

Abstract
Rationale:
Apert syndrome (AS) is an autosomal dominant inheritance pattern of the most severe craniosynostosis syndrome. AS is characterized by synostosis of cranial sutures and acrocephaly, including brachycephaly, midfacial hypoplasia, and syndactyly of the hands and feet. Patients with AS often present with craniosynostosis, severe syndactyly, and skin, skeletal, brain, and visceral abnormalities.
Patient concerns:
A pregnant Chinese woman presented with a fetus at 23 + 5 weeks of gestation with suspected AS in a prenatal ultrasound examination. Following ultrasound, the pregnancy underwent spontaneous abortion. Gene sequencing was performed on the back skin of the dead fetus.
Diagnosis:
The diagnosis of AS was confirmed on the basis of clinical manifestations of the fetus, and a de novo mutation in the fibroblast growth factor receptor 2 (FGFR2) gene was identified.
Interventions:
The couple finally chose to terminate the pregnancy based on the ultrasonic malformations and the risk of the parents having a neonate with AS in the future is small. However, any future pregnancy must be assessed by prenatal diagnosis.
Outcomes:
The dead fetus presented with bilateral skull deformation. Additionally, there were bilateral changes to the temporal bone caused by inwards movement leading to concave morphology, a “clover” sign, and syndactyly from the index finger/second toe to the little finger/little toe. AS was diagnosed by genetic testing, which showed a p.S137W (c.410C>G, chr10:123279677) mutation in the FGFR2 gene.
Lessons:
Clinicians should be aware that there are a variety of ultrasound findings for AS. Therefore, genetic testing should be used when appropriate to confirm diagnosis of AS.
Keywords: Apert syndrome, autosomal dominant inheritance, de novo mutation, fibroblast growth factor receptor 2 (FGFR2), syndactyly
1. Introduction
Apert syndrome (AS) is a rare genetic disorder, which was first reported by Wilkie et al and characterized by craniosynostosis, severe syndactyly, and a variety of skin, skeletal, brain, and visceral abnormalities.[1] AS exhibits an autosomal dominant inheritance pattern and the incidence of AS has been reported to be 1 in 65,000 individuals.[2,3]
The primary cause of AS is mutation in the fibroblast growth factor receptor 2 (FGFR2) gene. In fact, more than 98% of AS cases are caused by de novo FGFR2 mutations, referred to as S252W and P253R.[1,4] The mechanism underlying the exquisite genotype/phenotype correlation associated with AS mutations needs to be understood in terms of the biology of fibroblast growth factor/receptor signaling and the structural pathophysiology of FGFR2 mutations.[5] The FGFR2 gene is located on chromosome 10q26. FGFR2 encodes a transmembrane receptor with an extracellular region comprising 3 immunoglobulin-like domains (IgI, IgII, and IgIII), a hydrophobic transmembrane segment, and a cytoplasmic tyrosine-kinase1 domain. The immunoglobulin domains are encoded by exons 8, 9, and 10 of the FGR2 gene in which 25 to 75 of patients with AS have mutations.[6]
In this study, we report the case of a Chinese fetus at 23 + 5 weeks of gestation with unclear prenatal ultrasound findings (acrocephalosyndactyly) and abnormal physical characteristics. The fetus was subsequently diagnosed with AS on the basis of DNA sequencing analysis of FGFR. We detected a novel mutation in the FGR2 gene, p.S137W (c.410C>G chr10:123279677).
2. Case report
On 29 December 2016, a 32-year-old pregnant Chinese woman was admitted to the Prenatal Diagnosis Department of the First Hospital of Changchun, Jilin Province, northeastern China. She had previously undergone routine prenatal screening of her fetus (23 + 5 weeks of age). However, prenatal ultrasound findings were not clear. The woman had experienced a spontaneous abortion during the first trimester (8 weeks of gestation) of a previous pregnancy but without any known cause. She had not been exposed orally to harmful or hazardous substances during her pregnancies. During the second pregnancy, the pregnant woman had a febrile illness and received oral cold medication, although no specific records were available to confirm this. Following ultrasound, the second pregnancy underwent spontaneous abortion. The parents wanted to know the reason for the spontaneous abortion and brought the fetus to our department for analysis. There was no history of consanguineous marriage in the family and no family history of genetic disorders.
The study protocol was approved by the Ethics Committee of the First Hospital of Jilin University (No. 2016-365). Informed written consent was obtained from the parents for publication of this case report and accompanying images.
