

Abstract
Objectives:
To assess the early physiologic response to angiotensin-II treatment in patients with coronavirus disease 2019–induced respiratory failure and distributive shock.
Design:
Retrospective consecutive-sample cohort study.
Setting:
Three medical ICUs in New York during the coronavirus disease 2019 outbreak.
Patients:
All patients were admitted to the ICU with respiratory failure and were receiving norepinephrine for distributive shock.
Interventions:
The treatment groups were patients who received greater than or equal to 1 hour of angiotensin-II treatment. Time-zero was the time of angiotensin-II initiation. Controls were identified using a 2:1 hierarchical process that matched for 1) date and unit of admission; 2) specific organ support modalities; 3) age; 4) chronic lung, cardiovascular, and kidney disease; and 5) sex. Time-zero in the control group was 21 hours post vasopressor initiation, the mean duration of vasopressor therapy prior to angiotensin-II initiation in the treated group.
Measurements and Main Results:
Main outcomes were trajectories of vasopressor requirements (in norepinephrine-equivalent dose) and mean arterial pressure. Additionally assessed trajectories were respiratory (Pao2/Fio2, Paco2), metabolic (pH, creatinine), and coagulation (d-dimer) dysfunction indices after time-zero. We also recorded adverse events and clinical outcomes. Trajectories were analyzed using mixed-effects models for immediate (first 6 hr), early (48 hr), and sustained (7 d) responses. Twenty-nine patients (n = 10 treated, n = 19 control) were identified. Despite matching, angiotensin-II–treated patients had markedly greater vasopressor requirements (mean: 0.489 vs 0.097 µg/kg/min), oxygenation impairment, and acidosis at time-zero. Nonetheless, angiotensin-II treatment was associated with an immediate and sustained reduction in norepinephrine-equivalent dose (6 hr model: β = –0.036 µg/kg/min/hr; 95% CI: –0.054 to –0.018 µg/kg/min/hr, pinteraction=0.0002) (7 d model: β = –0.04 µg/kg/min/d, 95% CI: –0.05 to –0.03 µg/kg/min/d; pinteraction = 0.0002). Compared with controls, angiotensin-II–treated patients had significantly faster improvement in mean arterial pressure, hypercapnia, acidosis, baseline-corrected creatinine, and d-dimer. Three thrombotic events occurred, all in control patients.
Conclusions:
Angiotensin-II treatment for coronavirus disease 2019–induced distributive shock was associated with rapid improvement in multiple physiologic indices. Angiotensin-II in coronavirus disease 2019–induced shock warrants further study.
Keywords: angiotensin II, coronavirus disease 2019, norepinephrine, severe acute respiratory syndrome coronavirus-2 infection, shock, vasoconstrictor agents
Patients with critical illness induced by severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) infection frequently develop distributive shock (1, 2). Coronavirus disease 2019 (COVID-19) is frequently associated with cardiomyopathy (3), troponinemia, and acute kidney injury (4), as well as lymphopenia (1) and an “immunoparalysis” phenotype (5). Norepinephrine is recommended as the first-line vasopressor to treat distributive shock in COVID-19 (weak recommendation, low-quality evidence) (6). However, norepinephrine produces cardiovascular strain, exerts immunosuppressive effects, and often requires profoundly high doses when used as a single agent (7, 8). Therefore, alternative therapies to limit norepinephrine exposure for patients COVID-19 are important to explore.
Angiotensin-II is an U.S. Food and Drug Administration-approved treatment for distributive shock that reduces exogenous catecholamine requirements (8, 9). In contrast to norepinephrine, angiotensin-II enhances T-lymphocyte and natural killer cell proliferation and function (10, 11). Angiotensin-II is also associated with improved survival in distributive shock patients who require renal placement therapy (12) and in those with elevated plasma renin levels (13). Indeed, some has proposed angiotensin-II deficiency arising from SARS-CoV-2–induced endovascular damage, and angiotensin-converting enzyme (ACE)–1 “shedding” contributes to the pathogenesis of COVID-19–induced shock (14). These properties suggest that angiotensin-II is an appropriate alternative to norepinephrine for the treatment of distributive shock in the setting of COVID-19.
However, there are concerns about angiotensin-II use in COVID-19. In rodent models, angiotensin-II signaling increases pulmonary inflammation and lung injury (15). Further, angiotensin-II has prothrombotic properties (16), and COVID-19 appears to induce a hypercoagulable state (17). Therefore, observational studies to assess safety and efficacy signals of angiotensin-II in COVID-19–associated shock are warranted prior to undertaking randomized trials. To meet this need, we conducted a retrospective matched-cohort study of angiotensin-II use in critically ill patients with COVID-19 and vasodilatory shock. We sought to characterize the immediate and subsequent physiologic response to treatment and the frequency of adverse events.
METHODS
Design
We undertook a retrospective consecutive-sample matched-cohort study of adult critically ill patients with COVID-19 treated at three hospitals in New York between February 27 and April 24, 2020. We sought to characterize immediate and subsequent physiologic responses to angiotensin-II exposure, as well as to assess the development of adverse events. The study was approved by the local Institutional Review Board.
