

Abstract
Aims
Excessive activation of Ca/calmodulin‐dependent kinase II (CaMKII) is of critical importance in heart failure (HF) and atrial fibrillation. Unfortunately, lack of selectivity, specificity, and bioavailability have slowed down development of inhibitors for clinical use. We investigated a novel CaMKIIδ/CaMKIIɣ‐selective, ATP‐competitive, orally available CaMKII inhibitor (RA608) on right atrial biopsies of 119 patients undergoing heart surgery. Furthermore, we evaluated its oral efficacy to prevent deterioration of HF in mice after transverse aortic constriction (TAC).
Methods and results
In human atrial cardiomyocytes and trabeculae, respectively, RA608 significantly reduced sarcoplasmic reticulum Ca leak, reduced diastolic tension, and increased sarcoplasmic reticulum Ca content. Patch‐clamp recordings confirmed the safety of RA608 in human cardiomyocytes. C57BL6/J mice were subjected to TAC, and left ventricular function was monitored by echocardiography. Two weeks after TAC, RA608 was administered by oral gavage for 7 days. Oral RA608 treatment prevented deterioration of ejection fraction. At 3 weeks after TAC, ejection fraction was 46.1 ± 3.7% (RA608) vs. 34.9 ± 2.6% (vehicle), n = 9 vs. n = 12, P < 0.05, ANOVA, which correlated with significantly less CaMKII autophosphorylation at threonine 287. Moreover, a single oral dose significantly reduced inducibility of atrial and ventricular arrhythmias in CaMKIIδ transgenic mice 4 h after administration. Atrial fibrillation was induced in 6/6 mice for vehicle vs. 1/7 for RA608, P < 0.05, 'n − 1' χ 2 test. Ventricular tachycardia was induced in 6/7 for vehicle vs. 2/7 for RA608, P < 0.05, 'n − 1' χ 2 test.
Conclusions
RA608 is the first orally administrable CaMKII inhibitor with potent efficacy in human myocytes. Moreover, oral administration potently inhibits arrhythmogenesis and attenuates HF development in mice in vivo.
Keywords: Inhibition, Heart failure, Contractility, SR Ca leak, Arrhythmias
Introduction
The prevalence of heart failure (HF) is high and is expected to increase due to demographic changes. 1 Despite the advances in HF therapy, morbidity and mortality amongst HF patients are still high. 2 HF is accompanied by contractile dysfunction caused by abnormalities in intracellular Ca handling. 3 A hallmark of HF is a reduction of sarcoplasmic reticulum (SR) Ca content, which leads to reduced intracellular Ca transients with impaired systolic contractility. 3
There is an overwhelming body of evidence that Ca/calmodulin‐dependent protein kinase II (CaMKII) is involved in the pathogenesis of HF. 4 , 5 , 6 CaMKIIδ and CaMKIIɣ are the predominant isoforms in the heart. CaMKII has been shown to phosphorylate key Ca‐handling proteins like SR Ca release channel ryanodine receptor 2 (RyR2), phospholamban, L‐type Ca channel, and also Na and K channels, thereby regulating SR Ca content. 7 , 8 , 9 , 10 CaMKIIδ‐overexpressing transgenic (TG) mice have been shown to develop severe HF. 11 , 12 Increased CaMKII activity was shown to induce RyR2‐mediated SR Ca leak, which results in reduced SR Ca load and contractile dysfunction. 7 , 13 , 14 Moreover, increased CaMKII‐dependent SR Ca leak has been shown to result in delayed after‐depolarizations, important triggers for severe cardiac arrhythmias. 13 Thus, CaMKII is increasingly being regarded as a promising therapeutic target for both HF and cardiac arrhythmias. 15 , 16
However, CaMKII inhibitors with sufficient selectivity, specificity, and oral bioavailability are currently not available. Here, we evaluate RA608, a recently developed orally active, ATP‐competitive CaMKII inhibitor with high selectivity for CaMKIIδ and CaMKIIɣ, the predominant cardiac isoforms. Beside its high isoform selectivity, the ATP competitiveness of RA608 solves issues pertinent to previous research tools, such as the allosteric CaMKII inhibitor KN93, which could only inhibit inactive CaMKII, while RA608 can inhibit CaMKII regardless of its activation status. RA608 also shows vastly improved bioavailability and, importantly, tissue permeability, which prevented previous ATP‐competitive CaMKII inhibitors (e.g. AIP) from testing in multicellular preparations and, first and foremost, from testing in vivo. Therefore, we performed a large cross‐sectional observational study using right atrial biopsies from 119 patients to investigate the efficacy and safety of RA608 in human cardiac tissue for the first time. We accompanied the human cardiac tissue studies by in vivo studies in mice with HF to test the oral efficacy of RA608 to prevent the deterioration of contractile function and arrhythmias.
