

Abstract
Hypoglycemia is a detrimental complication of rigorous management of type 1 diabetes mellitus. Moderate hypoglycemia (MH) preconditioning of male rats partially affords protection from loss of vulnerable brain neurons to severe hypoglycemia (SH). Current research investigated whether MH preconditioning exerts sex-dimorphic effects on hippocampal CA1 neuron bio-energetic and anti-oxidant responses to SH. SH up-regulated CA1 glucose or monocarboxylate transporter proteins in corresponding hypoglycemia-naïve male versus female rats; precedent MH amplified glucose transporter expression in SH irrespective of sex. Sex-differentiating SH effects on glycolytic and tricarboxylic pathway markers correlated with elevated tissue ATP content and diminished CA1 5’-AMP-activated protein kinase (AMPK) activation in females. MH preconditioned-suppression of mitochondrial energy pathway enzyme profiles and tissue ATP in SH rats coincided with amplified CA1 AMPK activity in both sexes. Anti-oxidative stress enzyme protein responses to SH were primarily sex-contingent; preconditioning amplified most of these profiles, yet exacerbated expression of lipid and protein oxidation markers in SH male and female rats, respectively. Results show that MH preconditioning abolishes female CA1 neuron neuroprotection of positive energy balance through SH, resulting in augmented CA1 AMPK activity and oxidative injury and diminished tissue ATP in hypoglycemia-conditioned versus - naïve rats in each sex. It is unclear if SH elicits differential rates of CA1 neuronal destruction in the two sexes, or how MH may impact sex-specific cell loss. Further research is needed to determine if molecular mechanism(s) that maintain female CA1 neuron metabolic stability in the absence of MH preconditioning can be leveraged for therapeutic prevention of hypoglycemic nerve cell damage.
Keywords: laser-catapult microdissection, Western blot, glucose transporter-3, phosphofructokinase, 5’-AMP-activated protein kinase, nitrotyrosine
Introduction:
In type I diabetes mellitus patients, stabilization of blood glucose within the normal range is imperative for prevention or delay of microvascular and other complications of hyperglycemia. In reality, this therapeutic goal is impeded by fear of hypoglycemia, which occurs on a regular basis amidst standard insulin treatment regimens. Diabetes patients experience frequent episodes of moderate hypoglycemia (MH; 2.3 to 3.6 mM), which predisposes these individuals to hypoglycemia-associated autonomic failure (HAAF). HAAF reflects mal-adaptive desensitization to hypoglycemia [1], manifesting as hypoglycemia unawareness and glucose counter-regulatory dysfunction which is capable of exacerbating hypoglycemia decrements and capacity to damage the brain [2]. Severe hypoglycemia (SH; < 2.0 mM) can elicit cell death among vulnerable brain neuron populations, including the hippocampal CA1 region, dentate gyrus (DG), and cerebral cortical (CC) layers 2 and 3 [3]. As the hippocampus has a critical role in memory and learning, hypoglycemic injury or destruction of this structure has an associated risk of impairment of these vital neurological functions.
Recent studies show that prior exposure to MH, specifically one episode per day for three consecutive days, can significantly attenuate hippocampal CA1 nerve cell loss due to SH [4]. The mechanisms that underlie beneficial preconditioning remain unclear. Other studies show that harm by SH is exacerbated by exposure to MH for seven days, indicating that duration of preconditioning is a critical determinant of gain or loss of protection [5]. Precedent MH may confer protection, in part, through induction of adaptive neuronal mechanisms that prevent a shift toward negative intracellular energy balance due to systemic fuel deficiency. We theorize that antecedent MH may shield CA1 cells from SH injury through induction of adaptive mito-protective mechanisms that include augmented mitochondrial bio-energetic and anti-oxidant activity. Studies performed here utilized laser-catapult microdissection to obtain pure CA1 nerve cell samples from hypoglycemia-naïve versus MH-exposed rats to address the hypothesis that MH preconditioning may up-regulate substrate fuel transporter and/or key glycolytic (ATP-regulated kinase phosphofructokinase; PFKL), tricarboxylic (isocitrate dehydrogenase; IDH2), and oxidative respiratory (ATP synthase; ATPs) pathway marker protein responses to SH, coincident with diminished activity of the ultra-sensitive energy gauge 5’-AMP-activated protein kinase (AMPK). The ovarian steroid estradiol protects the female brain from bio-energetic insults due to hypoxia, stroke, and degenerative diseases through mechanisms that include energy-producing metabolic pathway stimulation, including augmentation of glycolytic, tricarboxylic acid cycle, and respiratory chain enzyme function and oxidative respiration [6–9]. Current research utilized a validated experimental model that achieves estradiol replacement in ovariectomized (OVX) female rats within physiological range to investigate whether antecedent MH exerts sex-specific effects on CA1 neuron energy pathway enzyme protein and AMPK activational responses to SH.
Oxidative damage is a key factor in the nerve cell death-inducing cascade of events triggered by hypoglycemia [10, 11]. It is plausible that hypoglycemic preconditioning may augment CA1 nerve cell defenses against deleterious downstream consequences of inadequate energy production, namely free radical production and oxidative stress, by increasing anti-oxidant enzyme expression. Superoxide dismutase (SOD) is a vital defense against oxidative stress by scavenging superoxide radicals, e.g. superoxide anion, a common type of reactive oxygen species (ROS) generated in cells, and converting those to hydrogen peroxide (H2O2). The three SOD enzyme variants are encoded by different genes and exhibit distinctive protein structures and subcellular distributions, as SOD1 (CuZnSOD) and −2 (MnSOD) are cytosolic or mitochondrial, respectively, in location, whereas SOD3 is exported from the cell. H2O2 is a potent agent of oxygen toxicity, and is removed by action of catalase and glutathione peroxidase (GPx). GPx4 catalyzes reduction of H2O2, organic hydroperoxides, and lipid peroxides to protect cells against oxidative stress; it differs from other GPx enzymes in its ability to reduce lipid-hydroperoxides inside biological membranes. Here, CA1 neuron lysates were analyzed by Western blot to ascertain the impact of hypoglycemia preconditioning on patterns of expression of SOD1, SOD2, catalase, GPx4, and protein markers of oxidative lipid (4-hydroxynonenal; 4-HNE) and protein (nitrotyrosine; NT) damage in SH male versus female rats.
Methods and materials:
Experimental Design:
Adult male and female Sprague Dawley (3–4 months of age; Envigo, Houston, TX) were housed in shoe-box cages, 2–3 per cage, according to sex, under a 14 hr light/10 hr dark cycle (lights on at 05.00 a.m.). Animals had free access to standard laboratory rat chow and water, and were acclimated to daily handling. All surgical and experimental protocols were conducted in accordance with NIH guidelines for care and use of laboratory animals, and approved by the ULM Institutional Animal Care and Use Committee. Seven days prior to experimentation, female rats were bilaterally OVX, and implanted with a subcutaneous (sc) silastic capsule (i.d. 0.062 in/o.d. 0.125 in.; 10 mm/100 g bw) containing 30 ug 17β estradiol-3-benzoate/mL safflower oil, under ketamine/xylazine (0.1 mL/100 g bw; 90 mg ketamine:10 mg xylazine/mL; Henry Schein Inc., Melville, NY) anesthesia. This steroid replacement regimen yields approximate plasma estradiol concentrations of 22 pg/ml [12], replicating circulating hormone levels characteristic of the estrous cycle stage of metestrus in 4-day cycling, ovary-intact rats [13]. On study day 1, testes-intact male rats and estradiol-replaced OVX female rats were randomly assigned to three treatment groups (summarized in Table 1): Group 1: intraperitoneal (ip) injection of vehicle (V; sterile insulin diluent; Eli Lilly & Co., Indianapolis, IN) at 09.00 a.m. (time zero, to) on days 1–4 (V1−4; n=4 males; n=5 females); Group 2: ip V injection on days 1–3, followed by ip neutral protamine Hagedorn insulin (NPH; 30.0 U/kg bw) on day 4 (V1−3/SH4; n=4 males; n=5 females); Group 3: ip injection of 5.0 U INS/kg bw on days 1–3, followed by ip 30.0 U NPH/kg bw on day 4 (MH1−3/SH4; n=5 males; n=5 females). On day 10, blood samples were collected via tail vein immediately before to and at +20, +60, +90 and +120 min after injection treatments for measurement of plasma glucose levels by glucometer, as described [14]. The 5.0 U NPH/kg dosage administered on days 1–3 to group 3 was observed in dose-titration to lower plasma glucose to approximately 67.3 mg/dL by +120 min. After sacrifice at 11.00 a.m., trunk blood was collected for storage at −20°C and brains were individually snap-frozen in liquid nitrogen-cooled isopentane for preservation at −80°C.