Study Timeline
For angiotensin-II patients, “time-zero” (T0) was defined as the moment immediately prior to angiotensin-II administration. For controls, T0 was set at 21 hours after the initiation of vasopressor therapy. This time period was chosen because it was the average duration of vasopressor therapy prior to angiotensin-II initiation in the treatment group. Data were abstracted hourly for all patients for the 6 hours following T0 and daily for the next 7 days. Adverse events and outcomes were recorded for the entire hospitalization.
Screening and Eligibility Criteria
All patients were admitted to the ICU with COVID-19–induced acute respiratory insufficiency and were receiving norepinephrine for clinically diagnosed distributive shock. A list of all admissions associated with an angiotensin-II order during the study-period was generated from the electronic medical record. The list was systematically screened by two reviewers (F.M., G.F.). Patients were included if angiotensin-II was administered for greater than or equal to 1 hour. To identify controls, a list of all admissions to the same ICU within 5 days of an angiotensin-II–treated patient’s admission date were systematically reviewed. We excluded patients with active do-not-resuscitate orders prior to T0 or with evidence of purely cardiogenic or hypovolemic shock at T0.
Matching Procedure
A propensity-score matching approach would have been poorly suited to this study given the high likelihood of endogenous treatment allocation (18). We employed a 2:1 hierarchical process to identify controls, with matching criteria selected and ranked a priori. Eligible records were reviewed consecutively until two controls were identified for each treated patient or the list was exhausted. The ranked criteria were as follows:
1) Unit and date of ICU admission: Eligible controls were admitted to the same ICU within 5 days of a treated patient. We prioritized these criteria due to progressive resource limitation, expansion of ICU care into non-ICU spaces, and rapidly fluctuating practice patterns over the study period. We reasoned that these secular changes in practice and environment would be hardest to identify and effectively account for if not matched as precisely as possible.
2) Organ support: Potential controls were then exactly matched to treated patients with respect to the following binary variables at T0: invasive mechanical ventilation, vasopressors, renal replacement therapy (RRT), and extracorporeal membrane oxygenation.
3) Age: Eligible controls were aged within 5 years of treated patients older than 50 years old and within 10 years of treated patients younger than 50 years old.
4) Comorbidities: Eligible controls–matched treated patients on diagnoses of chronic lung (composite of chronic obstructive pulmonary disease [COPD] or asthma), cardiovascular (composite of diabetes, hypertension, or coronary artery disease [CAD]), or chronic kidney disease (CKD). We selected these comorbidities because we reasoned these were highly relevant to physiologic outcomes in relation to both COVID-19 and angiotensin-II administration.
5) Sex: Although criteria 1–4 were obligate matching criteria, if a list of potential controls was exhausted without identifying two matches, the list was rereviewed for a sex-agnostic match. This occurred for two treated patients. If a second match was not identified after relaxing sex-match requirements, then only one control was included. This occurred for one treated patient.
Outcomes
The main outcomes of interest were indicators of physiologic responses to angiotensin-II. To assess cardiovascular response, we compared the amount of vasopressor support—measured in norepinephrine-equivalent dose (8)—and the mean arterial pressure (MAP). We assessed immediate (first 6 hr from T0), early (48 hr), and prolonged responses (7 d). Additional physiologic trajectories of interest were as follows: Pao2 to Fio2 ratio (P/F ratio), Paco2, arterial pH, serum creatinine, and Sequential Organ Failure Assessment (SOFA) score. For all measurements, the “worst” value (e.g., lowest MAP, highest Paco2, etc.) recorded from the time period was used.
We additionally recorded several inflammatory and immune markers. Specifically, we abstracted serum levels of d-dimer, C-reactive protein (CRP), ferritin, and troponin, as well as lymphocyte and monocyte counts.
Elevated plasma renin levels in patients with distributive shock are both indicative of ACE-1 dysfunction and predictive of response to angiotensin-II treatment (13, 19). For this reason, we also recorded direct plasma renin levels if drawn within 24 hours prior to T0.
As exploratory analyses we compared adverse event frequency and patient outcomes. The only complication that was greater among angiotensin-II–treated patients in the Angiotensin II for the Treatment of High-Output Shock trial was the frequency of thromboembolic complications (8, 9). Therefore, the primary adverse event in this study was the development of any thromboembolic complication, operationalized as a composite of deep-vein thrombosis, pulmonary embolism, limb ischemia, myocardial infarction, ischemic stroke, mesenteric ischemia, or circuit thrombosis for patients receiving RRT. Additional adverse events were positive blood cultures drawn greater than 48 hours after T0 and presence of a secondary infection. Secondary infections were considered present on the basis of antibiotic administration for a clinically documented presumed source of infection. Clinical outcomes were mortality, vasopressor-free, ventilator-free, and RRT-free days, where “free” indicates free of both organ support and death.
Data Collection and Validation
A single author (F.M,) abstracted data from the chart into a standardized electronic data collection form according to a prespecified protocol. This form was piloted and fine-tuned prior to data collection. It featured hard-stops for impossible values and confirmation requests for missing values. The abstractor was not blinded to the study hypothesis, but outcomes often had not yet occurred at the time of initial data collection. To ensure data fidelity, one author (D.E.L.) reviewed each completed form in blinded fashion for data that were possible but unlikely (e.g., a missing Paco2 level when a Pao2 level was recorded) and returned the forms to the abstractor with these queries highlighted.
