

Supplemental Digital Content is available in the text
Keywords: methylation, deoxyribonucleic acid, large for gestational age, placenta
Abstract
To investigate the molecular mechanisms of later metabolic health changes in large for gestational age (LGA) newborns by analyzing deoxyribonucleic acid (DNA) methylation patterns in the placenta of LGA and appropriate for gestational age (AGA) newborns.
A total of 6 placentas of LGA and 6 placentas of AGA newborns were enrolled as LGA group and AGA group. DNA methylation was analyzed using the Illumina Infinium Human MethylationEPIC BeadChip microarrays and verified via pyrosequencing and reverse transcription-quantitative real-time polymerase chain reaction. Functional enrichment analysis were constructed by gene ontology and Kyoto Encyclopedia of Genes and Genomes pathway analysis based on the differentially methylated regions between LGA and AGA groups.
Clinical investigation showed that LGA newborns had significantly lower hemoglobin and blood glucose compared to AGA newborns. Birth weight was negatively correlated to hemoglobin and blood glucose. Genome-wide DNA methylation analysis identified 17 244 methylation variable positions achieving genome-wide significance (adjusted P < .05). 34% methylation variable positions were located in the gene promoter region. A total of 117 differentially methylated regions were revealed by bump hunting analysis, which mapped to 107 genes. Function analysis showed 13 genes enriched in “adhesion and infection process, endocrine and other factor-regulated calcium reabsorption, calcium signaling pathway and transmembrane transport”. Four genes linked to type II diabetes mellitus. Among the 13 genes, we selected GNAS and calcium voltage-gated channel subunit alpha1 G for independent verification of pyrosequencing, and the messenger ribonucleic acid levels of guanine nucleotide binding protein, calcium voltage-gated channel subunit alpha1 G, DECR1, and FK506 binding protein 11 were verified by reverse transcription-quantitative real-time polymerase chain reaction.
DNA methylation variation and gene expression differences in placental samples were associated with LGA newborns, which linking the effect of intrauterine environment to regulation of the offspring's gene expression. Furthermore, pathway analysis suggested that intrauterine environment affecting fetal growth might had a functional impact on multiple signaling pathways involved in fetal growth, metabolism, and inflammation. Further studies were required to understand the differences of methylation patterns.
1. Introduction
With the development of socioeconomic levels in China, the prevalence of metabolic syndrome increased dramatically. Although some genetic factors have been associated with the risks of metabolic syndrome, which cannot be explained entirely by the changes of Mendelian inheritance. Sustained changes in metabolism following an adverse fetal or early neonatal environment have been connected to the mechanisms involving environmental factors regulation of gene expressions.[1] Environmental factors can trigger long-lasting changes through epigenetic processes, which regulate gene expressions without affecting the genetic sequences.[2–4] Notably, specific epigenetic changes have been linked to growth restriction.[5] Recently, impaired fetal growth decreased expressions of molecular regulators of β-cell mass and function, shown in some cases to be due to epigenetic changes initiated by an adverse fetal environment.[6,7] However, with the development of socioeconomic levels, the prevalence of small for gestational age (SGA) infant was dramatically declined while large for gestational age (LGA) was dramatically increased in China.
There is widespread knowledge that maternal obesity, diabetes and hyperinsulinemia are main causes of an infant being LGA. But now, women without apparent pregnancy complications also common have LGA newborns. LGA is regarded to be a well-established risk factor for metabolic syndrome as well.[8–10] Nevertheless, there is a mismatch between the given attentions versus the increasing number of LGA babies.
Epigenetic modifications, an important mechanism for diseases, can alter the expression patterns of genes, followed with the influence of glucose metabolism, fetal growth and even later life. Recently, methylation status of both coding and non-coding regions of the genome from cord blood was reported to be linked with birth weight.[11,12] Factors which contribute to reduced birth weight, such as maternal stress and depression, have also been associated with gene-specific promoter methylation patterns.[13] Epigenetic changes in placenta may have a great impact on fetal development and fetal adulthood, but less related research. Gene specific changes in methylation have also been reported in human placenta from SGA infants,[14] but not found in LGA.
Deoxyribonucleic acid (DNA) methylation is arguably the major epigenetic modification,[15] which controls a variety of cellular and developmental processes. During the last decade, more and more evidences have demonstrates that this process was involved in the occurrence and development of the human diseases.[16] Until now, the effect of LGA on DNA methylation and metabolic syndrome is poorly understood. Therefore, we analyzed the differences of DNA methylation in placenta to investigate the epigenetic mechanism of later metabolic changes in LGA newborns.
2. Methods
2.1. Subjects
In this study, we confirmed that all methods were carried out in accordance with relevant guidelines and regulations, all experimental protocols were approved by the Medical Ethical Committee of the Children Hospital of Zhejiang University School of Medicine (Approval Number: 2015-HP-025) and informed consent was obtained from all subjects or, if subjects are under 18, from a parent and/or legal guardian.
A case-control study was designed. A total of 150 newborns and their parents were recruited in the Women and Children's Hospital of Shaoxing (Zhejiang Province of China) from January to April 2015. Data of newborns and their parents were recorded before and after the newborn delivery.
