

Abstract
Three studies reveal that the presence in tumours of two key immune components — B cells and tertiary lymphoid structures — is associated with favourable outcomes when individuals undergo immunotherapy.
Current immunotherapies aim to reinvigorate immune cells called killer T cells to fight cancer, but only 20% of individuals with the disease see a lasting clinical benefit from this type of treatment1. Focusing on other immune cells in patients’ tumours might help us to improve these outcomes. Three studies, by Cabrita et al.2 (page 561), Petitprez et al.3 (page 556) and Helmink et al.4 (page 549), now demonstrate that the presence of B cells in human tumours in compartments called tertiary lymphoid structures (TLS) is associated with a favourable response to immunotherapy. These complementary studies add to the immunotherapy toolbox by providing new ways of predicting prognosis.
The presence of B cells in tumours has been considered to be a predictor of increased patient survival5,6, but there are reports of both anti- and pro-tumour roles for B cells7. These differing reports reflect the multiple roles that B cells can have in tumours. One component of the antitumour function of B cells is B-cell activation. This process involves the binding of tumour-derived proteins to the B-cell receptor protein on the cell surface and the subsequent processing of these tumour-derived proteins into smaller fragments called antigens. Further co-factors are also involved in activation. Activated B cells can release antibodies that tag tumour cells for attack by other cellular players of the immune system (a process known as antibody-dependent cell death)8, and can ‘educate’ T cells by presenting them with tumour antigens, enabling the T cells to target tumour cells effectively9. However, B cells in tumours can produce inhibitory factors that hinder the function of immune cells (Fig. 1). These might be signalling molecules that suppress the immune system7,10,11 or inhibitory molecules on the surfaces of B cells that limit the body’s ability to target and kill tumour cells.
Figure 1 |. Multifaceted B cells in the tumour microenvironment.
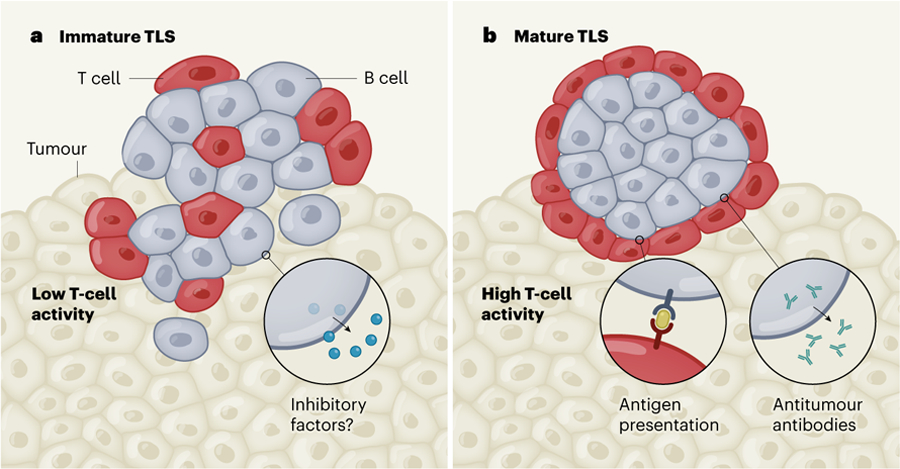
B cells are thought to have multiple roles in suppressing or promoting the immune system’s ability to kill tumour cells, depending on whether they are located in immature or mature compartments called tertiary lymphoid structures (TLS), which also contain T cells. a, In poorly structured, immature TLS, one hypothesis is that B cells generate inhibitory factors. These might be molecules released from B cells that dampen the response of other immune cells, or molecules on the surfaces of B cells that hinder the targeting and destruction of tumour cells. Both of these inhibitory mechanisms might arise if B cells have less interaction with T cells and more interaction with the malignant tumour. Three studies2–4 now provide indirect evidence that immature TLS are associated with low activity of T cells in tumours. b, By contrast, B cells in well-structured, mature TLS can release antibodies that couldtarget tumours, and B cells can present a tumour-derived protein called an antigen (yellow) to T cells in the tumour, activating the T cells. The studies suggest that the presence of B cells in mature TLS is correlated with increased T-cell activity, improving the immune system’s ability to target tumour cells, and increasing the likelihood that the tumour will respond to immunotherapy.
TLS are aggregates of immune cells (mostly T and B cells) that arise in response to immunological stimuli. Mature TLS nurture B-cell development and function in an inner region of the structure called the germinal centre, whereas immature TLS do not contain proper germinal centres, and might not nurture full B-cell function. The presence of TLS in a tumour also correlates with increased patient survival in many cancer types12. The three current studies confirm this trend in the context of immunotherapy, demonstrating that infiltration of B cells into a tumour, along with the presence of TLS, is associated with an improved response to this type of treatment.