Statistical Analysis
Continuous variables are reported as means (sd) or medians (interquartile range), as appropriate. Categorical variables are reported as frequencies (percentages). We built mixed-effects generalized linear models to compare physiologic trajectories over time. Each patient was entered as a random-effect to account for within-subject correlation. Fixed-effect independent variables were treatment group, time of measurement, the interaction-effect of treatment by time, and a class variable for treated control matches. Given that physiologic trajectory could reflect natural disease course evolution, we prespecified that the interaction effect of time by treatment would be the primary measure of comparison because this coefficient reflects how the rate-of-change differed between treatment groups.
Each model was iterated on two time-horizons: once over the entire 7-day study period and once with all observations after 48 hours censored. The latter was to account for the possibility of a Neyman bias and to focus on the early response to treatment. For norepinephrine-equivalents and MAP, models were iterated a third time with all observations censored after 6 hours to capture the immediate cardiovascular response to treatment. The creatinine models were adjusted for patients’ pre hospital baseline creatinine. When a baseline creatinine level could not be directly ascertained, it was estimated using the modified diet in renal disease equation as recommend by the Acute Dialysis Quality Initiative guidelines (20).
There were no missing data for cardiovascular, arterial blood gas, creatinine, or categorical outcomes. However, inflammatory markers were not always measured daily. Therefore, we interpolated missing values as the point along the slope of the line between the most recent and the subsequent measurement. Variables that had an excess of missing data were not analyzed. We considered an excess of missing data to be greater than 10% of patients in either treatment group with greater than 20% missing data. All analyses were performed in SAS: University-Edition (SAS Institute, Cary, NC), and figures were produced with Prism-8 (GraphPad, San Diego, CA).
RESULTS
The final analysis included 29 patients: 10 who received angiotensin-II and 19 matched-controls. The groups were balanced in terms of demographics, comorbidities, and home medications (Table 1). However, the angiotensin-II group appeared to be more severely ill at T0 (Table 2). Greater cardiovascular dysfunction was evidenced by nearly five-fold greater norepinephrine-equivalent dose (mean: 0.49 vs 0.01 µg/kg/min) and lower MAP (69.2 vs 83.2 mm Hg) at T0. The angiotensin-II group also had worse baseline gas-exchange (P/F ratio: 165 vs 215; Paco2: 60 vs 46 mm Hg), acidosis (pH: 7.21 vs 7.33), and SOFA scores (11.3 vs 10.2). In contrast, creatinine at T0 was similar between groups (1.8 vs 2.0 mg/dL). The average duration of angiotensin-II treatment was 2.7 (sd: 1.5) days. All angiotensin-II–treated patients received greater than 6 hours of treatment.
TABLE 1.
Demographic and Baseline Variables
| Variables | Total (n = 29) | Angiotensin-II (n = 10) | Controls (n = 19) |
|---|---|---|---|
| n | 29 | 10 | 19 |
| Demographics | |||
| Age (yr), mean (sd) | 56 (14) | 54 (15) | 57 (33) |
| Male, n (%) | 19 (66) | 7 (70) | 12 (63) |
| Body mass index, mean (sd) | 32.5 (7.1) | 32.1 (9.1) | 32.7 (6.1) |
| Baseline comorbidities, n (%) | |||
| Diabetes mellitus | 14 (48) | 4 (40) | 10 (53) |
| Coronary artery disease | 5 (17) | 2 (20) | 3 (16) |
| Hypertension | 10 (34) | 9 (90) | 9 (47) |
| Asthma | 4 (14) | 1 (10) | 3 (16) |
| Chronic kidney disease | 2 (7) | 1 (10) | 2 (11) |
| Baseline creatinine (dg/mL), median (interquartile range) | 1.06 (0.88–1.13) | 1.09 (0.93–1.15) | 1.06 (0.87–1.10) |
| Malignancy | 2 (7) | 0 | 2 (11) |
| Chronic obstructive pulmonary disease | 1 (3) | 1 (10) | 0 |
| HIV | 1 (3) | 0 | 1 (5) |
| Solid organ transplant | 1 (3) | 1 (10) | 0 |
| Cirrhosis | 0 | 0 | 0 |
| Chronic heart failure | 0 | 0 | 0 |
| Home medications, n (%) | |||
| Renin angiotensin aldosterone system blockade | 7 (24) | 3 (30) | 4 (21) |
| Angiotensin receptor blocker | 1 (3) | 1 (10) | 0 |
| Angiotensin-converting enzyme-1 inhibitor | 6 (21) | 2 (20) | 4 (21) |
| Beta blocker | 6 (21) | 3 (30) | 3 (16) |
| Calcium channel blocker | 4 (14) | 2 (20) | 2 (11) |
| Other antihypertensive | 5 (17) | 3 (30) | 2 (11) |
| Factor-directed anticoagulation | 2 (7) | 1 (10) | 1 (5) |
| Antiplatelet therapy | 3 (10) | 1 (10) | 2 (11) |
| Corticosteroids | 2 (7) | 1 (10) | 1 (5) |
| Immune modulator | 3 (10) | 2 (7) | 1 (5) |
TABLE 2.