Methods
An extended version of the Methods section can be found in the Supporting Information.
Statement on the use of human and murine tissue
All investigations conformed to directive 2010/63/EU of the European Parliament. The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85‐23, revised 1985) and to local institutional guidelines. The investigation conforms with the principles outlined in the Declaration of Helsinki. Written consent was given by all donors of human tissue after approval had been given by the local ethics committee.
Acquisition of human myocardium and human myocyte isolation
Patients planned for heart surgery were enrolled in a cross‐sectional observational study to investigate the efficacy of RA608 in human myocardium. Inclusion criteria were planned open heart surgery requiring extracorporal circulation. Exclusion criterion was lack of sufficient amounts of biomaterial obtained during surgery. After informed consent, right atrial biopsies, which were excised on a routine basis during implantation of extracorporal circulation, were acquired and immediately transferred to the lab. In addition, clinical data of the patients were collected from clinical files at presentation. For some patients, experiments were only possible for a subset of experimental groups, meaning that, for example, due to cell yield, not all groups could be investigated in total in each patient. Wherever possible, allocation to the experimental groups remained blinded and at random. However, for some normalized data (e.g. trabeculae experiments), vehicle control was essential for the experiment and thus applied unblinded.
Preparation of RA608 solutions and storage conditions
RA608 is a novel orally administrable ATP‐competitive CaMKII inhibitor developed by Sanofi. 17 For oral administration of RA608, a stock solution of 15 mg/mL was generated once every day and kept at 4°C until used; 15 mg of the compound was solubilized in 800 μL of acidic cyclodextrin (captisol) and HCl mixture (10% in HCl 0.1 N). Thereafter, the pH was adjusted to 4 with NaOH, and the final volume of 1 mL was adjusted by addition of neutral cyclodextrin (10% in H2O). The gavage mass for in vivo experiments was 30 mg/kg body weight (BW).
Ca/calmodulin‐dependent kinase II enzymatic assays
Ca/calmodulin‐dependent kinase II inhibition was measured by capillary electrophoresis mobility shift assay technology from Caliper Life Sciences (PerkinElmer) using purified human CaMKII protein isoforms: CaMKIIα (Carna Biosciences, 02‐109), CaMKIIβ (Millipore, 14‐718), CaMKIIɣ (Millipore, 14‐719), and CaMKIIδ (Millipore, 14‐723) as previously described. 18
Measurement of Ca sparks
Myocytes on laminin‐coated recording chambers were loaded with 10 μmol/L Fluo‐4 acetoxymethyl ester in the presence of 0.02% (w/v) pluronic acid (Molecular Probes; 15 min incubation), mounted on stage of a laser‐scanning confocal microscope (LSM 7, Zeiss), and superfused with normal Tyrode solution. Ca sparks were assessed at room temperature as described previously. 19 For some experiments, CaMKII was inhibited with RA608 at concentrations of 1, 3, or 10 μM. For comparison, the CaMKII inhibitor AIP (in its myristoylated form) was also investigated in some experiments (2 μM).
In vitro electrophysiology
Human atrial cardiomyocytes or Purkinje fibres were mounted on the stage of a microscope. Ruptured‐patch whole‐cell current clamp at room temperature was used to measure cardiac membrane potential and action potentials (APs) as described previously. 9
Genotoxicity assessment
The Ames and micronucleus in vitro tests were conducted according to ICH S2 guideline to assess the early genotoxic potential of RA608.
Western blots
Explanted hearts from mice after transverse aortic constriction (TAC) or sham operation were used for Western blotting as described previously. 9
Measurements of contractility in human trabeculae from right atrial appendage
Biopsies of human right atrial appendages of patients undergoing aorto‐coronary bypass grafting were used, and trabeculae were dissected for isometric force recordings. After an equilibration period of approximately 30 min, the trabeculae were gradually stretched until the maximum steady‐state twitch force was achieved. Post‐rest behaviour was assessed by using rest intervals 10, 30, and 120 s between beats at a basal stimulation frequency of 1 Hz. RA608 (1, 3, and 10 μM) and the CaMKII inhibitor KN93 (5 μM) were washed in for 20 min before force measurements were performed.
Mouse echocardiography
The animals were initially anaesthetized with 3% isoflurane, while temperature‐controlled, respiration‐controlled, and electrocardiogram (ECG)‐controlled anaesthesia was maintained with 1.5% isoflurane.
