

Summary
Background
Isocitrate dehydrogenase 1 (IDH1) mutations occur in approximately 13% of patients with intrahepatic cholangiocarcinoma, a relatively uncommon cancer with a poor clinical outcome. The aim of this international phase 3 study was to assess the efficacy and safety of ivosidenib (AG-120)—a small-molecule targeted inhibitor of mutated IDH1—in patients with previously treated IDH1-mutant cholangiocarcinoma.
Methods
This multicentre, randomised, double-blind, placebo-controlled, phase 3 study included patients from 49 hospitals in six countries aged at least 18 years with histologically confirmed, advanced, IDH1-mutant cholangiocarcinoma who had progressed on previous therapy, and had up to two previous treatment regimens for advanced disease, an Eastern Cooperative Oncology Group performance status score of 0 or 1, and a measurable lesion as defined by Response Evaluation Criteria in Solid Tumors version 1.1. Patients were randomly assigned (2:1) with a block size of 6 and stratified by number of previous systemic treatment regimens for advanced disease to oral ivosidenib 500 mg or matched placebo once daily in continuous 28-day cycles, by means of an interactive web-based response system. Placebo to ivosidenib crossover was permitted on radiological progression per investigator assessment. The primary endpoint was progression-free survival by independent central review. The intention-to-treat population was used for the primary efficacy analyses. Safety was assessed in all patients who had received at least one dose of ivosidenib or placebo. Enrolment is complete; this study is registered with ClinicalTrials.gov, NCT02989857.
Findings
Between Feb 20, 2017, and Jan 31, 2019, 230 patients were assessed for eligibility, and as of the Jan 31, 2019 data cutoff date, 185 patients were randomly assigned to ivosidenib (n=124) or placebo (n=61). Median follow-up for progression-free survival was 6·9 months (IQR 2·8–10·9). Progression-free survival was significantly improved with ivosidenib compared with placebo (median 2·7 months [95% CI 1·6–4·2] vs 1·4 months [1·4–1·6]; hazard ratio 0·37; 95% CI 0·25–0·54; one-sided p<0·0001). The most common grade 3 or worse adverse event in both treatment groups was ascites (four [7%] of 59 patients receiving placebo and nine [7%] of 121 patients receiving ivosidenib). Serious adverse events were reported in 36 (30%) of 121 patients receiving ivosidenib and 13 (22%) of 59 patients receiving placebo. There were no treatment-related deaths.
Interpretation
Progression-free survival was significantly improved with ivosidenib compared with placebo, and ivosidenib was well tolerated. This study shows the clinical benefit of targeting IDH1 mutations in advanced, IDH1-mutant cholangiocarcinoma.
Introduction
Isocitrate dehydrogenase 1 (IDH1) mutations are detected in approximately 13% (9%) of intrahepatic cholangiocarcinomas globally,1 with varying frequency.2–4 Preclinical data show the role of IDH mutations in cholangio carcinoma pathogenesis through their effect on liver progenitor cell differentiation and proliferation.5 Ivosidenib (AG-120) is an oral, potent, targeted inhibitor of mutated IDH1 approved for patients with newly diagnosed acute myeloid leukaemia who are ineligible for intensive chemotherapy, and for relapsed or refractory acute myeloid leukaemia.6–8 In a phase 1 dose-escalation and expansion study, ivosidenib showed a median progression-free survival of 3·8 months; 6-month progression-free survival of 40·1% and 12-month progression-free survival of 21·8%; a median overall survival of 13·8 months; and a favourable safety profile in patients with previously treated, IDH1-mutant, advanced cholangiocarcinoma.9 We report herein the results of a randomised, phase 3 study investigating the efficacy and safety of ivosidenib in this population after progression on standard chemotherapy.
Methods
Study design and participants
This multicentre, randomised, double-blind, placebo-controlled, phase 3 study was done across 49 hospitals in six countries (France, Italy, South Korea, Spain, the UK, and the USA; appendix pp 3–4). Eligible patients were aged 18 years or older with histologically confirmed, advanced, IDH1-mutant cholangiocarcinoma (appendix p 9). Up to two previous treatment regimens for advanced disease (unresectable or metastatic), with one gemcitabine-based or fluorouracil-based chemotherapy and no previous mutant IDH inhibitor therapy, were required. Progression at inclusion was determined and confirmed by the investigator on the basis of available medical history or imaging report. Additional key eligibility criteria included a life expectancy of at least 3 months; an Eastern Cooperative Oncology Group (ECOG) performance status score of 0 or 1;10 a measurable lesion as defined by Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1;11 and adequate haematological, hepatic, and renal function (appendix pp 9, 17). IDH1 mutation status was confirmed centrally by next-generation sequencing on formalin-fixed, paraffin-embedded tumour tissue (from a banked tumour sample collected preferably within the last 3 years or a fresh tumour biopsy) by means of the Oncomine Focus Assay (Thermo Fisher Scientific, Waltham, MA, USA) in a Clinical Laboratory Improvement Amendments-certified laboratory (appendix p 5).
Patients who had received previous local therapy (including but not limited to embolisation, chemo-embolisation, radiofrequency ablation, or radiotherapy) were eligible provided measurable disease fell outside of the treatment field, or within the field but had shown at least 20% growth in tumour size since the post-treatment assessment. Patients were excluded if they had received systemic anticancer therapy or an investigational agent less than 2 weeks before day 1 (washout from previous immune-based anticancer therapy being 4 weeks); had received radiotherapy to metastatic sites of disease less than 2 weeks before day 1; or had undergone hepatic irradiation, chemo embolisation, and radio frequency ablation less than 4 weeks before day 1. Patients with the following comorbidities were not permitted: active cardiac disease within 6 months before the start of study treatment; myocardial infarction; unstable angina or stroke; active hepatitis B or C viral infections; known positive HIV antibody results, or AIDS-related illness. This study was done according to the International Conference on Harmonisation of Good Clinical Practice guidelines and the principles of the Declaration of Helsinki.12,13 Approval from the institutional review board and international ethics committee was obtained at each study site. Patients provided written, informed consent before participating in the study. The complete study protocol is available in the appendix.
Randomisation and masking
Patients were enrolled and treated by the investigators at participating study centres on an outpatient basis. Patients were randomly assigned (2:1) to ivosidenib or matched placebo, with a block size of 6, and stratified by number of previous systemic treatment regimens for advanced disease (one vs two). Randomisation into the two treatment groups was implemented by an interactive web-based response system and generated by an independent statistical group. Ivosidenib and placebo were packaged and labelled identically to ensure that study personnel remained masked to treatment assignment. Patients, investigators and their teams, and designated individuals from the sponsor were masked to study treatment until disease progression as assessed by the investigator (appendix pp 4–5).
Procedures
Ivosidenib 500 mg or placebo was given orally once daily in continuous 28-day cycles (plus or minus 2 days), starting on cycle 1 day 1. Study visits were done every other week during cycles 1–3 (days 1 and 15) and on day 1 of subsequent cycles. Treatment was to continue until disease progression as determined by investigator, development of other unacceptable toxicity, confirmed pregnancy, death, withdrawal of consent, loss to follow-up, or study unblinding or ending. Continuation of treatment after radiographic disease progression was permitted, provided that the investigator deemed that there was a clinical benefit. A post-treatment follow-up visit for safety occurred 28 days (no more than 33 days) after the last dose of study drug. Dose modifications of ivosidenib or placebo from 500 mg to 250 mg were permitted in the study for management of adverse events. If more than one adverse event occurred that required a dose modification, on resolution of all adverse events to baseline or grade 1, ivosidenib or placebo dose was reduced to 250 mg. Re-escalation was allowed with approval from the medical monitor.
