

Abstract
Aims
Dendritic cells (DCs) are central mediators of adaptive immunity, and there is growing evidence of their role in myocardial inflammatory disease. We hypothesized that plasmacytoid and myeloid DCs are involved in the mechanisms of myocarditis and analysed these two main subtypes in human myocarditis subjects, as well as in a murine model of experimental autoimmune myocarditis (EAM).
Methods and results
Circulating DCs were analysed by flow cytometry in patients with acute myocarditis, dilated cardiomyopathy, and controls. Myocardial biopsies were immunostained for the presence of DCs and compared with non‐diseased controls. In a mouse model of acute myocarditis induced through synthetic cardiac myosine peptide injection, effects of immunomodulation including DC inhibition through MCS‐18 versus placebo treatment were tested at the peak of inflammation (Day 21), as well as 1 week later (partial recovery). Circulatory pDCs and mDCs were significantly reduced in myocarditis patients compared with controls (P < 0.01 for both) and remained so even after 6 months of follow‐up. Human myocarditis biopsies showed accumulation of pDCs (two‐fold CD304+/three‐fold CD123+, all P < 0.05) compared with controls. Myocardial pDCs and mDCs accumulated in EAM (P for both <0.0001). MCS‐18 treatment reduced pDC levels (P = 0.009), reduced myocardial inflammation (myocarditis score reduction from 2.6 to 1.8, P = 0.026), and improved ejection fraction (P = 0.03) in EAM at Day 21 (peak of inflammation). This effect was not observed during the partial recovery of inflammation on Day 28.
Conclusions
Circulating DCs are reduced in human myocarditis and accumulate in the inflamed myocardium. MCS‐18 treatment reduces DCs in EAM, leading to amelioration of inflammation and left ventricular remodelling during the acute phase of myocarditis. Our data further elucidate the role of DCs and their specific subsets in acute inflammatory cardiomyopathies.
Keywords: Myocarditis, Dilated cardiomyopathy, Dendritic cell, Heart failure, Experimental autoimmune myocarditis
Introduction
Myocarditis, a common condition generally attributed to cardiotropic viruses, is an inflammation of the myocardium and a major cause of non‐ischaemic heart failure (HF), as it often results in dilated cardiomyopathy (DCM). 1 , 2 Despite progress in medical and device therapy, there is still a major unmet need for curative therapies in myocarditis and DCM due to incomplete understanding of underlying pathomechanisms in these conditions. Special attention has been directed at inflammation and autoimmunity, as inflammation in myocardial biopsies has been identified as prognostic marker. 3 , 4 , 5 , 6 Inflammatory mediators such as tumour necrosis factor and interleukin‐6 have been shown to have a prognostic impact on the progression of HF. 7 Identifying key inflammatory players in HF might elucidate the pathology of disease and potentially lead to novel therapeutic strategies.
Dendritic cells (DCs) are antigen presenting cells that play a central role in adaptive and innate immunity. 8 These key inflammatory cells are subject to intensive research in different organ systems, as understanding of their function has led, among others, to novel cancer therapies. 9 , 10 While their role in HF is not sufficiently understood, there is growing evidence of their role in myocarditis and DCM. 11 , 12 , 13 For instance, it is possible to induce myocarditis and DCM in mice by activating autoantibodies through immunization with cardiac myosine unleashing a DC‐dependent T‐cell reaction. 14 , 15 , 16 , 17 Also, mice can be made resistant to experimental autoimmune myocarditis (EAM), if manipulated DCs are inable to activate T lymphocytes. 12 Our study group also found that circulating DCs decrease in the context of an inflammatory response during an acute myocardial infarction, while they accumulate into the myocardia of patients with acute myocardial infarction. 18
Despite the above evidence (mainly in animal models), the role of DCs in human myocarditis and cardiomyopathies needs further clarification and deeper understanding. In several experimental animal studies of ischaemic and non‐ischaemic HF, DCs were either shown to enhance disease progression, or in the contrary, to mediate protection from disease. 18 , 19 , 20 , 21 , 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 We previously showed DCs to be significantly reduced in the myocardium of patients with DCM. 32 Our research on circulating blood DCs also found the myeloid subtype (mDCs) to be significantly reduced in patients with chronic HF due to DCM or ischaemic cardiomyopathy (ICM). 33 In both studies, the reduction of these cells corresponded to the severity of cardiomyopathies.
In the present study, we focused on the role of DCs in myocarditis. First, circulating DCs of both the myeloid and the plasmacytoid subtype (mDCs and pDCs) were measured in patients with acute myocarditis and compared with control subjects at the time of diagnosis, as well as after a follow‐up period. The same analysis was also performed in patients with newly diagnosed DCM, in order to possibly reveal differencies between DCM and myocarditis. We then analysed myocardial DCs in biopsies of patients with acute myocarditis and compared them with non‐diseased myocardium. Furthermore, we assessed myocardial DCs in a murine model of EAM and analysed the effect of immune modulation through MCS‐18. This substance, which is extracted from the plant helleborus purpurascens, has immunomodulatory properties and has been previously used for preventing experimental autoimmune encephalitis, 34 as well as autoimmune diabetes 35 in animal models. Although not a specific DC inhibitor, it has been shown that MCS‐18 inhibits the production of functionally important DC surface proteins, as well as DC‐mediated T‐cell production. 36
Methods
Patient cohorts
This study was designed in a multi‐step fashion using two different patient cohorts, as well as a murine model of EAM, thus analysing DCs in the peripheral blood of patients with myocarditis and DCM (circulating DCs in human myocarditis), in myocardial biopsies of patients with myocarditis (myocardial DCs in human myocarditis), as well as in the myocardium of EAM mice (DCs in EAM and inhibition through MCS‐18).
Circulating dendritic cells in human myocarditis
Patients with acute myocarditis as well as newly diagnosed DCM were prospectively recruited between April 2010 and April 2013 shortly after admission at the University Hospital Jena, Germany. Myocarditis was clinically suspected and diagnosed according to usual clinical criteria such as typical anginal symptoms, elevated cardiac enzymes, unspecific ST segment elevation on electrocardiogram (EKG), then confirmed in cardiac magnetic resonance tomography via positive late gadolinium enhancement. DCM was first diagnosed clinically via echocardiography then confirmed on histopathological and immunohisochemical examination of endomyocardial biopsies, in accordance to the World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. 37 All patients with DCM underwent coronary angiography in order to exclude coronary artery disease (CAD, defined as ≥50% stenosis in at least one coronary artery). The control group of this cohort consisted of individuals with excluded structural heart disease who underwent a cardiac assessment including coronary angiography (CAD excluded for all these controls), mainly because of suspected angina pectoris and CAD. Excluded were all patients with systemic or malignant disease, active infection, immunosuppressive therapy, advanced kidney failure (glomerular filtration rate < 30 mL/min) or with other reasons for HF such as valvular, hypertensive, tachycardia related, or other forms of cardiomyopathy.
Fluorescence‐activated cell sorting (FACS) analyses were undertaken at the time of recruitment (baseline), as well in a 6 month follow‐up visit at our outpatient clinic. Patients with DCM were also analysed in a second 12‐month follow‐up. For each measurement, clinical assessment included an evaluation of symptoms and standard echocardiography. In each echocardiogram, left ventricular (LV) end‐diastolic dimension were measured by M‐mode in the parasternal long‐axis view using the leading‐ edge method, whereas ejection fraction (EF) was measured using the planimetric (Simpson) method. All patients and controls gave written informed consent, and the study was approved by the local Ethics Committee and performed in accordance with the Declaration of Helsinki.