Transthoracic echocardiography was performed blinded using a Vevo 3100 (VisualSonics, Toronto, Canada) system with a 30 MHz centre frequency transducer and performed as described previously. 20
Animal models of heart failure
Heart failure was induced in mice by TAC as described previously 20 or TG overexpression of CaMKIIδ. 11 , 12 Aortic flow (gradients) was assessed 3 days after the TAC procedure. A TAC with a gradient of less than 5 mmHg was deemed insufficient to induce HF, and data were excluded from later analysis. At the end of the experiment, mice were killed under isoflurane anaesthesia (5%) by cervical dislocation, and hearts were freshly frozen and stored for further analysis.
In vivo electrophysiological studies in mice
Twelve‐week‐old male CaMKIIδ TG mice were treated with either vehicle control or a single oral dose of RA608 (30 mg/kg BW) <4 h prior to the experiment. A Millar 1.1F octapolar EP catheter (EPR‐800; Millar Instruments) was inserted via the right jugular vein, as previously described. A computer‐based data acquisition system (PowerLab 16/35; AD Instruments) was used to record a one‐lead body surface ECG and four intracardiac bipolar electrograms (LabChart Pro software, Version 7; AD Instruments). Right ventricular pacing was performed using 2 ms current pulses delivered by an external stimulator (STG‐3008FA; Multi Channel Systems). Inducibility of ventricular arrhythmias was tested by decremental burst pacing. Burst pacing was repeated for a total of three times 1 min after the previous burst concluded or the termination of arrhythmias. Ventricular arrhythmias were considered significant if their duration was longer than 1 s. In parallel, atrial burst pacing was conducted to induce atrial fibrillation. Burst stimulation was repeated 10 min after intraperitoneal injection of isoproterenol (ISO) (2 mg/kg BW).
Pharmacokinetic profiling
The pharmacokinetics of RA608 was evaluated in mice after single dose at 3 mg/kg intravenous (i.v.) and 10 or 30 mg/kg per os (p.o.) with n = 3 male C57BL/6 mice per time point. A non‐serial sampling schedule was applied. After extraction, RA608 concentrations were determined in individual plasma samples, and in a pool of three mice per time point for the brain, muscle, kidney, and heart tissues by LC/MS/MS method using an API 4000 instrument (SCIEX) in positive electrospray ionization mode and Analyst software (Version 1.6.2). The pharmacokinetic analysis was performed using a non‐compartmental model and WinNonlin software (Phoenix, Version 6.4).
Statistical analysis
Statistical tests are referenced in the figure legends for each data set to improve legibility of the main text. Statistical analyses were performed using SAS 9.4. Graphs were generated using GraphPad Prism Version 8 (GraphPad Software, San Diego, CA, USA).
Results
Pharmacological and pharmacokinetic profile of RA608
RA608, chemical structure in Figure 1 A (molecular weight of the free base = 539.69 kDa), was identified from a screening campaign at Sanofi R&D using an internal library of small molecules against CaMKIIδ, followed by medicinal chemistry optimization in order to improve potency, selectivity, and drug‐like properties of the molecule identified. The in vitro potency of RA608 was assessed on all four human CaMKII isoforms with enzymatic calliper assays (Figure 1 B ). The geometric mean of IC50 for inhibition of CaMKIIδ, CaMKIIɣ, CaMKII⍺, and CaMKIIβ, respectively, were 22, 51, 121, and 1135 nM, respectively (Figure 1 C, n = 5). A shift of IC50 between 1 and 10 K m ATP was observed for each isoform (Figure 1 C ), which is consistent with an ATP‐competitive mechanism of action. The selectivity of RA608 was also measured for a broad panel of 304 human recombinant kinases (Supporting Information, Table S1 ), representative of the full human kinome using P33 radioactive catalogue assays (Eurofins) at 1× Michaelis constant (1× K m) ATP concentrations and RA608 at 100 nM. RA608 inhibited five kinases more than 50%: CaMKIIδ (99%), CaMKIIɣ (98%), CaMKIIβ (70%), CaMKIδ (61%), and activated CDC42 kinase 1 (68%). To further elucidate the selectivity and potential patient safety of RA608, ion currents were measured by whole‐cell patch clamp after specific expression of rapid rectifying K current (I Kr by human ether‐a‐go‐go‐related gene, hERG), cardiac Na current (I Na by SCN5A), transient outward potassium current (I to by KCND3), or Ca current (I Ca by CACNA1C). This profiling on human ion channels was performed by whole‐cell patch clamp on CHO recombinant cell line (hERG), human embryonic kidney recombinant cell line (Nav1.5 and Kv4.3), and Cor4U® human stem cell‐derived cardiomyocytes (Cav1.2) using full dose–response experiments for hERG, Nav1.5 and Kv4.3 (n = 3–4 cells per concentration, 4–5 concentrations) or 10 μM (n = 16) and 30 μM (n = 5) for Cav1.2. IC50 was estimated using the four‐parameter logistic model according to Ratkovsky and Reedy 21 . The adjustment was obtained by nonlinear regression using the Levenberg–Marquardt algorithm in SAS v9.4 software. RA608 inhibited I Kr with an IC50 of 21.4 [18.1; 25.3] μM. The IC50 for inhibition of I to (>100 μmol/L), I Na (> 30 μmol/L), or I Ca [14 ± 2% inhibition at 10 μM (n = 16) and 45 ± 7% inhibition at 30 μM (n = 5) with IC50 > 30 μM] was even greater, indicating a high selectivity of RA608. Representative original traces and mean data can be found in Supporting Information, Figure S1A–S1C .