3. Materials and methods
Sequencing analysis was conducted on the dead fetus and the parents. First, the DNA was disrupted and a library was prepared. DNA of the targeted gene coding region and the adjacent cleavage region was then captured and enriched by a chip. Finally, detection of mutations was performed using a high-throughput sequencing platform.
4. Results
During prenatal ultrasound, a number of anomalies were detected. These included a wide angle after the left ventricle (1.03 cm), bilateral skull deformation, bilateral temporal bone abnormalities showing inwards movement and a concave appearance, a “clover” sign, and syndactyly from the index finger/second toe to the little finger/little toe (Fig. 1).
Figure 1.
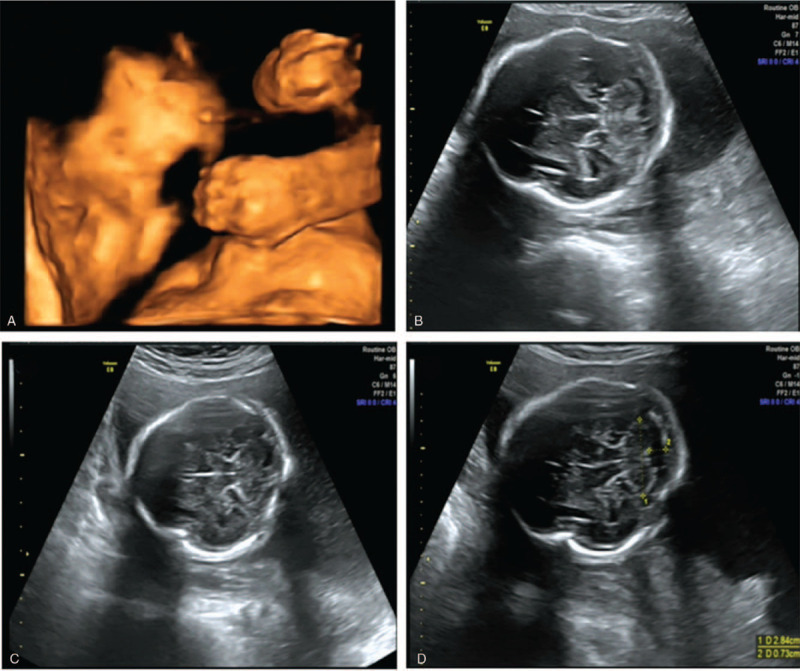
Prenatal ultrasound findings. (A) Syndactyly from the index finger to the little finger. (B–D) Physical characteristics of acrocephalia. The left ventricle was wider than normal.
The physical characteristics of the dead fetus were consistent with the prenatal ultrasound findings (Fig. 2). The dead fetus presented with the physical characteristics of acrocephalosyndactyly and hypertelorism. X-ray results showed that there was no craniosynostosis. Because of the prenatal ultrasound findings and the clinical manifestations evident upon physical examination of the dead fetus, we initially suspected that the fetus may have had AS.
Figure 2.
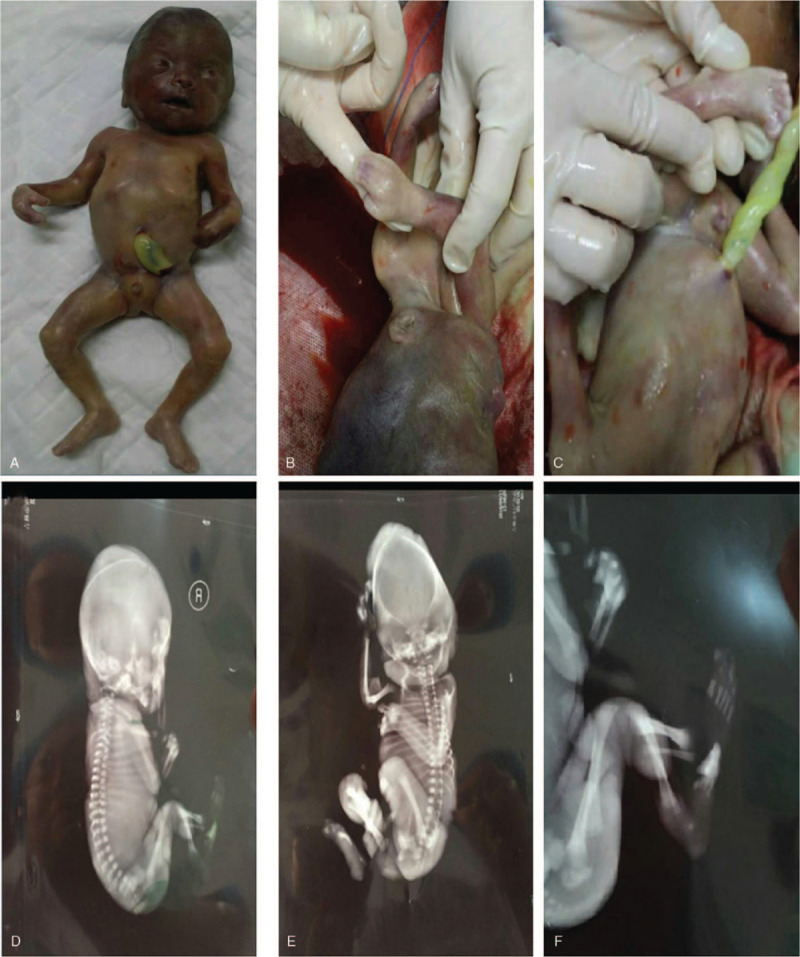
The dead fetus after induced labor. (A–C) Physical characteristics of acrocephalosyndactyly. (D–F) Presentation of acrocephalia as shown by X-ray. Craniosynostosis was not evident.
