

Abstract
Motivation
X-ray crystallography has facilitated the majority of protein structures determined to date. Sequence-based predictors that can accurately estimate protein crystallization propensities would be highly beneficial to overcome the high expenditure, large attrition rate, and to reduce the trial-and-error settings required for crystallization.
Results
In this study, we present a novel model, BCrystal, which uses an optimized gradient boosting machine (XGBoost) on sequence, structural and physio-chemical features extracted from the proteins of interest. BCrystal also provides explanations, highlighting the most important features for the predicted crystallization propensity of an individual protein using the SHAP algorithm. On three independent test sets, BCrystal outperforms state-of-the-art sequence-based methods by more than 12.5% in accuracy, 18% in recall and 0.253 in Matthew’s correlation coefficient, with an average accuracy of 93.7%, recall of 96.63% and Matthew’s correlation coefficient of 0.868. For relative solvent accessibility of exposed residues, we observed higher values to associate positively with protein crystallizability and the number of disordered regions, fraction of coils and tripeptide stretches that contain multiple histidines associate negatively with crystallizability. The higher accuracy of BCrystal enables it to accurately screen for sequence variants with enhanced crystallizability.
Availability and implementation
Our BCrystal webserver is at https://machinelearning-protein.qcri.org/ and source code is available at https://github.com/raghvendra5688/BCrystal.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
X-ray crystallography is a key method that is used to determine the structure of a protein. This method is quite expensive and has a high attrition rate due to the need for well-diffracting crystals. The total percentage of successful attempts for X-ray crystallography ranges between 2% and 10% (Terwilliger et al., 2009), whereas failed attempts attain >70% of the total cost (Service, 2005). Several in silico machine learning and statistical models have been developed to predict protein crystallization propensities, including CrystalP2 (Kurgan and Mizianty, 2009), PPCpred (Charoenkwan et al., 2013), PredPPCrys (Wang et al., 2014), XtalPred-RF (Jahandideh et al., 2014), TargetCrys (Hu et al., 2016), Crysalis (Wang et al., 2016), Crysf (Wang et al., 2017) and fDETECT (Meng et al., 2018).
These methods primarily rely on extracting physio-chemical, sequence-based and functional features from the raw protein sequences. However, the identification of novel biological features that can accurately estimate protein crystallization propensity still remains a significant challenge. Moreover, a majority of these methods follow a two-step process: (i) feature engineering and selection; (ii) protein crystallization propensity prediction i.e. distinguishing diffraction quality crystals from yet to be crystallized proteins, referred as non-crystallizable proteins in literature Wang et al. (2017), Hu et al. (2016) and Charoenkwan et al. (2013). Recently, a deep learning technique called DeepCrystal (Elbasir et al., 2019) showcased that by just using the raw protein sequences as input, it can outperform all other state-of-the-art sequence-based crystallization predictors. DeepCrystal utilizes complex non-linear features from the raw protein sequences. These features can be associated with the frequencies of k-mers and sets of k-mers of different lengths. However, it is not straightforward to pinpoint what are the specific k-mers in a given protein sequence driving its crystallization propensity and thus determine their biological relevance as mentioned in Elbasir et al. (2019).
To overcome the limitations of existing methods, we propose BCrystal (acronym for ‘Be Crystal’), an XGBoost based model (Chen and Guestrin, 2016) using several well-known physio-chemical and sequence-derived features. Additionally, BCrystal uses several novel secondary structure and disorder features extracted from the SCRATCH suite (Cheng et al., 2005) and DISOPRED version 3.16 (Ward et al., 2004), respectively. XGBoost is an optimized version of the gradient boosting machine (GBM; Friedman, 2001), which has been shown to perform very well on several bioinformatics problems, such as gene regulatory network reconstruction (Mall et al., 2017, 2018a, b), protein solubility (Rawi et al., 2017), transmembrane protein crystallization (Varga and Tusnády, 2018) and so on. Moreover, BCrystal has the unique attribute that can provide explanation for the predicted class label for each test protein based on its corresponding feature values using the SHapley Additive exPlanations (SHAP) (Lundberg and Lee, 2017) algorithm. Our primary contributions include:
Extraction of novel structure and disorder features from the protein sequence using SCRATCH suite and DISOPRED, respectively.
Usage of an XGBoost model enables BCrystal to outperform existing methods for various evaluation metrics on three independent test sets.
Provide an interpretation for BCrystal’s output by showing the most important features driving the model predictions towards diffraction quality crystals or non-crystallizable proteins.
A user-friendly public webserver for academic usage and availability of source code for further enhancements.
Figure 1 provides the steps undertaken by the proposed BCrystal model and graphical representation of the modeling techniques utilized by BCrystal.
Fig. 1.

(A) The flowchart of the proposed BCrystal model is shown. (B) The working mechanism of XGBoost model is illustrated. At each iteration a tree is fitted to all the samples and for a particular sample the final crystallization propensity is the average of all predictions for that instance across all the trees. In XGBoost, the primary task is identification of the optimal tree structure which is explained in detail in Section 2. (C) How Shapley value for a particular feature (Feature 1 here) is estimated by the SHAP model is demonstrated. SHAP model works by considering the tree structure with or without Feature 1 and all such possible combinations in case of multiple features. (Cii and iv) The importance of Feature 1 versus an empty set of features (Ci) and feature set with just Feature 2 (Ciii), respectively for predicting the crystallization propensity of protein P1 is illustrated. The prediction score for P1 in each of the four cases (i, ii, iii, and iv) would be 0.725, 0.5, 0.725, and 0.1 (math not shown), respectively and thus the Shapley value for Feature 1 would be (0.5 − 0.725)/2 + (0.1 − 0.725)/2 = −0.425 according to Equation 3. This indicates that Feature 1 drives BCrystal model prediction to non-crystallizable class (negative Shapley value) for protein P1 which aligns with the propensity score of 0.1 estimated by BCrystal for protein P1. A detailed description of SHAP method for generating explanations is provided in the Section 3
2 Materials and methods
2.1 Overview
Our task is a binary classification problem i.e. distinguishing crystallizable proteins from non-crystallizable ones. Our goal is to learn a function (H), which takes features engineered from a protein sequence i.e. as input and outputs a prediction score between i.e. . In this work, H is an XGBoost model (Chen and Guestrin, 2016), a white-box non-linear tree-based interpretable boosting machine that exploits the interactions between the engineered features.