Radiographic assessment (CT or MRI) for evaluation of disease response was done by the investigator from cycle 1, day 1 every 6 weeks (plus or minus 5 days) through week 48, and every 8 weeks (plus or minus 5 days) thereafter. The central independent radiology centre (IRC) did not perform real-time confirmation of locally determined radiographic progression. Quality of life (QOL) was assessed by means of the European Organisation for Research and Treatment of Cancer Quality of Life Question naire Core 30 (EORTC QLQ-C30) and cholangio carcinoma and gallbladder cancer module (EORTC QLQ-BIL21), Patient Global Impression (PGI) questions adapted from the National Institute of Mental Health PGI of change (PGI-C) for three prespecified domains of interest (physical functioning, pain, and appetite loss), and the 5-level EuroQol 5-Dimension (EQ-5D-5L) for future health economic modelling.14–17 A detailed QOL assessment schedule is provided in the appendix (p 6). Safety and tolerability were assessed from the first dose of study treatment by the incidence of treatment-emergent adverse events; by severity and type of adverse event (per the National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.03); and by evaluation of vital signs, ECOG performance status, clinical laboratory test results, and electrocardiograms (because ivosidenib-treated patients can develop QT prolongation).6 Adverse events are reported for patients before crossover unless otherwise specified.
Patients who discontinued treatment for reasons other than disease progression or withdrawal of consent entered progression-free survival follow-up (every 6 weeks through week 48, and every 8 weeks thereafter) until documented disease progression or the start of new cancer treatment. On the basis of investigator-confirmed radiographic progression, unmasking was permitted and eligible patients receiving placebo were permitted to receive open-label ivosidenib. Follow-up for overall survival occurred every 12 weeks after the end of treatment, unless the patient was in follow-up for progression-free survival, and continued after the primary endpoint was reached.
Blood samples were drawn before and after dosing to establish circulating plasma concentrations of ivosidenib and D-2-hydroxyglutarate (2-HG), an oncometabolite that accumulates as a result of IDH mutations.18
Outcomes
The primary endpoint was progression-free survival as assessed by the central IRC based on RECIST version 1.1 assessment. Progression-free survival was defined as the time from the date of randomisation to the date of first docu mentation of disease progression or death owing to any cause, whichever occurred first.
Secondary endpoints were overall survival; objective response rate by RECIST version 1.1; duration of response and time to response (assessed by the investigator and IRC); progression-free survival (by investigator review); pharmacokinetics and pharma codynamics; QOL assessed by EORTC QLQ-C30 and EORTC QLQ-BIL21 change from baseline and PGI-C anchor questions; and EQ-5D-5L for health economic modelling. EQ-5D-5L findings based on final data will be reported elsewhere.
Statistical analysis
Assuming a hazard ratio (HR) of 0·5 for progression-free survival, a total of 131 progression-free survival events would be required to provide 96% power at a one-sided α level of significance of 0·025 to reject the null hypothesis. Overall survival analyses were done once at the time of the final analysis for progression-free survival and will be done again at the occurrence of 150 overall survival events, approximately 24 months after the last patient has been randomised. Assuming an HR of 0·67 for overall survival, a total of 150 deaths will provide 64% power at a one-sided α level of significance of 0·025.
The intention-to-treat population, comprising all randomly assigned patients within the designated treatment group, was used for primary efficacy analyses and other analyses unless otherwise specified. The safety analysis population included all patients who received at least one dose of study treatment, with the actual treatment received before crossover as the treatment group unless otherwise specified. The crossover population included a subset of placebo patients who crossed over and received open-label ivosidenib upon radiographic disease progression (appendix p 5).
A Cox regression model stratified by the randomisation stratification factor was used to estimate the HR and the 95% CI for the progression-free and overall survival comparison of the ivosidenib and placebo groups as well as the overall survival analyses. A log-rank test stratified by the randomisation stratification factor was used to assess significance. 95% CIs for the survival rate estimates were calculated via log–log transformation. Patients starting treatment with a new anticancer therapy before IRC-assessed progression or death were censored at the last adequate assessment before the new anticancer therapy. Patients alive without a post-baseline assessment were censored at the randomisation date. Patients who did not progress or die by the data cutoff date were censored at the last adequate assessment date. Patients with progression or death following a long gap (≥2 consecutive scheduled assessments missing) were censored at the date of the last adequate assessment before the gap. For overall survival, patients without docu mentation of death at the time of the data cutoff date were censored at the date the patient was last known to be alive or the data cutoff date, whichever was earlier.
The rank-preserving structural failure time (RPSFT) method was used to reconstruct the survival curve (prespecified exploratory analysis) for patients receiving placebo as if crossover had never occurred (appendix p 5).19 RPSFT assumes that the treatment effect is the same for all patients, regardless of when the treatment is given.
Subgroup analyses by previous line of therapy, sex, extent of disease at screening, cholangiocarcinoma type, ECOG performance status score, and geographical region were performed on progression-free survival per IRC and overall survival, and included Kaplan-Meier summaries, unstratified log-rank test, p values, and HRs from Cox regression models. The proportional hazard assumption was met on the basis of graphic check.
Mixed-effect models with repeated measurements (with baseline score, treatment, visit, and treatment-by-visit as fixed effects and patient as random effect) were used on change scores from baseline to cycle 2 day 1 for subscales of the EORTC QLQ-C30 and QLQ-BIL21 corresponding to the three domains of interest (physical functioning, pain, and appetite loss; appendix p 6).14,15 Clinically meaningful change thresholds on these subscales were estimated by means of the respective PGI-C ratings as anchors (appendix p 6). The focus was on cycle 2 day 1, considering the availability of QOL data. QOL analyses were exploratory in nature; therefore, type 1 error control for multiplicity was not considered.
All time-to-event endpoints were estimated by means of Kaplan-Meier methods. Descriptive statistics were used to summarise safety data, response rates, QOL data, and pharmacokinetic and pharmacodynamic data. All reported p values are one-sided unless otherwise specified. Statistical analyses were done with SAS software (version 9.4).
An independent data and safety monitoring board regularly reviewed the data to ensure treatment safety and proper study conduct. This study is registered with ClinicalTrials.gov, NCT02989857.
Role of the funding source
The funder had a role in study design, data collection, data analysis, and data interpretation. Medical writing support was provided by the funder. The first and last authors had full access to all of the data in the study and had final responsibility for the decision to submit for publication.