Table 1.
Experimental Design
| Study Days | ||||
|---|---|---|---|---|
| Treatment Groups | 1 | 2 | 3 | 4 |
| Group 1 (n=4 males, n=5 females) | V sca | V sc | V sc | V sc |
| Group 2 (n=4 males, n=5 females) | V sc | V sc | V sc | INS scb |
| Group 3 (n=5 males, n=5 females) | INS scc | INS scc | INS scc | INS scb |
vehicle administered by subcutaneous route
30.0 U neutral protamine Hagedorn insulin (INS)/kg bw sc
5.0 U neutral protamine Hagedorn insulin (INS)/kg bw sc
Brain Tissue Collection:
Forebrains were cut into alternating series of 200 μm- (2 ×100 μm sections) or 10 μm- (20×10 sections) thick frozen sections over a predetermined length of the hippocampal CA1 region (−4.45 to −6.0 mm), and from over specified lengths of other neural structures characterized by vulnerability to hypoglycemic damage, e.g. DG (−3.2 to −4.2 mm) and CC layer 2 (−6.0 to −7.0 mm) [15]. Tissue sections were also harvested from the arcuate hypothalamic nucleus (ARH; −2.00 to −3.00 mm), which is not characterized by SH-associated nerve cell demise. 10 μm-thick sections were mounted on polyethylene naphtholate (PEN) membrane-covered slides (Carl Zeiss Microscopy, Thornwood, NY) for histological delineation of nerve cell perikarya. CA1, DG, CC-2, and ARH tissues were bilaterally micropunch-dissected from 200 μm-thick sections using calibrated hollow punch tools (Stoelting Co., Wood Dale, IL) and pooled by collection into lysis buffer (2% sodium dodecyl sulfate [SDS], 0.05M dithiothreitol, 10% glycerol, 1mM EDTA, 60mM Tris-HCl, pH 7.2) for ATP measurements.
Western Blot Analysis of Laser-Catapult Microdissected CA1 Neurons:
PEN slide-mounted sections were processed by immersion in fresh 1.0% aqueous methylene blue [2.0 mL methylene blue stock solution (SigmaAldrich Co., St Louis, MO); 2.0 mL 0.1M citric acid 0.2M phosphate buffer, pH 5.6; 5.0 mL acetone; 28.0 mL distilled water] for 15 min., followed by distilled water washing for 15 min. Individual methylene blue-stained CA1 neurons were harvested using a Zeiss P.A.L.M. UV-A microlaser IV system and collected into lysis buffer (described above). Individual proteins were analyzed in triplicate pools of n=50 CA1 nerve cell lysates prepared per treatment group for separation on Bio-Rad TGX 10–12% stain-free gels [16]. After electrophoresis, gels were activated for 1 min. by UV light in a ChemiDoc TM Touch Imaging System (Bio-Rad, Hercules, CA) prior to protein transfer (30 V, overnight at 4°C; Towbin buffer) to 0.45-μm PVDF membranes (Thermo Fisher Scientific; Waltham, MA). Membranes were blocked for 2 hr with Tris-buffer saline, pH 7.4 (TBS) containing 0.1% Tween-20 and 2% bovine serum albumin ahead of overnight (4°C) incubation with primary antisera raised in rabbit [glucose transporter-3 (GLUT3; NBP2–66872, 1:1500; Novus Biologicals, LLC, Centennial, CO), glucose transporter-4 (GLUT4; NBP1–19533SS, 1:1200; Novus Biol.), monocarboxylate transporter-2 (MCT2; AB3540P, 1:2000; MilliporeSigma, Burlington, MA), PFKL (CA-30117, 1:1500; ProSci Inc., Poway, CA ), SOD1 (NBP2–24915, 1:1500; Novus Biol.), SOD2 (NB100–1992, 1:1200; Novus Biol.), GPx4 (NBP1–5491, 1:1500, Novus Biol.), catalase (NBP2–24916, 1:1250; Novus Biol.), AMPK (2532; 1:2000; Cell Signaling Technology, Danvers, MA) and phospho-AMPK (pAMPK; 2531S; 1:2000; Cell Signal. Technol.), goat [pyruvate dehydrogenase kinase component E1 (PDK1; CA-46155, 1:1250; ProSci.), IDH2 (CA-42704, 1:1500; ProSci.), ATPs (CA-42704, 1:1500; ProSci.), 4-HNE (NB100–63093, 1:3000, Novus Biol.), or mouse [NT (MAB3248; 1:2000; R&D Research, MN)], Membranes were incubated for 1 hr with peroxidase-conjugated rabbit anti-goat (AP106P, 1:5000; EMD Millipore, Billerica, MA), goat anti-rabbit (NEF812001EA, 1:5000; PerkinElmer, Boston, MA), or goat anti-mouse (NEF822001EA; 1:5000; PerkinElmer) secondary antibodies before exposure to SuperSignal West Femto chemiluminescent substrate (34095; ThermoFisherScientific, Rockford, IL). Optical density (O.D.) signals were detected and quantified by Image Lab software in a ChemiDoc MP Imaging System, and normalized within individual lanes by Bio-Rad Stain-Free Imaging Technology
Micropunch-Dissected Tissue ATP Content:
For each neural structure evaluated, tissue punches obtained from individual subjects were pooled within treatment groups. Aliquots of pooled sample lysates were homogenized (Model Q55 QSONICA, QSonica, LLC, Newtown, CT; 30 sec.) on ice, heated to 95 °C (10 min.) for ATP extraction, and centrifuged (8 × 1000 rpm; 1 min.). Tissue ATP content was measured using ATP Bioluminescent Assay Kit luciferin–luciferase system reagents, as described by the manufacturer (prod. no. FLAA; Sigma Aldrich Inc., MO). Briefly, ATP standard was diluted with sterile water for preparation of standard curve (2.0 −0.00390625 μmole/mL) by serial dilution. ATP Assay mix (50 μL) was pipetted into individual extraction plate wells; after 3 min., 50 μL standard or sample aliquots were added for incubation. Emitted light per well was measured using a Synergy HT microplate luminometer (BioTek Instruments, Inc., Winooski, VT).
Statistical Analyses:
Plasma glucose concentrations were analyzed by two-analysis of variance for repeated measures and Duncan’s multiple range test. Mean normalized protein O.D. values were evaluated among treatment groups within each sex by one-way analysis of variance and Duncan’s multiple range test, using Graph pad prism 5.0 and IBM SPSS Statistics 22.0. Mean tissue ATP content was analyzed among male and female treatment groups by two-way analysis of variance and Duncan’s multiple range test. Differences were considered significant at p < 0.05. In each Figure, statistical differences between specific pairs of treatment groups are denoted with the following symbols: *p<0.05; **p<0.01; ***p<0.001.
Results:
Effects of MH precondititioning on CA1 nerve cell glucose and monocarboxylate transporter protein responses to SH in male versus female rats.
Data presented in Table 2 show that plasma glucose concentrations were significantly decreased at +2 hr after onset of SH, and that MH preconditioning did not modify this decrement in either sex [F(8,56) = 4.39, p < 0.0001]. Figure 1 depicts GLUT3 (Figure 1A), GLUT4 (Figure 1B), and MCT2 profiles (Figure 1C) protein profiles in V1−4 (solid white bars), V1−3/SH4 (solid gray bars), and MH1−3/SH4 (diagonal-striped gray bars) groups of male (left side) versus female (right side) rats. Data show that male [F(2,6) = 9.06, p = 0.003], but not female [F(2,6) = 8.45, p = 0.003] hypoglycemia-naïve rats exhibited significant up-regulation of CA1 GLUT3 protein expression in response to SH (Figure 1A). Prior exposure to MH enhanced this profile during SH in female, but not male rats (Table 3). SH increased CA1 GLUT4 protein levels in male rats, but this stimulatory response was abolished by precedent MH [F(2,6) = 10.37, p = 0.003] (Figure 1B). GLUT4 expression was refractory to SH in hypoglycemia-naïve female rats, but was increased by SH in MH-exposed animals [F(2,6) = 8.39, p = 0.005]. CA1 nerve cell MCT2 protein expression was not affected by SH in male rats [F(2,6) = 5.32, p = 0.02], but was up-regulated by SH in females (F(2,6) = 15.72, p = 0.0002). MCT2 profiles in both males and females were significantly lower in MH1−3/SH4 versus V1−3/SH4 animals.