We took a sample of skin from the back of the dead fetus for gene sequencing. We found that the fetus had a heterozygous p.S137W (c.410C>G, chr10:123279677) mutation in the FGFR2 gene (Fig. 3A). However, further analysis showed that neither of the parents carried this mutation (Fig. 3B and C). The diagnosis of AS was confirmed on the basis of clinical manifestations of the fetus and identification of a de novo mutation in the FGFR2 gene.
Figure 3.
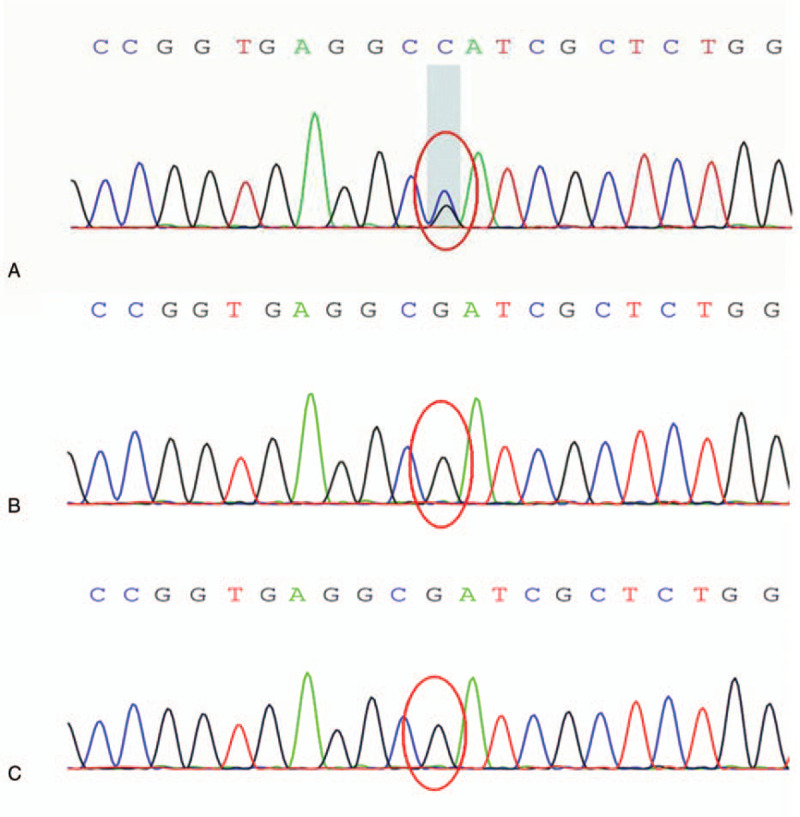
Sanger sequencing results for (A) the fetus, (B) the fetus's father, and (C) the fetus's mother. We identified the presence of a p.S137W (c.410C>G chr10:123279677) mutation in the FGFR2 gene (red circle) in the fetus and the absence of a mutation at c.410C (red circles) in the parents.
5. Discussion
We report here a new case of the rare genetic syndrome AS. AS is primarily characterized by cutaneous and osseous symmetric syndactyly in the hands and feet, with variable presentations in the bones, brain, skin, and other internal organs. AS is one of the most severe craniosynostosis syndromes.