Blood samples were collected in EDTA tubes and immediately cooled (4–10°C). Samples were analysed by flow cytometry for circulating DC precursors within 8 h after collection. The same blood samples were used for routine analyses, in particular leukocyte count. Routine blood analyses were performed by standardized techniques in the central laboratories of the University Hospital of Jena. Using the Blood Dendritic Cell Enumeration kit™ (BDCA kit; MiltenyiBiotec), circulating mDCs and pDCs were analysed by four‐colour staining and FACS analyses in fresh blood samples collected in tubes containing EDTA. Circulating mDCs and pDCs were identified according to their expression of BDCA‐1 and BDCA‐2, respectively, and the absence of the expression of other peripheral blood mononuclear cell markers. Thus, the cells were classified according to CD303, CD1c, CD14, CD19, and CD141. For this purpose, 300 μL of blood were mixed with 20 μL of the control cocktail for isotype control and 300 μL of blood were mixed with 20 μL of the anti‐BDCA cocktail for cell staining. In order to discriminate dead cells, 10 μL of a fluorescent cell‐impermanent dye (which binds to nucleic acids of dead cells) were added, and samples were incubated for 10 min under 60 W light bulb. After cell staining, erythrocytes were lysed using the red blood cell lysis solution from the Blood Dendritic Cell Enumeration kit™ (BDCA kit; MiltenyiBiotec). Cells were washed and fixed using fix solution. Another solution was added to the samples for optimal dead cell discrimination even after prolonged storage (MiltenyiBiotec). Finally, cells were analysed using FACSCalibur flow cytometer with CellQuest software (Becton Dickinson). As circulating DCs comprise only 0.1–1% of white blood cells (WBC), a special gating strategy (Figure 1 E ) to exactly analyse the number of mDCs and pDCs was used. In Region R1 2*105 WBC were registered defined by forward scatter and side scatter. In Region R2, granulocytes were excluded according to their high side scatter, and lymphocytes, monocytes, and dead cells were excluded according to their CD19, CD14, and propidium iodide staining, respectively. Circulating mDCs and pDCs were detected due to their specific staining for BDCA‐1 and BDCA‐2 in regions R3 and R4 as described previously. 38 The relative cell numbers of circulating DCs were thus determined as percentage of WBC.
Figure 1.
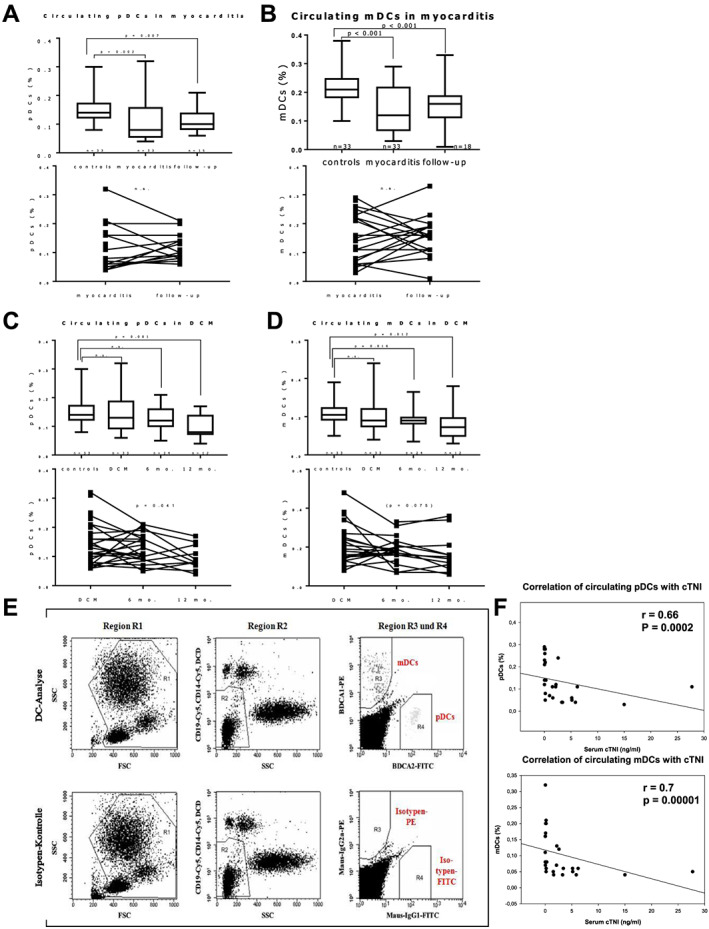
Flow cytometry analysis of circulating plasmacytoid (p) and myeloid (m) dendritic cells (DCs), as percentage of total white blood cells. (A–D) Boxplots showing relative circulating m and pDCs at baseline and follow‐up compared with controls; P‐values indicate significant difference to controls (Mann–Whitney U test); error bars indicate minimal and maximal values. Both DC subsets are reduced in myocarditis at baseline compared with controls (A and B). In patients with dilated cardiomyopathy (DCM), circulating DCs are reduced gradually during the course of the disease, reaching statistical significance after 6 or 12 months (C, D). Individual longitudinal (paired) patient data of myocarditis and DCM at baseline and follow‐up are shown in the respective graphs below. P values in longitudinal DCM data indicate significance (or near significance) in non‐parametric variance analysis (Friedman test). (E) Typical gating strategy for flow cytometry analysis of circulating DCs. (F) Circulating DCs correlate with serum cardiac Troponin I (cTNI) in myocarditis patients (Spearman rank order test).
Myocardial dendritic cells in human myocarditis
Left ventricular endomyocardial biopsies of 18 patients with acute myocarditis were previously obtained for diagnostic purposes and stored in TissueTek medium at −80°C at our tissue bank. Endomyocardial biopsies were performed when at least one of the specific clinical scenarios stated in ESC guidelines were present. 39 Active myocarditis was diagnosed using histological criteria. 40 As controls, myocardial specimens from the left ventricle of 10 accident or suicide victims were embedded in TissueTek (Sakura, Finetek) and crash‐frozen in liquid nitrogen during autopsy and subsequently preserved at −80°C. In all subjects, no past history of cardiac or systemic disease was known. Autopsy showed no macroscopical signs of cardiac structural abnormalities, ischaemia or coronary lesions.
Immunohistochemistry of dendritic cells
Acetone‐fixed cryosections of biopsies and control specimens were stained with antibodies specific for pDCs (CD123 and CD304) and mDCs (fascin and CD209), as well as for T‐cells and macrophages (see Table 2 for a dateiled description of used antibodies) using the catalysed signal amplification technique (CSA System™, DAKO, Hamburg, Germany) as previously described. 23 The sources, applications, and specificities of the antibodies used for immunohistochemical analyses are listed in Table 2. Digital images of immunostained sections were taken with a CCD camera (Zeiss AxioCamHRc, Jena, Germany) at a magnification of 100×. For each analysis, the colour threshold for immunostained cells was manually adjusted in the images until the computerized detection matched the visual interpretation. Stained cells were counted digitally in a defined area of 0.2 mm2, using a digital image processing software (ImageJ 1.43u, Wayne Rasband, NIH, USA). If biopsy sections were smaller than or shaped in a way that they could not fully cover the prespecified area, the cell count was proportionally corrected for tissue coverage.
Table 2.
(A) Antibodies used for immunohistochemical staining of human myocarditis and control biopsies and (B) their respective results in mean values ± standard error of the mean
| (A) | |||||||
| Antibody | Clone | Against | Host | Ig‐type | Dilution | Source | Specificity |
| CD 68 | PG‐M1 | Human | Mouse | IgG3 | I:500 | DAKO | Macrophages |
| CD 69 | FN50 | Human | Mouse | IgG1 | I:50 | Acris | Activated T cells |
| CD 209 (DC‐sign) | DCN46 | Human | Mouse | IgG2b | I:50 | BD Pharmingen | Immature mDCs |
| CD 304 (BDCA‐4/neutropilin‐1) | AD5‐17F6 | Human | Mouse | IgG1 | I:100 | Miltenyi Biotec | pDCs |
| CD 123 | 6H6 | Human | Mouse | IgG1 | I:100 | Serotec | pDCs |
| Fascin | 55 K‐2 | Human | Mouse | IgG1 | I:100 | DAKO | mDCs |
| (B) | |||||||
| Epitome (target cells) | Cells/0.2 mm2 area | P value | |||||
| Myocarditis | Controls | ||||||
| Fascin (mDCs) | 82 ± 11.8 | 94.8 ± 19 | n.s. | ||||
| CD 209 (mDCs) | 30.4 ± 2.8 | 33.7 ± 4.6 | n.s. | ||||
| CD 304 (pDCs) | 76.1 ± 6.8 | 43.2 ± 10.5 | 0.011 | ||||
| CD 123 (pDCs) | 62.2 ± 7.1 | 26.2 ± 5 | 0.001 | ||||
| CD 69 (activated T cells) | 10.1 ± 2.7 | 3.1 ± 0.6 | 0.025 | ||||
| CD 68 (macrophages) | 13.5 ± 2.6 | 6.2 ± 1.9 | 0.036 | ||||
P‐values indicate significant differences (Student's t‐test) between myocarditis and controls. MDCs, myeloid dendritic cells; pDCs, plasmacytoid dendritic cells.