Laboratory and Treatment Variables at Time-Zero
| Variables | Total | Angiotensin-II | Controls |
|---|---|---|---|
| n | 29 | 10 | 19 |
| Vasopressor duration before T0 (hr), median (interquartile range) | Not applicable | 21.30 (6–32) | 21 (21–21) |
| Mean arterial pressure (mm Hg), median (interquartile range) | 78.3 (67–89) | 69.2 (60–81) | 83.2 (68–95) |
| Norepinephrine equivalent dose (µg/kg/hr), median (interquartile range) | 0.232 (0.04–0.22) | 0.489 (0.06–0.81) | 0.097 (0.03–0.15) |
| Norepinephrine, n (%) | 28 (97) | 10 (100) | 18 (95) |
| Vasopressin, n (%) | 2 (6.9) | 2 (20) | 0 |
| Phenylephrine, n (%) | 1 (3) | 0 | 2 (5) |
| Epinephrine, n (%) | 0 | 0 | 0 |
| Right heart failure, n (%) | 8 (28) | 4 (40) | 4 (21) |
| Invasive mechanical ventilation, n (%) | 29 (100) | 10 (100) | 19 (100) |
| Baseline Fio2, median (interquartile range) | 0.59 (0.40–0.80) | 0.66 (0.40–0.80) | 0.55 (0.40–0.80) |
| Pao2 (mm Hg), median (interquartile range) | 103 (76–116) | 99 (73–119) | 105.32 (82–114) |
| Paco2 (mm Hg), median (interquartile range) | 50.8 (39–55) | 60 (46–75) | 46 (39–50) |
| pH, median (interquartile range) | 7.29 (7.19–7.37) | 7.21 (7.14–7.32) | 7.33 (7.26–7.39) |
| Pao2:Fio2, median (interquartile range) | 198 (134–232) | 165 (119–207) | 215 (142–290) |
| Tidal volume (mL/kg)a, median (interquartile range) | 6.3 (5.5–6.8) | 5.8 (5.2–6.2) | 6.5 (5.9–7.0) |
| Respiratory rate, median (interquartile range) | 25.5 (20–32) | 24 (16–28) | 26 (20–34) |
| Positive end-expiratory pressure (cm H2O), median (interquartile range) | 13.3 (10–16) | 14.6 (14–18) | 12.6 (10–16) |
| Mean airway pressure (cm H2O), median (interquartile range) | 21.5 (16.5–23) | 22 (17–23) | 19.75 (16–24) |
| Peak pressure (cm H2O), median (interquartile range) | 32.7 (28–37.5) | 34.6 (32–37) | 31.7 (27–38) |
| Creatinine (mg/dL), median (interquartile range) | 1.95 (1.18–2.16) | 1.82 (1.31–2.14) | 2.00 (0.61–2.88) |
| Bicarbonate (mmol/L), median (interquartile range) | 22.28 (20–24) | 20.50 (18–24) | 23.21 (20–25) |
| Base excess (mmol/L), median (interquartile range) | –2.5 (–5.7 to 0.9) | –4.50 (–8.5 to –0.4) | –1.38 (–5.1 to 0.9) |
| Renal replacement therapy, n (%) | 2 (7) | 1 (10) | 2 (11) |
| Positive blood culture prior to T0, n (%) | 1 (3) | 0 | 1 (5) |
| Sequential Organ Failure Assessment, median (interquartile range) | 10.6 (9–12) | 11.3 (9–13) | 10.16 (8–11) |
| Lymphocyte count (K/μL), median (interquartile range) | 1.26 (0.71–1.63) | 1.46 (0.76–1.63) | 1.16 (0.68–1.67) |
| Monocyte count (K/μL), median (interquartile range) | 0.58 (0.23–0.84) | 0.65 (0.17–0.84) | 0.56 (0.23–0.88) |
| Platelet count (K/μL), median (interquartile range) | 319 (218–380) | 362 (174–433) | 314 (218–342) |
| d-dimer (ng/mL), median (interquartile range) | 1,687 (722–3,652) | 1,981 (1,083–2,392) | 1,394 (675– 3,861) |
| Ln(d-dimer), mean (sd) | 7.31 (0.86) | 7.45 (0.74) | 7.23 (0.94) |
| C-reactive protein (mg/L), median (interquartile range) | 96.7 (63.9–241.7) | 142.8 (87.2–270.8) | 77.0 (51.2–196.3) |
| Ferritin (ng/mL), median (interquartile range) | 895 (636–1,272) | 843 (534–1,019) | 895 (751–1,463) |
| ECMO before T0 | 2 (7) | 1 (10) | 2 (10) |
| ECMO after T0 | 1 (3) | 0 | 1 (5) |
ECMO = extracorporeal membrane oxygenation.
aTidal volume is reported as mL/kg of ideal body weight.