Table 2.
Effects of Moderate Hypoglycemia Preconditioning (MH) on Neutral Protamine Hagedorn Insulin (NPH; 30.0 U/kg bw) – Induced Hypoglycemia in Male versus Female Rats
| Time Points | |||||||
|---|---|---|---|---|---|---|---|
| Treatment Groups | Time Zero (to) | +20 min. | +60 min. | +90 min. | +120 min. | ||
| Male | Vl−4 a | (n=4) | 106.8 ± 3.2 | 113.3 ± 4.0 | 115.3 ± 2.3 | 112.3 ± 3.4 | 113.0 ± 3.9 |
| Vl−3/SH4 b | (n=4) | 105.5 ± 4.9 | 83.5 ± 3.2d | 67.5 ± 3.2d | 51.0 ± 3.4d | 39.2 ± 1.6d | |
| MH1−3/SH4 c | (n=5) | 102.8 ± 2.7 | 88.7 ± 2.3d | 56.7 ± 2.0d | 50.7 ± 1.0d | 46.7 ± 0.9d | |
| Female | Vl−4 | (n=5) | 108.2 ± 2.3 | 115.0 ± 3.0 | 111.8 ± 2.6 | 113.8 ± 1.9 | 107.8 ± 1.4 |
| V1−3/SH4 | (n=5) | 112.2 ± 3.7 | 81.4 ± 5.0d | 52.2 ± 2.4d | 41.4 ± 3.7d | 36.2 ± 2.8d | |
| MH1−3/SH4 | (n=5) | 109.2 ± 1.4 | 80.2 ± 3.6d | 57.2 ± 3.1d | 41.2 ± 1.8d | 27.6 ± 3.3de | |
intraperitoneal (ip) vehicle (V) injection on days 1–4
ip V injection on days 1–3; ip NPH (30.0 U/kg bw) injection on day 4
ip NPH (5.0 U/kg bw) injection on days 1–3; ip NPH (30.0 U/kg bw) injection on day 4
p<0.05 versus V1−4 at same time point
p<0.05 versus MH1−3/SH4 males at same time point
Figure 1. Effects of Moderate Hypoglycemia (MH) Preconditioning on Hippocampal CA1 Nerve Cell Glucose and Monocarboxylate Transporter Protein Expression during Severe Insulin-Induced Hypoglycemia (SH) in Male versus Female Rats.
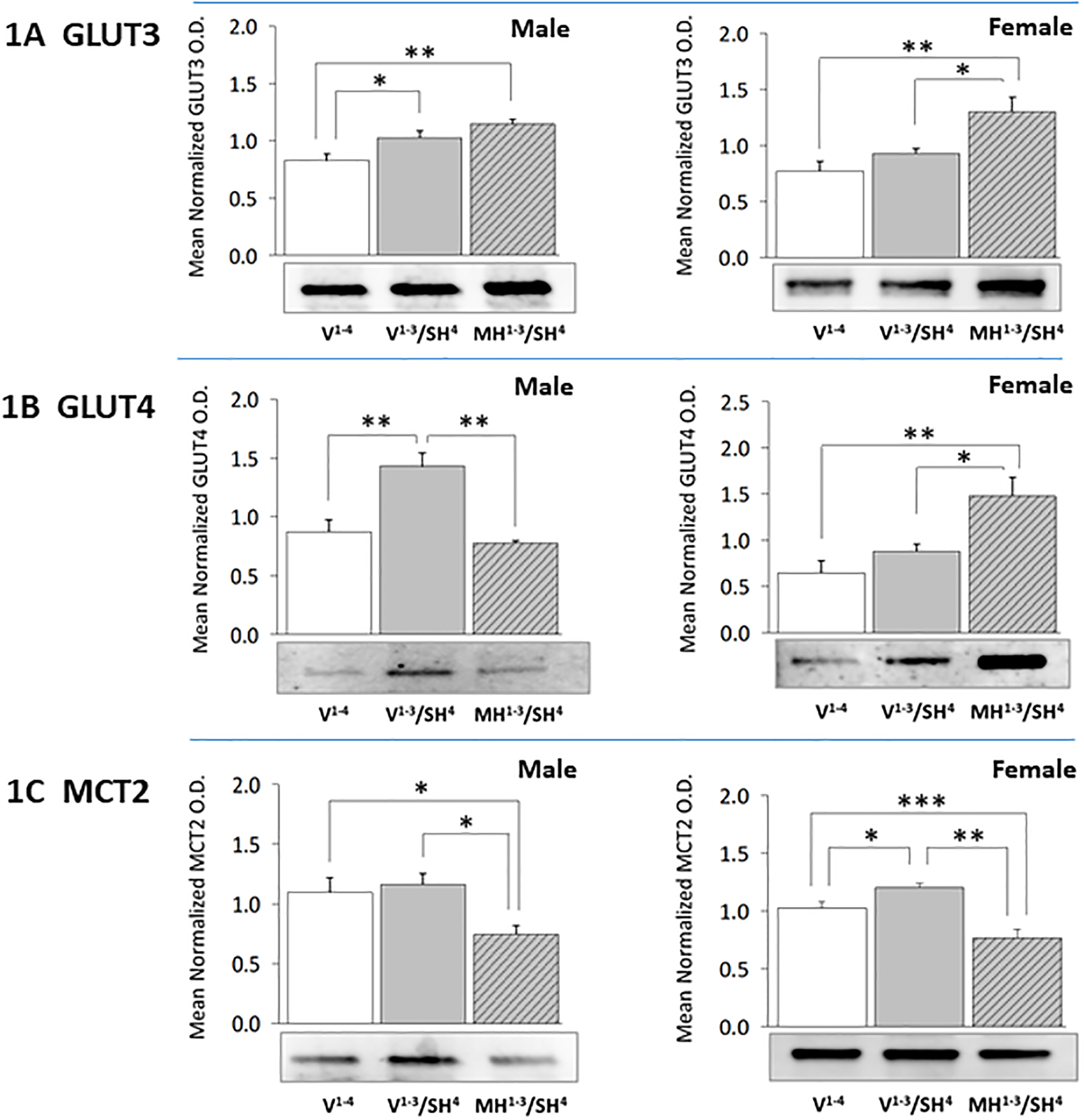
Groups of testes-intact and estradiol-replaced ovariectomized female rats were injected intraperitoneally (ip) on days 1–3 with vehicle (V) (groups 1 and 2, n=4 males and n=5 females each) or 5.0 U neutral protamine Hagedorn insulin (NPH)/kg bw (group 3, n=5 males, n=5 females), followed by ip injection on day 4 with V (group 1) or 30.0 U NPH/kg bw (groups 2 and 3). Methylene blue-stained CA1 neurons were harvested by laser catapult-microdissection from 10 μm-thick frozen sections cut at regular intervals through each hippocampus between −4.45 to −6.0 mm relative to bregma. For each treatment group, CA1 cell lysate aliquots from individual subjects were pooled (n=50 neurons/group) in triplicate for Western blot analysis of each target protein, e.g. glucose transporter-3 (GLUT3; Figure 1A: male data at left, female data at right), glucose transporter-4 (GLUT4; Figure 1B: male data at left, female data at right), or monocarboxylate transporter-2 (MCT2; Figure 1C: male data at left, female data at right). Bars depict mean normalized protein optical density (O.D.) measures ± S.E.M. for male and female V1−4 (solid white bars), V1−3/SH4 (solid gray bars), and MH1−3/SH4 (cross-hatched gray bars) treatment groups. *p<0.05; **p<0.01; ***p<0.001.
Table 3.