Results
Between Feb 20, 2017, and Jan 31, 2019, 230 patients were assessed for eligibility and as of Jan 31, 2019 (the analysis cutoff date based on investigator-assessed progression-free survival), 185 (80%) patients were randomly assigned to ivosidenib (n=124) or placebo (n=61; figure 1). 44 (19%) patients were considered ineligible, mainly owing to not having documented IDH1-mutant disease or having an ECOG performance status score of 2 or greater. Baseline demographic and disease characteristics were similar in the ivosidenib and placebo groups; among all 185 patients, R132C was the most prevalent IDH1 mutation (129 [70%]), 171 (92%) had metastatic disease, and 86 (46%) had received two previous lines of therapy (table 1). Most patients (173 [94%] of 185) had received a previous platinum-based therapy (118 in the ivosidenib group and 55 in the placebo group). At the data cutoff, 35 (57%) of 61 patients in the placebo group had crossed over to receive open-label ivosidenib. Of the remaining 26 patients, 13 (50%) had died, eight (31%) were still receiving placebo, two never received study drug, two withdrew consent, and one received another treatment. Among the 121 patients who received ivosidenib, 14 (12%) were permitted to continue treatment beyond radiographic progression, as determined by the local investigator.
Figure 1: Trial profile.

ITT=intention-to-treat. *As of data cutoff, Jan 31, 2019.
Table 1:
Demographic and baseline characteristics
| Ivosidenib (n=124) | Placebo (n=61) | |
|---|---|---|
| Sex | ||
| Female | 80 (65%) | 37 (61%) |
| Male | 44 (35%) | 24 (39%) |
| Age, years | 61 (33–80) | 63 (40–83) |
| Previous lines of therapy | ||
| One | 66 (53%) | 33 (54%) |
| Two | 58 (47%) | 28 (46%) |
| Eastern Cooperative Oncology Group performance status | ||
| 0 | 49 (40%) | 19 (31%) |
| 1 | 74 (60%) | 41 (67%) |
| 2 | 0 | 1 (2%) |
| 3 | 1 (1%) | 0 |
| Cholangiocarcinoma type at diagnosis | ||
| Intrahepatic | 111 (90%) | 58 (95%) |
| Extrahepatic | 1 (1%) | 1 (2%) |
| Perihilar | 4 (3%) | 0 |
| Unknown | 8 (6%) | 2 (3%) |
| Extent of disease at screening | ||
| Local–regional | 9 (7%) | 5 (8%) |
| Metastatic | 115 (93%) | 56 (92%) |
| Liver cirrhosis at screening | ||
| Yes | 6 (5%) | 3 (5%) |
| Hepatitis B | 1 (1%) | 0 |
| Hepatitis C | 0 | 1 (2%) |
| Alcohol | 1 (1%) | 0 |
| Other | 4 (3%) | 2 (3%) |
| No | 118 (95%) | 58 (95%) |
| IDH1 mutation | ||
| R132C | 84 (68%) | 45 (74%) |
| R132L | 21 (17%) | 7 (11%) |
| R132G | 17 (14%) | 6 (10%) |
| R132S | 2 (2%) | 1 (2%) |
| R132H | 0 | 2 (3%) |
| CA19-9 concentration at | 42.0 | 39.0 |
| baseline,* units per mL | 0–18 560·0)† | (0·1–11 529·0)† |
Data are median (range) or n (%). IDH1=Isocitrate dehydrogenase 1.
From patients included in the safety analysis set, before crossover.
Placebo, n=59; ivosidenib, n=121.
The median follow-up for progression-free survival by IRC assessment was 6·9 months (IQR 2·8–10·9). 76 (61%) of 124 patients in the ivosidenib group and 50 (82%) of 61 patients in the placebo group had progression-free survival events by IRC assessment. Progression-free survival by IRC assessment was longer for patients in the ivosidenib group (median 2·7 months [95% CI 1·6–4·2]) than for those in the placebo group (1·4 months [1·4–1·6]; HR 0·37; 95% CI 0·25–0·54; one-sided p<0·0001; figure 2A). Progression-free survival at 6 months was 32% (95% CI 23–42) and 22% (13–32) at 12 months for ivosidenib; no patients in the placebo group were free from progression for 6 months or more. Progression-free survival benefit according to subgroups is shown in figure 2B. Progression-free survival by investigator review was similar to that observed by IRC assessment (median 2·7 months [95% CI 1·6–3·6] for ivosidenib vs 1·4 months [1·4–2·5] for placebo; HR 0·47; 95% CI 0·33–0·68; p<0·0001), with an overall concordance of 77% for progression-free survival status between investigator and IRC assessment (appendix p 10). Progression-free survival by investigator assessment for patients who crossed over from placebo to ivosidenib is shown in the appendix (p 11).
Figure 2: Progression-free survival assessed by the independent radiology centre before crossover in the intention-to-treat population.

(A) The Kaplan-Meier plot of the probability of progression-free survival among patients receiving ivosidenib compared with those receiving placebo. Scans after local disease progression per investigator assessment were not submitted to the independent radiology centre for evaluation and thus were excluded from this analysis. Cross marks indicate censored observations. (B) Forest plot of progression-free survival HRs for key subgroups. Scans after local disease progression per investigator assessment were not submitted to the independent radiology centre for evaluation and thus were excluded from this analysis. The HR for the overall subgroup was calculated from the stratified Cox regression model and for each subgroup from the unstratified Cox regression model. The number of previous lines of therapy was based on the actual previous lines that patients received per eligibility, reviewed by the sponsor’s medical monitor. If patients had both local and metastatic status, disease was considered to be metastatic. Perihilar disease was included as extrahepatic disease. The baseline ECOG performance status measurement was defined as the most recent measurement before the first dose of study drug. If patients did not receive study drug, the latest assessment was considered to be the baseline assessment. ECOG=Eastern Cooperative Oncology Group. HR=hazard ratio.
Median overall survival (intention-to-treat population) was 10·8 months (95% CI 7·7–17·6) for the ivosidenib group versus 9·7 months (4·8–12·1) for the placebo group (HR 0·69 [95% CI 0·44–1·10]; p=0·060) based on 78 deaths (29 in the placebo group and 49 in the ivosidenib group) and crossover of 35 patients from the placebo group (figure 3). The 6-month overall survival rate for ivosidenib was 67% (95% CI 56–75) and the 12-month rate was 48% (36–59), versus 59% (44–71) and 38% (22–54), respectively, for placebo. Overall survival by subgroup is reported in the appendix (p 12). The RPSFT-adjusted median overall survival was 6·0 months (95% CI 3·6–6·3) for the placebo group (HR 0·46 [95% CI 0·28–0·75]; p=0·0008).
Figure 3: Overall survival in the intention-to-treat population.

Cross marks indicate censored observations. RPSFT=rank-preserving structural failure time.
The objective response rate per IRC assessment for ivosidenib was three (2%) of 124 patients, comprising three partial responses (appendix pp 7, 13). 63 (51%) of 124 patients in the ivosidenib group had stable disease (appendix p 18). No patients in the placebo group had an objective response and 17 (28%) of 61 patients in the placebo group had stable disease (appendix p 18). Best overall response per investigator assessment before and after crossover are in the appendix (pp 14, 19). Characteristics of patients receiving ivosidenib achieving a confirmed partial response per IRC before unmasking are described in the appendix (p 20).