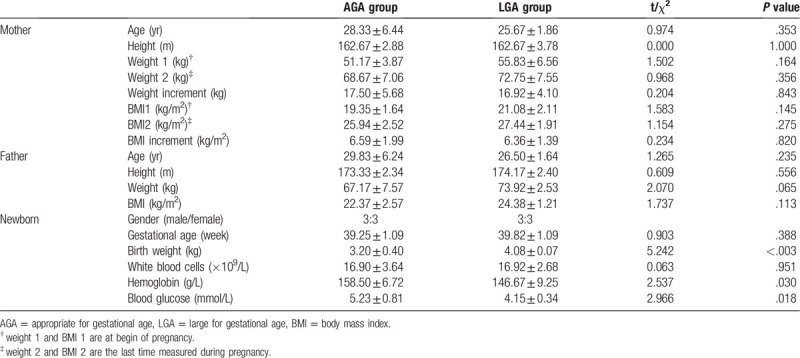
According to the gender, gestational age, birth weight, and delivery, 6 LGA and 6 appropriate for gestational age (AGA) newborns were enrolled for further study. All the newborns were natural labor full-term. Each group included 3 boys and 3 girls. The diagnosis of LGA and AGA was according to the 9th edition of Chinese pediatrics textbook. Mother with hyperglycemia, infection, chronic diseases of the heart, lung, liver and kidney, immunologic diseases or other diseases and smoking, or children with asphyxia, meconium-stained amniotic fluid, premature rupture of membranes, production process extension, precipitates, analgesics or anesthetics administration was excluded. The physical parameters of the newborns and their parents were listed in Table 1. The differences of gender distribution and gestational age were not significant between these 2 groups (t = 0.91, P = .39, respectively).
Table 1.
Characteristics of the newborns and their parents.
2.2. Placental samples collection
Two 1 × 2 cm placenta samples near the umbilical cord were collected immediately after delivery. After decidua and blood removal, then saline wash and frozen in liquid nitrogen for 1-5 hourr. Then, these samples were stored in -80 °C for further measurement.
2.3. Biochemical parameters
Baseline blood samples were obtained from the heel of newborns and immediately measured. White blood cells and hemoglobin of whole blood were determined with a blood cell automated analyzer (Sysmex XS-500i). Blood glucose was measured by the glucose oxidase method.
2.4. DNA extraction and DNA methylation data collection
Genomic DNA was purified with QIAamp DNA Mini Kit (Qiagen, Catalog No: 51304) and bisulfite treated with EZ DNA Methylation Kit (Zymo Research, Catalog No: D5001&D5002) according to the manufacturer's instructions. DNA methylation was then analyzed using the Illumina Infinium Human Methylation EPIC BeadChips (850K) (Illumina, San Diego, CA, USA) following the protocol guide (infinium-hd-methylation-experienced-user-card-manual-15019522-01) by Genergy Boi Technology Inc. (Shanghai, China). All samples were run on the microarray together within the same batch and quality control (QC) for obtained DNA methylation data was performed with the Minfi R package tools.
2.5. DNA methylation data analysis
In order to obtain good quality probes on the microarray, the 853 307 methylation probes were filtered on the following 4 conditions as previous report[17]: first were probes in 65 internal control SNPs; second were probes with SNP and probes that align to multiple locations; third were probe methylated loci with a P-value >.01; fourth were probes with low bead count (<3 beads). Additionally, a sample will be excluded in the further analysis if the ratio of filter out probes more than 5% or zero signal. The ratio of fluorescent signals was then computed from the two alleles using the β value. Beta Mixture Quantile dilation Beta method was used to standardize of β value.[18] Differentially methylated CpG sites were found by Limma R package tools with a P-values <0.05. Differentially methylated regions (DMRs) were found based on methylation variable positions (MVPs) by Probe Lasso method.[19]
2.6. Validation of DNA methylation status by pyrosequencing
In the validations of HM850K results, based on the analysis results and methylation levels observed, we chose to verify the methylation level of specific CpG sites with bisulfite pyrosequencing in the placenta of another 8 LGA and 7 AGA in the following genes guanine nucleotide binding protein (GNAS) and calcium voltage-gated channel subunit alpha1 G (CACNA1G). The forward, biotinylated-reverse and sequencing primers were designed using Pyro-Mark Assay Design 2.0 and were listed in Supplemental Table S1. All procedures were performed according to the manufacturer's protocols. Pyrosequencing was performed on a PyroMark Q24 system (Qiagen) and cytosine methylation was quantified using PyroMark Q241.010 software.