Cardiovascular Response
Despite baseline differences in cardiovascular function, angiotensin-II initiation was associated with an immediate cardiovascular response. There was no change in norepinephrine-equivalents for the control group over the initial 6 hours (0.00 µg/kg/min/hr). In contrast, mean norepinephrine-equivalent dose in the angiotensin-II group fell nearly 50% by 6 hours to 0.23 µg/kg/min (rate of change: –0.04 µg/kg/min/hr; 95% CI vs controls: –0.05 to –0.02 µg/kg/min/hr; pinteraction = 0.0002) (Fig. 1). When the modeling time-horizon was expanded to 48 hours, there was a significant reduction in norepinephrine-equivalents among controls (–0.03 µg/kg/min/d; 95% CI: –0.06 to 0.00 µg/kg/min/d). However, the rate of norepinephrine-equivalent dose reduction was significantly greater in the angiotensin-II group (–0.13 µg/kg/min/d; difference: –0.01 µg/kg/min/d; 95% CI: –0.15 to –0.05 µg/kg/min/d; pinteraction = 0.0004). Similarly, although MAP in control patients did not change over the 6-hour time-horizon, MAP increased by 7.7 mm Hg (1.3 mm Hg/hr; 95% CI: 0.01–2.9 mm Hg/hr; pinteraction = 0.0454) over the first 6 hours in angiotensin-II–treated patients. The groups did not significantly differ in MAP trajectory over 48 hours, in the setting of MAP equilibrating between groups in the first hour after T0 (Fig. 1B). Trajectories over the 7-day study period were more favorable in the angiotensin-II group for both norepinephrine-equivalent dose (–0.04 µg/kg/min/d; 95% CI: –0.05 to –0.03 µg/kg/min/d; pinteraction = 0.0002) and MAP (1.3 mm Hg/d; 95% CI: 0.3–2.4 mm Hg/d; pinteraction = 0.0162).
Figure 1.
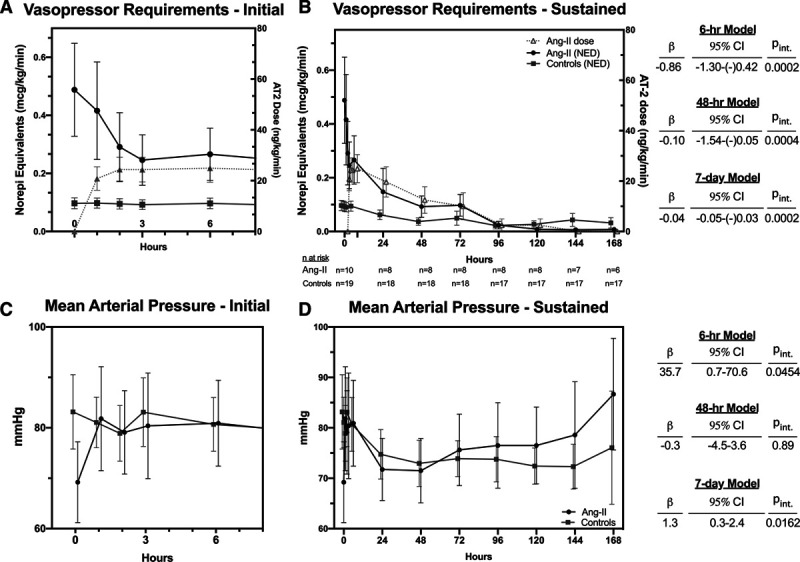
Vasopressor requirements in norepinephrine-equivalent dose (NED) (A and B) and mean arterial pressure (C and D) over time in angiotensin-II (Ang-II)–treated patients versus controls. A and B, Also shows the angiotensin-II dose in Ang-II–treated patients. Markers and error bars indicate mean and sd. Righthand tables indicate interaction coefficients (β), their 95% CIs, and interaction p values from each of the mixed-effects models for time by treatment group. All coefficients are standardized to reflect the difference in rates of change per day, with the control group as the referent. For example, β equals to –0.10 in (A) indicates that the NED decreased in the Ang-II by 0.10 µg/kg/min more per day than it did in the control group over 48 hours. AT2 = angiotensin-II, pint = p value of the interaction coefficient.
Physiologic Trajectories
We depict the trajectories of additional physiologic variables in Figure 2. Despite greater severity at baseline, both Paco2 and pH improved to a greater degree and more rapidly in angiotensin-II-treated patients than in controls. This effect was most pronounced in the 48-hour time-horizon models. These models adjusted for minute ventilation, suggesting the rapid improvement in hypercapnia and acidosis was not attributable to simultaneous changes in ventilator management. Indeed, minute ventilation over the initial 48 hours post T0 did not change in either group. However, although there was no change in positive end-expiratory pressure (PEEP) among controls over 48 hours, there was a significant reduction in PEEP among angiotensin-II–treated patients (difference: –1.8 cm H2O/d; 95% CI: –3.0 to –0.6 cm H2O/d; pinteraction = 0.0035). The P/F ratio increased 7.6 U/d more in the angiotensin-II group than was noted for controls, but this difference was not statistically significant (95% CI: –5.2 to 20.4 U/d; pinteraction = 0.24).
Figure 2.