The median duration of treatment was 2·6 months (IQR 1·4–6·0) for ivosidenib and 1·6 months (1·1–2·7) for placebo (figure 4; see appendix p 14 for duration by investigator assessment). All grade 3–5 treatment-emergent adverse events that occurred before crossover, and their corresponding grade 1–2 treatment-emergent adverse events, are shown in table 2. An expanded list of adverse events, including treatment-emergent adverse events reported in the ivosidenib population after crossover are in the appendix (p 21). The most common grade 3 or worse adverse event in both treat ment groups was ascites (four [7%] of 59 patients who received placebo and nine [7%] of 121 patients who received ivosidenib; table 2). Treatment-related adverse events are shown in the appendix (p 22). Serious adverse events were reported for 36 (30%) of 121 patients receiving ivosidenib, and were deemed treatment related in three (2%) patients (grade 4 hyper bilirubinaemia, grade 3 jaundice cholestatic, grade 2 electrocardiogram QT prolonged, and grade 3 pleural effusion; hyperbilirubinaemia and jaundice cholestatic were recorded for the same patient). Serious adverse events were reported in 13 (22%) of 59 patients receiving placebo; none were deemed treatment related. 14 (12%) patients receiving ivosidenib and ten (17%) patients receiving placebo died within 30 days of receiving the last dose. Four (3%) of 121 patients receiving ivosidenib had an adverse event leading to death (pneumonia, sepsis, intestinal obstruction, and pulmonary embolism; n=1 each), none of which were assessed by the investigator as treatment related (appendix p 23), and ten died owing to progressive disease. No treatment-emergent adverse events leading to death were reported in the placebo group, with all ten deaths due to progressive disease.
Figure 4: Treatment duration and response assessed by the independent radiology centre before crossover in the intention-to-treat population.
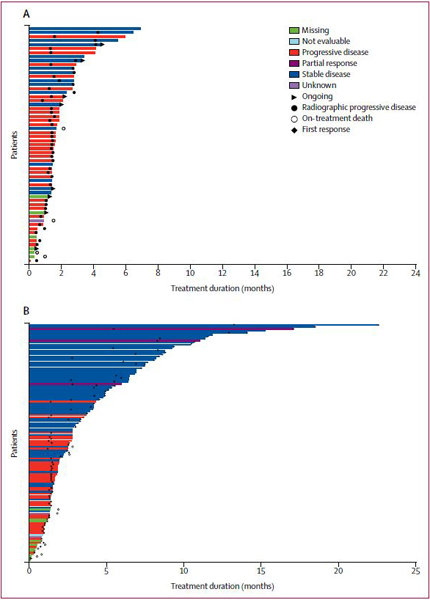
(A) Patients receiving placebo. (B) Patients receiving ivosidenib. Partial response required confirmation per Response Evaluation Criteria in Solid Tumors version 1.1. Stable disease occurring <38 days from the randomisation date was deemed to be unknown.
Table 2:
Treatment-emergent adverse events (TEAEs)
| Ivosidenib (n=121) |
Placebo (n=59) |
|||||||
|---|---|---|---|---|---|---|---|---|
| Grade 1–2 | Grade 3 | Grade 4 | Grade 5 | Grade 1–2 | Grade 3 | Grade 4 | Grade 5 | |
| Nausea | 40 (33%) | 3 (2%) | 0 | 0 | 14 (24%) | 1 (2%) | 0 | 0 |
| Diarrhoea | 37 (31%) | 0 | 0 | 0 | 9 (15%) | 0 | 0 | 0 |
| Fatigue | 28 (23%) | 4 (3%) | 0 | 0 | 9 (15%) | 1 (2%) | 0 | 0 |
| Cough | 25 (21%) | 0 | 0 | 0 | 5 (8%) | 0 | 0 | 0 |
| Abdominal pain | 23 (19%) | 3 (2%) | 0 | 0 | 7 (12%) | 1 (2%) | 0 | 0 |
| Decreased appetite | 21 (17%) | 2 (2%) | 0 | 0 | 11 (19%) | 0 | 0 | 0 |
| Vomiting | 20 (17%) | 3 (2%) | 0 | 0 | 10 (17%) | 0 | 0 | 0 |
| Ascites | 16 (13%) | 9 (7%) | 0 | 0 | 5 (8%) | 4 (7%) | 0 | 0 |
| Asthenia | 15 (12%) | 0 | 0 | 0 | 6 (10%) | 2 (3%) | 0 | 0 |
| Constipation | 15 (12%) | 0 | 0 | 0 | 10 (17%) | 0 | 0 | 0 |
| Oedema peripheral | 15 (12%) | 0 | 0 | 0 | 6 (10%) | 0 | 0 | 0 |
| Pyrexia | 15 (12%) | 0 | 0 | 0 | 6 (10%) | 0 | 0 | 0 |
| Anaemia | 14 (12%) | 4 (3%) | 0 | 0 | 3 (5%) | 0 | 0 | 0 |
| Headache | 13 (11%) | 0 | 0 | 0 | 4 (7%) | 0 | 0 | 0 |
| Dyspnoea | 12 (10%) | 1 (1%) | 0 | 0 | 7 (12%) | 2 (3%) | 0 | 0 |
| Abdominal distension | 10 (8%) | 1 (1%) | 0 | 0 | 5 (8%) | 0 | 0 | 0 |
| Electrocardiogram QT prolonged | 10 (8%) | 1 (1%) | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Back pain | 10 (8%) | 0 | 0 | 0 | 4 (7%) | 1 (2%) | 0 | 0 |
| Alanine aminotransferase increased | 8 (7%) | 2 (2%) | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Hypokalaemia | 8 (7%) | 1 (1%) | 0 | 0 | 2 (3%) | 0 | 1 (2%) | 0 |
| Insomnia | 8 (7%) | 1 (1%) | 0 | 0 | 3 (S%) | 0 | 0 | 0 |
| Aspartate aminotransferase increased | 7 (6%) | 6 (5%) | 0 | 0 | 2 (3%) | 1 (2%) | 0 | 0 |
| Blood alkaline phosphatase increased | 7 (6%) | 3 (2%) | 0 | 0 | 3 (5%) | 3 (5%) | 0 | 0 |
| Hypoalbuminaemia | 7 (6%) | 0 | 0 | 0 | 3 (5%) | 1 (2%) | 0 | 0 |
| Hyponatraemia | 6 (5%) | 4 (3%) | 2 (2%) | 0 | 1 (2%) | 5 (8%) | 1 (2%) | 0 |
| White blood cell count decreased | 6 (5%) | 2 (2%) | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Arthralgia | 6 (5%) | 1 (1%) | 0 | 0 | 4 (7%) | 0 | 0 | 0 |
| Weight decreased | 6 (5%) | 1 (1%) | 0 | 0 | 1 (2%) | 1 (2%) | 0 | 0 |
| Hypertension | 6 (5%) | 0 | 0 | 0 | 1 (2%) | 1 (2%) | 0 | 0 |
| Blood bilirubin increased | 5 (4%) | 7 (6%) | 0 | 0 | 3 (5%) | 1 (2%) | 0 | 0 |
| Pleural effusion | 4 (3%) | 1 (1%) | 1 (1%) | 0 | 2 (3%) | 0 | 0 | 0 |
| Confusional state | 4 (3%) | 1 (1%) | 0 | 0 | 4 (7%) | 0 | 0 | 0 |
| Pruritus | 4 (3%) | 1 (1%) | 0 | 0 | 2 (3%) | 0 | 0 | 0 |
| Urinary tract infection | 4 (3%) | 1 (1%) | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Hyperkalaemia | 3 (2%) | 3 (2%) | 0 | 0 | 3 (5%) | 2 (3%) | 0 | 0 |
| Hyperbilirubinaemia | 3 (2%) | 1 (1%) | 2 (2%) | 0 | 0 | 0 | 0 | 0 |
| Platelet count decreased | 2 (2%) | 3 (2%) | 0 | 0 | 3 (5%) | 0 | 0 | 0 |
| Fall | 2 (2%) | 2 (2%) | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Hypercalcaemia | 2 (2%) | 1 (1%) | 0 | 0 | 5 (8%) | 1 (2%) | 0 | 0 |
| Rash maculo-papular | 2 (2%) | 1 (1%) | 0 | 0 | 3 (5%) | 0 | 0 | 0 |
| Thrombocytopenia | 2 (2%) | 1 (1%) | 0 | 0 | 2 (3%) | 0 | 0 | 0 |
| Dysphagia | 2 (2%) | 0 | 1 (1%) | 0 | 2 (3%) | 0 | 0 | 0 |
| Lymphocyte count decreased | 2 (2%) | 0 | 0 | 0 | 0 | 2 (3%) | 0 | 0 |
| Dehydration | 1 (1%) | 4 (3%) | 0 | 0 | 1 (2%) | 1 (2%) | 0 | 0 |
| Hypophosphataemia | 1 (1%) | 3 (2%) | 0 | 0 | 0 | 3 (5%) | 0 | 0 |
| Pneumonia | 1 (1%) | 2 (2%) | 0 | 1 (1%) | 0 | 1 (2%) | 0 | 0 |