Dendritic cells in experimental autoimmune myocarditis and immune modulation through MCS‐18
Experimental autoimmune myocarditis was induced in 6‐ to 8‐week‐old male Balb/c mice (Janvier Labs, France) on Day 1 through subcutaneous injection of 150 μg synthetic myhc‐α peptide (CASLO, Denmark). A second boost injection was followed on Day 7. For the inhibition of DCs, 100 μg of MCS‐18 were injected intraperitoneally on Days 0, 2, and 4. Control mice were injected with PBS. MCS‐18 was isolated as previously described at the University of Erlangen, Germany. 34
Echocardiography was performed on Day 21 (peak of inflammation) and Day 28 (partial recovery) on anaesthesized mice, which were consequently sacrificed. Animals were anesthetized with isoflurane (5% for induction, 0.5–2% for maintenance). Chests were shaved, and the mice were examined in supine position with a 30 MHz scanhead and a high‐resolution micro‐ultrasound imaging system especially designed for mice (Vevo 770, VisualSonics, Ontario, Canada). All measurements were averaged from three separate recordings. Two‐dimensional short‐axis views of the left ventricle at the papillary muscle level were obtained. Two‐dimensional guided M‐mode tracings were recorded with a sweep speed of 100 mm/s. Ejection fraction was determined according to the Teichholz method.
After sacrifice, hearts were sectioned and fixed with formaldehyde and embedded in paraffin for histological evaluation or embedded in TissueTek and snap frozen in liquid nitrogen for immunohistochemistry of DCs. Haematoxylin and eosin stained heart sections were scored according to a semi‐quantitative scale (0, indicated no inflammatory infiltrates; 1, small foci of inflammatory cells between myocytes; 2, larger foci of >100 inflammatory cells; 3, <10% of a cross‐section involved; 4, >30% of a cross‐section involved), as previously described. 41 , 42 Immunohistochemistry was performed using specific antibodies for pDCs and mDCs (CD304 and CD11c, respectively, see supplement section for information of sources, applications, and specificities) as described above. All animal experiments were approved by the local state authority for animal experimentation.
Statistical analysis
Data were tested for normal distribution. The results are presented either as mean values ± standard error of the mean in the case of continuous data or as median values with their respective 25th and 75th percentiles in the case of non‐continuous data. All comparisons between groups with unpaired data were performed using Student's t‐test when normal distributed, otherwise with the Mann–Whitney U test. For the FACS analysis, besides comparison of controls with respective myocarditis and DCM data, a further analysis of longitudinal paired data within groups was performed using either the Friedman test (analysis of variance) when at least three data sets were available (i.e. for the three DCM measurements at baseline and follow‐ups 1 and 2), or the Wilcoxon signed‐rank test when less than three data sets were available (i.e. for the two myocarditis measurements at baseline and the single follow‐up). Correlation analyses were performed using the Spearman rank order test. For all tests, a P‐value <0.05 was considered statistically significant. Correlation analysis including correlation graphs were performed with Sigma Plot Software Version 13.0 (Systat Software Inc.) All other statistical analysis and graphs were performed using GraphPad Prism Version 7.03 (GraphPad Software Inc., La Jolla, CA, USA).
Results
Circulating dendritic cells are reduced in human myocarditis with distinct kinetic patterns
The demographical and clinical characteristics of the 99 recruited eligible patients with either acute myocarditis (n = 33) or DCM (n = 33), as well as controls (n = 33) are shown in Table 1. Both myocarditis and DCM groups were significantly younger than controls, with patients with myocarditis being the youngest (mean age 38.5 ± 2.6 years) and mostly male (79%). Among the cardiovascular risk factors, hypertension was more predominant in the control group (78%, P = 0.005), while there were more smokers in the myocarditis and DCM groups (P = 0.005 and 0.27, respectively). The main reason for hospital admission of patients with myocarditis was angina pectoris (73%), whereas patients with DCM presented mainly with HF symptoms including severe shortness of breath (77% NYHA III or IV) and peripheral oedema (30%). Accordingly, patients with DCM had severe LV impairment and dilation in echocardiography (EF 28.1 ± 1.5%, LV end‐diastolic diameter 62 ± 1.7 mm). LV dilation was not present in acute myocarditis, but this group showed a moderately reduced ejection fraction (mean EF 49.1 ± 3.4%). Patients with DCM received significantly more HF medication than the other two groups (100% on betablockers and ACE inhibitors/ARB, 79% on diuretics and aldosterone antagonists).
Table 1.
Clinical characteristics of myocarditis, DCM, and control subjects
| Myocarditis1 | DCM2 | Controls3 | P‐value | P‐value | P‐value | |
|---|---|---|---|---|---|---|
| 1 vs. 2 | 2 vs. 3 | 1 vs. 3 | ||||
| n | 33 | 33 | 33 | |||
| Age (years) | 38.5 ± 2.6 | 46.5 ± 2.4 | 61.2 ± 1.9 | 0.024 | <0.001 | <0.001 |
| Sex (male/female) | 26/7 | 25/8 | 12/21 | 0.77 | 0.001 | <0.001 |
| BMI (kg/m2) | 29.2 ± 0.9 | 27.4 ± 0.7 | 26.6 ± 0.7 | 0.13 | 0.42 | 0.027 |
| Cardiovascular risk factors (n (%)) | ||||||
| Hypertension | 14 (42.4) | 14 (42.4) | 25 (75.8) | 1 | 0.005 | 0.005 |
| Diabetes | 1 (3) | 2 (6.1) | 5 (15.2) | 0.56 | 0.237 | 0.089 |
| Smoking | 22 (66.7) | 21 (63.6) | 12 (36.4) | 0.36 | 0.027 | 0.005 |
| Dyslipidaemia | 9 (27.3) | 9 (27.3) | 12 (36.4) | 1 | 0.436 | 0.436 |
| Medication, n (%): | ||||||
| Betablockers | 13 (39.4) | 33 (100) | 19 (57.6) | <0.001 | <0.001 | 0.144 |
| ACE inhibitors/ARB | 13 (39.4) | 33 (100) | 25 (75.8) | <0.001 | 0.002 | 0.003 |
| Diuretics | 11 (33.3) | 26 (78.8) | 17 (51.5) | <0.001 | 0.02 | 0.139 |
| Aldosterone antagonists | 9 (27.3) | 26 (78.8) | 0 | <0.001 | <0.001 | 0.001 |
| Clinical presentation, n (%): | ||||||
| Angina pectoris | 24 (72.7) | 8 (24.2) | 18 (54.5) | <0.001 | 0.011 | 0.129 |
| Dyspnoea on exertion, NYHA Class: | ||||||
| I | 8 (24.2) | 3 (9.1) | 26 (78.8) | 0.102 | <0.001 | <0.001 |
| II | 12 (36.4) | 8 (24.3) | 5 (15.2) | 0.29 | 0.36 | 0.05 |
| III | 9 (27.3) | 14(42.4) | 2 (6.1) | 0.202 | <0.001 | 0.021 |
| IV | 4 (12.1) | 8 (24.3) | 0 | 0.207 | 0.002 | 0.04 |
| Peripheral oedema | 6 (18.2) | 10 (30.3) | 0 | 0.26 | <0.001 | 0.01 |
| Palpitations | 10 (30.3) | 9 (27.3) | 5 (15.2) | 0.78 | 0.14 | 0.146 |
| Echocardiography: | ||||||
| LVEF (%) | 49.1 ± 3.4 | 28.1 ± 1.5 | 67.8 ± 1.7 | <0.001 | <0.001 | <0.001 |
| LVEDd (mm) | 54.4 ± 1.6 | 62 ± 1.7 | 46.1 ± 1.5 | 0.002 | <0.001 | 0.002 |
Data are presented as mean values ± standard error of the mean or number (%) of subjects. BMI, body‐mass index; ACE, angiotensin‐converting enzyme; ARB, angiotensin‐receptor blocker; NYHA, New York Heart Association; LVEF, left ventricular ejection fraction; LVEDD, left ventricular end‐diastolic diameter.