Summary of Moderate Hypoglycemia Preconditioning (MH) Effects on Hippocampal CA1 Neuron Glycolytic, Tricarboxylic Acid, Oxidative Respiratory, and Anti-Oxidative Stress Enzyme Responses to Severe Hypoglycemia (SH) in Male versus Female Rats
| MALE | FEMALE | ||||||
|---|---|---|---|---|---|---|---|
| Day 4 | Vehiclea | SHb | Vehicle | SH | |||
| Days 1–3 | Vehicle n=4 |
Vehicle n=4 |
MHc n=5 |
Vehicle n=5 |
Vehicle n=5 |
MH n=5 |
|
| Glucose Transporter-3 (GLUT3) | ↑d vs V | N.D.e vs SH | N.D. vs V | ↑ vs SH | |||
| Glucose Transporter-4 (GLUT4) | ↑ vs V | ↓ vs SH (N)f | N.D. vs V | ↓ vs SH (N) | |||
| Monocarboxylate Transporter-2 (MCT2) | N.D. vs V | ↓ vs SH | ↑ vs V | ↓ vs SH, ↓ vs V | |||
| Phosphofructokinase (PFKL) | ↓ vs V | ↑ vs SH (N) | N.D. vs V | ↑ vs SH | |||
| Pyruvate Dehydrogenase Component E1 (PDK1) | N.D. vs V | ↑ vs SH | ↓ vs V | ↑ vs SH (N) | |||
| Isocitrate Dehydrogenase (IDH2) | N.D. vs V | ↓ vs SH | ↑ vs V | ↓ vs SH (N) | |||
| ATP Synthase (ATPS) | ↑ vs V | ↓ vs SH (N) | ↑ vs V | ↓ vs SH (N) | |||
| Superoxide Dismutase-1 (SOD1) | ↓ vs V | N.D. vs SH | ↓ vs V | ↑ vs SH (N) | |||
| Superoxide Dismutase-2 (SOD2) | ↑ vs V | N.D. vs SH | N.D. vs V | ↑ vs SH | |||
| Catalase | ↓ vs V | ↑ vs SH (N) | ↑ vs V | ↑ vs SH | |||
| Glutathione Peroxidase (GPx4) | ↓ vs V | ↑ vs SH (N) | N.D. vs V | N.D. vs SH | |||
| 5’-AMP-Activated Protein Kinase (AMPK) | ↓ vs V | N.D. vs SH | ↓ vs V | N.D vs SH | |||
| Phospho-AMPK (pAMPK) | ↑ vs V | N.D. vs SH | ↓ vs V | ↑ vs SH, ↑ vs V | |||
| 4-Hydroxynonenal (4-HNE) | ↑ vs V | N.D. vs SH | N.D. vs V | ↑ vs SH | |||
| Nitrotyrosine (NT) | ↑ vs V | ↑ vs SH | ↑ vs V | N.D. vs SH | |||
intraperitoneal;
30 U neutral protamine Hagedorn insulin (INS)/kg bw ip;
5 U INS/kg bw ip;
arrows indicate a significant difference of at least p<0.05;
N.D. indicates that data are not different;
normalized relative to V.
Effects of SH on glycolytic, tricarboxylic, and oxidative respiratory pathway marker protein expression on CA1 neurons in hypoglycemia-naïve versus hypoglycemia-preconditioned male versus female rats.
Figure 2 illustrates CA1 PFKL (Figure 2A), PKD1 (Figure 2B), IDH2 (Figure 2C), and ATPS (Figure 2D) protein responses in rats exposed to SH with or without prior exposure to MH. SH diminished expression of the rate-limiting glycolytic pathway enzyme protein PFKL in male (left side) [F(2,6) = 4.56, p = 0.03], but not female (right side) [F(2,6) = 10.47, p = 0.001] rat CA1 neurons. Prior exposure to MH normalized PFKL profiles in males and up-regulated PFKL expression in females after induction of SH. CA1 PDK1 levels were unaffected by SH in males (left side) [F(2,6) = 6.28, p = 0.01], but were down-regulated in female (right side) [F(2,6) = 4.28, p = 0.034] rats. This profile was higher in MH1−3/SH4 males and females relative to V1−3/SH4 groups. CA1 IDH2 protein levels were refractory to SH in males [F(2,6) = 5.71, p = 0.002], but were augmented by this treatment in females [F(2,6) = 13.48, p = 0.002]. Precedent MH significantly decreased this profile compared to V1−3/SH4 male and female rats. SH elevated CA1 ATPS protein expression in both sexes [male: F(2,6) = 5.25, p = 0.02; female: F(2,6) = 9.54, p = 0.003], but this response was prevented by precedent MH.
Figure 2. Effects of Precedent MH on Hippocampal CA1 Nerve Cell Glycolytic Pathway, Tricarboxylic Acid Cycle, and Respiratory Chain Marker Protein Expression in SH Male versus SH Female Rats.
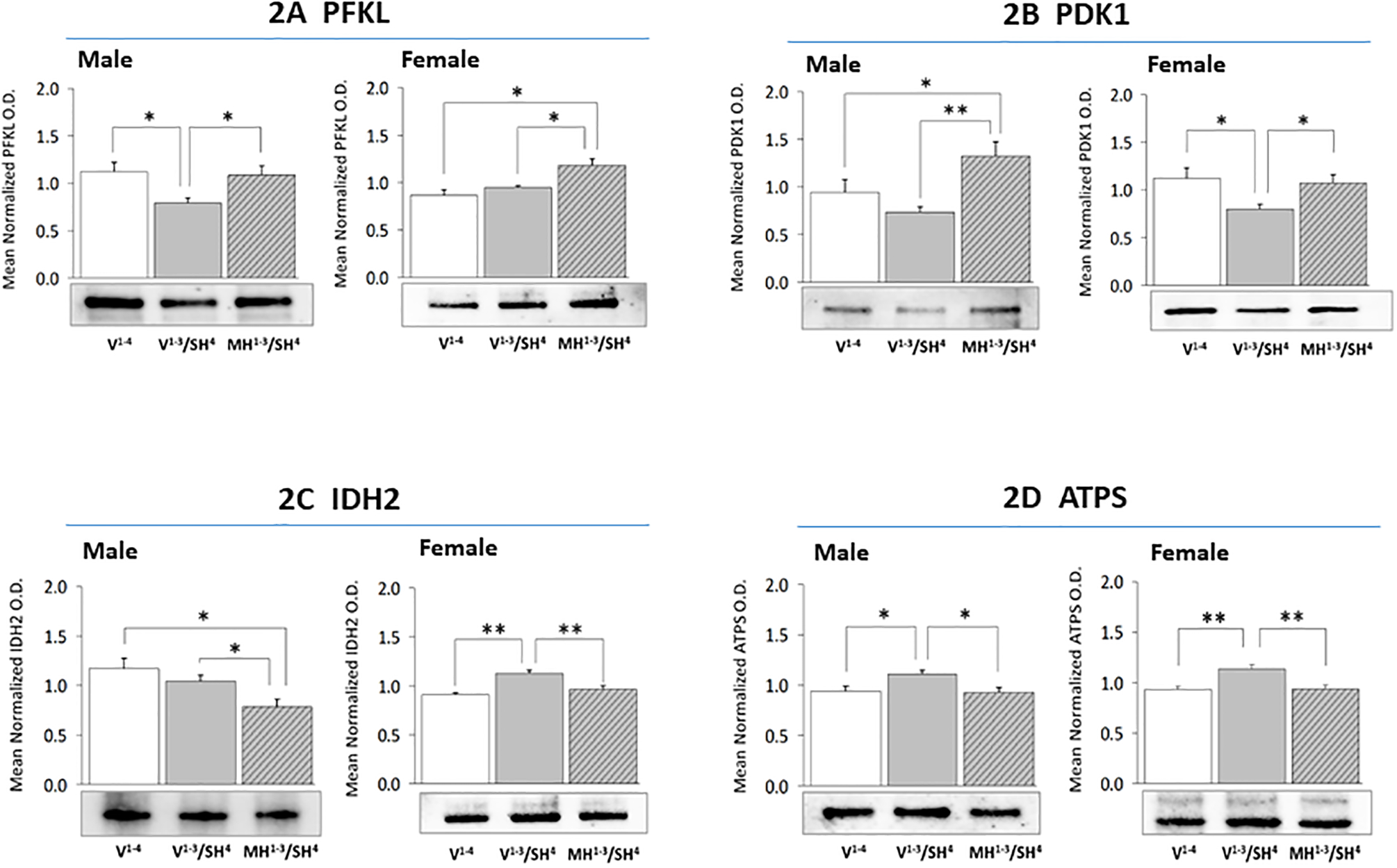
For each treatment group, triplicate CA1 lysate pools were created for Western blot analysis of ATP-regulated kinase phosphofructokinase (PKFL; Figure 2A: male data at left, female data at right), pyruvate dehydrogenase (acetyl-transferring)] kinase isozyme 1 (PDK1; Figure 2B), isocitrate dehydrogenase (IDH2; Figure 2C), or ATP synthase (ATPs; Figure 2D). Data illustrate mean normalized protein O.D. measures ± S.E.M. for male and female V1−4 (solid white bars), V1−3/SH4 (solid gray bars), and MH1−3/SH4 (cross-hatched gray bars) treatment groups. *p<0.05; **p<0.01; ***p<0.001.