2.2 Data information
All our experiments are performed on publicly available datasets. The training dataset is obtained from Wang et al. (2014) which has five class labels including diffraction-quality crystals, cloning failure, material production failure, purification failure and crystallization failure. A total of 28 731 proteins, including 5383 diffraction-quality crystals (positive class), are present in the dataset. We consider all the remaining 23 348 protein sequences as non-crystallizable ones (negative class). The authors in Wang et al. (2014) highlighted that all the sequences in individual classes were passed through a filter of >25% sequence similarity to de-bias and remove highly similar protein sequences within each class.
We randomly divide this training dataset into two parts: and . Here, consists of 891 crystallizable and 897 non-crystallizable proteins forming a fairly balanced test set for evaluation. We use two other independent test sets which were generated in Wang et al. (2017) for comprehensive comparison with state-of-the-art web-servers including DeepCrystal, fDETECT, Crysf, Crysalis I and II, TargetCrys, XtralPred-RF, PPCPred and CrystalP2. The two independent test sets are obtained from SwissProt and Trembl databases and are named SP_final and TR_final, respectively. In the SP_final dataset, we have 148 proteins belonging to the positive class and 89 protein sequences are non-crystallizable whereas in the TR_final dataset, there are 374 crystallizable and 638 proteins belonging to the negative class.
We perform a more stringent filtering by removing all proteins from belonging to positive class and having sequence similarity >15% with the crystallizable proteins in fairly balanced set, SP_final and TR_final using CD-HIT method Fu et al. (2012) resulting in 2880 proteins corresponding to the positive class in the final training set . We perform the same operation for proteins belonging to the negative class in to obtain a total of 8474 protein sequences associated with the non-crystallizable class in . In total, has a total of 11 354 protein sequences. We perform 5-fold cross-validation to obtain the optimal set of hyper-parameters. We use the optimal hyper-parameters with to build the XGBoost classifier.
2.3 Feature engineering
One of our main contributions is designing and engineering features which are useful for discriminating diffraction quality crystals from non-crystallizable ones. We devise three groups of features which are then used to train the BCrystal model (see Fig. 2). The first set is composed of features based on global properties of the protein including sequence length (), molecular weight ((Mol-Weight)), fraction of turn-forming residues, the average of hydropathicity (Gravy) and aliphatic indices, along with the total absolute charge. The second group of features are derived directly from the protein sequence and consist of frequencies of mono- (single amino acids, denoted AA), di- (two consecutive AAs) and tri-peptides (three consecutive AAs) within the protein sequences.
Fig. 2.

Different sets of features engineered for the BCrystal model. The number of features for each component is shown within parentheses
The third group of features are structural information obtained from the protein sequence using SCRATCH (Cheng et al., 2005) and DISOPRED (Ward et al., 2004). It has been shown previously (Hou et al., 2018) that SCRATCH based features are useful for protein fold prediction. We predict 3- and 8-state secondary structure (SS) information as well as the fraction of exposed residues (FER) with 20 different relative solvent accessibility (RSA) cutoffs (≥ 0% to ≥ 95% cutoffs at 5% intervals). From the 3-state SS obtained via SCRATCH, we extract mono- (1 state i.e. turn, strand or coil), di- (two consecutive states) and tri-state (three consecutive states) frequencies for a given protein sequence. We follow a similar procedure for the more granular 8-state SS information as shown in Figure 2. Additionally, we multiply the FER by the average hydrophobicity of these exposed residues at different RSA cutoffs. From DISOPRED, we obtain information about which AAs in the protein sequences are part of disordered regions as well as which AAs from the protein binding sites (PBS) of a protein are part of disordered regions. Given this information, we engineer features such as the fraction of the protein sequence which is disordered, frequency of each of the AAs (out of the 20 AAs) in disordered regions, number of disordered regions, number of disordered regions of length <5 AAs, number of disordered regions of length between 5 and 10 AAs, and number of disordered regions of length >10 AAs in a protein sequence. A similar set of features are extracted from the PBS of a protein.
In total, we include 9139 features for each protein sequence. In contrast to other sequence-based predictors, we do not perform a feature selection step to exclude features, rather we rely on the XGBoost model to prioritize the most important features and filter out irrelevant ones.
2.4 Methods
2.4.1 Gradient boosting machine
In this work, we utilized an optimized version of the white-box, non-linear, ensemble GBM (Friedman, 2001; Schapire, 2003) called XGBoost (Chen and Guestrin, 2016) for building our BCrystal model. Gradient boosting is a machine-learning technique based on a constructive strategy by which the learning procedure will additively fit new models, typically decision trees and repetitively leverage the patterns in residuals to provide a more accurate estimate of the response variable (crystallizable versus non-crystallizable proteins). A brief explanation of GBM is provided in Supplementary Material.
2.4.2 XGBoost algorithm
Tree boosting is a learning technique to improve the classification of weaker classifiers by repeatedly adding new decision trees to the ensembles. XGBoost (Chen and Guestrin, 2016) is a scalable machine learning technique for tree boosting. It was shown in Chen and Guestrin (2016) that its performance is better than other boosting algorithms.
The main components of XGBoost algorithm are the objective function and its iterative solution. The objective function is initialized to describe the model’s performance. Given the training dataset, where , d = 9139, and N denotes the total number of training samples. The predicted output obtained from the ensemble model can be represented as , where represents the prediction score of the tth decision tree for the ith protein sequence in the training dataset. If the decision trees are allowed to grow unregulated, then the resulting model is bound to overfit (Chen and Guestrin, 2016). Hence, the following objective has to be minimized:
| (1) |
where L is the loss function and Ω(⋅) is the penalty that is used to prevent overfitting which is defined as: , where γ and λ are the parameters which control the penalty for the number of leaf nodes (A) and leaf weights (w), respectively in the decision tree Ht.
The objective function can be re-written as follows . After applying a Taylor expansion and expanding Ω(Ht), we obtain:
where and are the first and second order gradient statistics on the loss function. For a fixed tree structure , where is an instance of leaf node j, the optimal weight for leaf node j is:
The corresponding optimal objective function then becomes:
| (2) |
Equation 2 can be used as a scoring function to measure the quality of a tree structure Ht at the tth iteration. This score is equivalent to the impurity score used for evaluating decision trees in random forests (Breiman, 2001). The authors in Chen and Guestrin (2016), come up with a fast, greedy and iterative algorithm to identify these optimal tree structures.
2.5 Training
We train our XGBoost classifier on top of physio-chemical (global), sequence and structural features extracted from the protein sequence as mentioned earlier. Since our training set is imbalanced, we weight the samples belonging to crystallizable class by α, where and Nn is the total number of non-crystallizable proteins and Np is the total number of diffraction quality crystals in the training set.