Fluorescence‐activated cell sorting analyses of patients with myocarditis showed significantly reduced relative numbers of both subsets of circulating DCs (% of WBC) compared with controls (for pDCs P = 0.002, mDCs P < 0.001), while there was no difference between WBC counts (7.27 × 109/L ± 0.49 × 109/L vs. 7.17 × 109/L ± 0.26 × 109/L, P = 0.8). After 6 months of follow‐up period, circulating DCs remained significantly lower than controls (P < 0.01 for both DC subsets) (see Figure 1 ). Paired data analysis did not reveal a significant trend between the two measurements at baseline and at 6 months of follow‐up in myocarditis.
We saw a different time kinetic pattern in DCM: while there was no significant difference with controls at baseline, both DC subsets deteriorated continuously during the 12 month follow‐up period. Analysis of variance showed a significant reduction of pDCs from baseline to the 6 and 12 month follow‐up measurements (P = 0.041). Such trend was statistically not significant for mDCs (P = 0.075), but of note, only 12 out of initially 33 patients of the DCM group were FACS‐measured after 12 months. Compared with controls, both DC subsets were significantly lower at 12 months from baseline (both P < 0.01). Such result is consistent with our previous observation of mDCs being severely reduced in patients with chronic DCM and ICM. 33 In all groups, circulating mDCs varied between 0.03% and 0.45% of the WBC of peripheral blood, comparable with the results from previous publications. 38 , 43
Correlation analyses revealed a strong correlation between the reduction of peripheral DCs of both subsets and serum cardiac troponin I (r = 0.66, P = 0.0002 for pDCs and r = 0.7, P = 0.00001 for mDCs) in patients with myocarditis (Figure 1 ). No other significant correlations were found, including the LV function parameters.
Increased expression of myocardial dendritic cells in human myocarditis
Biopsies from patients with myocarditis (n = 18) showed clear cellular infiltration of macrophages and T‐cells (see immunostaining results at Table 2 B ). We saw a significant increase of pDCs with both used antibodies (>2‐fold for CD304 and >3‐fold for CD123, both P < 0.05, see Figure 2 ). Myeloid DCs on the other hand did not show any significant differences compared with controls. The marked increased expression of pDCs is in stark contrast with our previous observation on human DCM biopsies, 32 as we found a significant decrease of both subunits.
Figure 2.
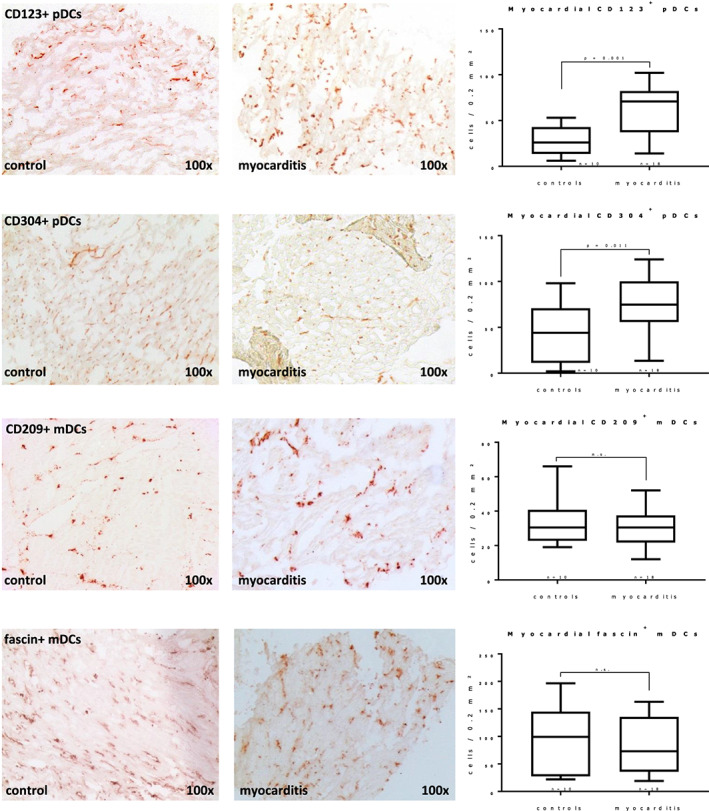
Immunohistochemistry of endomyocardial biopsies shows increased plasmacytoid dendritic cells (pDCs) in human myocarditis, while myeloid DCs (mDCs) do not differ significantly from controls. Significant differences (Student's t‐test) are indicated with the corresponding P values in the box plots; error bars indicate minimal and maximal values. See Table 2 for a full description of used antibodies and staining results.
Myocardial accumulation of dendritic cells in experimental autoimmune myocarditis and the impact of inhibition by MCS‐18
Mice immunized with cardiac myosin peptide (n = 10) showed distinct histological and echocardiographical features of EAM (mean myocarditis score 2.6 ± 0.3, mean EF reduced from 63.3% to 49.9%; both P < 0.001), with massive T‐cell infiltration in HE histology and LV impairment on echocardiography (see Figure 3 ). Immunostaining with antibodies for pDCs and mDCs showed myocardial pDCs to be significantly increased compared with controls (P < 0.0001). MDCs were also significantly increased, but pDCs were much more abundant (mean 104 ± 10 vs. 10 ± 1.6 cells per immunostained area).
Figure 3.
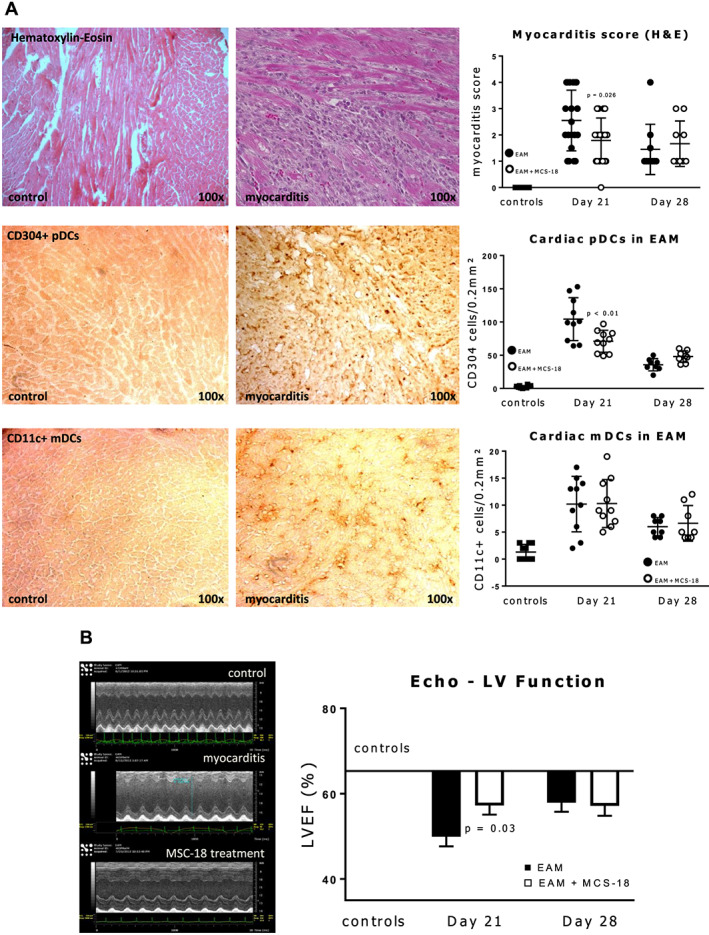
(A) Haematoxylin and eosin (H&E) and immunohistochemistry staining with antibodies for the two subtypes of dendritic cells (DCs) of myocarditis mouse heart sections showing massive T cell infiltration and accumulation of plasmacytoid (p) and myeloid (m) dendritic cells (DCs) in myocarditis compared to controls. Scatter plots on the right show myocarditis scores for H&E sections and numbers of stained DCs at peak of inflammation (Day 21) and at recovery phase (Day 28). T‐cell inflammation and infiltration with pDC (but not mDC) at the acute phase is milder in animals treated with MCS‐18. Scattered plots indicate individual values, as well as mean and standard deviations. P values indicate significant differences (Student's t‐test) between MSC‐18 treated and untreated groups. See Methods section for a full description of the myocarditis score. Used antibodies for pDCs: CD304, Neuropilin‐1; for mDCs: CD11c, Integrin α‐X. (B) Left: typical M‐mode mouse echocardiography showing dilation and reduced ejection fraction of the left ventricle (LVEF) in experimental autoimmune myocarditis (EAM) compared with controls; right: graphic representation of LVEF reduction in EAM mice with and without MCS‐18 treatment. Horizontal bar shows mean LVEF of control mice. MCS‐18 treatment ameliorates EF reduction in peak myocarditis at Day 21 but not at recovery (Day 28). P value indicates significant difference between treated and untreated mice (Student's t‐test).