Figure 1.
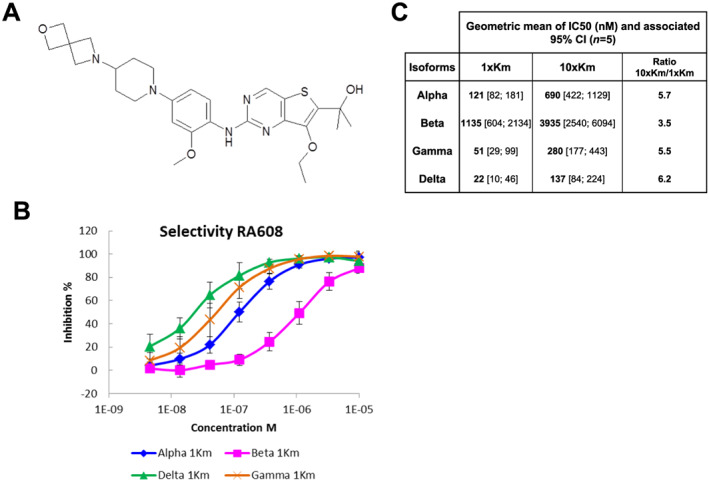
Chemical structure and pharmacodynamic profile of RA608. The chemical structure of RA608 (molecular weight of the free base 539.69 kDa) is depicted in (A). Dose–response effect of RA608 (mean ± standard deviation, n = 5 observations) using 30 μM ATP for CaMKIIδ (green), CaMKIIɣ (orange), CaMKII⍺ (blue), and CaMKIIβ (pink) (B). Geometric mean and 95% confidence interval (CI) of IC50 (n = 5, monoplicate for each isoform) shown in (C). IC50 was estimated for each experiment and each isoform using the four‐parameter logistic model with bottom fixed at 0 and if necessary top fixed at 100 (B, C).
Assessment of mutagenicity in a bacterial reverse gene mutation test (Ames) tested up to 1.85 mM of RA608 and chromosomal aberrations (micronucleus) tested up to 1 mM of RA608 were both negative.
The pharmacokinetic properties of RA608 were assessed following i.v. administration at 3 mg/kg BW, or p.o. administration at 10 or 30 mg/kg in male C57BL/6 mice (n = 3 mice per time point and administration route). RA608 displayed a low clearance (0.71 L/h/kg), a large volume of distribution at steady state (8.63 L/kg), and a long terminal elimination half‐life (t 1/2) close to 11 h. Following oral administration at 10 or 30 mg/kg BW, maximal RA608 concentrations of 1.2 μg/mL (~2.2 μM) and 2.4 μg/mL (~4.4 μM) were reached at 1 or 2 h after dosing in plasma, with long terminal elimination half‐life of 7.4 and 6.9 h, respectively (Supporting Information, Figure S2A–S2C ). The estimated absolute bioavailability was high (around 100%) regardless of the dose. Given this pharmacological profile, we concluded that RA608 was a good candidate for further functional studies.
RA608 significantly reduces sarcoplasmic reticulum Ca leak in human atrial cardiomyocytes
The effect of RA608 on cardiac function was tested in isolated human atrial cardiomyocytes or human atrial trabeculae from 119 patients undergoing aorto‐coronary bypass grafting (clinical data in Table 1 ). These patients represent a typical ischaemic heart disease collective (96% coronary heart disease). They were mainly older men (82%, mean age 68 years) with increased body mass index (mean body mass index 29) and a large prevalence of cardiovascular risk factors (arterial hypertension, hypercholesterolaemia, and diabetes mellitus). A third of these patients also showed impaired systolic left ventricular function (ejection fraction <45%), and about 14% had atrial fibrillation.