| Acute kidney injury | 1 (1%) | 2 (2%) | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Jaundice | 1 (1%) | 2 (2%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Pain | 1 (1%) | 2 (2%) | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Hypotension | 1 (1%) | 1 (1%) | 0 | 0 | 1 (2%) | 1 (2%) | 0 | 0 |
| Rectal haemorrhage | 1 (1%) | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Transaminases increased | 1 (1%) | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Abdominal pain lower | 1 (1%) | 0 | 0 | 0 | 0 | 1 (2%) | 0 | 0 |
| Hepatic cirrhosis | 1 (1%) | 0 | 0 | 0 | 0 | 1 (2%) | 0 | 0 |
| Jaundice cholestatic | 0 | 2 (2%) | 1 (1%) | 0 | 0 | 0 | 0 | 0 |
| Cholangitis | 0 | 2 (2%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Hepatic failure | 0 | 2 (2%) | 0 | 0 | 0 | 1 (2%) | 0 | 0 |
| Abdominal infection | 0 | 1 (1%) | 1 (1%) | 0 | 0 | 0 | 0 | 0 |
| Intestinal obstruction | 0 | 1 (1%) | 0 | 1 (1%) | 0 | 0 | 0 | 0 |
| Neutrophil count decreased | 0 | 0 | 2 (2%) | 0 | 0 | 0 | 0 | 0 |
| Bile duct obstruction | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Bile duct stenosis | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Biliary sepsis | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Cachexia | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Cholangitis acute | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Cognitive disorder | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Device-related infection | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Encephalopathy | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Escherichia coli bacteraemia | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Failure to thrive | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Gastroenteritis | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Gastrointestinal haemorrhage | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Hip fracture | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Malnutrition | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Mental status changes | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Muscular weakness | 0 | 1 (1%) | 0 | 0 | 2 (3%) | 0 | 0 | 0 |
| Parainfluenza virus infection | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Portal vein thrombosis | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Restlessness | 0 | 1 (1%) | 0 | 0 | 1 (2%) | 0 | 0 | 0 |
| Staphylococcal infection | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Upper gastrointestinal haemorrhage | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Vascular access complication | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Arterial injury | 0 | 0 | 1 (1%) | 0 | 0 | 0 | 0 | 0 |
| γ-glutamyltransferase increased | 0 | 0 | 1 (1%) | 0 | 0 | 0 | 1 (2%) | 0 |
| Pulmonary embolism | 0 | 0 | 0 | 1 (1%) | 0 | 0 | 0 | 0 |
| Sepsis | 0 | 0 | 0 | 1 (1%) | 0 | 0 | 2 (3%) | 0 |
Data are n (%). TEAE is defined as any adverse event that occurred between the first dose of any study drug and 28 days following the last dose. Grade 1–2 adverse events reported in ≥10% of patients are shown, and all grade 3–4 are shown. TEAEs that occurred after crossover from the placebo group to the ivosidenib group are reported in the appendix (p 21). TEAEs are sorted in descending frequency based on the grade 1–2 column for ivosidenib.
Treatment-emergent adverse events requiring a dose reduction occurred in four (3%) of 121 patients receiving ivosidenib versus none receiving placebo. Treatment-emergent adverse events leading to treatment discontinuation occurred in seven (6%) of 121 patients receiving ivosidenib versus five (8%) receiving placebo. Treatment-related adverse events leading to treatment discontinuation occurred in two (2%) of 121 patients receiving ivosidenib (grade 2 generalised oedema and grade 4 hyperbilirubinaemia).
At baseline, 113 (91%) of 124 patients in the ivosidenib group and 52 (85%) of 61 patients in the placebo group completed the EORTC QLQ-C30 assessment, and 107 (86%) and 51 (84%) completed the QLQ-BIL21 assessment. At cycle 2 day 1, EORTC QLQ-C30 change scores from baseline were available for 62 (55%) of 113 patients in the ivosidenib group and 20 (38%) of 52 patients in the placebo group; and QLQ-BIL21 change scores from baseline were available for 60 (56%) of 107 patients in the ivosidenib group and 19 (37%) of 51 patients in the placebo group. The decline from baseline at cycle 2 day 1 on the EORTC QLQ-C30 physical functioning subscale (higher score denoting better functioning) was significantly less for patients in the ivosidenib group (n=62; least squares mean −3·4 [SE 1·81]) than for patients in the placebo group (n=20; –13·1 [3·04]; difference 9·8 [95% CI 2·8–16·7]; p=0·0059; appendix pp 15, 24). The decline was clinically meaningful in the placebo arm only (appendix pp 7–8, 25). Differences in change from baseline for pain and appetite loss subscales were not significant between groups, and clinically meaningful changes could not be established owing to data availability (appendix p 24).
Pharmacokinetic and pharmacodynamic parameters observed in this study were consistent with previous findings.9,20 At the cycle 2 day 1 visit, samples were collected from 126 patients receiving ivosidenib (99 active ivosidenib patients and 27 crossover patients) for pharmacokinetic and pharmacodynamic analyses. After one cycle of ivosidenib, mean trough plasma 2-HG decreased by up to 97% from baseline to cycle 2 day 1, to concentrations similar to those observed in healthy individuals, versus a 47% increase with placebo from baseline to cycle 2 day 1 (two-sided p<0·0001). This decrease was main tained throughout continued ivosidenib dosing (up to 19 cycles), whereas plasma 2-HG remained elevated for patients receiving placebo during the observation period (appendix p 16).