During craniofacial development at different stages of embryonic formation, signaling pathways involving fibroblast growth factors, their receptors (FGFRs), and specifically, FGFR2, regulate the balance between proliferation and differentiation of progenitor osteogenic cells on the neural crest.[7] Later in development, these pathways are also involved in formation of cartilage, skull bones, and the maxilla, as well as migration of the plates that give rise to the palate and lips.[8]
The S252W and P253R mutations were originally described by Wilkie et al[1]and are associated specifically with AS. These mutations present with more severe syndactyly in patients with the Pro253Arg mutation compared with the Ser252Trp mutation. However, the fetus described above showed no association of these mutations and syndactyly. Instead, the fetus had a novel mutation, which, to the best of our knowledge, has not been reported previously. Molecular analysis detected a p.S137W (c.410C>G chr10:123279677) mutation in the FGFR2 gene. Because the parents of the fetus did not have this mutation, the fetus had a de novo heterozygous mutation, which led to AS.
Early surgery is currently advocated by many craniofacial centers to prevent complications arising from AS.[9] Generally, the most important management protocol for patients with AS involves immediate craniotomy, hand and feet surgery, and long-term follow-up. However, in most developing countries, early intervention of AS is hampered by late diagnosis, a lack of facilities, and financial constraints.[10]
6. Conclusion
In conclusion, clinicians should be aware that there are a variety of ultrasound findings for AS. In the present study, the fetus showed abnormal prenatal ultrasound findings (acrocephalosyndactyly). However, craniosynostosis was not clearly indicative of AS. Therefore, genetic testing should be used when appropriate to confirm diagnosis of AS. Our data indicate that the case of fetal AS was caused by a new mutation. The risk of the parents having a neonate with AS in the future is small. However, any future pregnancy must be assessed by prenatal diagnosis.
Author contributions
Conceptualization: Yanhong Liu.
Data curation: Qingyang Shi.
Funding acquisition: Ruizhi Liu.
Investigation: Qingyang Shi, Ruixue Wang.
Methodology: Xiaowei Yu.
Software: Jili Jing.
Writing – original draft: Rulin Dai.
Writing – review & editing: Qingyang Shi, Yanhong Liu.
Footnotes
Abbreviations: AS = Apert syndrome, FGFR2 = fibroblast growth factor receptor 2.
How to cite this article: Shi Q, Dai R, Wang R, Jing J, Yu X, Liu R, Liu Y. A novel FGFR2 (S137W) mutation resulting in Apert syndrome: A case report. Medicine. 2020;99:39(e22340).
This work was kindly supported by the Finance Department Health Special Project of Jilin Province, China (JLSCZD2019- 022).
The authors report no conflicts of interest.
All data generated or analyzed during this study are included in this published article [and its supplementary information files].
References
- [1].Wilkie AO, Slaney SF, Oldridge M, et al. Apert syndrome results from localized mutations of FGFR2 and is allelic with Crouzon syndrome. Nat Genet 1995;9:165–72. [DOI] [PubMed] [Google Scholar]
- [2].Freiman A, Tessler O, Barankin B. Apert syndrome. Int J Dermatol 2006;45:1341–3. [DOI] [PubMed] [Google Scholar]
- [3].Park WJ, Theda C, Maestri NE, et al. Analysis of phenotypic features and FGFR2 mutations in Apert syndrome. Am J Hum Genet 1995;57:321–8. [PMC free article] [PubMed] [Google Scholar]
- [4].Coomaralingam S, Roth P. Apert syndrome in a newborn infant without craniosynostosis. J Craniofac Surg 2012;23:e209–11. [DOI] [PubMed] [Google Scholar]
- [5].Aimee LF, Sarah CB, Regan EM. A deletion of FGFR2 creating a chimeric IIIb/IIIc exon in a child with Apert syndrome. BMC Med Genet 2011;12:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Eswarakumar VP, Lax I, Schlessinger J. Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 2005;16:139–49. [DOI] [PubMed] [Google Scholar]
- [7].Chao L, Yazhou C, Jing L, et al. The molecular and cellular basis of Apert syndrome. Intractable Rare Dis Res 2013;2:115–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bhatt S, Diaz R, Trainor PA. Signals and switches in mammalian neural crest cell differentiation. Cold Spring Harb Perspect Biol 2013;5:pii: a008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Marucci DD, Dunaway DJ, Jones BM, et al. Raised intracranial pressure in Apert syndrome. Plast Reconstr Surg 2008;122:1162–8. [DOI] [PubMed] [Google Scholar]
- [10].Mundhofir FE, Sistermans EA, Faradz SM, et al. p.Ser252Trp and p.Pro253Arg mutations in FGFR2 gene causing Apert syndrome: the first clinical and molecular report of Indonesian patients. Singapore Med J 2013;54:e72–5. [DOI] [PubMed] [Google Scholar]