The BCrystal classifier is based on several parameters, such as maximum depth of a tree (M), the learning rate (ν), the minimum child weight of a leaf node (wj), the sampling rate for features (r) and the subsample ratio of the training set (s). We keep the regularization parameter, γ, on the number leaf nodes to the default value of 0. We then performed a hyper-parameter optimization procedure by varying these parameters over a grid of combinations. In particular with and . We performed a five-fold cross-validation for each of these combinations (variance in the results is highlighted in Supplementary Fig. S1). Finally, we selected the XGBoost classifier which had the maximum 5-fold cross-validation area under the curve, which was obtained corresponding to the parameters M = 5, , wj = 6, r = 0.7 and s = 0.6. The final BCrystal classifier had a maximum training accuracy of 93.7% and a maximum Matthew’s correlation-coefficient (MCC) of 0.867. The first and the last tree of the BCrystal model are highlighted in the Supplementary Figure S2.
2.6 Evaluation metrics
The BCrystal method was compared against different in silico sequence-based crystallization predictors using several well-known metrics such as recall (REC), precision (PRE), accuracy (ACC), MCC, F-score (F) and negative predictive value (NPV). All these evaluation metrics are based on true positives (TP), true negatives (TN), false positives (FP) and false negatives (FN). The set TP represents the proteins which produce diffraction quality crystals (class Label 1) and for which BCrystal predicts . Similarly, the set TN consists of those proteins which are non-crystallizable (class Label 0) and for which BCrystal predicts . The set FP represents those proteins whose true label is non-crystallizable i.e. 0 but BCrystal predicts and the set FN comprises proteins whose true label is crystallizable i.e. 1 but BCrystal estimates . A detailed definition of these sets and importance of each of these evaluation metrics are provided in Khurana et al. (2018), Elbasir et al. (2019) and Rawi et al. (2017).
3 Results
The performance of BCrystal was evaluated on three independent test sets, including a fairly balanced test, the SP_final and TR_final test sets. A comprehensive comparison was done against several state-of-the-art sequence-based protein crystallization predictors, including DeepCrystal, Crysf, Crysalis I and II, fDETECT, TargetCrys, XtalPred-RF, PPCPred and CrystalP2. The comparison with Crysf was conducted only on the SP_final and TR_final datasets as Crysf required Uniprot ids as input which were available only for these two datasets.
3.1 Fairly balanced set
The fairly balanced test set was composed of 1787 proteins with 896 non-crystallizable proteins and 891 crystallizable proteins. The prediction accuracy achieved by BCrystal was 95.4%, an increase of 12% over than the-state-of-art, DeepCrystal that achieved an accuracy of 82.8%. Moreover, the prediction accuracy of BCrystal was 15%, 17%, 27%, 30%, 30%, 32%, 36% higher than Crysalis II (80.4%), Crysalis I (77.7%), PPCPred (67.2%), XtalPred-RF (65%), fDETECT (64.6%), TargetCrys (62.7%) and CrysalP2 (58.5%), respectively. Noteworthly, BCrystal achieved an MCC score of 0.908, which was 25% higher than the MCC score obtained by DeepCrystal (0.658), 29% and 35% higher than second and third-best competitor, Crysalis II (0.61) and Crysalis I (0.556), respectively. As depicted in Table 1 and Figure 3, on the balanced test set BCrystal achieved an area under curve (AUC) score of 0.981 that was 7.8% higher than DeepCrystal (0.903), 9% higher than Crysalis II (0.89) and 11% higher than Crysalis I (0.865), respectively. In addition, BCrystal was at least 20% better than other competitors in terms of AUC, fDETECT (0.78), PPCPred (0.75), TargetCrys (0.64) and CrystalP2 (0.61), respectively. BCrystal attained a score of 0.939 and 0.970 w.r.t. precision and recall scores, which reflected the capability of BCrystal to accurately identify and discriminate between both crystallizable and non-crystallizable proteins.
Table 1.
BCrystal outperforms eight other protein crystallization predictors on the balanced test data
| Models | Accuracy | MCC | AUC | F-score | Recall | Precision | NPV |
|---|---|---|---|---|---|---|---|
| PPCpred | 0.672 | 0.359 | 0.754 | 0.616 | 0.528 | 0.740 | 0.635 |
| fDETECT | 0.646 | 0.355 | 0.778 | 0.504 | 0.36 | 0.840 | 0.593 |
| Crysalis I | 0.777 | 0.556 | 0.865 | 0.767 | 0.738 | 0.799 | 0.758 |
| Crysalis II | 0.804 | 0.61 | 0.888 | 0.796 | 0.767 | 0.828 | 0.784 |
| XtalPred-RF | 0.650 | 0.301 | 0.710 | 0.654 | 0.663 | 0.645 | 0.655 |
| TargetCrys | 0.627 | 0.255 | 0.637 | 0.637 | 0.656 | 0.619 | 0.593 |
| CrystalP2 | 0.585 | 0.177 | 0.608 | 0.627 | 0.700 | 0.568 | 0.613 |
| DeepCrystal | 0.828 | 0.658 | 0.903 | 0.822 | 0.795 | 0.851 | 0.809 |
| BCrystal | 0.954 | 0.908 | 0.981 | 0.954 | 0.970 | 0.939 | 0.969 |
Note: Best results are highlighted in bold.
Fig. 3.

The AUC plots for the three different test sets. (A) AUPR for balanced test set. (B) AUPR for SP final test set. (C) AUPR for TR final test set
3.2 SP_final test set
This test set contained 237 proteins which had very low sequence similarly with the training set. On all of the evaluation metrics except precision, BCrystal was superior when compared to state-of-the-art crystallization models. BCrystal achieved an MCC score of 0.773 which was 24%, 26% and 33% higher than DeepCrystal, Crysalis II and Crysf, respectively as depicted in Table 2. BCrystal obtained a prediction accuracy of 89.4%, that was 13% better than current state-of-the-art, DeepCrystal (75.9%), 14%, 17%, 19%, 22%, 23%, 27%, 27%, 43% higher than Crysalis II (75.1%), Crysalis I (72.5%), Crysf (70.0%), PPCPred (66.6%), CrystalP2 (65.8%), fDETECT (61.6%), TargetCrys (61.1%) and XtalPred-RF (45.1%), respectively. Remarkably, BCrystal could detect diffraction quality crystals better than other sequence-based predictors with a F-score of 0.919 while DeepCrystal achieved a F-score of 0.788, Crysalis II obtained 0.783, Crysalis I obtained 0.763, Crysf attained a score of 0.727, while PPCPred, fDETECT and CrystalP2 obtained F-scores of 0.675, 0.580 and 0.734, respectively. BCrystal comprehensively outperformed all sequence-based predictors w.r.t. the recall evaluation metric. BCrystal obtained a recall value (0.966), while it achieved a precision score of (0.877), making it the method with the highest ability to correctly identify crystallizable proteins. Even though fDETECT had the higher precision score (0.913), it obtained a very low recall value (0.425), diminishing its ability to correctly detect crystallizable proteins. Finally, BCrystal obtained an area under precision-recall (AUPR) score of (0.957) which was 8% higher than DeepCrystal (0.877), 10% higher than Crysf (0.853) and 7% higher than Crysalis II (0.883), its three nearest competitors as observed in Figure 4.