Discussion
This randomised, phase 3 study shows the clinical benefit of targeting mutant IDH1 in patients with advanced, IDH1-mutant cholangiocarcinoma. The increase in progression-free survival for ivosidenib compared with placebo is clinically meaningful. Although the absolute improvement in median progression-free survival seems modest, the statistical strength of the HR reflects a high reduction in risk of progression, along with a substantial improvement in the proportion of patients progression free at 6 and 12 months. The benefit is independent of number of previous therapies and is consistent across most subgroups. This improvement in progression-free survival is important in the context of a favourable safety and tolerability profile in the chemotherapy-refractory setting. The disease control rate associated with ivosidenib was primarily driven by stable disease, reflecting the mechanism of action of ivosidenib, which is specific to epigenetic modifications promoting cellular differentiation rather than a direct cytotoxic mechanism.
Although the crossover design enabled 35 (57%) of 59 patients receiving placebo to receive ivosidenib at disease progression, there was still a favourable overall survival result for ivosidenib versus placebo in the intention-to-treat population. Use of RPSFT modelling to adjust for the effect of placebo–ivosidenib crossover resulted in a significant improvement in overall survival, with a difference of 4·8 months in median overall survival between ivosidenib and placebo. The RPSFT-adjusted overall survival results from the placebo group are consistent with survival outcomes from historical and recent data for patients managed with best supportive care, active symptom control, or second-line chemotherapy.21–24 Despite small post-baseline sample sizes, the clinical benefit of ivosidenib was further supported by EORTC QLQ-C30 physical functioning subscale scores, indicating that patients receiving placebo had a significantly greater decline in physical functioning than did patients receiving ivosidenib at cycle 2 day 1. Moreover, a favourable pharma cokinetic and pharmacodynamic profile was observed in patients with advanced IDH1-mutant cholangiocarcinoma who received once-daily 500 mg ivosidenib. Detailed pharma cokinetic and pharma codynamic data for this patient population will be published elsewhere.
Ivosidenib was well tolerated; the most common treatment-emergent adverse events in patients receiving ivosidenib were low-grade diarrhoea, nausea, and fatigue. The rates of treatment discontinuation or dose reduction were low. Although the findings reported here are specific to patients with IDH1-mutant, advanced cholangio carcinoma, representing a relatively small subset of the disease popu lation,1 the incidence of intrahepatic cholangiocarcinoma is increasing inter nationally25,26 and represents an area of growing unmet need.1,9,27
The study has some limitations. Although median overall survival in patients receiving ivosidenib was longer than in those receiving placebo, the difference was not significant; this might be partly attributed to the effect of the placebo–ivosidenib crossover and the data not being mature at the time of primary analysis (42% of events). Despite this, there was a significant improve ment in overall survival for ivosidenib versus RPSFT-adjusted data for placebo. Without established efficacious alternatives, there was no justification for withholding ivosidenib from patients receiving placebo. Additionally, the limited patient-reported outcome data collection prevented a thorough evaluation of relevant QOL parameters in this specific population.
In conclusion, ivosidenib therapy significantly improved progression-free survival and overall survival after adjusting for crossover, with a favourable safety profile, in patients with advanced, IDH1-mutant cholangiocarcinoma who had progressed on standard chemotherapy. This study shows the feasibility and clinical benefit of targeting a molecularly defined subgroup of cholangiocarcinoma and warrants tumour mutation profiling as a new standard of care in this heterogeneous disease.28–30
Supplementary Material
Research in context.
Evidence before this study
The prognosis for intrahepatic cholangiocarcinoma is poor, with 5-year survival rates below 10%, and global incidence of the disease is increasing. Surgery is the only curative option for localised cholangiocarcinoma, although rates of recurrence are high. For unresectable or metastatic disease, chemotherapy remains the primary treatment strategy, with gemcitabine plus cisplatin being the standard of care. We searched PubMed for manuscripts published between June 20, 2006, and Feb 1, 2016, with no language restrictions, using the terms “metastatic cholangiocarcinoma AND treatment”, and “IDH1 AND cholangiocarcinoma”. We identified several reports describing mutations in the gene for the metabolic enzyme isocitrate dehydrogenase 1 (IDH1) in approximately 20% of patients with intrahepatic cholangiocarcinoma. Moreover, we evaluated preclinical and clinical work published between June 20, 2006, and Feb 1, 2016, including that presented at scientific congresses, to understand the biological effect of the disease, as well as the outcomes among previously treated patients with advanced biliary tract cancers receiving chemotherapy. Ivosidenib is a potent, oral inhibitor of mutated IDH1. In a phase 1 dose-escalation and expansion study, ivosidenib showed promising progression-free survival and overall survival outcomes, combined with a favourable safety and tolerability profile, in previously treated patients with IDH1-mutant, advanced cholangiocarcinoma.
Added value of this study
This study establishes the efficacy and safety of ivosidenib in patients with IDH1-mutant cholangiocarcinoma who had progressed on previous standard chemotherapy. Ivosidenib treatment resulted in a significant improvement in progression-free survival, with a favourable safety and tolerability profile.
Implications of all the available evidence
With no approved targeted therapies, and modest survival outcomes with chemotherapy in patients with unresectable or metastatic cholangiocarcinoma, there is an urgent need for new therapies. Although cholangiocarcinoma-associated genetic alterations are now better defined, there are still no approved targeted therapies in this disease. This study of ivosidenib shows a benefit of targeting mutant IDH1 in patients with advanced, IDH1-mutant cholangiocarcinoma, and highlights the clinical relevance of tumour mutation profiling in the management of this rare cancer with poor outcomes. Results from this study and the follow-up mature overall survival data will be used to support an application for regulatory approval of the drug to the US Food and Drug Administration and other agencies in the future.
Acknowledgments
This study was supported by Agios Pharmaceuticals. Medical writing assistance was provided by Vanessa Ducas, of Excel Medical Affairs, supported by Agios Pharmaceuticals.
Funding Agios Pharmaceuticals.