Sex-specific impact of precedent MH on SH-associated CA1 nerve cell anti-oxidative stress enzyme protein expression.
Figure 3 presents SOD1 (Figure 3A), SOD2 (Figure 3B), catalase (Figure 3C), and GPx4 (Figure 3D) protein profiles in hypoglycemia-naïve versus hypoglycemia-preconditioned male versus female rats. Data indicate that SH reduces SOD1 protein expression in male (left side) [F(2,6) = 4.45, p = 0.03] and female (right side) [F(2,6) = 8.95, p = 0.004] rats. This decline was averted in females, but not males by prior induction of MH. CA1 SOD2 profiles were enhanced in male [F(2,6) = 15.60, p = 0.0005], but not female [F(2,6) = 45.66, p < 0.0001] rats. Prior exposure to MH elevated SOD2 expression relative to V1−3/SH4 females, but not males. Catalase protein expression was significantly reduced during SH in male [F(2,6) = 7.40, p = 0.03], but elevated in female [F(2, 6) = 33.91, p < 0.0001] CA1 neurons. This profile was augmented in MH1−3/SH4 versus V1−3/SH4 male and female rats. CA1 GPx4 protein was diminished by SH in males [F(2,6) = 5.87, p = 0.02], but not females [F(2,6) = 5.53, p = 0.02]. GPx4 expression was higher in MH1−3/SH4 versus V1−3/SH4 males and females (although not statistically significant in the latter sex).
Figure 3. Effects of MH Preconditioning on Hippocampal CA1 Neuron Anti-Oxidation Enzyme Protein Expression during SH in Male versus Female Rats.
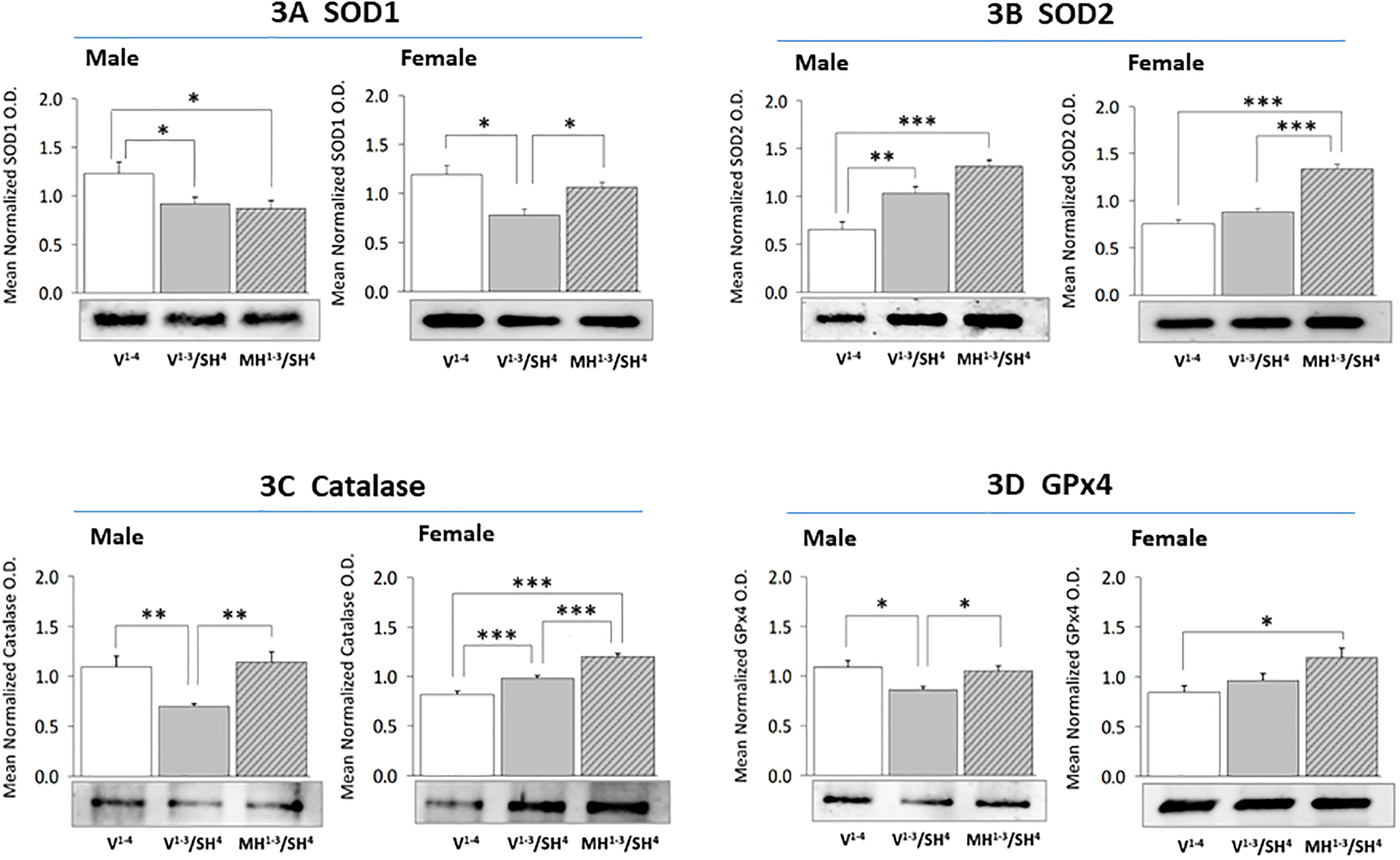
For each treatment group, triplicate CA1 lysate pools were created for Western blot analysis of superoxide dismutase-1 (SOD1; Figure 3A: male data at left, female data at right), superoxide dismutase-2 (SOD2; Figure 3B), catalase (Figure 3C), or glutathione peroxidase-4 (GPx4; Figure 3D). Data illustrate mean normalized protein O.D. measures ± S.E.M. for male and female V1−4 (solid white bars), V1−3/SH4 (solid gray bars), and MH1−3/SH4 (cross-hatched gray bars) treatment groups. *p<0.05; **p<0.01; ***p<0.001.
Effects of SH on CA1, ARH, DG, and CC tissue ATP content with or without MH preconditioning.
Data in Figure 4A show that baseline CA1 tissue ATP levels did not differ between males (left side) versus females (right side). SH did not modify CA1 tissue ATP content in male rats, but elevated levels in females [F(5,12) = 40.35, p = 0.006]. CA1 ATP levels were lower in MH1−3/SH4 versus V1−3/SH4 males (not statistically significant) and female (statistically significant) rats; in the latter sex, CA1 ATP levels were equivalent between V1−4 and MH1−3/SH4 treatment groups. As shown in Figure 4B, ARH ATP levels were significantly decreased by SH in each sex [F(5,12) = 16.06, p = 0.002]; prior exposure to MH reversed this inhibitory SH in females. SH significantly reduced male and female DG ATP content (Figure 4C) [F(5,12) = 20.65, p = 0.001]; MH preconditioning suppressed this response (albeit not significantly) to SH in male, but not female rats. Data in Figure 4D indicate that SH decreased CC ATP content in each sex [ [F(5,12) = 40.35, p = 0.006]; the magnitude of this response was unaffected by MH preconditioning in males and females. Notably, outcomes disclose sex differences in baseline (ARH, CC) and hypoglycemic (CA1, ARH) patterns of tissue ATP accumulation.