2.7. Validation of gene expression level by reverse transcription-quantitative real-time polymerase chain reaction (RT-qPCR)
To confirm the DNA methylation microarray analysis results, RT-qPCR was performed to investigate messenger ribonucleic acid (RNA) levels of FK506 binding protein 11 (FKBP11), 2,4-dienoyl-CoA reductase 1(DECR1), GNAS and CACNA1G in the placenta of another 8 LGA and 8 AGA. Total RNA was purified with RNeasy Protect Mini Kit (Qiagen, Catalog No: 74124). Reverse transcription of total RNA into complementary DNA (cDNA) use PrimeScript 1st Strand cDNA Synthesis Kit (TaKaRa, Catalog No: 6110A). The gene-specific primers were designed based on the cDNA sequences (Supplemental Table S2). RT-qPCR was performed using GoTaq qPCR SYBR Green Master Mix (Promega, Catalog No: A6001) and ABI StepOne PlusTM Sequence Detection System (Applied Biosystems; Thermo Fisher Scientific Inc.). The relative expression of each gene was determined using the 2-ΔCT method.
2.8. Gene ontology analysis and functional enrichment
To identify prominent clinical mechanisms affecting and/or affected in LGA, genes with evidence of differential methylation were investigated with respect to their functions and major biological roles. Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) (http://www.kegg.jp/kegg/pathway.html) pathway analysis were constructed based on the DMRs between LGA and AGA. The P value threshold was limited to 10−3.
2.9. Statistics analyses
Anthropometric and clinical characteristics of individuals are expressed as mean and standard deviation. Differences in these data between AGA and LGA placenta were tested by unpaired Student t test. The methylation level of each cytosine was expressed as a β value calculated as the fluorescence intensity ratio of the methylated to the unmethylated versions of the probes. β values with detection P-values >.01 were considered to fall below the minimum intensity and threshold, and these values were consequently removed from further analysis.
Differences in DNA methylation levels detected by pyrosequencing and expression of selected genes between AGA and LGA placenta were assessed by the nonparametric Mann–Whitney T test using the SPSS version 16.0 software (SPSS Inc, Chicago). P-values ≤.05 were considered statistically significant.
3. Results
Among 853 307 methylation probes, the ratios of filter out probes of 12 samples were all less than 0.001%. Hence, all samples were enrolled in the further analysis. Following quality filtering and normalization 803 146 individual probes were included in downstream data analyses. Density plots showed a typical pattern for each of the samples and highly comparable distributions indicating success with the Mixture Quantile dilation Beta normalization procedure (Supplemental Fig S1).
3.1. Genome-wide DNA methylation patterns
Analysis the methylation levels with 803 146 probes, methylation levels in 17 244 MVPs were significantly different (P < .05, respectively) between LGA and AGA, which accounted for 2.02% probes (Fig. 1). Among these 17 244 MVPs, 705 were hypermethylated (> 1.7-fold) and 351 were hypomethylated (< 0.5-fold). Among 705 hypermethylated loci, the average β values of MVPs were 0.17 ± 0.13 (LGA) and 0.08 ± 0.06 (AGA) with significant difference (t = 18.09, P < .001), as shown in Figure 2A. Among 351 hypomethylated loci, the average β values of MVPs were 0.04 ± 0.03 (LGA) versus 0.96 ± 0.08 (AGA) with significant difference (t = 12.58, P < .001), as shown in Fig. 2B.
Figure 1.

Heat map of methylation variable regions across LGA and AGA groups. Each probe set is represented by a single row of colored bars. Red color denotes higher methylation level. Blue color denotes low methylation levels. While yellow color denotes no change of methylation levels. Each line represents a placentas sample from AGA (n = 6) and LGA (n = 6) groups. AGA = appropriate for gestational age, AGA = appropriate for gestational age, LGA = large for gestational age.
Figure 2.

MVPs between the LGA and AGA groups. (A). The average β value of 705 hypermethylated locis between the LGA and AGA groups; (B). The average β value of 351 hypomethylated locis between the LGA and AGA groups (Independent t test; Data: mean ± SD; ∗∗: P < .01). AGA = appropriate for gestational age, LGA = large for gestational age.
The identified MVPs between AGA and LGA were mainly located in CpG islands. Moreover, these MVPs were also enriched within the promoter regions. Among these 17 244 MVPs, 2 357 overlapped the transcription start site 1500 promoter regions (909 in GpG island, 44 in shelf and 1014 in shore), 1 897 overlapped the transcription start site 200 promoter regions (1 374 in GpG island, 15 in shelf and 282 in shore), 1 598 overlapped the 5’UTR (890 in GpG island, 104 in shelf and 271 in shore), 1 452 overlapped the exon one (1 233 in GpG island, 9 in shelf and 78 in shore), 561 overlapped the 3’UTR (136 in GpG island, 50 in shelf and 104 in shore) and 5 416 in the body (1 867 in GpG island, 525 in shelf and 1 064 in shore), as shown in Table 2.
Table 2.
The location distribution of MVPs on the gene.

To reduce data dimensionality and to demonstrate differential methylation integrated over several gene areas, we analyzed our methylation data for differentially methylated regions (DMR). We identified a total of 117 statistically significant DMRs between LGA and AGA groups, including 54 hypermethylated and 63 hypomethylated. Among these, the greatest mean Δ was 0.2121, and 25 of the DMRs had a mean Δ≥0.10. The top DMRs resided in genes such as NTN4, FKBP11, PDX1, DECR1, ACTC1 and SMOC2, with many of them related with metabolism. Most of the above mentioned DMRs resided within gene promoters.