Markers and error bars indicate mean and sd. Annotations indicate interaction coefficients (β), their 95% CIs, and interaction p values from each of the mixed-effects models for time by treatment group. β reflect the difference in daily rates of change with the control group as the referent. For example, β equals to –0.2 in (C) indicates creatinine decreased in the angiotensin-II group by 0.2 mg/dL/d more than in controls. Paco2 is adjusted for minute ventilation. Creatinine is adjusted for the patient’s prehospitalization baseline. d-dimer was log-transformed to correct violation of the homoscedastic error assumption for modeling. AT2 = angiotensin-II, pint = p value of the interaction coefficient, P/F ratio = ratio of Pao2 to Fio2, SOFA = Sequential Organ Failure Assessment.
A significantly greater reduction in creatinine (0.18 vs 0.05 mg/dL/d) was observed over the study period in angiotensin-II–treated patients than in controls (95% CI of difference: –0.09 to –0.28 mg/dL/d; pinteraction = 0.0001). In contrast to the angiotensin-II group, creatinine increased over this period in the controls (0.05 mg/dL/d; –0.00 to 0.10 mg/dL/d; 95% CI: 0.09–0.28 mg/dL/d; p = 0.051). In contrast to pH and Paco2, the change in creatinine trajectories was more pronounced after the initial 48 hours. There was no difference in creatinine level or trajectory in the first 48 hours between groups. SOFA scores fell more rapidly in angiotensin-II–treated patients than in controls. Similarly, d-dimer levels progressively declined in the angiotensin-II–treated group but not in the control group.
We did not analyze troponin, CRP, ferritin, or lymphocyte counts because the quantity of missing data exceeded what could be reasonably imputed.
Renin Levels
Four of the 10 patients in the angiotensin-II group had a direct plasma renin level measured between 24 and 0 hours before angiotensin-II treatment was initiated. The levels were markedly elevated in all four and were 963.0, 820.0, 105, and 73.5 pg/mL, respectively (reference range: 2.5–45.7pg/mL). None of these patients had prior exposure to ACE-1 inhibitor or angiotensin-II receptor blocker therapy.
Adverse Events and Patient Outcomes
We identified three adverse thrombotic events (16%), all in control patients, one of which occurred while the patient was receiving therapeutic anticoagulation (Table 3). Blood cultures after 48 hours were positive in five control patients (29%) and one angiotensin-II–treated patient (10%). Hospital mortality was similar between groups: six of 10 angiotensin-II–treated patients (60%), and nine of 19 control patients (47%) died. Other clinical outcomes were generally comparable between groups.
TABLE 3.
Treatments, Adverse Events, and Outcomes
| Variables | Total | Angiotensin-II | Controls |
|---|---|---|---|
| n | 29 | 10 | 19 |
| Other treatments | |||
| Hydroxychloroquine | 25 (86) | 6 (60) | 19 (100) |
| Azithromycin | 13 (45) | 3 (30) | 10 (53) |
| Remdesivir | 0 | 0 | 0 |
| Corticosteroids | 18 (62) | 4 (40) | 14 (74) |
| Interleukin-6 antagonist | 8 (28) | 2 (20) | 6 (32) |
| Interleukin-1 antagonist | 7 (24) | 1 (10) | 6 (32) |
| Therapeutic anticoagulation (excluding initiation after a thrombotic event if one occurred) | 15 (52) | 7 (70) | 8 (42) |
| Antiplatelet agent | 5 (17) | 3 (30) | 2 (11) |
| Adverse events | |||
| Thrombosis | |||
| Any thrombotic event | 3 (10) | 0 | 3 (16) |
| Deep vein thrombosis | 0 | 0 | 0 |
| Pulmonary embolism | 0 | 0 | 0 |
| Ischemic stroke | 0 | 0 | 0 |
| Myocardial infarction | 1 (3) | 0 | 1 (5) |
| Critical limb ischemia | 1 (3) | 0 | 1 (5) |
| Mesenteric ischemia | 1 (3) | 0 | 1 (5) |
| Continuous renal replacement therapy with filter clot (n = 7 at risk) | 0 | 0 | 0 |
| Secondary infections | |||
| Any secondary infection | 10 (34) | 3 (30) | 7 (37) |
| Positive blood culture drawn > 48 hr after T0 | 6 (21) | 1 (10) | 5 (29) |
| Gram-positive organism | 2 | 0 | 2 |
| Gram-negative organism | 4 | 1 | 3 |
| Outcomes | |||
| Discharged alive | 14 (48) | 4 (40) | 10 (53) |
| Vasopressor-free days | 3.5 (2.5) | 2.8 (2.49) | 3.9 (2.5) |
| Ventilator-free days | 0.2 (0.5) | 0.3 (0.7) | 0.1 (0.5) |
| Renal replacement therapy–free days | 5.0 (3.1) | 4.3 (3.5) | 5.4 (2.9) |
All continuous variables are presented as means (sd) unless otherwise indicated. All categorical variables are presented as frequency (proportion).
DISCUSSION
In this retrospective cohort study of COVID-19–associated distributive shock, angiotensin-II treatment was associated with an immediate and sustained reduction in vasopressor requirements, an increase in MAP, and improvement in several physiologic indices. Similar changes were not observed in matched controls. This greater improvement was observed despite the angiotensin-II group’s markedly higher severity-of-illness at T0.