Declaration of interests
GKA-A is a consultant for 3DMedcare, Agios, Alignmed, Amgen, Antengene, Aptus, ASLAN, Astellas, AstraZeneca, Bayer, Beigene, Bioline, Bristol-Myers Squibb, Boston Scientific, Bridgebio, Carsgen, Celgene, Casi, Cipla, CytomX, Daiichi, Debio, Delcath, Eisai, Exelixis, Flatiron, Genoscience, Halozyme, Hengrui, Incyte, Inovio, Ipsen, Jazz, Jansen, Klus, Kyowa Kirin, LAM, Lilly, Loxo, Merck, Mina, Novella, Onxeo, PCI Biotech, Pfizer, Pieris, QED, Redhill, Sanofi, Servier, Silenseed, SillaJen, Sobi, Targovax, Tekmira, Twoxar, Vicus, Yakult, and Yiviva; has received research grants or funding from ActaBiologica, Agios, Array, AstraZeneca, Bayer, Beigene, Bristol-Myers Squibb, Casi, Celgene, Exelixis, Genentech, Halozyme, Incyte, Lilly, Mabvax, Novartis, OncoQuest, Polaris Puma, QED, and Roche. TM is on advisory boards for Baxalta, Celgene, H3B, QED, and Shire; has received honoraria from Genzyme, Roche, Sanofi, Shire, and Tesaro; has participated in speaker bureaus for Celgene, Sanofi, and Shire; has received research grants or funding (to institution) from Agios, ASLAN, AstraZeneca, Baxalta, Bayer, Genentech, Halozyme, Immunomedics, Lilly, Merrimack, Millennium, Novartis, Novocure, Pfizer, Pharmacyclics, and Roche; has received travel and accommodation funding from Bayer, H3B, Merck, and Sanofi. MMJ is on advisory boards for EDO, More Health, and OrigiMed; has received honoraria from Merck, Seattle Genetics, and Taiho; has received research grants or funding from ArQule (to institution), Lilly (to institution), Meclun (to individual), Novartis (to individual), and QED (to individual). RKK is on advisory boards for Agios (funding to institution), AstraZeneca (funding to institution), Bristol-Myers Squibb (funding to institution), Genentech–Roche (Independent Data Monitoring Committee; advisory board and funding to individual), and Ipsen (funding to individual); has received travel and accommodation funding from Ipsen; has received research grants or funding (to institution) from Agios, AstraZeneca, Bayer, Bristol-Myers Squibb, Exelixis, Lilly, MedImmune, Merck, Merck Serono, Novartis, Partner Therapeutics, QED, and Taiho. SJL is a consultant for Farcast Biosciences. JMC is on advisory boards for Agios and Bristol-Myers Squibb; has received honoraria from Agios; has received travel and accommodation funding from Agios, Bristol-Myers Squibb, and Roche; has received research grants or funding from Merck and Tesaro. DVC has received honoraria from Astellas, Bristol-Myers Squibb, Daiichi Sankyo, Five Prime, Foundation Medicine, Genentech–Roche, Guardant, Lilly, Merck, Taiho, and Tempus. MJB holds stock in AVEO, Intercept, and OncBioMune; has received honoraria from ADC, Exelixis, G1, Immunovative, Inspyr, Lynx Group, Western Oncolytics; has received travel and accommodation funding from AstraZeneca; has received research grants or funding (to institution) from Adaptimmune, Agios, ARIAD, Basilea, Bioline, Boston Biomedical, Celgene, Dicerna, Halozyme, Incyte, Isis, MedImmune, Merck Serono, Mirna, Novartis, Pieris, PUMA, QED, Redhill, Senhwa, SillaJen, Sun, Taiho, and Toray. JB is on advisory boards for AstraZeneca, Basilea, Bayer, Incyte, Merck Serono, Roche, and Servier; has received honoraria from Merck Serono and Servier; has received travel or accommodation funding from Bristol-Myers Squibb, Bristol-Myers Squibb–Medarex, Merck, Sharp & Dohme, Merck Serono, and Servier; has participated in speaker bureaus for Amgen and Celgene; has received research grants and funding from Amgen. WPH is on advisory boards for Bayer, Bristol-Myers Squibb, Eisai, Exelixis, Neo Therma, QED, and Zymeworks; has received research grants or funding from Agios, ArQule, Bayer, Bristol-Myers Squibb, BTG, Eisai, Exelixis, Halozyme, MedImmune, and Merck. AGM has received research grants or funding from Bristol-Myers Squibb. D-YO is on advisory boards from ASLAN, AstraZeneca, Bayer, Celgene, Genentech–Roche, Halozyme, Merck Serono, Novartis, Taiho, and Zymeworks; has research grants or funding (to institution) from Array, AstraZeneca, Lilly, and Novartis. MAL is on advisory boards for Agios, and Roche; has participated in speaker bureaus for Novartis; has received travel funding from Ipsen. LG is on advisory boards for Agios, Alentis, Debiopharm, Klus, Pieris, QED, Sirtex, and Taiho; is a consultant for Alentis, H3B, Incyte; has received travel funding from Taiho; is an Independent Data Monitoring Committee member for AstraZeneca. RTS is on advisory boards for Agios, Clovis, Debiopharm, Exelixis, Incyte, Merck, QED, and Seattle Genetics; has received research grants or funding from Agios, Exelixis, Halozyme, Merck, Pieris, and Taiho. ABE-K is on advisory boards for Agenus, Agios, Bayer, Bristol-Myers Squibb, CytomX, Eisai, Exelixis, Gilead, Merck, Merck Serono, Pieris, and Roche–Genentech (consultant and advisory board); has received research grants or funding from Astex (to institution), AstraZeneca (to institution), and Merck (to institution). BF, CXC, LJ, CG, and SSP are employees of and hold stock in Agios Pharmaceuticals. BW is an employee of, holds stock in, and holds patents, royalties, and other intellectual property with Agios Pharmaceuticals. JWV is on advisory boards for Agios, AstraZeneca, Debiopharm, Delcath, Genoscience, Imaging Equipment Limited, Incyte, Ipsen, Keocyt, Merck, Mundipharma, Novartis, NuCana, PCI, Pieris, Pfizer, QED, Servier, and Wren Laboratories; has received travel or accommodation funding from Ipsen, Novartis, and NuCana; has participated in speaker bureaus for Ipsen, Novartis, and Pfizer. AXZ is on advisory boards for Bayer, Eisai, Exelixis, Lilly, Merck, Roche–Genentech, and Sanofi. JA and JW declare no competing interests.
Data sharing
The data collected for the study will not be made available to others. We encourage investigators interested in data sharing and collaboration to contact the corresponding author.
Contributor Information
Prof Ghassan K Abou-Alfa, Department of Medicine, Memorial Sloan Kettering, Cancer Center, New York, NY, USA.
Teresa Macarulla, Vall d´Hebron University Hospital and Vall d´Hebron Institute of Oncology (VHIO), Barcelona, Spain.
Prof Milind M Javle, Department of Gastrointestinal, Medical Oncology, MD, Anderson Cancer Center, Houston, TX, USA.
Robin K Kelley, Helen Diller Family Comprehensive, Cancer Center, University of, California San Francisco, San Francisco, CA, USA.
Sam J Lubner, Department of Medicine, University of Wisconsin Carbone Cancer Center, Madison, WI, USA.
Jorge Adeva, Department of Medical Oncology, Hospital Universitario 12 de Octubre, Madrid, Spain.
James M Cleary, Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, USA.
Daniel V Catenacci, Department of Medicine, University of Chicago Medical Center, Chicago, IL, USA.
Mitesh J Borad, Department of Hematology– Oncology, Mayo Clinic Cancer Center, Phoenix, AZ, USA.
Prof John Bridgewater, Department of Medical Oncology, UCL Cancer Institute, London, UK.
William P Harris, Department of Medicine, University of Washington, Seattle, WA, USA.
Adrian G Murphy, Department of Oncology-Gastrointestinal Cancer, Johns Hopkins University, Baltimore, MD, USA.
Prof Do-Youn Oh, Department of Internal Medicine, Seoul National University Hospital, Cancer Research Institute, Seoul National University College of Medicine, Seoul, Korea.
Jonathan Whisenant, Medical Oncology and Hematology, Utah Cancer Specialists, Salt Lake City, UT, USA.
Prof Maeve A Lowery, Trinity St James Cancer Institute, Trinity College Dublin, Dublin, Ireland.
Lipika Goyal, Department of Medicine, Massachusetts General Hospital Cancer Center, Harvard Medical School, Boston, MA, USA.
Rachna T Shroff, Department of Medicine, University of Arizona Cancer, Center, Tucson, AZ, USA.
Anthony B El-Khoueiry, Department of Medicine, University of, Southern California Norris, Comprehensive Cancer Center, Los Angeles, CA, USA.
Bin Fan, Agios Pharmaceuticals, Cambridge, MA, USA.
Bin Wu, Agios Pharmaceuticals, Cambridge, MA, USA.