After analyzing the methylation levels of 22 pair chromosomes, we noted that the chromosome 6 had lots of DMRs (9 hypermethylated and 9 hypomethylated), accounting for 15% of DMRs. In contrast, no DMR was found in the chromosome 9. Meanwhile, highest rate of hypermethylated/hypomethylated (6 hypermethylated and 1 hypomethylated) was noticed in chromosome 12 while lowest rate of hypermethylated/hypomethylated (1 hypermethylated and 6 hypomethylated) was in chromosome 17 (Table 3).
Table 3.
The location distribution of DMR on chromosome.

3.2. GO and KEGG pathway analysis of DMRs
These hypermethylated DMRs matched 54 genes while hypomethylated DMRs matched 53 genes (Supplemental Table S3). A GO analysis showed that most DMRs were related to adhesion and fetal developmental process (Biological Processes), plasma membrane part (Cellular Components), calcium, metal ion, other ion, cation, protein, microtubule binding, and transmembrane transporter (molecular functions) (Fig. 3A). A KEGG pathway analysis showed that DMRs might mainly involved in 9 pathways, such as sulfur relay system, type 2 diabetes mellitus, endocrine and other factor-regulated calcium reabsorption and so on (Fig. 3B). These DMRs matched the promoters of 13 genes, including dynamin 2 (DNM2), GNAS, pancreatic and duodenal homeobox 1 (PDX1), CACNA1G, potassium voltage-gated channel subfamily Q member 1, adenomatous polyposis coli 2, RAN binding protein 1, solute carrier family 25 member 4, major histocompatibility complex, class I, G, dedicator of cytokinesis 1, cytosolic thiouridylase subunit 2, DECR1 and cyclin dependent kinase inhibitor 1B (Table 4).
Figure 3.
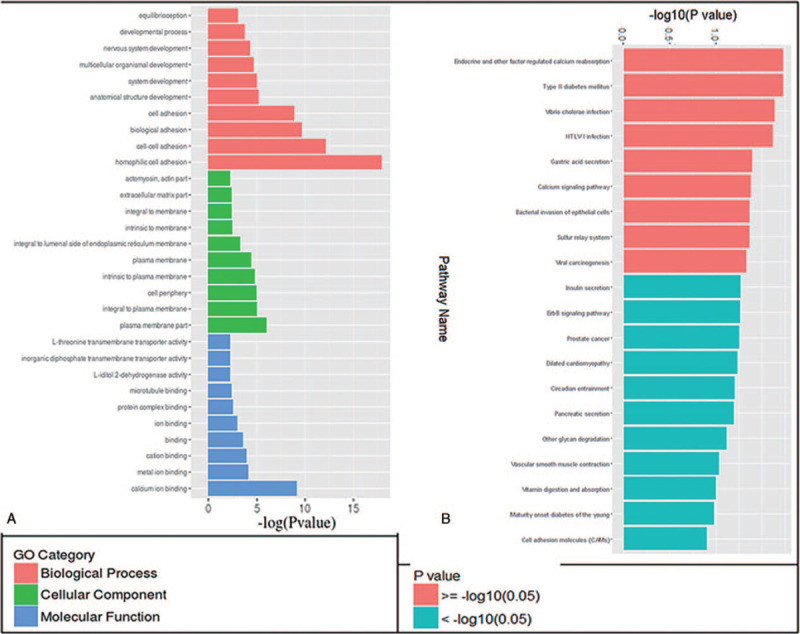
GO and KEGG pathway analysis for the differentially Methylated Regions (DMRs) between LGA and AGA groups. (A) GO analysis showed that LGA associated with biological process (red), cellular component (green) and molecular function (blue); (B) KEGG pathway analysis showed 9 differentiate pathways (red). GO = gene ontology. KEGG = Kyoto Encyclopedia of Genes and Genomes. AGA = appropriate for gestational age, LGA = large for gestational age.
Table 4.
KEGG pathway analysis showed 9 differentiate pathway between AGA and LGA group.

3.3. FKBP11, DECR1, GNAS and CACNA1G validation
A further analysis was performed by searching for these DMRs located in promoter regions with the ratio of β values ≥ 1.0 or ≤0.70 and P-values ≤.01. Under these conditions, 58 DMRs sites were detected. Among those, GNAS and CACNA1G were chosen for validation by targeted bisulfite pyrosequencing and RNA detection by RT-PCR because they were involved in type 2 diabetes mellitus pathways and GNAS paternal mutations could led to severe intrauterine growth retardation.[20] Another 2 genes, FKBP11 and DECR1, were chosen for validation by RT-PCR because they exhibited the largest methylation differences in microarray analysis.
We used pyrosequencing to evaluate further DNA methylation levels for GNAS and CACNA1G in an independent cohort (validation cohort, n = 15). The results for 3 CpG sites and total CpGs within the GNAS DMRs and 5 CpG sites and total CpGs within the CACNA1G DMRs were showed in Figure 4. These results suggested that the changes in methylation levels of GNAS and CACNA1G were consistent with the microarray results.