Although this study is too small to provide meaningful evidence concerning binary patient outcomes, the marked decrease in vasopressor requirements and increase in MAP within hours of treatment initiation suggests that patients with COVID-19 and distributive shock are highly responsive to angiotensin-II. Further, the rapid improvement in noncardiovascular measures could suggest that the effects of angiotensin-II are not limited to the cardiovascular system. Metabolic and respiratory improvement was most pronounced in the period that directly overlapped with when angiotensin-II treatment was being administered for all indices except creatinine, which is a marker that tends to lag behind acute changes in renal function. Meaning, these diverse physiologic changes were pronounced versus nontreated controls and appeared to occur in close temporal association to the time of angiotensin-II infusion.
A prior case-series from Italy described angiotensin-II use in COVID-19–associated shock and similarly reported improvement in cardiovascular and respiratory status after treatment (21). However, this report lacked a comparator group, limiting inference.
The potential role for renin-angiotensin-aldosterone system (RAAS) modulation in COVID-induced critical illness remains unclear. This uncertainty arises because elements of the RAAS, in particular angiotensin-II, modulate different, and potentially discrepant, pathways that may contribute to the pathobiology of COVID-19. Indeed, some have advocated for and initiated trials of RAAS blockade in COVID-19, whereas others have suggested trials of angiotensin-II therapy (14, 22). The putative mechanisms by which RAAS-blockade would benefit COVID-19–induced critical illness include dampening angiotensin-II–mediated pulmonary inflammation and antagonizing angiotensin-II–mediated platelet activation and factor-mediated thrombosis (22). However, angiotensin-II initiation was not associated with a decrease in P/F ratio in this study and was in fact associated with improvement in hypercapnia independent of minute ventilation. Further, d-dimer, which correlates with thrombosis and appears highly prognostic of mortality in COVID-19 (17), decreased over the course of the study in the treated patients but not in controls. All major thrombotic events in this study occurred in the control group.
Only a small number of patients had a plasma renin-level measured before treatment initiation. However, renin was highly elevated in all patients assessed, none of whom were previously taking RAAS-blocking medications. This is consistent with the immediate cardiovascular response to angiotensin-II treatment we observed, as elevated renin both suggests ACE-1 dysfunction and is associated with a “hyper-responsive” phenotype to angiotensin-II treatment in distributive shock (13, 19). The combination of highly elevated renin, a marked cardiovascular and metabolic response, temporally associated with treatment initiation that was not observed in controls, and the absence of signs of worsening coagulation or lung function supports equipoise for a randomized trial of angiotensin-II versus norepinephrine in COVID-19 patients with shock.
This study has important limitations. First, retrospective design limits causal-inference and is prone to bias (23). We attempted to adhere to recommendations by Kaji et al (24) for reducing bias in chart-review with systematic review of a consecutive sample by a single abstractor following protocolized collection procedure and by employing multiple a priori and post hoc validation practices. Second, this small study would have been inadequately powered to detect all but the largest differences in dichotomous outcomes. We particularly stress our adverse event, and clinical outcomes analyses are exploratory; we urge restraint in angiotensin-II use for COVID-19 until randomized evidence becomes available. Third, a Neyman bias could impact 7-day trajectory modeling on the basis of early death. For this reason, we regard the shorter time-horizon models as more reliable. Fourth, the matching process identified controls that were markedly less ill at T0 than the treated patients. This difference may have reflected the use of inadequately granular data for matching or treatment-endogeneity where experimental therapy was more readily applied in a nonresearch context to patients with worse prognoses. Either scenario would be expected to bias results in favor of the control group. Alternatively, this could also indicate T0 for some angiotensin-treated patients overlapped with initial resuscitation, overstating their severity of illness relative to controls. Fifth, we noted a higher frequency of therapeutic anticoagulation among patients who received angiotensin-II, which might explain why all thrombotic events occurred in the controls. Two of the events occurred in patients not receiving anticoagulant therapy at the time and one in a patient receiving an infusion of unfractionated heparin. Given the emerging evidence regarding thrombosis in COVID-19, anticoagulation therapy even in the absence of thrombosis may become more prevalent. Sixth, two patients in the angiotensin-II group were also receiving vasopressin at T0, which could be a confounder.
Larger, future studies should consider strategies to account for these key confounders. However, given the numerous potential sources of bias and the limited use of angiotensin-II in COVID-19 to date, small, randomized studies may provide a more efficient and informative approach than larger observational analyses.
CONCLUSIONS
Angiotensin-II treatment in COVID-induced distributive shock is associated with rapid reduction in vasopressor requirements and improvement in multiple physiologic indices. Angiotensin-II in COVID-19 shock warrants further study.
Footnotes
Drs. Leisman and Mastroianni contributed equally as co-first authors. Drs. Taylor and Deutschman contributed equally as co-senior authors.
Dr. Leisman discloses that La Jolla Pharmaceutical Company, the manufacturer of Giapreza, provided a sample of their drug free of charge for mouse experiments that are not related to the present study, and La Jolla Pharmaceutical Company had no access to any of that data or any role in its analysis, interpretation, or eventual dissemination and has had no role whatsoever in any aspect of the present study. Dr. Taylor discloses grant support from the National Institutes of Health (NIH). Dr. Deutschman discloses grant support from the NIH and consulting fees from Enlivex Therapeutics. The remaining authors have disclosed that they do not have any potential conflicts of interest.