Figure 4. Effects of MH Preconditioning on Hippocampal CA1 (Figure 4A), Hypothalamic Arcuate Nucleus (ARH; Figure 4B), Dentate Gyrus (DG; Figure 4C), and Cerebral Cortex-lamina II (CC; Figure 4D) Tissue ATP Content in SH Male versus SH Female Rats.
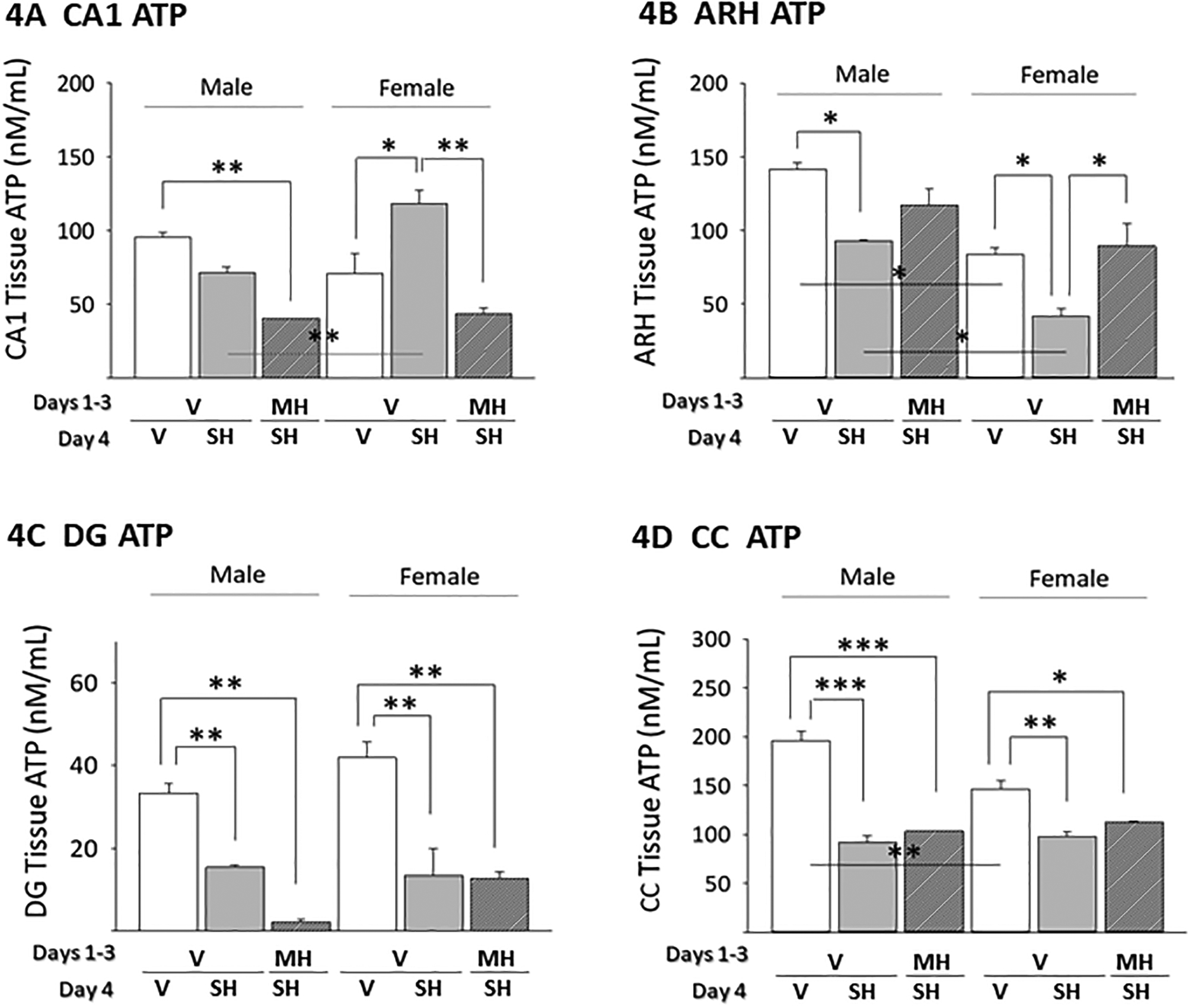
For each animal, individual neural structures were micropunch-dissected from 100 μm-thick frozen sections cut at regular intervals over their respective lengths. For each structure, triplicate tissue lysate pools were created for each treatment group for measurement of ATP content. Data depict mean tissue ATP levels ± S.E.M. for male and female V1−4 (solid white bars), V1−3/SH4 (solid gray bars), and MH1−3/SH4 (cross-hatched gray bars) treatment groups. *p<0.05; **p<0.01; ***p<0.001.
Effects of MH preconditioning on SH patterns of CA1 neuron energy sensor activity and lipid/protein oxidation marker expression.
Figure 5 depicts AMPK (Figure 5A), pAMPK (Figure 5B), 4-HNE (Figure 5D), and NT (Figure 5E) protein expression in male versus female rat CA1 neurons after induction of SH with or without precedent MH. As shown in Figure 5A, SH elicited opposite adjustments in AMPK levels in male (down-regulated) [F(2,6) = 5.298; p = 0.0473] and female (up-regulated) [F (2,6) = 5.198; p =0.0237] CA1 cells. Prior MH normalized AMPK expression in SH-exposed animals of each sex. Data in Figure 5B reveal that SH elevated or suppressed CA1 pAMPK profiles in male [F(2,6) = 14.72; p = 0.0015] versus female [F(2,6) =49.72; p =0.0002] rats, respectively. Precedent MH did not modify this stimulatory response in males, but normalized pAMPK expression in females exposed to SH. Figure 5C depicts effects of hypoglycemia preconditioning on the mean ratio of pAMPK/AMPK protein expression in CA1 neurons. This ratio was respectively up- or down-regulated by SH in males [F(2,6) = 23.50; p = 0.0003] versus females [F(2,6) = 9.58; p =0.003]. Hypoglycemia preconditioning did not affect the mean pAMPK/AMPK value in males, but normalized this ratio in females. CA1 HNE protein expression was increased or refractory to SH in male [F(2,6) = 23.41; p <0.0001] versus female [F(2,6) = 5.579; p =0.0266] rats; this profile was similar in MH1−3/SH4 versus V1−3/SH4 males, but was elevated in MH1−3/SH4 versus V1−3/SH4 females (Figure 5D). Data in Figure 5E show that SH augmented NT protein expression in male [F(2,6) =12.12; p = 0.0007] and female [F(2,6) = 13.85; p = 0.0008] CA1 neurons. This stimulatory response was further augmented in MH1−3/SH4 males, but equivalent between MH1−3/SH4 versus V1−3/SH4 female rats.
Figure 5. Effects of MH Preconditioning on Hippocampal CA1 Neuron 5’-AMP-Activated Protein Kinase (AMPK), Phospho-AMPK (pAMPK), 4-Hydroxynonenal (4-HNE), and Nitrotyrosine (NT) Protein Expression in SH Male versus SH Female Rats.