Table 2.
BCrystal surpassed nine other protein crystallization predictors on the SP_final set
| Models | Accuracy | MCC | AUC | F-score | Recall | Precision | NPV |
|---|---|---|---|---|---|---|---|
| Crysf | 0.700 | 0.426 | 0.811 | 0.727 | 0.641 | 0.840 | 0.572 |
| PPCpred | 0.666 | 0.403 | 0.784 | 0.675 | 0.554 | 0.863 | 0.535 |
| fDETECT | 0.616 | 0.381 | 0.837 | 0.580 | 0.425 | 0.913 | 0.494 |
| Crysalis I | 0.725 | 0.448 | 0.835 | 0.763 | 0.709 | 0.826 | 0.609 |
| Crysalis II | 0.751 | 0.505 | 0.851 | 0.783 | 0.722 | 0.856 | 0.633 |
| XtalPred-RF | 0.451 | 0.149 | 0.449 | 0.548 | 0.553 | 0.564 | 0.288 |
| TargetCrys | 0.611 | 0.223 | 0.641 | 0.659 | 0.601 | 0.729 | 0.486 |
| CrystalP2 | 0.658 | 0.257 | 0.696 | 0.734 | 0.756 | 0.713 | 0.55 |
| DeepCrystal | 0.759 | 0.53 | 0.874 | 0.788 | 0.716 | 0.876 | 0.637 |
| BCrystal | 0.894 | 0.774 | 0.951 | 0.919 | 0.966 | 0.877 | 0.932 |
Note: Best results are highlighted in bold.
Fig. 4.

The AUPR plots for the three different test sets. (A) AUPR for balanced test set. (B) AUPR for SP final test set. (C) AUPR for TR final test set
3.3 TR_final test set
The final experiment was performed on the TR_final test set. BCrystal outperformed all sequence-based predictors for every evaluation metric. BCrystal obtained a prediction accuracy score of 96.3% which was 12% higher than state-of-the-art, DeepCrystal (84.1%). It was also better than Crysf (84.1%), Crysalis II (81.6%), Crysalis I (78.7%) and PPCPred (74.8%). In addition, BCrystal was also superior w.r.t. AUC, F-score and MCC metrics. Figure 3C illustrated how BCrystal performed well w.r.t. AUC. BCrystal achieved an AUC value of 0.988, which was 8%, 9%, 10%, 11%, 14%, 16.9%, 46% higher than DeepCrystal (0.91), Crysalis II (0.892), Crysf (0.887), Crysalis I (0.87), fDETECT (0.847), PPCPred (0.819) and XtalPred-RF (0.525), respectively. For the MCC evaluation metric, BCrystal outperformed all in silico methods (see Table 3). BCrystal obtained an MCC of 0.922, which was better by 25%, 26%, 31.9%, 37%, 47% than Crysf (0.663), DeepCrystal (0.657), Crysalis II (0.603), Crysalis I (0.546) and PPCPred (0.448), respectively. On the other hand, in terms of recall, precision and NPV, BCrystal attained maximum values of 0.970, 0.933 and 0.982, respectively, when compared with other crystallization predictors.
Table 3.
BCrystal outperformed nine other protein crystallization predictors on the TR_final set
| Models | Accuracy | MCC | AUC | F-score | Recall | Precision | NPV |
|---|---|---|---|---|---|---|---|
| Crysf | 0.841 | 0.663 | 0.887 | 0.747 | 0.631 | 0.918 | 0.817 |
| PPCpred | 0.748 | 0.448 | 0.819 | 0.64 | 0.606 | 0.677 | 0.782 |
| fDETECT | 0.75 | 0.447 | 0.847 | 0.548 | 0.411 | 0.823 | 0.733 |
| Crysalis I | 0.787 | 0.546 | 0.87 | 0.715 | 0.724 | 0.707 | 0.836 |
| Crysalis II | 0.816 | 0.603 | 0.892 | 0.748 | 0.74 | 0.756 | 0.849 |
| XtalPred-RF | 0.451 | 0.04 | 0.525 | 0.452 | 0.537 | 0.39 | 0.651 |
| TargetCrys | 0.634 | 0.325 | 0.693 | 0.614 | 0.788 | 0.503 | 0.733 |
| CrystalP2 | 0.581 | 0.241 | 0.673 | 0.577 | 0.775 | 0.460 | 0.78 |
| DeepCrystal | 0.841 | 0.657 | 0.910 | 0.781 | 0.762 | 0.800 | 0.864 |
| BCrystal | 0.963 | 0.922 | 0.988 | 0.951 | 0.970 | 0.933 | 0.982 |
Note: Best results are highlighted in bold.
3.4 Model interpretation
An advantage of tree-based non-linear machine learning techniques, in contrast to black-box modeling techniques like support vector machines Drucker et al. (1997) and artificial neural networks Fausett et al. (1994), is that we can easily obtain feature/variable importance scores for all input features. The importance of a feature is the sum of information gained when splits (tree branching) are performed using that variable. A distinct benefit of using an XGBoost classifier is that out of all the 9139 features used during training, variables which are not used for optimal tree splits in the BCrystal model are pruned automatically and get a feature importance score of 0. We observe from Table 4 that only 127 features have non-zero feature importance scores. A list of all these features with their individual feature importance score is available in Supplementary Table S1.
Table 4.