In animals injected with MCS‐18, myocardial inflammatory infiltration measured by the myocarditis score, as well as LV function on echocardiography were both improved compared with untreated EAM mice at the peak of inflammation on Day 21 (P = 0.026 for myocarditis score, P = 0.03 for LVEF). Interestingly, the expression of pDCs was reduced (compared with uninhibited EAM, P = 0.009) but not of mDCs. Such could be explained by the smaller numbers of mDCs, but it is consistent with the aforementioned findings in human biopsies.
At the partial recovery phase of EAM at Day 28, as both the myocarditis score and LVEF improved, MCS‐18 treated mice showed paradoxically less improvement than untreated animals (see Figure 3 , P not significant). The same was observed for pDC expression but again not for mDCs. Thus, DC inhibition leads to an amelioration of myocarditis but not to better recovery in mice.
Discussion
This study is a systematic investigation of circulating and cardiac DCs in myocarditis, in human subjects and in an animal model of EAM. We designed the study starting from the hypothesis that peripheral DCs would be reduced in patients with myocarditis, as they would infiltrate into the myocardium (Figure 4 ). We could show both major subtypes of circulating DCs to be decreased at the time of diagnosis of myocarditis with persistently reduced levels over an intermediate follow‐up period. This differed from newly diagnosed DCM subjects in whom we found initially normal but progressively decreasing circulating DCs during the course of the disease. Along with this finding, we found DCs of the plasmacytoid type to be increased in the myocardium of patients with myocarditis. Both DC subtypes were overexpressed in the experimental mouse model, but pDCs were predominant. The inhibition of DCs led to an amelioration of myocarditis in terms of myocardial infiltration and LV ejection fraction at the acute phase but not during recovery. Such finding confirms the presumption that DCs play a major role in myocarditis during the acute inflammatory reaction and is in line with earlier histological findings of increased myocardial DCs in human myocarditis. 44 , 45
Figure 4.
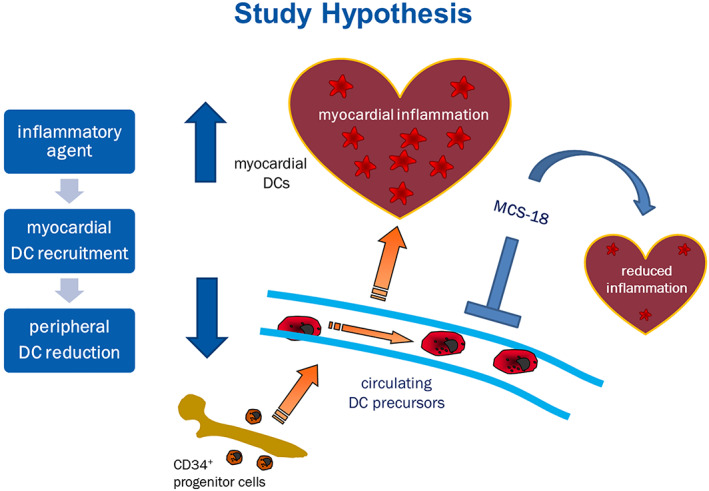
Study hypothesis. DC percursors originate from CD34+ progenitor cells in the bone marrow. Following antigen contact, circulating DC precursors maturate and migrate to the site of inflammation, in this case the heart, unleashing myocardial inflammation through T‐cell activation. MCS‐18 treatment causes immune modulation and DC inhibition, resulting in milder inflammation.
The role of DCs in human myocarditis is also in line with autoimmune processes in inflammatory heart disease. 46 DCs are known key mediators of autoimmunity. As DCs can initiate myocarditis via an autoimmune response in mice, such role in human hearts remains speculative but becomes more plausible with our present findings. DCs on the other hand are not solely linked to autoimmunity, as they are not only known to be activated through viruses but also modulate immune responses. 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31
The notable fact that mDCs remained significantly reduced even after 6 months following the acute myocarditis episode should be further evaluated. Although such finding does not come as a surprise, as previous systematical studies recognized a viral persistence in human myocarditis following the resolution of disease. 2 , 47 It is therefore possible that inflammatory processes persist long after clinical symptoms resolve in human myocarditis.
The involvement of DCs in myocarditis should not necessarily mean a pro‐inflammatory role, but as it has been amply reported in animal experiments, DCs often modulate inflammation, with the net effect of amelioration of myocarditis. 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 However, it will be a fruitfull attempt to analyse in a larger study cohort whether patients who develop DCM after myocarditis have different changes of circulating DCs compared with patients with full recovery. Whether future techniques involving vaccination with modified DCs can alter the course of human disease remains to be seen. 47 , 48
Another novel finding of this study was that in newly diagnosed patients with DCM circulating DCs were not different from controls. Their ‘delayed’ and gradual reduction did not resemble the time kinetic of patients with myocarditis. As circulating DCs in patients with DCM continued to decline in the short‐term and medium‐term follow‐ups, they reached levels known from our previous study on patients with chronic HF (DCM and ICM). 33 This difference to myocarditis is an interesting finding, because it is thought that myocarditis often precedes DCM and the two conditions often clinically overlap. 1 , 2 , 49 , 50
In our experimental mouse model of myocarditis, we could reduce the myocardial presence of DCs using MCS‐18, a compound that has previously been used succesfully to prevent autoimmune encephalitis and diabetes in mice via modulation of inflammation, including inhibition (though not specific) of DC maturation. 34 , 36 To the best of our knowledge, the current study is the first application of this substance in a myocarditis model. Besides the significant reduction of myocardial DCs, we could show that MCS‐18 treatment succesfully reduces the acute inflammation in EAM. Furthermore, its effect on myocardial T‐cell infiltration and LV function confirms the general assumption that EAM is a DC‐dependent disease. 11 , 12 , 13 , 14 , 15 , 16 , 17 It is tempting to speculate on possible effects of DC inhibition of substances such as MCS‐18 beyond the animal EAM model. Yet the end‐effect on myocardial inflammatory disease through DC inhibition or stimulation is far from obvious. We did not see a difference in myocardial DC expression or indeed inflammation and LV function at the partial recovery phase of the EAM. Although statistically not significant, LV impairment, inflammation, and DC expression tended to increase after the peak of myocarditis in MCS‐treated mice. It is also unknown what effect DC stimulation has on the late phase of EAM. In a previous publication, Blyszczuk et al. succesfully stimulated DCs with GM‐CSF in EAM mice but could not change the course of myocarditis or tissue fibrosis. 19
The discrepancy of the results concerning plasmacytoid and myeloid DCs in our study is not surprising. These subsets differ greatly in their immune responses, resulting from different Toll‐like receptor expression profiles and antigen presenting properties. 51 PDCs possibly play a greater role than mDCs in innate immunity as a response to viral infections and are capable of activating T‐cells through presentation of endogenous antigens, rather than extracellular substances like mDCs. 52 , 53 , 54 Such key distinctions of pDCs might make them more capable to be involved in both antiviral and autoimmune responses in human, as well as autoimmune murine myocarditis.