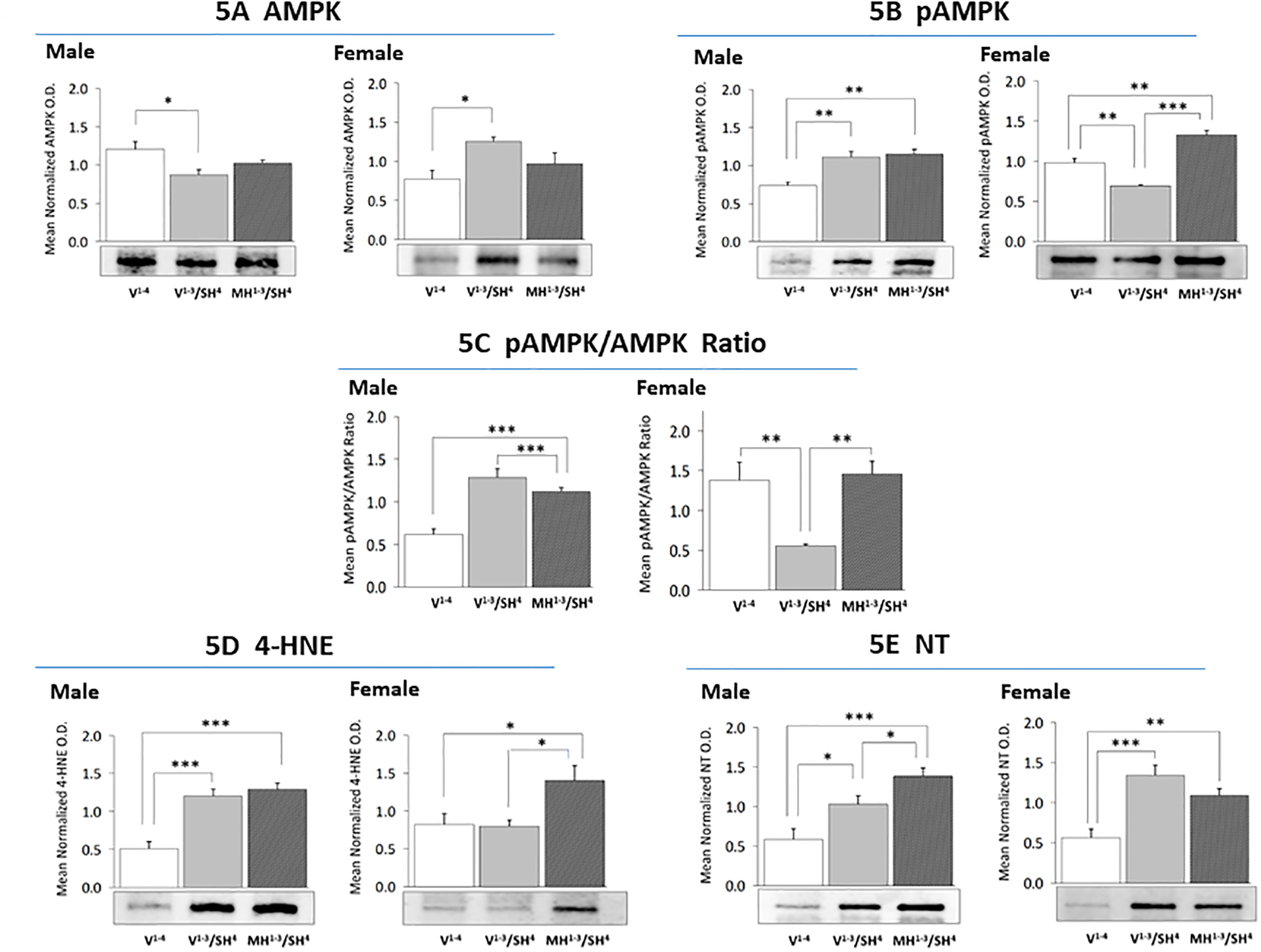
For each treatment group, triplicate CA1 lysate pools were created for Western blot analysis of AMPK (Figure 5A: male data at left, female data at right), pAMPK (Figure 5B), 4-HNE (Figure 5D), or NT (Figure 5E). Figure 5C presents mean pAMPK/AMPK ratios in male versus female CA1 neurons. Data illustrate mean normalized protein O.D. measures ± S.E.M. for male and female V1−4 (solid white bars), V1−3/SH4 (solid gray bars), and MH1−3/SH4 (cross-hatched gray bars) treatment groups. *p<0.05; **p<0.01; ***p<0.001.
Discussion:
Current research examined the hypothesis that MH preconditioning may impact CA1 nerve cell mitochondrial respiratory efficiency during SH by sex-specific regulation of substrate fuel uptake, metabolic pathway function, and/or anti-oxidant enzyme expression. A correlated assumption was that preconditioned responses may correlate with a positive shift in neuronal energy state and diminished oxidative stress in one or both sexes. Data show that male and female CA1 neurons exhibit respective up-regulation of glucose or monocarboxylate transporter proteins during SH, but that precedent MH augmented GLUT3, while causing suppression of MCT2 profiles in each sex. MH preconditioning did not alter augmenting effects of SH on CA1 AMPK activity in males, and significantly increased activity of this sensor beyond levels measured in SH females. In females, precedent MH caused mal-adaptive loss of CA1 neuron gain in positive energy balance during SH. It is unclear if decrements in CA1 lactate uptake contribute to diminished mitochondrial energy pathway activity and tissue ATP in MH1−3/SH4 versus SH groups. Preconditioned augmentation of anti-oxidative enzyme protein expression coincided with elevated SH-induced lipid/protein oxidation damage, implying that MH may intensify oxygen radical production and/or diminish net enzyme activity. MH caused sex-dimorphic effects of SH on ATP levels in CA1, but not DG and CC areas. Unlike hypoglycemia-vulnerable structures examined here, the ARH exhibited augmented ATP content after MH1−3/SH4 versus SH treatment. It remains to be determined if SH causes sex-dimorphic CA1 nerve cell loss, and how MH may impact such loss. Additional effort is also required to determine if molecular mechanisms that sustain female CA1 neuron metabolic stability in the absence of MH preconditioning, including estradiol signaling, may be potential therapeutic targets for alleviation of hypoglycemic neuron injury and destruction. Given the consensus opinion that estradiol benefits mitochondrial energy metabolism, it would be informative to learn if neuroprotection against SH alone involves estradiol-dependent actions and how precedent MH may influence CA1 nerve cell receptivity to this hormone.
Present outcomes demonstrate differential energy substrate, e.g. glucose versus lactate preferences of male versus female rat CA1 neurons during SH, as GLUT3 and −4 proteins were up-regulated in males whereas MCT2 profiles were increased in females. MH preconditioning did not further augment CA1 GLUT3 expression in SH males, but elevated GLUT3 and −4 profiles in SH females, while suppressing MCT2 protein levels below control values in both sexes. MH thus likely amplifies CA1 cell reliance on glucose during SH, signifying a shift from lactate to glucose utilization in the female. It is not known if preconditioned reductions in lactate transporter expression during SH reflect, in part, decreased astrocyte lactate provision in one or both sexes. Sex-specific effects of SH on glycolytic and tricarboxylic acid cycle markers correlated with dimorphic adjustments in CA1 tissue ATP (unaltered in males; elevated in females) and CA1 pAMPK protein expression (increased in males; decreased in females). While our working assumption was that precedent MH would improve CA1 mitochondrial energy metabolism in SH rats to a variable extent between the sexes, outcomes instead documented preconditioned-diminution of mitochondrial energy pathway protein markers (IDH2, ATPs) and tissue ATP (statistically non-significant in males) in V1−3/SH versus MH1−3/SH rats of each sex, coincident with elevated CA1 AMPK activation relative to V1−4 and MH1−3/SH groups in males and MH1−3/SH animals in females. Impaired tricarboxylic acid cycle and oxidative respiratory function after MH1−3/SH4 versus SH treatment, despite intensified glucose uptake in the former condition, may be a consequence of preconditioned-stimulation expression of PDK1, a kinase that inactivates pyruvate dehydrogenase. There remains a need to elucidate the molecular mechanisms that sustain female CA1 neuron metabolic stability in the absence of MH preconditioning, as such insight could reveal potential therapeutic targets for alleviation of hypoglycemic neuron injury and destruction. It is interesting to note that while preconditioning decreases ATP in the female rat CA1 cell area during SH, ATP levels were not different from non-hypoglycemic controls; thus, it is possible that augmented pAMPK expression in those cells may reflect, to some extent, enhanced sensitivity to endocrine and neurotransmitter signals that up-regulate sensor activity independent of cellular energy status.
Current results provide novel evidence for differential SH regulation of tissue ATP levels in hypoglycemia-vulnerable structures in the female rat brain. For example, DG and CC ATP content was significantly decreased, in contrast to the CA1 area, in SH female rats. Moreover, MH preconditioning did not modify SH-associated diminution of ATP in those two locations. On the other hand, male MS1−3/SH4 rats exhibited greater, albeit not statistically significant reductions in CA1 and DG, but not CC ATP compared to SH alone; thus potential SH damage to the former two loci may likely be exacerbated by MH preconditioning in males. The ARH is not associated with hypoglycemic nerve cell injury or death. Here, ARH tissue ATP content was diminished by SH in both sexes; yet, precedent MH normalized ARH ATP levels in males and females exposed to SH. ARH mechanisms that mediate preconditioned- attenuation of SH depletion of local ATP remains to be elucidated. Measurable decrements in tissue ATP content are likely indicative of negative shift in supply versus demand ratio, but do not shed light on how SH may influence the magnitude of local ATP production and use, or affect cellular energy partitioning.