Variable importance percentages grouped by feature classes for the BCrystal model highlighting the feature contributions in order as depicted in Figure 2
| Feature class | Total features | Total features (>0) | Total imp (%) |
|---|---|---|---|
| Log(L) | 1 | 1 | 0.052 |
| Log(MW) | 1 | 1 | 0.032 |
| Turn Freq | 1 | 0 | 0.000 |
| Gravy index | 1 | 1 | 0.017 |
| Aliphatic index | 1 | 0 | 0.000 |
| Total charge | 1 | 0 | 0.000 |
| Mono Freq SS3 | 3 | 1 | 0.068 |
| Di Freq SS3 | 9 | 0 | 0.000 |
| Tri Freq SS3 | 27 | 3 | 0.063 |
| Mono Freq SS8 | 8 | 4 | 0.664 |
| Di Freq SS8 | 64 | 8 | 1.051 |
| Tri Freq SS8 | 512 | 18 | 1.001 |
| FER at RSA cutoffs | 20 | 10 | 81.08 |
| FER at RSA cutoff × HP | 20 | 12 | 11.53 |
| Disorder features | 25 | 5 | 1.906 |
| PBS in disorder features | 25 | 2 | 0.099 |
| Mono Freq AA | 20 | 7 | 0.479 |
| Di Freq AA | 400 | 31 | 1.018 |
| Tri Freq AA | 8000 | 23 | 0.937 |
| All features | 9139 | 127 | 100.00 |
Note: Here, total features (>0) represents the total number of variables from a feature class with non-zero variable importance scores.
We then analyzed the total feature importance contribution of all features according to their feature types/classes as shown in Figure 2 and Table 4. At the highest level, we had three macro classes of features including global, sequence, and structure derived features contributing 0.101%, 2.434%, and 97.465%, respectively in the overall variable importance scores. From Table 4, we can observe that the maximum feature importance is associated with FER at different RSA cutoffs (81.08%, more details in Supplementary Fig. S4), followed by FER at different RSA cutoffs multiplied by average hydrophobicity of exposed residues (11.53%). Thus, RSA features (see Fig. 2) account for ≈ 92% of the total variable importance in the BCrystal model. Figure 5 showcases the difference between the feature values for these Top 3 variables in case of crystallizable versus non-crystallizable proteins.
Fig. 5.
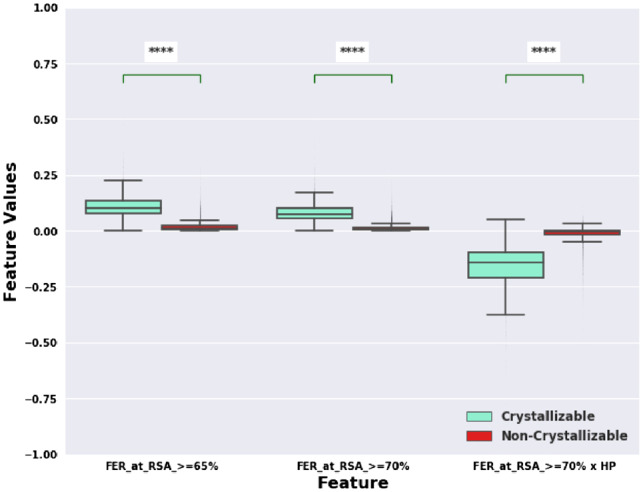
Frequency distribution of Top 3 features with relative importance higher than 5% for crystallizable and non-crystallizable training protein sequences shown as box plots (****P value < 0.0001)
One of the disadvantages of the inherent feature importance scores obtained from the XGBoost model is that the directionality is not apparent i.e. when a particular feature for a protein sample takes a high value, does that correspond to high or low feature importance score. Moreover, at the test phase, it is not straightforward for traditional white-box, tree-based, machine learning techniques to provide information about, say, the Top 5 features driving the prediction to be diffraction quality crystals or non-crystallizable class.
Recently, several techniques such as the LIME (Ribeiro et al., 2016) and SHAP (Lundberg and Lee, 2017) methods have been proposed to overcome the aforementioned limitations. These methods have the ability to interpret feature importance scores from complex training models as well provide interpretable predictions for a test sample by grounding their reasoning on the top k features for that particular test instance. In our work, we use the SHAP (SHapley Additive exPlanations) method, a unified framework for interpretating predictions, as it was shown in (Lundberg and Lee, 2017) to outperform LIME and they demonstrated that its predictions are better aligned with human intuitions.
The SHAP method belongs to the class of additive feature attribution methods where a test instance prediction is composed as a linear function of features and satisfies three critical properties comprised of local accuracy, missingness and consistency. The explicit SHAP regression values comes from a game-theory framework (Lipovetsky and Conklin, 2001; Shapley, 1953) and can be computed as:
| (3) |
Here, Q represents the set of all d features and S represents the subsets obtained from Q after removing the feature and is an estimate of the importance of feature i in the model. In order to refrain from undergoing differences to estimate , the SHAP method approximates the Shapley value by either performing Shapley sampling (Štrumbelj and Kononenko, 2014) or quantitative input influence (Datta et al., 2016). A detailed description of the SHAP method for model interpretation is available in Lundberg and Lee (2017).
We passed our BCrystal model along with the training set to the SHAP method as shown in Figure 1 to obtain importance of features based on Shapley values. Figure 6 highlights the Top 25 training features based on Shapley values. Moreover, it also provides directionality i.e. when a feature attains ‘high’ or ‘low’ values, the corresponding Shapley values are positive or negative. The positive Shapley values drive the predictions towards crystallizable class, whereas the negative Shapley values influence the predictions to move toward the non-crystallizable class. From Figure 6, we can observe that when top features such as FER at RSA cutoffs and take high values, the corresponding Shapley values are positive driving model prediction to diffraction quality crystals, whereas when these features take low values (i.e. closer to 0), the corresponding Shapley values are negative. Similarly, Figure 6 illustrates that when top features such as fraction of the sequence which is disordered, number of disordered regions comprising >10 AAs and frequency of coils in protein sequences are high, the corresponding Shapley values are negative driving the model prediction towards non-crystallizable class.
Fig. 6.