Limitations
For the analysis of human peripheral and myocardial DCs, we used different patient collectives and, for obvious ethical reasons, different control groups (patients and post‐mortem accident victims). The intermediate longitudinal character of the FACS analysis falls short of the impact of a long‐term observational study with survival data. Although our patients were recruited as soon as their diagnosis was suspected, we cannot exclude a previous ‘silent’ disease. There were also age and gender differences between the groups, with patients with myocarditis representing the youngest collective. Presently, there is no consensus regarding peripheral DCs and aging (some authors observed similar numbers between young and old humans, others found a decline of either mDCs or pDCs with advancing age 55 , 56 , 57 ). Yet we are not aware of any reporting of increase of peripheral DCs with age; hence, our results (reduced peripheral DCs in myocarditis and DCM) are unlikely due to age difference between groups. We measured the relative, as opposed to absolute number of DCs. It is therefore possible that other leukocytes were increased, thus artificially reducing the relative DC count. Although we did not notice an increase of blood leukocytes in our patients with myocarditis compared with controls, we are aware of the limitation and possible bias in our measurement method. We also lacked sufficient virus PCR data from our myocarditis biopsies, which could have possibly given our results an important prospective in relation to myocarditis ethiology. The protocols and specificity of the antibodies for DCs in human myocardial tissue were previously established and tested (including double staining) in one of our previous publications. 32 Yet we can not exclude some unspecific staining of endothelial cells, and such must be regarded as a limitation. In this study, we used two different antibodies for each DC subset, yielding similar results.
In the myocarditis mouse model, LV function was only mildly reduced in myocarditis (EF from 63% to 50%). Such is in line with previous observations, 42 while EF was even found to remain constant in another EAM study. 36 Although we found significant changes in EF in our model, other parameters such as fibrosis and haemodynamics should be considered in future studies.
Finally, although MCS‐18 was shown to modulate the immune response in experimental animal models, it is by no means a specific DC inhibitor. We could show a reduction of myocardial DCs, as well as inflammation and myocardial damage in the acute phase of myocarditis. Yet MCS‐18 is also known to inhibit B cell proliferation, as well as cytokine induced immunoglobuline production. 58 The reduction of inflammation and myocardial dysfunction in our model could have therefore resulted in part from other anti‐inflammatory properties of MCS‐18.
In conclusion, we report a reduction in circulating and increase in myocardial DCs in acute human myocarditis, suggesting an association between DC myocardial recuitment and acute myocarditis in humans. The presence of DCs in myocarditis was confirmed in the animal experiment, and we further showed that immune modulation and inhibition of DCs led to myocardial DC reduction and amelioration of the acute phase of myocarditis in mice. We further report differences between myocarditis and DCM in circulating DCs during the course of disease. From the DC subsets, pDCs might play a specific role in myocarditis. Our study contributes to the growing evidence that DCs play a major role in myocarditis, in animal experiment, and in humans.
Conflict of interest
None declared.
Funding
This study was supported by the Else‐Kröner‐Fresenius Foundation (PCS).
Acknowledgements
We thank Prof. Steinkasserer of the University of Erlangen for the kind assistance in providing MCS‐18 for this research. We also thank Mr. Christian Hellriegel, embedded ZEISS consultant at Harvard Center for Biological Imaging, for his kind technical assistance.
Pistulli, R. , Andreas, E. , König, S. , Drobnik, S. , Kretzschmar, D. , Rohm, I. , Lichtenauer, M. , Heidecker, B. , Franz, M. , Mall, G. , Yilmaz, A. , and Schulze, P. C. (2020) Characterization of dendritic cells in human and experimental myocarditis. ESC Heart Failure, 7: 2305–2317. 10.1002/ehf2.12767.
References
- 1. Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB, American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention . Contemporary definitions and classification of the cardiomyopathies: An American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation 2006; 113: 1807–1816. [DOI] [PubMed] [Google Scholar]
- 2. Mahrholdt H, Wagner A, Deluigi CC, Kispert E, Hager S, Meinhardt G, Vogelsberg H, Fritz P, Dippon J, Bock CT, Klingel K, Kandolf R, Sechtem U. Presentation, patterns of myocardial damage, and clinical course of viral myocarditis. Circulation 2006; 114: 1581–1590. [DOI] [PubMed] [Google Scholar]
- 3. Heymans S, Hirsch E, Anker SD, Aukrust P, Balligand JL, Cohen‐Tervaert JW, Drexler H, Filippatos G, Felix SB, Gullestad L, Hilfiker‐Kleiner D, Janssens S, Latini R, Neubauer G, Paulus WJ, Pieske B, Ponikowski P, Schroen B, Schultheiss HP, Tschöpe C, Van Bilsen M, Zannad F, McMurray J, Shah AM. Inflammation as a therapeutic target in heart failure? A scientific statement from the Translational Research Committee of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail 2009; 11: 119–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Caforio AL, Pankuweit S, Arbustini E, Basso C, Gimeno‐Blanes J, Felix SB, Fu M, Helio T, Heymans S, Jahns R, Klingel K, Linhart A, Maisch B, McKenna W, Mogensen J, Pinto YM, Ristic A, Schultheiss HP, Seggewiss H, Tavazzi L, Thiene G, Yilmaz A, Charron P, Elliott PM, European Society of Cardiology Working Group on Myocardial and Pericardial Diseases . Current state of knowledge on aetiology, diagnosis, management, and therapy of myocarditis: a position statement of the European Society of Cardiology – Working Group on Myocardial and Pericardial Diseases. Eur Heart J 2013; 34: 2636–2648. [DOI] [PubMed] [Google Scholar]
- 5. Mason JW. Myocarditis and dilated cardiomyopathy: an inflammatory link. Cardiovasc Res 2003; 60: 5–10. [DOI] [PubMed] [Google Scholar]
- 6. Kindermann I, Kindermann M, Kandolf R, Klingel K, Bültmann B, Müller T, Lindinger A, Böhm M. Predictors of outcome in patients with suspected myocarditis. Circulation 2008; 118: 639–648 Erratum in: Circulation 2008; 118: e493. [DOI] [PubMed] [Google Scholar]
- 7. Deswal A, Petersen NJ, Feldman AM, Young JB, White BG, Mann DL. Cytokines and cytokine receptors in advanced heart failure: an analysis of the cytokine database from the Vesnarinone trial (VEST). Circulation 2001; 103: 2055–2059. [DOI] [PubMed] [Google Scholar]
- 8. Steinman RM. Dendritic cells: understanding immunogenicity. Eur J Immunol 2007; 37: S53–S60. [DOI] [PubMed] [Google Scholar]
- 9. Constantino J, Gomes C, Falcão A, Cruz MT, Neves BM. Antitumor dendritic cell‐based vaccines: lessons from 20 years of clinical trials and future perspectives. Transl Res 2016; 168: 74–95. [DOI] [PubMed] [Google Scholar]