REFERENCES
- 1.Cummings MJ, Baldwin MR, Abrams D, et al. Epidemiology, clinical course, and outcomes of critically ill adults with COVID-19 in New York City: A prospective cohort study. Lancet. 2020; 395:1763–1770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu C, Chen X, Cai Y, et al. Risk factors associated with acute respiratory distress syndrome and death in patients with Coronavirus disease 2019 pneumonia in Wuhan, China. JAMA Intern Med. 2020; 180:934–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arentz M, Yim E, Klaff L, et al. Characteristics and outcomes of 21 critically ill patients with COVID-19 in Washington State. JAMA. 2020; 323:1612–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirsch JS, Ng JH, Ross DW, et al. ; Northwell COVID-19 Research Consortium; Northwell Nephrology COVID-19 Research Consortium. Acute kidney injury in patients hospitalized with COVID-19. Kidney Int. 2020; 98:209–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanco-Melo D, Nilsson-Payant BE, Liu WC, et al. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020; 181:1036–1045.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alhazzani W, Møller MH, Arabi YM, et al. Surviving sepsis campaign: Guidelines on the management of critically ill adults with coronavirus disease 2019 (COVID-19). Crit Care Med. 2020; 48:e440–e469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stolk RF, van der Poll T, Angus DC, et al. Potentially inadvertent immunomodulation: Norepinephrine use in sepsis. Am J Respir Crit Care Med. 2016; 194:550–558 [DOI] [PubMed] [Google Scholar]
- 8.Khanna A, English SW, Wang XS, et al. ; ATHOS-3 Investigators. Angiotensin II for the treatment of vasodilatory shock. N Engl J Med. 2017; 377:419–430 [DOI] [PubMed] [Google Scholar]
- 9.Senatore F, Jagadeesh G, Rose M, et al. FDA approval of angiotensin II for the treatment of hypotension in adults with distributive shock. Am J Cardiovasc Drugs. 2019; 19:11–20 [DOI] [PubMed] [Google Scholar]
- 10.Jurewicz M, McDermott DH, Sechler JM, et al. Human T and natural killer cells possess a functional renin-angiotensin system: Further mechanisms of angiotensin II-induced inflammation. J Am Soc Nephrol. 2007; 18:1093–1102 [DOI] [PubMed] [Google Scholar]
- 11.Crowley SD, Rudemiller NP. Immunologic effects of the renin-angiotensin system. J Am Soc Nephrol. 2017; 28:1350–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tumlin JA, Murugan R, Deane AM, et al. ; Angiotensin II for the Treatment of High-Output Shock 3 (ATHOS-3) Investigators. Outcomes in patients with vasodilatory shock and renal replacement therapy treated with intravenous angiotensin II. Crit Care Med. 2018; 46:949–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bellomo R, Forni L, Busse L, et al. Renin and survival in patients given angiotensin II for catecholamine-resistant vasodilatory shock. Am J Respir Crit Care Med. 2020. July 1. [online ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Leisman DE, Deutschman CS, Legrand M. Facing COVID-19 in the ICU: Vascular dysfunction, thrombosis, and dysregulated inflammation. Intensive Care Med. 2020; 46:1105–1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imai Y, Kuba K, Rao S, et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005; 436:112–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ereso AQ, Ramirez RM, Sadjadi J, et al. Angiotensin II type 2 receptor provides an endogenous brake during inflammation-induced microvascular fluid leak. J Am Coll Surg. 2007; 205:527–533 [DOI] [PubMed] [Google Scholar]
- 17.Iba T, Levy JH, Levi M, et al. Coagulopathy of Coronavirus disease 2019. Crit Care Med. 2020; 48:1358–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Leisman DE. Ten pearls and pitfalls of propensity scores in critical care research: A guide for clinicians and researchers. Crit Care Med. 2019; 47:176–185 [DOI] [PubMed] [Google Scholar]
- 19.Chawla LS, Chen S, Bellomo R, et al. Angiotensin converting enzyme defects in shock: Implications for future therapy. Crit Care. 2018; 22:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McCullough PA, Shaw AD, Haase M, et al. Diagnosis of acute kidney injury using functional and injury biomarkers: Workgroup statements from the tenth acute dialysis quality initiative consensus conference. Contrib Nephrol. 2013; 182:13–29 [DOI] [PubMed] [Google Scholar]
- 21.Zangrillo A, Landoni G, Beretta L, et al. ; COVID-BioB Study Group. Angiotensin II infusion in COVID-19-associated vasodilatory shock: A case series. Crit Care. 2020; 24:227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.South AM, Tomlinson L, Edmonston D, et al. Controversies of renin-angiotensin system inhibition during the COVID-19 pandemic. Nat Rev Nephrol. 2020; 16:305–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maslove DM, Leisman DE. Causal inference from observational data: New guidance from pulmonary, critical care, and sleep journals. Crit Care Med. 2019; 47:1–2 [DOI] [PubMed] [Google Scholar]
- 24.Kaji AH, Schriger D, Green S. Looking through the retrospectoscope: Reducing bias in emergency medicine chart review studies. Ann Emerg Med. 2014; 64:292–298 [DOI] [PubMed] [Google Scholar]