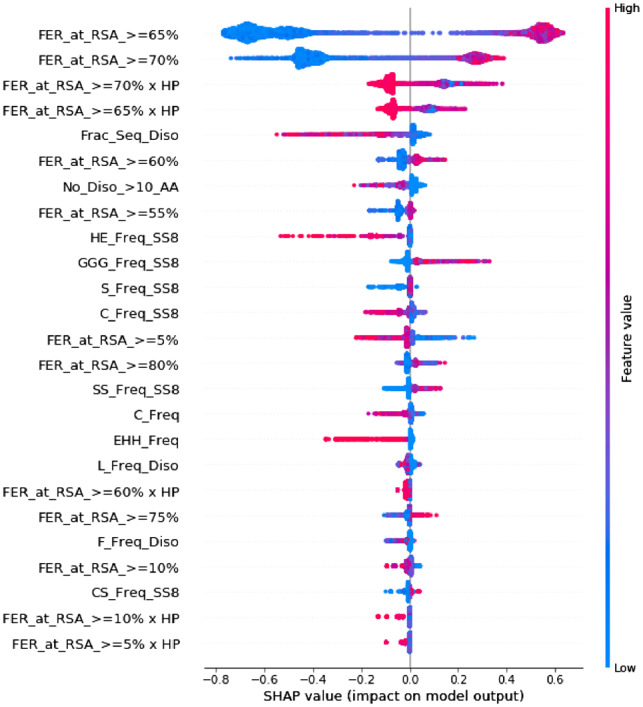
Top 25 features from SHAP (Lundberg and Lee, 2017)
4 Case study
We perform a truncation analysis i.e. removing one amino acid at a time from N-terminal of full length (l = 680) of PF21A human protein (Protein ID: Q96BD5). We generate features for each such construct and pass it to BCrystal model for predicting the crystallization propensity. The same analysis is performed for all constructs obtained by performing truncation from the C-terminal one amino acid at a time as depicted in Figure 7A. In the truncation analysis, we consider only those constructs whose lengths are greater than 10 AAs.
Fig. 7.

Crystallization propensity analysis for serial truncation of a very homologous protein (hard to crystallize protein) complimented with secondary structure analysis. (A) Crystallization propensities for serial truncation experiments. (B) Predicted secondary structure for PF21A human protein
The crystallization propensity of full length PF21A protein remains at 0.216, whereas serial truncation from N-terminal more or less maintains this value upto amino acid 200. After AA at position 200, the crystallization propensity increases but still remains below 0.5 till position 477. However, several constructs comprising AAs starting from positions [478, 500] and ending at position 680 are crystallizable. These constructs include majority of the AAs from the functional Zinc finger domain (PDB ID: 2PUY, made of AAs between position 486 and 543 of PF21A protein) which has a known crystal structure (BCrystal prediction score: 0.777) as depicted in Supplementary Figure S3. The functional domain in the full length protein was predicted using SMART (Schultz et al., 1998) method.
Similarly, serial truncation from C-terminal result in constructs with low crystallization propensities (start position is 1) till end position reaches 70. All constructs of smaller lengths obtained from truncation of one amino acid at time from the C-terminal are crystallizable. This is due to the fact that these residues primarily belong to the helical regions. We predicted the secondary structure of PF21A using PSIPRED (McGuffin et al., 2000) which indicated that majority of the residues are part of helixes between the positions [1, 70] as shown in Figure 7B. The helical regions are usually the best-folded regions of a protein, providing stability and thereby more probability of forming crystal contacts (Deller et al., 2016). Thus, our BCrystal model accurately suggests that small sized constructs containing the functional Zinc finger domain (with known crystal structure) and constructs with primarily helical secondary structures are crystallizable for the protein PF21A (Protein ID: Q96BD5).
We perform additional analysis on two other proteins to showcase that the Top 10 features obtained via SHAP algorithm for BCrystal prediction are biologically relevant. Acinetobacter baylyi pyrimidine nucleoside phosphorylase (Protein ID: ACIAD0356, PDB ID: 3HQX) attained a relatively high BCrystal prediction score of 0.776, while the human AT-rich interactive domain-containing protein 3A’s (Protein ID: ARID3A, PDB ID: 4LJX) got a score of 0.75. The first protein’s Top 10 features had almost exclusively positive Shapley values, driving BCrystal prediction to be closer to 1. In particular, features such as FER at different RSA cutoffs were the primary driving force for the high BCrystal output score as observed in Figure 8B and C. For the second example protein, the Top 10 features included both, features with positive and negative Shapley values, lowering the BCrystal prediction score. In Figure 8, we illustrated the two proteins crystal structures in cartoon (Figure 8A and D) as well as in surface representation (Figure 8B and E) with the RSA mapped onto the structures. It was readily apparent that the phosphorylase has more RSA amino acids (depicted with light colors) than the second example protein ARID3A. Furthermore, ARID3A had 2 disordered regions at its terminal regions, which were not visible in the crystal structure, illustrated by dashed lines (Figure 8D) that had slight antagonistic effect on the protein crystallization propensity. We provide additional information about the two proteins in Supplementary Figure S4.
Fig. 8.

Top predictive features—relative solvent accessibilities and disordered regions—correlate with BCrystal scores. (A) Crystal structure of pyrimidine/purine nucleoside phosphorylase with predicted RSA values mapped onto the structure in cartoon illustration (PDB ID: 3HQX, 108 residues). (B) Predicted RSA values shown on structure in surface representation. (C) Top 10 SHAP-features with corresponding Shapely values. (D) Crystal structure of human AT-rich interactive domain-containing protein 3A (Protein ID: ARID3A, PDB ID: 4LJX, 145 residues) in cartoon illustration. (E) Predicted RSA values mapped onto structure shown in surface representation. (F) Top 10 SHAP-features with corresponding Shapely values
Finally, we perform assessment of crystallizability of all proteins in the human proteome. We downloaded all the proteins from TargetTrack database which maintains crystallization status of proteins. We filter to keep only those proteins associated with humans and have low sequence similarity (¡15%) with the training proteins. We then categorize these proteins into ‘Some success in crystallization’ and ‘No evidence in crystallization’ classes based on their working status and run BCrystal to obtain the crystallization propensity scores. The predictive power of BCrsytal for these hard real-world protein targets is depicted in Supplementary Table S2. Additionally, probing the proteins which are misclassified can help to prioritize human targets that warrant further investigation as highlighted in Supplementary Figure S5.
5 Discussion
The development of in silico sequence-based protein crystallization prediction tools with high accuracy continues to be highly sought after. In this study, we introduce BCrystal, a crystallization predictor that uses the XGBoost modeling technique and features that represent physio-chemical, sequence as well as structural properties of proteins. BCrystal outperforms, to the best of our knowledge, all existing sequence-based crystallization predictors by >12.5% in accuracy and 0.25 in MCC.