Figure 4.

Validation of candidate LGA regions by bisulfite pyrosequencing. Bar charts representing the DNA methylation levels at five CpG sites and total CpGs overlapping CACNA1G (A), and three CpG sites and total CpGs overlapping GNAS (B) in LGA (red color) and controls (blue color) (Nonparametric Mann–Whitney T test; Data: mean ± SD; ∗: P < .05; ∗∗: P < .01). LGA = large for gestational age, DNA = deoxyribonucleic acid, GNAS = guanine nucleotide binding protein, alpha stimulating.
DNA methylation has previously been associated with transcription repression. Accordingly, LGA samples showed significantly (P = .0074) higher expression levels of the gene CACNA1G (11.5679 ± 4.6735) and (P = .0059; P = .0019; P = .0043) lower expression levels of the genes FKBP11 (13.59 ± 7.55), DECR1 (10.99 ± 6.90) and GNAS (2.77 ± 1.65) than AGA (4.06 ± 3.28; 61.06 ± 34.93; 50.58 ± 10.65; 8.06 ± 3.23), and these expressions were associated with the DNA methylation levels of CACNA1G, FKBP11, DECR1 and GNAS (Fig. 5). These results suggested that the epigenetic regulation of these genes could have a relevant biologic function.
Figure 5.
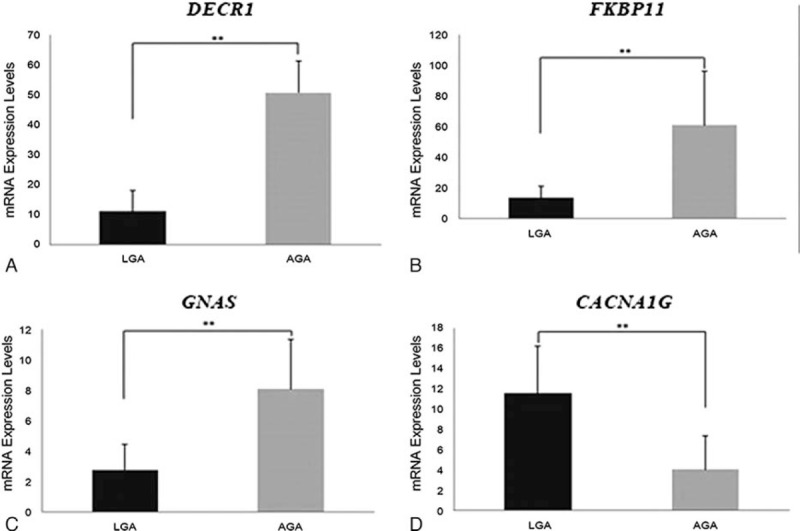
Validation the microarray results of LGA candidate genes by qRT-PCR. Bar charts representing the mRNA expression levels at DECR1(A), FKBP11(B), GNAS (C), and CACNA1G (D) in LGA (black color) and controls (yellow color) (Independent t test; Data: mean ± SD; ∗∗: P < .01). LGA = large for gestational age, DECR1 = 2,4-dienoyl-CoA reductase 1. GNAS = guanine nucleotide binding protein, alpha stimulating, mRNA = messenger RNA.
3.4. Functional enrichment
We also analyzed the enrichment of the 107 DMRs associated genes. Enrichment analysis showed a significant over-representation of functional groups highly relevant to the LGA phenotype. The functional categories detected were related to transmembrane transporter, developmental processes (eg, anatomical structure development, nervous system development) and cell adhesion (eg, homophilic cell adhesion, biological adhesion). Among 107 genes, 41 were involved in “anatomical structure development” and 23 genes were associated with “nervous system development.” As many as 79 genes, or 73.8% of our list, were annotated as “binding.” Other enriched functional groups were related to cell adhesion, developmental processes, and regulation of transcription.
4. Discussion
Environmental and lifestyle factors are recognized as influence in an individual's DNA methylation patterns. So we hypothesized that LGA might correlated with changes in placental DNA methylation and expressions of genes conferring risk for metabolic disease. As far as we are aware, our study was the first time to compare the DNA methylation level between AGA and LGA neonates of women without medical or obstetric complications at term, using the Illumina Infinium Human Methylation EPIC BeadChips (850K), and identify potential DNA methylation signatures.
Although, epigenetics changes have been associated with the intrauterine growth and later health, previous studies were mostly focus on the epigenetics changes of newborn genome, but not the placenta. To our knowledge, whether epigenetics play a role in the placenta of LGA neonates has remained unknown, and this is the first report. Our study revealed DNA methylation changes in LGA group, which have 17 244 significantly different MVPs, including hypermethylated 705 MVPs and 351 hypomethylated MVPs, between the LGA and AGA groups. These significantly different MVPs were mainly located in the promoter region (42.4%) and GpG island (42.6%). It is well established that DNA methylation patterns are associated with gene expression levels, especially in regions defining enhancers and promoters. In fact, the differentially MVPs identified are preferentially located within regulatory regions of the genome, the CpG islands, as these are known to have important implications on transcriptional regulation. Based on these results, we proposed that some intrauterine environment resulting in LGA might alter the genomic methylation, thereby affecting the expression levels, or even later health.