- 10. Anguille S, Smits EL, Lion E, van Tendeloo VF, Berneman ZN. Clinical use of dendritic cells for cancer therapy. Lancet Oncol 2014; 15: e257–e267 Review. [DOI] [PubMed] [Google Scholar]
- 11. Eriksson U, Ricci R, Hunziker L, Kurrer MO, Oudit GY, Watts TH, Sonderegger I, Bachmaier K, Kopf M, Penninger JM. Dendritic cell‐induced autoimmune heart failure requires cooperation between adaptive and innate immunity. Nat Med 2003. Dec; 9: 1484–1490. Erratum in: Nat Med 2004 10(1):105. [DOI] [PubMed] [Google Scholar]
- 12. Marty RR, Dirnhofer S, Mauermann N, Schweikert S, Akira S, Hunziker L, Penninger JM, Eriksson U. MyD88 signaling controls autoimmune myocarditis induction. Circulation 2006; 113: 258–265. [DOI] [PubMed] [Google Scholar]
- 13. Marty RR, Eriksson U. Dendritic cells and autoimmune heart failure. Int J Cardiol 2006; 112: 34–39. [DOI] [PubMed] [Google Scholar]
- 14. Eriksson U, Kurrer MO, Sonderegger I, Iezzi G, Tafuri A, Hunziker L, Suzuki S, Bachmaier K, Bingisser RM, Penninger JM, Kopf M. Activation of dendritic cells through the interleukin 1 receptor 1 is critical for the induction of autoimmune myocarditis. J Exp Med 2003; 197: 323–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Griffin GK, Lichtman AH. Two sides to every proinflammatory coin: new insights into the role of dendritic cells in the regulation of T‐cell driven autoimmune myocarditis. Circulation 2013; 127: 2257–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Blyszczuk P, Kania G, Dieterle T, Marty RR, Valaperti A, Berthonneche C, Pedrazzini T, Berger CT, Dirnhofer S, Matter CM, Penninger JM, Lüscher TF, Eriksson U. Myeloid differentiation factor‐88/interleukin‐1 signaling controls cardiac fibrosis and heart failure progression in inflammatory dilated cardiomyopathy. Circ Res 2009; 105: 912–920. [DOI] [PubMed] [Google Scholar]
- 17. Blyszczuk P, Behnke S, Lüscher TF, Eriksson U, Kania G. GM‐CSF promotes inflammatory dendritic cell formation but does not contribute to disease progression in experimental autoimmune myocarditis. Biochim Biophys Acta 2013; 1833: 934–944. [DOI] [PubMed] [Google Scholar]
- 18. Kretzschmar D, Betge S, Windisch A, Pistulli R, Rohm I, Fritzenwanger M, Jung C, Schubert K, Theis B, Petersen I, Drobnik S, Mall G, Figulla HR, Yilmaz A. Recruitment of circulating dendritic cell precursors into the infarcted myocardium and pro‐inflammatory response in acute myocardial infarction. Clin Sci (Lond) 2012; 123: 387–398. [DOI] [PubMed] [Google Scholar]
- 19. Anzai A, Anzai T, Nagai S, Maekawa Y, Naito K, Kaneko H, Sugano Y, Takahashi T, Abe H, Mochizuki S, Sano M, Yoshikawa T, Okada Y, Koyasu S, Ogawa S, Fukuda K. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 2012; 125: 1234–1245. [DOI] [PubMed] [Google Scholar]
- 20. Maekawa Y, Mizue N, Chan A, Shi Y, Liu Y, Dawood S, Chen M, Dawood F, de Couto G, Li GH, Suzuki N, Yeh WC, Gramolini A, Medin JA, Liu PP. Survival and cardiac remodeling after myocardial infarction are critically dependent on the host innate immune interleukin‐1 receptor‐associated kinase‐4 signaling: a regulator of bone marrow‐derived dendritic cells. Circulation 2009; 120: 1401–1414. [DOI] [PubMed] [Google Scholar]
- 21. Fukui D, Yasukawa H, Sugi Y, Oba T, Nagata T, Kyogoku S, Futamata N, Yokoyama T, Yokoyama S, Kai H, Ueno T, Kage M, Imaizumi T. Transient reduction and activation of circulating dendritic cells in patients with acute myocardial infarction. Int J Cardiol 2012; 160: 216–219. [DOI] [PubMed] [Google Scholar]
- 22. Carvalheiro T, Velada I, Valado A, Mendes F, Martinho A, António N, Gonçalves L, Providência L, Pais ML, Paiva A. Phenotypic and functional alterations on inflammatory peripheral blood cells after acute myocardial infarction. J Cardiovasc Transl Res 2012; 5: 309–320. [DOI] [PubMed] [Google Scholar]
- 23. Machino‐Ohtsuka T, Tajiri K, Kimura T, Sakai S, Sato A, Yoshida T, Hiroe M, Yasutomi Y, Aonuma K, Imanaka‐Yoshida K. Tenascin‐C aggravates autoimmune myocarditis via dendritic cell activation and Th17 cell differentiation. J Am Heart Assoc 2014; 3: e001052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Weinzierl AO, Szalay G, Wolburg H, Sauter M, Rammensee HG, Kandolf R, Stevanović S, Klingel K. Effective chemokine secretion by dendritic cells and expansion of cross‐presenting CD4−/CD8+ dendritic cells define a protective phenotype in the mouse model of coxsackievirus myocarditis. J Virol 2008; 82: 8149–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cai G, Wang H, Qin Q, Zhang J, Zhu Z, Liu M, Shen Q. Amelioration of myocarditis by HVEM‐overexpressing dendritic cells through induction of IL‐10‐producing cells. Cardiovasc Res 2009; 84: 425–433. [DOI] [PubMed] [Google Scholar]
- 26. Yang S, Li W, Liu W, Gao C, Zhou B, Li S, Li Y, Kong Y. IL‐10 gene modified dendritic cells induced antigen‐specific tolerance in experimental autoimmune myocarditis. Clin Immunol 2006; 121: 63–73. [DOI] [PubMed] [Google Scholar]
- 27. Li Y, Heuser JS, Kosanke SD, Hemric M, Cunningham MW. Protection against experimental autoimmune myocarditis is mediated by interleukin‐10‐producing T cells that are controlled by dendritic cells. Am J Pathol 2005; 167: 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee JH, Kim TH, Park HE, Lee EG, Jung NC, Song JY, Seo HG, Seung KB, Chang K, Lim DS. Myosin‐primed tolerogenic dendritic cells ameliorate experimental autoimmune myocarditis. Cardiovasc Res 2014; 101: 203–210. [DOI] [PubMed] [Google Scholar]
- 29. Kania G, Siegert S, Behnke S, Prados‐Rosales R, Casadevall A, Lüscher TF, Luther SA, Kopf M, Eriksson U, Blyszczuk P. Innate signaling promotes formation of regulatory nitric oxide‐producing dendritic cells limiting T‐cell expansion in experimental autoimmune myocarditis. Circulation 2013; 127: 2285–2294. [DOI] [PubMed] [Google Scholar]
- 30. Kania G, Blyszczuk P, Valaperti A, Dieterle T, Leimenstoll B, Dirnhofer S, Zulewski H, Eriksson U. Prominin‐1+/CD133+ bone marrow‐derived heart‐resident cells suppress experimental autoimmune myocarditis. Cardiovasc Res 2008; 80: 236–245. [DOI] [PubMed] [Google Scholar]
- 31. Valaperti A, Nishii M, Germano D, Liu PP, Eriksson U. Vaccination with Flt3L‐induced CD8α+ dendritic cells prevents CD4+ T helper cell‐mediated experimental autoimmune myocarditis. Vaccine 2013; 31: 4802–4811. [DOI] [PubMed] [Google Scholar]
- 32. Pistulli R, König S, Drobnik S, Kretzschmar D, Rohm I, Lichtenauer M, Fritzenwanger M, Mall G, Mall G, Figulla HR, Yilmaz A. Decrease in dendritic cells in endomyocardial biopsies of human dilated cardiomyopathy. Eur J Heart Fail 2013; 15: 974–985. [DOI] [PubMed] [Google Scholar]
- 33. Pistulli R, Hammer N, Rohm I, Kretzschmar D, Jung C, Figulla HR, Yilmaz A. Decrease of circulating myeloid dendritic cells in patients with chronic heart failure. Acta Cardiol 2016; 71: 165–172. [DOI] [PubMed] [Google Scholar]
- 34. Horstmann B, Zinser E, Turza N, Kerek F, Steinkasserer A. MCS‐18, a novel natural product isolated from Helleborus purpurascens, inhibits dendritic cell activation and prevents autoimmunity as shown in vivo using the EAE model. Immunobiology 2007; 212: 839–853. [DOI] [PubMed] [Google Scholar]