The superiority of BCrystal over other predictors is due to three factors. The first factor is the choice of the machine learning method XGBoost. The non-linear optimized gradient boosting technique, XGBoost, is able to capture non-linear relationships between the features and the dependent vector, which makes its performance comparable to non-linear methods like SVMs (Chang and Lin, 2011). Additionally, XGBoost reduces the bias of the model without increasing the variance, leading to better generalization performance. In addition, XGBoost has the ability to provide variable importance, making the model interpretable, which is a drawback of black-box methods like SVMs and deep learning (DeepCrystal) (LeCun et al., 1998). The second factor is the choice of features. We include several features that provide information about the physio-chemical, sequence and structural properties of the protein of interest. Previous tools such as DeepSF (Hou et al., 2018) have shown that features extracted from the SCRATCH suite are very helpful in correct protein fold recognition. We observe that the predicted FER at different RSA cutoffs and predicted average hydrophobicity of such residues determined via the SCRATCH suite plays a very vital role in protein crystallization propensity. An inherent advantage of the XGBoost model is that it performs regularization i.e. feature pruning automatically, reducing the risk of overfitting and including only those features which helps in discriminating the positive class from the non-crystallizable ones. Thus, it has an advantage over two-stage methods like CrystalP2, PPCpred, Cryalis and Crysf, which are susceptible to loss of information by explicitly performing feature selection. Finally, unlike other sequence-based crystallization predictors, BCrystal has the ability to provide a meaningful explanation for each test sample using the SHAP method. This empowers crystallographers to more quickly screen for good crystallization targets and to attempt mutations of initial targets for diffraction quality crystal production reliably and with more information to reason about the effects of proposed modifications.
From the BCrystal model, (see Figs 5 and 6), we observe that the features with the highest variable importance were FER at RSA cutoffs 65% and 70% and FER at RSA cutoff 70% × HP of corresponding residues. We notice from the training set that the FERs for the crystallizable set is significantly different than the FERs for the non-crystallizable set (see Fig. 5, P-value < 1e−4) at almost all RSA cutoff levels (see Supplementary Fig. S6), which is the reason that the FER is a dominant feature of the classifier.
Moreover, the RSA cutoff (0–100%) for a residue in a protein sequence is used to determine whether the residue is buried or exposed. In the past, several RSA cutoffs were used to divide the residues into the two classes. For example in Chen and Zhou (2005), RSA cutoffs 20–25% were used arbitrarily to get roughly balanced buried and exposed residues in order to determine the secondary structure of proteins. Similarly, in Tien et al. (2013), RSA cutoff of 0–5% were used arbitrarily to identify buried residues and the fraction of buried residues were correlated with properties like transfer energy from vapor to water, cyclohexane to water and so on. Intuitively, at least half of the residue would be available to the solvent at RSA cutoff of 50%. If only half of the residue is available to the solvent (i.e. RSA at 50%), the residue might still be quite rigid and thus there would be a lower probability that the residue lines up perfectly to create a crystal lattice Salemme et al. (1988); Zhang et al. (2009). So, a relatively higher RSA cutoff {60%, 65%, 70%} for a residue would imply that the residue which we consider exposed has enough flexibility to be part of the crystal lattice and hence enhances the chances of the protein to crystallize Zhang et al. (2015). This is further complemented by the difference between median (DBM) values of FER at RSA cutoffs: 60%, 65% and 70% for crystallizable vs non-crystallizable class (see Supplementary Fig. S6), where the median FER values at each of these cutoffs are much higher in crystallizable proteins in comparison to non-crystallizable ones (closer to 0). Additionally, a statistical measure based on ratio of Difference Between Median (DBM) and Overall Visible Spread (OVS) is highest for FER at RSA cutoffs: 60%, 65% and 70% (Supplementary Fig. S7). The higher this percentage, the larger is the difference in the FER feature values at that particular RSA cutoff between the crystallizable and non-crystallizable class, and stronger is the discrimination power of this feature Wild et al. (2011).
An important issue which can influence the BCrystal predictions is the presence of a recognizable homolog in the PDB as it would enhance the accuracy of the obtained RSA values provided by the SCRATCH suite. Since we enforce a strong sequence similarity criterion i.e. removal of all sequences within the training set having >15% intra-training sequence similarity as well as removal of sequences having >15% sequence similarity with test protein sequences, BCrystal model should be able to overcome this bias. Additionally, other features extracted by the SCRATCH suite (secondary structure features) would have benefitted from presence of a recognizable homolog and their feature importance would have been inflated, which is not the case as observed from Table 4.
Furthermore, from Figure 6, we also detect that the non-crystallizable proteins tend to have large disordered regions (Frac_Seq_Diso > 0 and No_Diso_>10_AA > 0) in the protein sequence, higher frequency coils in secondary structure and higher frequency of tri-peptides containing multiple histidines. Interestingly, positively charged surface residues and polyhistidine-tags have been previously (Chan et al., 2013; Woestenenk et al., 2004) correlated with protein insolubility, which explains that higher frequency of the tripeptide EHH in the protein sequence drives the model prediction to negative class (see Fig. 6).
Apart from the comprehensively outperforming all existing sequence-based protein crystallization predictors, BCrystal is the first sequence-based bioinformatics tool that provides meaningful interpretations for its prediction by using the SHAP method. BCrystal always attains a very high recall (average recall of 0.966) for the three independent test sets, suggesting that it can very efficiently select crystallizable proteins from an initial set of candidates, thereby, reducing the high attrition rate and the production cost. The ultimate goal of an in silico sequence-based crystallization predictor would be the ability to design protein sequence variants i.e. perform single/double point mutations in the protein sequence such that these mutations positively drive the protein crystallization propensity. Ideally, rendering the protein to be crystallizable (from non-crystallizable class) without changing the intrinsic functionality of the protein. In the future, we plan to provide wrapper software on top of BCrystal that will automatically generate/suggest mutants (with/without fixing specific residues; for instance functionally important ones) for crystallographers to use.
Conflict of Interest: none declared.