DMR analyses showed that a total of 117 DMRs, including 54 hypermethylated and 63 hypomethylated DMRs, were found in LGA groups. An analysis of DMRs of LGA compared to AGA also identified many hypomethylated/hypermethylated DMRs in LGA in metabolism-related genes, such as PDX1, CACNA1G, DECR1, GNAS and so on. It was interesting that the methylated changes were different among different chromosomes. Chromosome 6 had the most number of DMRs and the chromosome 9 had no change of DMRs. Moreover, the chromosome 12 has the highest rate of hypermethylated to hypomethylated (6:1) while the chromosome 17 has the lowest rate (1:6). These might be associated with the gene distributions and indicated that methylated changes caused by LGA were imbalance among different chromosomes. Certainly, further studies about these imbalances will need to be conducted to understand the accurate mechanisms of epigenetic regulation.
The functions of 107 genes associated with 117 DMRs were analyzed. They tend to be associated with developmental and adhesion biological processes. These biological processes could promote the survival and adaptation of LGA fetuses because the relevant genetic networks identified important pathways in fetal development. Major networks identified by multiple pathway and functional analyses highlighted the importance of ion binding, protein binding and cell-to-cell signaling, adhesion, transport, as well as system development and function. However, network analysis also interestingly identified methylation differences in genes that were important in the eventual risk of metabolic disease, such as endocrine and other factor-regulated calcium reabsorption, calcium signaling pathway and type 2 diabetes mellitus genes (e.g GNAS, DNM2, PDX1, CACNA1G, DECR1 and solute carrier family 25 member 4. In addition, several interesting genes that might played role in immunity were identified, including potassium voltage-gated channel subfamily Q member 1, adenomatous polyposis coli 2, RAN binding protein 1, major histocompatibility complex, class I, G, dedicator of cytokinesis 1, and DNM2. Although these findings implied that this epigenomic data set might reflect true pathophysiological processes, further mechanistic studies were needed to be carried out. Indeed, it was important to consider using pathway analysis to explore biological mechanisms affected by fetal growth-associated epigenetic variation.
We found that 3 differentially methylated genes (PDX1, DECR1 and GNAS) had been previously associated with SGA,[21,22] suggesting (at least in part) the changes of some genes in SGA and LGA were similar in some pathways and regulatory effects. In this study, the largest methylation difference was demonstrated in the promoter region of the FKBP11 gene. This gene encodes FKBP11, which belongs to the FKBP family of peptidyl-prolyl cis/trans isomerases, and catalyzes the folding of proline-containing polypeptides. The peptidyl-prolyl isomerase activity of FKBP proteins is inhibited by the immunosuppressant compounds FK506 and rapamycin. The C-terminal of this protein contains a putative transmembrane domain followed by a motif found in endoplasmic reticulum membrane proteins. Thus, FKBP11 has been conjectured to be involved in protein folding and secretion.[23] The different expression of FKBP11 gene in type 2 diabetes mellitus has been reported.[24] The next largest methylation difference identified was hypermethylation in the promoter of the DECR1 gene in the LGA group. This gene encodes DECR protein, the rate-limiting mitochondrial enzyme in unsaturated fatty acid beta oxidation, which is required for growth and neurodevelopment.[25] An association between unsaturated fatty acids and intrauterine growth has been previously reported in that prenatal supplementation of the unsaturated fatty acid, omega-3, which has been shown to increase birth weight.[26] Prior studies of Decr1−/− mice suggested that the enzyme was responsible for the induction of gluconeogenesis during fasting.[27] Whether this gene is related to metabolic syndrome requires further research and exploration. Another highly ranked candidate gene is the GNAS gene, which encodes, through the use of alternative first exons, the signaling proteins Gαs and XLαs and the NESP55. The GNAS locus furthermore gives rise to an AS transcript as well as the A/B transcript that contributes to the regulation of Gαs expression and may be translated into an amino-terminally truncated Gαs variant that appears to antagonize the actions of the full-length signaling protein.[28]GNAS is a complex imprinted locus involved in metabolic regulation. A recent study had further demonstrated that a single nucleotide polymorphism in GNAS in the maternal genotype of African American women (SNP rs6026576) was significantly associated with birth weight.[29] In other studies, it was also found that the expression level of GNAS in the placenta of SGA individuals was decreased,[20,30] but the importance of its reduced expression in SGA was not immediately clear. To date, our study was the first to link changes of FKBP11, DECR1 and GNAS and so on methylation with metabolic syndrome caused by LGA in humans. These could provide assistance in understand the development of potential epigenetic biomarkers and signals in altering fetal growth and arousing the risk for adult metabolic disease. However, the role and mechanism of FKBP11 DECR1 and GNAS changes in LGA-induced metabolic syndrome was still unclear and the further studies were required.
