-selectivity (85% conversion, 1:19 α:β). α- and β-Configurations were assigned based on 3JH1-H2 (0 Hz for α-products and 2.5–3.0 Hz for β-products) and 1JC1-H1 (ca. 170 Hz for α-products and ca. 160 Hz for β-products).
- (22). For selective hydrolysis of 4,6-acetonides, see. [Google Scholar]; (a) Combemale S; Assam-Evoung J-N; Houaidji S; Bibi R; Barragan-Montero V. Gold Nanoparticles Decorated with Mannose-6-phosphate Analogues. Molecules 2014, 19, 1120–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Neogi A; Majhi TP; Achari B; Chattopadhyay P. Palladium-Catalyzed Intramolecular C–O Bond Formation: An Approach to the Synthesis of Chiral Benzodioxocines. Eur. J. Org. Chem 2008, 2008, 330–336. For deprotection of bis-acetonide, see [Google Scholar]; (c) Cocinero EJ; Stanca-Kaposta EC; Scanlan EM; Gamblin DP; Davis BG; Simons JP Conformational Choice and Selectivity in Singly and Multiply Hydrated Monosaccharides in the Gas Phase. Chem. Eur. J 2008, 14, 8947–8955. [DOI] [PubMed] [Google Scholar]
- (23). Mannosylations with bis-acetonide donors under Lewis acidic conditions were generally α-selective or unselective. For examples, see. [Google Scholar]; (a) Mattson AL; Michel AK; Cloninger MJ Using In(III) as a Promoter for Glycosylation. Carbohydr. Res 2012, 347, 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang J; Cooper-Vanosdell C; Mensah EA; Nguyen HM Cationic Palladium(II)-Catalyzed Stereoselective Glycosylation with Glycosyl Trichloroacetimidates. J. Org. Chem 2008, 73, 794–800. [DOI] [PubMed] [Google Scholar]; (c) Crich D; Sun S. Direct Chemical Synthesis of β-Mannopyranosides and Other Glycosides via Glycosyl Triflates. Tetrahedron 1998, 54, 8321–8348. Mildly β-selective, silver-salt-promoted mannosylations with bis-acetonide donors have been reported. For examples, see [Google Scholar]; (d) Garegg PJ; Iversen T; Johansson R. Synthesis of Disaccharides Containing β-D-Mannopyranosyl Groups. Acta Chem. Scand 1980, B34, 505–508. [Google Scholar]; (e) Garegg PJ; Iversen T; Norberg T. A Practical Synthesis of p-Nitrophenyl β-d-Mannopyranoside. Carbohydr. Rer, 1979, 73, 313. [Google Scholar]; (f) Silver-salt-promoted rhamnosylations with 2,3-acetonide donors with moderate β-selectivity and limited acceptor scope have been reported. Iversen T; Bundle DR A New and Efficient Synthesis of β-l-Rhamnopyranosides. Carbohydr. Res 1980, 84, C13–C15. [Google Scholar]
- (24).(a) Whitfield DM Computational Studies of the Role of Glycopyranosyl Oxacarbenium Ions in Glycobiology and Glycochemistry. Adv. Carbohydr. Chem. Biochem 2009, 62, 83–159. [DOI] [PubMed] [Google Scholar]; (b) Ionescu AR; Whitfield DM; Zgierski MZ; Nukada T. Investigations into the Role of Oxacarbenium Ions in Glycosylation Reactions by ab initio Molecular Dynamics. Carbohydr. Res 2006, 341, 2912–2920. [DOI] [PubMed] [Google Scholar]; (c) Barnett CB; Wilkinson KA; Naidoo KJ Molecular Details from Computational Reaction Dynamics for the Cellobiohydrolase I Glycosylation Reaction. J. Am. Chem. Soc 2011, 133, 19474–19482. [DOI] [PubMed] [Google Scholar]
- (25).(a) Huang M; Garrett GE; Birlirakis N; Bohe L; Pratt DA; Crich D. Dissecting the Mechanisms of a Class of Chemical Glycosylation Using Primary 13C Kinetic Isotope Effects. Nat. Chem 2012, 4, 663–667. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Crich D; Chandrasekera NS Mechanism of 4,6-O-Benzylidene-Directed β-Mannosylation as Determined by α-Deuterium Kinetic Isotope Effects. Angew. Chem. Int. Ed 2004, 43, 5386–5389. [DOI] [PubMed] [Google Scholar]
- (26).(a) Sun P; Wang P; Zhang Y; Zhang X; Wang C; Liu S; Lu J; Li M. Construction of β-Mannosidic Bonds via Gold(I)-Catalyzed Glycosylations with Mannopyranosyl ortho-Hexynylbenzoates and Its Application in Synthesis of Acremomannolipin A. J. Org. Chem 2015, 80, 4164–4175. [DOI] [PubMed] [Google Scholar]; (b) Heuckendorff M; Bendix J; Pedersen CM; Bols M. β-Selective Mannosylation with a 4,6-Silylene-tethered Thiomannosyl Donor. Org. Lett 2014, 16, 1116–1119. [DOI] [PubMed] [Google Scholar]
- (27).(a) Moume-Pymbock M; Crich D. Stereoselective C-Glycoside Formation with 2-O-Benzyl-4,6-O-benzylidene Protected 3-Deoxy Gluco- and Mannopyranoside Donors: Comparison with O-Glycoside Formation. J. Org. Chem 2012, 77, 8905–8912. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Nukada T; Bérces A; Whitfield DM Can the Stereochemical Outcome of Glycosylation Reactions Be Controlled by the Conformational Preferences of the Glycosyl Donor? Carbohydr. Res 2002, 337, 765–774. [DOI] [PubMed] [Google Scholar]
- (28).The bis-acetonide-protected oxocarbenium intermediate 5e was found to be destabilized by 2.8 kcal/mol relative to 5d, suggesting that the high β-selectivities obtained with donor 2f even with relatively unreactive, hindered nucleophiles result from a combination of acceleration of β-selective and deceleration of α-selective pathways arising from 5e.