Cabrita et al. studied individuals who had a type of cancer called metastatic melanoma, and Petitprez et al. investigated people with sarcoma, a cancer of the bone. Both teams found that the presence of B cells in TLS in the tumour before treatment was associated with an increased chance that patients’ tumours would respond to immunotherapy. Helmink et al. corroborated these findings for metastatic melanoma, and reported the same pretreatment trend in renal cell carcinoma. These authors also demonstrated that, during treatment, TLS are more prevalent in people who have tumours that are responding to treatment than in those whose tumours are not. This timing is important — when present before treatment, TLS could be considered a predictor of patient response to immunotherapy, whereas the presence of TLS during treatment indicates that key combinations of immune cells are being manipulated to induce TLS formation. Identifying these cell combinations could help in establishing new and effective immune-based therapies.
The three groups found that the B-cell and TLS signature was often more pronounced in responders than in non-responders. Furthermore, the signature was more prominent than typical T-cell signatures currently used for understanding immunotherapy outcomes. This suggests that B cells and TLS could have a key role in antitumour immunity.
In addition to these synergistic results, each study highlights a unique role for B cells or TLS in antitumour immunity. First, Cabrita et al. demonstrate that B cells in TLS synergize with killer T cells that could ultimately target tumour cells. Second, Petitprez et al. describe signatures characteristic of mature TLS in sarcoma. This implies that mature TLS can exist in tumour sites that are not normally thought to be infiltrated by immune cells, a phenomenon that has not previously been shown. Third, Helmink et al. find increased diversity of B-cell receptors in responders compared with non-responders. This indicates that pools of B cells in responders might have a greater ability to specifically recognize tumour antigens than do the B cells of non-responders.
These papers are technologically savvy, use patient populations that are statistically robust and bring B cells and TLS to the forefront of antitumour immunity. However, there is much still to learn. First, more emphasis should be placed on understanding how TLS form in tumours. It is clear that these structures are variable, and can be immature or mature. What does this diversity mean for the function of B cells in TLS, and what causes the induction of one ‘flavour’ of TLS versus another? The contribution of environmental factors such as smoking or viral and bacterial infections should be considered, along with a person’s gender, age and tumour type.
Researchers should also ask whether mature TLS could be routinely induced to form in tumours, to maximize B-cell immunity. Addressing this issue will require investigation of B cells and TLS in individuals who have not yet undergone treatment, as well as proper modelling of the human tumour microenvironment. Current evidence indicates that B cells actually impede antitumour responses in most mouse models of cancer13–15. However, TLS formation is rare in these animals, and a lack of TLS might alter the fate and subsequent function of B cells. Indeed, more knowledge about B-cell function outside TLS is needed to provide a complete picture of B cells in the tumour microenvironment.
There is still a need to define the full range of functions that B cells perform in tumours. In addition to their known roles in producing tumour-specific antibodies and presenting antigens8,9, B cells are likely to have other functions — for instance, inducing antibody-dependent cell death8. It will also be necessary to link these functions to specific B-cell types and to determine whether such cells are found inside or outside TLS. There are clear biomarkers for B-cell subsets, but linking these subsets to functions in human tumours would allow us to design treatments that optimize specific antitumour activities. Furthermore, this knowledge would help us to understand whether subsets of B cells perform separate tasks, or if there is crosstalk between subsets. For example, can the same B cell both produce a tumour-specific antibody and present antigens to T cells? Some of these studies can be done in human tumours, but in-depth mechanistic studies will require physiologically relevant models that contain naturally occurring TLS.
With regard to clinical implications, the current studies suggest that therapeutics to enhance B-cell responses should be prioritized as a complement to T-cell-mediated immunotherapies. Researchers should now ask whether B cells could be engineered to target specific tumour antigens, similar to current efforts to engineer antigen-targeting T cells. More generally, could immunotherapies be improved by inducing B cells to form in TLS after a person has received T-cell-based immunotherapy?
Overall, the current studies should act as a springboard for future mechanistic studies of B cells and TLS in cancer. Understanding how current therapies can be combined with approaches to harness B cells and TLS will be crucial for the development of effective B-cell-specific immunotherapies.
References
- 1.Brahmer JR et al. J. Clin. Oncol 28, 3167–3175 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cabrita R et al. Nature 577, 561–565 (2020). [DOI] [PubMed] [Google Scholar]
- 3.Petitprez F et al. Nature 577, 556–560 (2020). [DOI] [PubMed] [Google Scholar]
- 4.Helmink BA et al. Nature 577, 549–555 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shimabukuro-Vornhagen A et al. Oncotarget 5, 4651–4664 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Germain C et al. Am. J. Respir. Crit. Care Med 189, 832–844 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Shalapour S et al. Nature 521, 94–98 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DeFalco J et al. Clin. Immunol 187, 37–45 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Bruno TC et al. Cancer Immunol. Res 5, 898–907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kessel A et al. Autoimmun. Rev 11, 670–677 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Khan AR et al. Nature Commun 6, 5997 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Sautès-Fridman C, Petitprez F, Calderaro J & Fridman WH Nature Rev. Cancer 19, 307–325 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Affara NI et al. Cancer Cell 25, 809–821 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shalapour S et al. Nature 551, 340–345 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ammirante M et al. Nature 464, 302–305 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]