There were several limitations of our study, although we obtained several significant results. First, the sample sizes were relatively small. Second, due to the placental tissue samples limitation, the samples used for microarray detection, pyrosequencing verification and messenger RNA verification did not come from the same placental tissue. Third, the consistency of placental samples might be biased because of the inability to fully control the state of the pregnant woman and the fetus. Further study with larger sample is required to investigate the associations between DNA methylation patterns and the mechanisms of LGA and adult metabolic syndrome. Certainly, this study had its strengths, which might be helpful in the development of potential epigenetic biomarkers and novel therapeutic targets in the prevention and treatment of abnormal fetal growth and adult metabolic disease.
5. Conclusion
In conclusion, our genome-wide DNA methylation study, carried out using paired AGA and LGA placenta tissues, demonstrated specific differential DNA methylation profiles in LGA cases. Furthermore, our results of epigenetic variation and gene expression differences in placenta of LGA infants suggested that intrauterine environment might be resulted in epigenetic and gene expression changes of placental genome, then the abnormal intrauterine growth and even later health. Based on the results obtained from this study might be helpful in the development of potential epigenetic biomarkers and novel therapeutic targets in the prevention and treatment of abnormal fetal growth and adult metabolic disease. Future studies including larger sample sizes to improve our findings and investigate the mechanisms are necessary.
Acknowledgments
We want to thank the patients and their parents for participating this study.
Author contributions
Professor Zou response for the design of the study, validation analysis, and drafted the manuscript. Mr Shen take participated in the design of the study, performed the bioinformatic analyses. Mrs Tang and Mrs Song take part in the recruited of subjects, data and sample collection. Mrs Shen performed the sample selection and methylation array interpretation. All authors read and approved the final manuscript.
Conceptualization: Chaochun Zou.
Data curation: Zheng Shen.
Funding acquisition: Zheng Shen, Chaochun Zou.
Investigation: Yanfei Tang.
Methodology: Yemei Song.
Project administration: Chaochun Zou.
Resources: Yanfei Tang.
Validation: Wenxia Shen.
Writing – original draft: Zheng Shen.
Writing – review & editing: Chaochun Zou.
Supplementary Material
Footnotes
Abbreviations: AGA = appropriate for gestational age, CACNA1G = calcium voltage-gated channel subunit alpha1 G, CDKN1B = cyclin dependent kinase inhibitor 1B, cDNA = complementary DNA, DECR1 = 2,4-dienoyl-CoA reductase 1, DMRs = differentially methylated regions, DNA = deoxyribonucleic acid, DNM2 = dynamin 2, DOCK1 = dedicator of cytokinesis 1, FKBP11 = FK506 binding protein 11, GNAS = guanine nucleotide binding protein, alpha stimulating, GO = gene ontology, KCNQ1 = potassium voltage-gated channel subfamily Q member 1, KEGG = Kyoto Encyclopedia of Genes and Genomes, LGA = large for gestational age, MVPs = methylation variable positions, PDX1 = pancreatic and duodenal homeobox 1, RNA = ribonucleic acid, RT-qPCR = reverse transcription-quantitative real-time polymerase chain reaction, SGA = small for gestational age.
How to cite this article: Shen Z, Tang Y, Song Y, Shen W, Zou C. Differences of DNA methylation patterns in the placenta of large for gestational age infant. Medicine. 2020;99:39(e22389).
This work was supported by the Natural Science Foundation of Zhejiang Province (grant number LY17H040001 to SZ), Zhejiang Medical and Health Scientific Research Fund (grant number 2016KYB311 to ZCC) and Zhejiang Provincial Program for the Cultivation of High-Level Innovative Health Talents (grant number 2014 to ZCC).
In this study, we confirmed that all methods were carried out in accordance with relevant guidelines and regulations, all experimental protocols were approved by the Medical Ethical Committee of the Children Hospital of Zhejiang University School of Medicine (Approval Number: 2015-HP-025) and informed consent was obtained from all subjects or, if subjects are under 18, from a parent and/or legal guardian.
I would like to declare on behalf of my co-authors that the work described was original research that has not been published previously, and not under consideration for publication elsewhere, in whole or in part. All the authors listed have approved the manuscript that is enclosed.
All data generated or analysed during this study are included in this published article.
The authors have no conflicts of interest to disclose.