Christina X Chamberlain, Agios Pharmaceuticals, Cambridge, MA, USA.
Liewen Jiang, Agios Pharmaceuticals, Cambridge, MA, USA.
Camelia Gliser, Agios Pharmaceuticals, Cambridge, MA, USA.
Shuchi S Pandya, Agios Pharmaceuticals, Cambridge, MA, USA.
Prof Juan W Valle, Division of Cancer Sciences, University of Manchester, Manchester, UK; Department of Medical Oncology, The Christie NHS Foundation Trust, Manchester, UK.
Prof Andrew X Zhu, Department of Medicine, Massachusetts General Hospital Cancer Center, Harvard Medical School, Boston, MA, USA; Jiahui International Cancer Center, Jiahui Health, Shanghai, China.
References
- 1.Boscoe AN, Rolland C, Kelley RK. Frequency and prognostic significance of isocitrate dehydrogenase 1 mutations in cholangiocarcinoma: a systematic literature review. J Gastrointest Oncol 2019; 10: 751–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Borger DR, Tanabe KK, Fan KC, et al. Frequent mutation of isocitrate dehydrogenase (IDH)1 and IDH2 in cholangiocarcinoma identified through broad-based tumor genotyping. Oncologist 2012; 17: 72–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kipp BR, Voss JS, Kerr SE, et al. Isocitrate dehydrogenase 1 and 2 mutations in cholangiocarcinoma. Hum Pathol 2012; 43: 1552–58. [DOI] [PubMed] [Google Scholar]
- 4.Wang P, Dong Q, Zhang C, et al. Mutations in isocitrate dehydrogenase 1 and 2 occur frequently in intrahepatic cholangiocarcinomas and share hypermethylation targets with glioblastomas. Oncogene 2013; 32: 3091–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saha SK, Parachoniak CA, Ghanta KS, et al. Mutant IDH inhibits HNF-4α to block hepatocyte differentiation and promote biliary cancer. Nature 2014; 513: 110–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Agios Pharmaceuticals TIBSOVO (ivosidenib). Highlights of prescribing information. 2019. https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211192s001lbl.pdf (accessed Aug 5, 2019).
- 7.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in ed relapsed or refractory AML. N Engl J Med 2018; 378: 2386–98. [DOI] [PubMed] [Google Scholar]
- 8.Popovici-Muller J, Lemieux RM, Artin E, et al. Discovery of AG-120 (ivosidenib): a first-in-class mutant IDH1 inhibitor for the treatment of IDH1 mutant cancers. ACS Med Chem Lett 2018; 9: 300–05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lowery MA, Burris HA 3rd, Janku F, et al. Safety and activity of ivosidenib in patients with IDH1-mutant advanced cholangiocarcinoma: a phase 1 study. Lancet Gastroenterol Hepatol 2019; 4: 711–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oken MM, Creech RH, Tormey DC, et al. Toxicity and response criteria of the Eastern Cooperative Oncology Group. Am J Clin Oncol 1982; 5: 649–55. [PubMed] [Google Scholar]
- 11.Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009; 45: 228–47. [DOI] [PubMed] [Google Scholar]
- 12.Baber N International conference on harmonisation of technical requirements for registration of pharmaceuticals for human use (ICH). Br J Clin Pharmacol 1994; 37: 401–04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.World Medical Association. 64th General Assembly. Declaration of Helsinki. Ethical principles for medical research involving human subjects. 2013. https://www.wma.net/policies-post/wmadeclaration-of-helsinki-ethical-principles-for-medical-researchinvolving-human-subjects/ (accessed Aug 5, 2019).
- 14.Aaronson NK, Ahmedzai S, Bergman B, et al. The European Organization for Research and Treatment of Cancer QLQ-C30: a quality-of-life instrument for use in international clinical trials in oncology. J Natl Cancer Inst 1993; 85: 365–76. [DOI] [PubMed] [Google Scholar]
- 15.Friend E, Yadegarfar G, Byrne C, et al. Development of a questionnaire (EORTC module) to measure quality of life in patients with cholangiocarcinoma and gallbladder cancer, the EORTC QLQ-BIL21. Br J Cancer 2011; 104: 587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guy W ECDEU assessment manual for psychopharmacology. Rockville, MD: US Department of Health, Education, and Welfare, Public Health Service, Alcohol, Drug Abuse, and Mental Health Administration, 1976. [Google Scholar]
- 17.Szende A, Janssen B, Cabases J, eds. Self-reported population health: an international perspective based on EQ-5D. Dordrecht, Netherlands: Springer, 2014. 10.1007/978-94-0077596-1. [DOI] [PubMed] [Google Scholar]
- 18.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009; 462: 739–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morden JP, Lambert PC, Latimer N, Abrams KR, Wailoo AJ. Assessing methods for dealing with treatment switching in randomised controlled trials: a simulation study. BMC Med Res Methodol 2011; 11: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan B, Mellinghoff IK, Wen PY, et al. Clinical pharmacokinetics and pharmacodynamics of ivosidenib, an oral, targeted inhibitor of mutant IDH1, in patients with advanced solid tumors. Invest New Drugs 2020; 38: 433–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Moik F, Riedl JM, Winder T, et al. Benefit of second-line systemic chemotherapy for advanced biliary tract cancer: a propensity score analysis. Sci Rep 2019; 9: 5548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lamarca A, Palmer DH, Wasan HS, et al. ABC-06 | A randomised phase III, multi-centre, open-label study of active symptom control (ASC) alone or ASC with oxaliplatin / 5-FU chemotherapy (ASC+mFOLFOX) for patients (pts) with locally advanced/metastatic biliary tract cancers (ABC) previously-treated with cisplatin/ gemcitabine (CisGem) chemotherapy. J Clin Oncol 2019; 37: 4003. [Google Scholar]
- 23.Lamarca A, Hubner RA, David Ryder W, Valle JW. Second-line chemotherapy in advanced biliary cancer: a systematic review. Ann Oncol 2014; 25: 2328–38. [DOI] [PubMed] [Google Scholar]
- 24.Ying J, Chen J. Combination versus mono-therapy as salvage treatment for advanced biliary tract cancer: a comprehensive meta-analysis of published data. Crit Rev Oncol Hematol 2019; 139: 134–42. [DOI] [PubMed] [Google Scholar]
- 25.Patel N, Benipal B. Incidence of cholangiocarcinoma in the USA from 2001 to 2015: a US cancer statistics analysis of 50 states. Cureus 2019; 11: e3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blechacz B Cholangiocarcinoma: current knowledge and new developments. Gut Liver 2017; 11: 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rizvi S, Khan SA, Hallemeier CL, Kelley RK, Gores GJ. Cholangiocarcinoma - evolving concepts and therapeutic strategies. Nat Rev Clin Oncol 2018; 15: 95–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Javle M, Bekaii-Saab T, Jain A, et al. Biliary cancer: Utility of next-generation sequencing for clinical management. Cancer 2016; 122: 3838–47. [DOI] [PubMed] [Google Scholar]
- 29.Le DT, Durham JN, Smith KN, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017; 357: 409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Javle M, Lowery M, Shroff RT, et al. Phase II study of BGJ398 in patients with FGFR-altered advanced cholangiocarcinoma. J Clin Oncol 2018; 36: 276–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.