- (29).Chromatographic separation of 4b from 3b was challenging, so accurate isolated yield could not be determined. Thus, 3b was not included in Figure 4.
- (30).(a) Chamni S; He Q-L; Dang Y; Bhat S; Liu JO; Romo D. Diazo Reagents with Small Steric Footprints for Simultaneous Arming/SAR Studies of Alcohol-Containing Natural Products via O–H Insertion. ACS Chem. Biol 2011, 6, 1175–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McPherson M; Yang Y; Hammond PW; Kreider BL Drug Receptor Identification from Multiple Tissues Using Cellular-Derived mRNA Display Libraries. Chem. Biol 2002, 9, 691–698. [DOI] [PubMed] [Google Scholar]
- (31).The anomeric configuration of 4k was assigned based on 1JC1-H1 (158.2 Hz) of deprotected compound 8b. No other glycosylated FK-506 derivatives were detectable in the crude NMR spectra or isolated upon chromatographic purification.
- (32).The TMSOTf-promoted glycosylation reactions were carried out using the procedures reported by Seeberger and Hashimoto:; (a) Plante OJ; Palmacci ER; Andrade RB; Seeberger PH Oligosaccharide Synthesis with Glycosyl Phosphate and Dithiophosphate Triesters as Glycosylating Agents. J. Am. Chem. Soc 2001, 123, 9545–9554. [DOI] [PubMed] [Google Scholar]; (b) Tsuda T; Nakamura S; Hashimoto S. A Highly Stereoselective Construction of 1,2-trans-β-Glycosidic Linkages Capitalizing on 2-azido-2-deoxy-d-Glycosyl Diphenylphosphates as Glycosyl Donors. Tetrahedron 2004, 60 10711–10737. [Google Scholar]
- (33).(a) Crich D; Picione J. Direct Synthesis of the β-l-Rhamnopyranosides. Org. Lett 2003, 5, 781–784. [DOI] [PubMed] [Google Scholar]; (b) Crich D; Vinod AU; Picione J. The 3,4-O-Carbonate Protecting Group as a β-Directing Group in Rhamnopyranosylation in Both Homogeneous and Heterogeneous Glycosylations As Compared to the Chameleon-like 2,3-O-Carbonates. J. Org. Chem 2003, 68, 8453–8458. [DOI] [PubMed] [Google Scholar]; (c) Zhu Y; Shen Z; Li W; Yu B. Stereoselective Synthesis of β-Rhamnopyranosides via Gold(I)-Catalyzed Glycosylation with 2-Alkynyl-4-nitro-benzoate Donors. Org. Biomol. Chem 2016, 14, 1536–1539. [DOI] [PubMed] [Google Scholar]; (d) Heuckendorff M; Pedersen CM; Bols M. Rhamnosylation: Diastereoselectivity of Conformationally Armed Donors. J. Org. Chem 2012, 77, 5559–5568. [DOI] [PubMed] [Google Scholar]; (e) Elferink H; Pedersen CM l-Rhamnosylation: The Solvent is the Solution. Eur. J. Org. Chem 2017, 53–59. For a recent review, see: [Google Scholar]; (f) Rai D; Kulkarni SS Recent Advances in β-l-Rhamnosylation. Org. Biomol. Chem 2020, 18, 3216–3228. [DOI] [PubMed] [Google Scholar]
- (34).The anomeric configuration of 7k was assigned based on 1JC1-H1 (158.2 Hz). We did not isolate any other glycosylated FK-506 derivatives after chromatographic purification, and no discernible peaks corresponding to other glycosylated FK-506 derivatives were present in the crude NMR. We therefore reported the selectivity as <1:50 α:β.
- (35).The stereoselective synthesis utilized 4,6-benzylidene acetal strategy for β-mannosylations. Crich D; Banerjee A; Yao Q. Direct Chemical Synthesis of the β-d-Mannans: The β -(1→2) and β -(1→4) Series. J. Am. Chem. Soc 2004, 126, 14930–14934. [DOI] [PubMed] [Google Scholar]
- (36).The reaction proceeded to 94% conversion after 22 h.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.