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
- [1].Waterland RA, Garza C. Potential mechanisms of metabolic imprinting that lead to chronic disease. Am J Clin Nutr 1999;69:179–97. [DOI] [PubMed] [Google Scholar]
- [2].Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science 2004;305:1733–6. [DOI] [PubMed] [Google Scholar]
- [3].Ozanne SE, Constancia M. Mechanisms of disease: the developmental origins of disease and the role of the epigenotype. Nat Clin Pract Endocrinol Metab 2007;3:539–46. [DOI] [PubMed] [Google Scholar]
- [4].Waterland RA, Michels KB. Epigenetic epidemiology of the developmental origins hypothesis. Annu Rev Nutr 2007;27:363–88. [DOI] [PubMed] [Google Scholar]
- [5].Banister CE, Koestler DC, Maccani MA, et al. Infant growth restriction is associated with distinct patterns of DNA methylation in human placentas. Epigenetics 2011;6:920–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Tabano S, Colapietro P, Cetin I, et al. Epigenetic modulation of the IGF2/H19 imprinted domain in human embryonic and extra-embryonic compartments and its possible role in fetal growth restriction. Epigenetics 2010;5:313–24. [DOI] [PubMed] [Google Scholar]
- [7].Einstein F, Thompson RF, Bhagat TD, et al. Cytosine methylation dysregulation in neonates following intrauterine growth restriction. PLoS One 2010;5:e8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Johnsson IW, Haglund B, Ahlsson F, et al. A high birth weight is associated with increased risk of type 2 diabetes and obesity. Pediatr Obes 2015;10:77–83. [DOI] [PubMed] [Google Scholar]
- [9].Mingrone G, Manco M, Mora ME, et al. Influence of maternal obesity on insulin sensitivity and secretion in offspring. Diabetes Care 2008;31:1872–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Chiavaroli V, Marcovecchio ML, de Giorgis T, et al. Progression of cardio-metabolic risk factors in subjects born small and large for gestational age. PLoS One 2014;9:e104278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fryer AA, Nafee TM, Ismail KM, et al. LINE-1 DNA methylation is inversely correlated with cord plasma homocysteine in man: a preliminary study. Epigenetics 2009;4:394–8. [DOI] [PubMed] [Google Scholar]
- [12].Fryer AA, Emes RD, Ismail KM, et al. Quantitative, high-resolution epigenetic profiling of CpG loci identifies associations with cord blood plasma homocysteine and birth weight in humans. Epigenetics 2011;6:86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Mulligan CJ, D’Errico NC, Stees J, et al. Methylation changes at NR3C1 in newborns associate with maternal prenatal stress exposure and newborn birth weight. Epigenetics 2012;7:853–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ferreira JC, Choufani S, Grafodatskaya D, et al. WNT2 promoter methylation in human placenta is associated with low birthweight percentile in the neonate. Epigenetics 2011;6:440–9. [DOI] [PubMed] [Google Scholar]
- [15].Schubeler D. Function and information content of DNA methylation. Nature 2015;517:321–6. [DOI] [PubMed] [Google Scholar]
- [16].Chen K, Zhao BS, He C. Nucleic acid modifications in regulation of gene expression. Cell Chem Biol 2016;23:74–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Fortin JP, Fertig E, Hansen K. shinyMethyl: interactive quality control of Illumina 450k DNA methylation arrays in R. F1000Res 2014;3:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Teschendorff AE, Marabita F, Lechner M, et al. A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 2013;29:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Butcher LM, Beck S. Probe Lasso: a novel method to rope in differentially methylated regions with 450K DNA methylation data. Methods 2015;72:21–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Richard N, Molin A, Coudray N, et al. Paternal GNAS mutations lead to severe intrauterine growth retardation (IUGR) and provide evidence for a role of XLalphas in fetal development. J Clin Endocrinol Metab 2013;98:E1549–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Wang Y, Zhu W, Chen L, et al. Early growth hormone intervention improves glucose metabolism in adult rats born small for gestational age. Exp Clin Endocrinol Diabetes 2018;128:125–32. [DOI] [PubMed] [Google Scholar]
- [22].Murphy R, Mackay D, Mitchell EA. Beckwith Wiedemann imprinting defect found in leucocyte but not buccal DNA in a child born small for gestational age. Bmc Med Genet 2012;13:99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rulten SL, Kinloch RA, Tateossian H, et al. The human FK506-binding proteins: characterization of human FKBP19. Mamm Genome 2006;17:322–31. [DOI] [PubMed] [Google Scholar]
- [24].Lu H, Yang Y, Allister EM, et al. The identification of potential factors associated with the development of type 2 diabetes: a quantitative proteomics approach. Mol Cell Proteomics 2008;7:1434–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Herrera E. Lipid metabolism in pregnancy and its consequences in the fetus and newborn. Endocrine 2002;19:43–55. [DOI] [PubMed] [Google Scholar]
- [26].Szajewska H, Horvath A, Koletzko B. Effect of n-3 long-chain polyunsaturated fatty acid supplementation of women with low-risk pregnancies on pregnancy outcomes and growth measures at birth: a meta-analysis of randomized controlled trials. Am J Clin Nutr 2006;83:1337–44. [DOI] [PubMed] [Google Scholar]
- [27].Miinalainen IJ, Schmitz W, Huotari A, et al. Mitochondrial 2,4-dienoyl-CoA reductase deficiency in mice results in severe hypoglycemia with stress intolerance and unimpaired ketogenesis. PLoS Genet 2009;5:e1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Puzhko S, Goodyer CG, Kerachian MA, et al. Parathyroid hormone signaling via Galphas is selectively inhibited by an NH(2)-terminally truncated Galphas: implications for pseudohypoparathyroidism. J Bone Miner Res 2011;26:2473–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Adkins RM, Krushkal J, Magann EF, et al. Association of maternally inherited GNAS alleles with African-American male birth weight. Int J Pediatr Obes 2010;5:177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Dunk CE, Roggensack AM, Cox B, et al. A distinct microvascular endothelial gene expression profile in severe IUGR placentas. Placenta 2012;33:285–93. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.