- 35. Seifarth C, Littmann L, Resheq Y, Rössner S, Goldwich A, Pangratz N, Kerek F, Steinkasserer A, Zinser E. MCS‐18, a novel natural plant product prevents autoimmune diabetes. Immunol Lett 2011; 139: 58–67. [DOI] [PubMed] [Google Scholar]
- 36. Mirna M, Paar V, Kraus T, Sotlar K, Wernly B, Pistulli R, Hoppe UC, Lichtenauer M. Autoimmune myocarditis is not associated with left ventricular systolic dysfunction. Eur J Clin Invest 2019; 49: e13132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Richardson P, McKenna W, Bristow M, Maisch B, Mautner B, O'Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I, Martin I, Nordet P. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996; 93: 841–842. [DOI] [PubMed] [Google Scholar]
- 38. Yilmaz A, Weber J, Cicha I, Stumpf C, Klein M, Raithel D, Daniel WG, Garlichs CD. Decrease in circulating myeloid dendritic cell precursors in coronary artery disease. J Am Coll Cardiol 2006; 48: 70–80. [DOI] [PubMed] [Google Scholar]
- 39. McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Bohm M, Dickstein K, Falk V, Filippatos G, Fonseca C, Gomez‐Sanchez MA, Jaarsma T, Køber L, Lip GY, Maggioni AP, Parkhomenko A, Pieske BM, Popescu BA, Rønnevik PK, Rutten FH, Schwitter J, Seferovic P, Stepinska J, Trindade PT, Voors AA, Zannad F, Zeiher A, Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology , Bax JJ, Baumgartner H, Ceconi C, Dean V, Deaton C, Fagard R, Funck‐Brentano C, Hasdai D, Hoes A, Kirchhof P, Knuuti J, Kolh P, McDonagh T, Moulin C, Popescu BA, Reiner Z, Sechtem U, Sirnes PA, Tendera M, Torbicki A, Vahanian A, Windecker S, McDonagh T, Sechtem U, Bonet LA, Avraamides P, Ben Lamin HA, Brignole M, Coca A, Cowburn P, Dargie H, Elliott P, Flachskampf FA, Guida GF, Hardman S, Iung B, Merkely B, Mueller C, Nanas JN, Nielsen OW, Orn S, Parissis JT, Ponikowski P, ESC Committee for Practice Guidelines . ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the EuropeanSociety of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail 2012; 14: 803–869. [DOI] [PubMed] [Google Scholar]
- 40. Aretz HT, Billingham ME, Edwards WD, Factor SM, Fallon JT, Fenoglio JJ Jr, Olsen EG, Schoen FJ. Myocarditis: a histopathologic definition and classification. Am J Cardiovasc Pathol 1987; 1: 3–14. [PubMed] [Google Scholar]
- 41. Eriksson U, Kurrer MO, Schmitz N, Marsch SC, Fontana A, Eugster HP, Kopf M. Interleukin‐6‐deficient mice244 resist development of autoimmune myocarditis associated with impaired upregulation of complement C3. Circulation 2003; 107: 320–325. [DOI] [PubMed] [Google Scholar]
- 42. Pistulli R, Quitter F, Andreas E, Rohm I, Kretzschmar D, Figulla HR, Yilmaz A, Jung C. Intravital microscopy—a novel tool in characterizing congestive heart failure in experimental autoimmune myocarditis. Clin Hemorheol Microcirc 2015; 63: 153–162. [DOI] [PubMed] [Google Scholar]
- 43. Kunitani H, Shimizu Y, Murata H, Higuchi K, Watanabe A. Phenotypic analysis of circulating and intrahepatic dendritic cell subsets in patients with chronic liver diseases. J Hepatol 2002; 36: 734–741. [DOI] [PubMed] [Google Scholar]
- 44. Suzuki K. A histological study on experimental autoimmune myocarditis with special reference to initiation of the disease and cardiac dendritic cells. Virchows Arch 1995; 426: 493–500. [DOI] [PubMed] [Google Scholar]
- 45. Yokoyama H, Kuwao S, Kohno K, Suzuki K, Kameya T, Izumi T. Cardiac dendritic cells and acute myocarditis in the human heart. Jpn Circ J 2000; 64: 57–64. [DOI] [PubMed] [Google Scholar]
- 46. Caforio AL, Mahon NG, Baig MK, Tona F, Murphy RT, Elliott PM, McKenna WJ. Prospective familial assessment in dilated cardiomyopathy: cardiac autoantibodies predict disease development in asymptomatic relatives. Circulation 2007; 115: 76–83. [DOI] [PubMed] [Google Scholar]
- 47. Wu F, Fan X, Yue Y, Xiong S, Dong C. A vesicular stomatitis virus‐based mucosal vaccine promotes dendritic cell maturation and elicits preferable immune response against coxsackievirus B3 induced viral myocarditis. Vaccine 2014; 32: 3917–3926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chai D, Yue Y, Xu W, Dong C, Xiong S. Mucosal co‐immunization with AIM2 enhances protective SIgA response and increases prophylactic efficacy of chitosan‐DNA vaccine against coxsackievirus B3‐induced myocarditis. Hum Vaccin Immunother 2014; 10: 1284–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kühl U, Pauschinger M, Noutsias M, Seeberg B, Bock T, Lassner D, Poller W, Kandolf R, Schultheiss HP. High prevalence of viral genomes and multiple viral infections in the myocardium of adults with ‘idiopathic’ left ventricular dysfunction. Circulation 2005; 111: 887–893. [DOI] [PubMed] [Google Scholar]
- 50. Mason JW, O'Connell JB, Herskowitz A, Rose NR, McManus BM, Billingham ME, Moon TE. A clinical trial of immunosuppressive therapy for myocarditis. The Myocarditis Treatment Trial Investigators. N Engl J Med 1995; 333: 269–275. [DOI] [PubMed] [Google Scholar]
- 51. Schreibelt G, Tel J, Sliepen KH, Benitez‐Ribas D, Figdor CG, Adema GJ, de Vries IJ. Toll‐like receptor expression and function in human dendritic cell subsets: implications for dendritic cell‐based anti‐cancer immunotherapy. Cancer Immunol Immunother 2010; 59: 1573–1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cella M, Jarrossay D, Facchetti F, Alebardi O, Nakajima H, Lanzavecchia A, Colonna M. Plasmacytoid monocytes migrate to inflamed lymph nodes and produce large amounts of type I interferon. Nat Med 1999; 5: 919–923. [DOI] [PubMed] [Google Scholar]
- 53. Villadangos JA, Young L. Antigen‐presentation properties of plasmacytoid dendritic cells. Immunity 2008; 29: 352–361. [DOI] [PubMed] [Google Scholar]
- 54. Tel J, Schreibelt G, Sittig SP, Mathan TS, Buschow SI, Cruz LJ, Lambeck AJ, Figdor CG, de Vries IJ. Human plasmacytoid dendritic cells efficiently cross‐present exogenous Ags to CD8+ T cells despite lower Ag uptake than myeloid dendritic cell subsets. Blood 2013; 121: 459–467. [DOI] [PubMed] [Google Scholar]
- 55. Jing Y, Shaheen E, Drake RR, Chen N, Gravenstein S, Deng Y. Aging is associated with a numerical and functional decline in plasmacytoid dendritic cells, whereas myeloid dendritic cells are relatively unaltered in human peripheral blood. Hum Immunol 2009; 70: 777–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gupta S. Role of dendritic cells in innate and adaptive immune response in human aging. Exp Gerontol 2014; 54: 47–52. [DOI] [PubMed] [Google Scholar]
- 57. Della Bella S, Bierti L, Presicce P, Arienti R, Valenti M, Saresella M, Vergani C, Villa ML. Peripheral blood dendritic cells and monocytes are differently regulated in the elderly. Clin Immunol 2007; 122: 220–228. [DOI] [PubMed] [Google Scholar]
- 58. Littmann L, Rössner S, Kerek F, Steinkasserer A, Zinser E. Modulation of murine bone marrow‐derived dendritic cells and B‐cells by MCS‐18 a natural product isolated from Helleborus purpurascens. Immunobiology 2008; 213: 871–878. [DOI] [PubMed] [Google Scholar]