Supplementary Material
References
- Breiman L. (2001) Random forests. Mach. Learn., 45, 5–32. [Google Scholar]
- Chan P. et al. (2013) Soluble expression of proteins correlates with a lack of positively-charged surface. Sci. Rep., 3, 3333.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C.-C., Lin C.-J. (2011) LIBSVM: a library for support vector machines. ACM Trans. Intell. Syst. Technol., 2, 27. [Google Scholar]
- Charoenkwan P. et al. (2013) SCMCRYS: predicting protein crystallization using an ensemble scoring card method with estimating propensity scores of P-collocated amino acid pairs. PLoS One, 8, e72368.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Zhou H.-X. (2005) Prediction of solvent accessibility and sites of deleterious mutations from protein sequence. Nucleic Acids Res., 33, 3193–3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T., Guestrin C. (2016) XGBoost: a scalable tree boosting system In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp. 785–794. ACM, San Francisco, CA. [Google Scholar]
- Cheng J. et al. (2005) Scratch: a protein structure and structural feature prediction server. Nucleic Acids Res., 33 (Suppl_2), W72–W76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta A. et al. (2016) Algorithmic transparency via quantitative input influence: theory and experiments with learning systems In: 2016 IEEE Symposium on Security and Privacy (SP), pp. 598–617. IEEE, San Jose, CA. [Google Scholar]
- Deller M.C. et al. (2016) Protein stability: a crystallographer’s perspective. Acta Crystallogr. F, 72, 72–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drucker H. et al. (1997) Support vector regression machines. In: Advances in Neural Information Processing Systems, pp. 155–161. MIT Press, Cambridge. [Google Scholar]
- Elbasir A. et al. (2019) DeepCrystal: a deep learning framework for sequence-based protein crystallization prediction. Bioinformatics, 35, 2216–2225. [DOI] [PubMed] [Google Scholar]
- Fausett L.V. et al. (1994) Fundamentals of Neural Networks: Architectures, Algorithms, and Applications. Vol. 3 Prentice-Hall, Englewood Cliffs, NJ, USA. [Google Scholar]
- Friedman J.H. (2001) Greedy function approximation: a gradient boosting machine. Ann. Stat., 29, 1189–1232. [Google Scholar]
- Fu L. et al. (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics, 28, 3150–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou J. et al. (2018) DeepSF: deep convolutional neural network for mapping protein sequences to folds. Bioinformatics, 34, 1295–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J. et al. (2016) TargetCrys: protein crystallization prediction by fusing multi-view features with two-layered SVM. Amino Acids, 48, 2533–2547. [DOI] [PubMed] [Google Scholar]
- Jahandideh S. et al. (2014) Improving the chances of successful protein structure determination with a random forest classifier. Acta Crystallogr. D, 70, 627–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khurana S. et al. (2018) DeepSol: a deep learning framework for sequence-based protein solubility prediction. Bioinformatics, 1, 9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurgan L., Mizianty M.J. (2009) Sequence-based protein crystallization propensity prediction for structural genomics: review and comparative analysis. Nat. Sci., 1, 93–106. [Google Scholar]
- LeCun Y. et al. (1998) Gradient-based learning applied to document recognition. Proc. IEEE, 86, 2278–2324. [Google Scholar]
- Lipovetsky S., Conklin M. (2001) Analysis of regression in game theory approach. Appl. Stoch. Models Bus. Ind., 17, 319–330. [Google Scholar]
- Lundberg S.M., Lee S.-I. (2017) A unified approach to interpreting model predictions. In: Advances in Neural Information Processing Systems, pp. 4765–4774. MIT Press, Cambridge. [Google Scholar]
- Mall R. et al. (2017) Differential community detection in paired biological networks In: Proceedings of the 8th ACM International Conference on Bioinformatics, Computational Biology, and Health Informatics, pp. 330–339. ACM, Boston, Massachusetts. [Google Scholar]
- Mall R. et al. (2018a) An unsupervised disease module identification technique in biological networks using novel quality metric based on connectivity, conductance and modularity. F1000Research, 7, 378. [Google Scholar]
- Mall R. et al. (2018b) RGBM: regularized gradient boosting machines for identification of the transcriptional regulators of discrete glioma subtypes. Nucleic Acids Res., 46, e39–e39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin L.J. et al. (2000) The PSIPRED protein structure prediction server. Bioinformatics, 16, 404–405. [DOI] [PubMed] [Google Scholar]
- Meng F. et al. (2018) fDETECT webserver: fast predictor of propensity for protein production, purification, and crystallization. BMC Bioinformatics, 18, 580.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawi R. et al. (2017) PaRSnIP: sequence-based protein solubility prediction using gradient boosting machine. Bioinformatics, 34, 1092–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro M.T. et al. (2016) Why should I trust you?: Explaining the predictions of any classifier In: Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, pp. 1135–1144. ACM, San Francisco, CA. [Google Scholar]
- Salemme F. et al. (1988) Molecular factors stabilizing protein crystals. J. Cryst. Growth, 90, 273–282. [Google Scholar]
- Schapire R.E. (2003) The boosting approach to machine learning: an overview In Nonlinear Estimation and Classification, pp. 149–171. Springer, New York. [Google Scholar]
- Schultz J. et al. (1998) Smart, a simple modular architecture research tool: identification of signaling domains. Proc. Natl. Acad. Sci. USA, 95, 5857–5864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Service R. (2005) Structural biology. Structural genomics, round 2. Science, 307, 1554.. [DOI] [PubMed] [Google Scholar]
- Shapley L.S. (1953) A value for n-person games. Contributions to the Theory of Games, 2, 307–317. [Google Scholar]
- Štrumbelj E., Kononenko I. (2014) Explaining prediction models and individual predictions with feature contributions. Knowl. Inform. Syst., 41, 647–665. [Google Scholar]
- Terwilliger T.C. et al. (2009) Lessons from structural genomics. Ann. Rev. Biophys., 38, 371–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien M.Z. et al. (2013) Maximum allowed solvent accessibilities of residues in proteins. PLoS One, 8, e80635.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga J.K., Tusnády G.E. (2018) TMCrys: predict propensity of success for transmembrane protein crystallization. Bioinformatics, 34, 3126–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. et al. (2014) PredPPCrys: accurate prediction of sequence cloning, protein production, purification and crystallization propensity from protein sequences using multi-step heterogeneous feature fusion and selection. PLoS One, 9, e105902.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. et al. (2016) Crysalis: an integrated server for computational analysis and design of protein crystallization. Sci. Rep., 6, 21383.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. et al. (2017) Critical evaluation of bioinformatics tools for the prediction of protein crystallization propensity. Brief. Bioinform., 19, 838–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward J.J. et al. (2004) The DISOPRED server for the prediction of protein disorder. Bioinformatics, 20, 2138–2139. [DOI] [PubMed] [Google Scholar]
- Wild C. et al. (2011) Towards more accessible conceptions of statistical inference. J. Royal Stat. Soc., 174, 247–295. [Google Scholar]
- Woestenenk E.A. et al. (2004) His tag effect on solubility of human proteins produced in Escherichia coli: a comparison between four expression vectors. J. Struct. Funct. Genomics, 5, 217–229. [DOI] [PubMed] [Google Scholar]
- Zhang H. et al. (2009) On the relation between residue flexibility and local solvent accessibility in proteins. Proteins, 76, 617–636. [DOI] [PubMed] [Google Scholar]
- Zhang X. et al. (2015) Character-level convolutional networks for text classification. In: Advances in Neural Information Processing Systems, pp. 649–657. MIT Press, Cambridge. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.