Male and female SH rats exhibited dissimilar patterns of CA1 neuron anti-oxidative enzyme protein expression, excepting SOD1 profiles which declined in both sexes. For example, CA1 SOD2 protein levels were elevated in SH males, but unaltered in SH females; SH respectively decreased or increased catalase content in male and female CA1 cells; and CA1 GPx4 protein profiles was diminished in SH male, but unchanged in SH female rats. At the same time, SH elevated 4-HNE protein expression in male, but not female rat CA1 neurons, but increased CA1 NT profiles in both sexes. These results infer that during SH, cellular anti-oxidation defenses are incapable of preventing lipid or protein oxidation in male CA1 neurons, whereas lipid, but not protein damage is averted in the female. MH preconditioning reversed SH inhibition of SOD1 expression in females, and in each sex up-regulated SOD2 (statistically non-significant in males), catalase, and GPx4 (statistically non-significant in females) relative to SH. Nevertheless, MH1−3/SH4 animals exhibited equivalent (males) or higher (females) 4-HNE levels compared to SH, and greater (males) or comparable (females) NT levels compared to SH rats. This evidence that CA1 oxidative damage occurs despite up-regulation of anti-oxidation enzyme protein profiles infers that such defenses may be inadequate due to preconditioned impairment of enzyme activity and/or intensification of radical oxygen species production in these cells. At present, analytical methods of sufficient sensitivity for measurement of enzyme activity in small-volume pure nerve cell samples, such as those obtained here, are not currently available; thus, speculation that MH preconditioning may exert divergent control of anti-oxidation enzyme protein expression versus activity lacks definitive proof. Further studies are needed to determine effects of SH with or without precedent MH on cellular levels of non-protein anti-oxidants, including glutathione, α-tocopherol, and ascorbic acid. Outcomes here bolster the need to investigate whether CA1 nerve cell demise owing to SH varies in magnitude between the sexes, and if MH preconditioning exerts similar or divergent effects on rates of neuron loss in the male versus female.
In summary, current research advances awareness of sex dimorphic brain responsiveness to bio-energetic insults with documentation of discriminating SH effects, with or without MH preconditioning, on hippocampal neuron energy pathway and anti-oxidant function in each sex. Laser-catapult microdissection was efficacious for procurement of pure CA1 neuron samples for Western blot analysis of energy substrate transporter, glycolytic/tricarboxylic pathway enzyme, and lipid and protein oxidation markers in this discrete hypoglycemia-vulnerable cell population. Sex-differentiating glycolytic and tricarboxylic pathway protein responses to SH correlated with elevated CA1 tissue ATP content and diminished CA1 AMPK activation in females. Yet, preconditioning abolished this acute neuroprotection in females, resulting in augmented energy sensor activity and oxidative injury and diminished CA1 tissue ATP in hypoglycemia-conditioned versus -naïve rats in females (statistically significant) and males (statistically non-significant). Expanded research is necessary to determine if SH elicits differential rates of CA1 destruction in the two sexes, or how MH may impact sex-specific cell loss.
Funding:
This research was supported by grants from the National Institutes of Health (DK 109382) and the BRF organization, 2031 Kings Highway, Shreveport, LA 71103
Funding Source: National Institutes of Health grant DK 109382 and BRF (Shreveport, LA)
Footnotes
Declaration of Conflicting Interests: The authors declare no potential conflicts of interest with respect to this research and/or publication of this article.
Research Involving Animals: We certify that the animal experiments reported were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80–23), revised 1996. We also certify that formal approval to conduct this research was obtained from the University of Louisiana at Monroe College of Pharmacy Institutional Animal Care and Use Committee, and that such approval can be provided upon request. We further attest that all efforts were made to minimize the numbers of animals used and their suffering.
References:
- 1.Smith D, Amiel SA (2002) Hypoglycaemia unawareness and the brain. Diabetologia 45:949–958. [DOI] [PubMed] [Google Scholar]
- 2.Cryer PE (2015) Hypoglycemia-Associated Autonomic Failure in Diabetes: Maladaptive, Adaptive, or Both? Diabetes 64:2322–2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Auer RN, Wieloch T, Olsson Y, Seisjö BK (1984) The distribution of hypoglycemic brain damage. Acta Neuropathol 64: 177–191. [DOI] [PubMed] [Google Scholar]
- 4.Puente EC, Silverstein J, Bree AJ, Musikantow DR, Wozniak DF, Maloney S, Daphna-Iken D, Fisher SJ. (2010) Recurrent moderate hypoglycemia ameliorates brain damage and cognitive dysfunction induced by severe hypoglycemia. Diabetes 59:1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Languren G, Montiel T, Ramírez-Lugo L, Balderas I, Sánchez-Chávez G, Sotres-Bayón F, Bermúdez-Rattoni F, Massieu L (2019) Recurrent moderate hypoglycemia exacerbates oxidative damage and neuronal death leading to cognitive dysfunction after the hypoglycemic coma. J Cereb Blood Flow Metab 39:808–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kostanyan A, Nazaryan K (1992) Rat brain glycolysis regulation by estradiol-17 beta. Biochim Biophys Acta 1133:301–316. [DOI] [PubMed] [Google Scholar]
- 7.Nilsen J, Irwin RW, Gallaher TK, Brinton RD (2007) Estadiol in vivo regulation of brain mitochondrial proteome. J Neurosci 27:14069–14077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Irwin RW, Yao J, Hamilton RT, Cadenas E, Brinton RD, Nilsen J (2008) Progesterone and estrogen regulate oxidative metabolism in brain mitochondria. Endocrinology 149:3167–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen JQ, Brown TR, Russo J (2009) Regulation of energy metabolism pathways by estrogens and estrogenic chemicals and potential implications in obesity associated with increased exposure to endocrine disruptors. Biochim Biophys Acta 1793:1128–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Suh SW, Hamby AM, Gum ET, Shin BS, Won SJ, Sheline CT, Chan PH, Swanson RA (2008) Sequential release of nitric oxide, zinc, and superoxide in hypoglycemic neuronal death. J Cereb Blood Flow Metab 28:1697–1706. [DOI] [PubMed] [Google Scholar]
- 11.Haces ML, Montiel T, Massieu L (2010) Selective vulnerability of brain regions to oxidative stress in a non-coma model of insulin-induced hypoglycemia. Neuroscience 165:28–38. [DOI] [PubMed] [Google Scholar]
- 12.Briski KP, Marshall ES, Sylvester PW (2001) Effects of estradiol on glucoprivic transactivation of catecholaminergic neurons in the female rat caudal brainstem. Neuroendocrinology 73: 369–377. [DOI] [PubMed] [Google Scholar]
- 13.Butcher RL, Collins WE, Fugo NW (1974) Plasma concentration of LH, FSH, prolactin, progesterone and estradiol-17beta throughout the 4-day estrous cycle of the rat. Endocrinology 94:1704–1708. [DOI] [PubMed] [Google Scholar]
- 14.Alhamami HN, Uddin MM, Mahmood ASMH, Briski KP (2018) Lateral but not medial hypothalamic AMPK activation occurs at the hypoglycemic nadir in insulin-injected male rats: Impact of caudal dorsomedial hindbrain catecholamine signaling. Neuroscience 379:103–114. [DOI] [PubMed] [Google Scholar]
- 15.Auer RN (2004) Hypoglycemia brain damage. Forensic Sci Int 146: 105–110. [DOI] [PubMed] [Google Scholar]
- 16.Shakya M, Shrestha PK, Briski KP (2018) Hindbrain 5’-monophosphate-activated protein kinase mediates short-term food deprivation inhibition of the gonadotropin-releasing hormone-luteinizing hormone axis: role of nitric oxide. Neuroscience 383:46–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. https://www.bio-rad.com/en-us/applications-technologies/stain-free-imaging-technology ?ID=NZ0G1815.