Table 1.
Clinical data of patients, from which atrial biopsies were obtained
| Parameter | Value |
|---|---|
| Number of patients | 119 |
| Male gender, n (%) | 97/119 (81.5) |
| Age (years), mean ± SD | 68.12 ± 9.07 (n = 119) |
| Body mass index (kg/m2), mean ± SD | 28.92 ± 4.47 (n = 92) |
| Type of surgery | |
| CABG, n (%) | 119 (100%) |
| Aortic valve replacement, n (%) | 14/119 (11.8) |
| Mitral valve repair, n (%) | 5/119 (4.2) |
| Cardiovascular risk factors | |
| Arterial hypertension, n (%) | 95/105 (90.5) |
| Hypercholesterolaemia, n (%) | 50/115 (43.5) |
| Diabetes mellitus, n (%) | 13/104 (12.5) |
| Sleep disordered breathing, n (%) | 8/29 (27.6) |
| Heart disease | |
| Coronary heart disease, n (%) | 112/117 (95.7) |
| Left ventricular hypertrophy, n (%) | 29/70 (41.4) |
| Dilated left atrium, n (%) | 35/59 (59.3) |
| History of myocardial infarction, n (%) | 38/117 (32.5) |
| Left ventricular assist device, n (%) | 1/119 (0.8) |
| Heart function | 104/119 (87.4) |
| LVEF (%), median (Q1, Q3), median (IQR) | 55 (45, 60) (n = 104) |
| LVEF ≤ 45%, n (%) | 33/104 (31.7) |
| Atrial fibrillation, n (%) | 16/112 (14.3) |
| GFR (mL/min), mean ± SD | 60.28 ± 19.98 (n = 111) |
CABG, coronary artery bypass grafting; GFR, glomerular filtration rate; IQR, inter‐quartile range; LVEF, left ventricular ejection fraction; SD, standard deviation.
Q1 is the 25th percentile, and Q3 is the 75th percentile. Sleep disordered breathing (apnoea/hypopnea index >15 per hour).
Figure 2 shows illustrative original traces of confocal line scans (A) and mean data (B) for Ca spark frequency (CaSpF) measured in isolated atrial myocytes of 82 patients. For some patients, these experiments were only possible for a subset of experimental groups (Figure 2 B ). Isolated cardiomyocytes in separate measurement chambers but from the same patients were exposed to either vehicle control (DMSO) or 1, 3, or 10 μM RA608, or the CaMKII inhibitor autocamtide‐2‐related autoinhibitory peptide. Compared with vehicle control, CaSpF was significantly reduced in the presence of 1, 3, and 10 μM RA608. AIP has been previously shown to reduce SR Ca leak. 15 , 22 RA608 at 1 μM inhibited CaSpF to a similar extent as AIP (2 μM). RA608 did not significantly alter the amplitude of Ca sparks, Ca spark width, or Ca spark duration (Supporting Information, Table S2 ).
Figure 2.
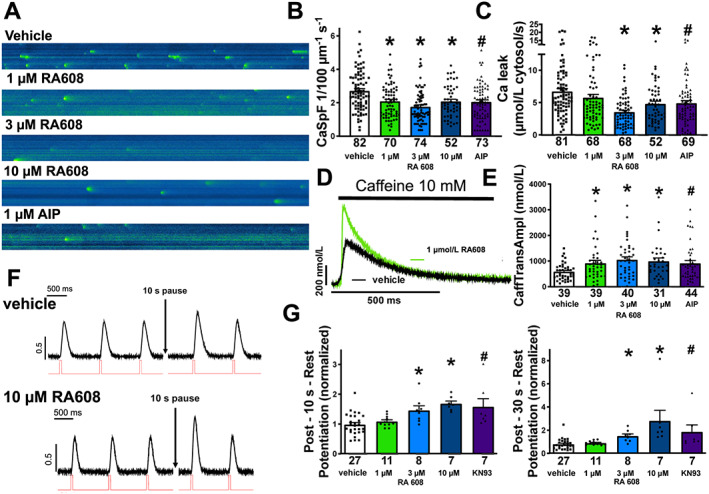
RA608 significantly reduces SR Ca leak in human atrial cardiomyocytes. Original confocal line scans (A) and mean ± SEM for CaSpF (B) and SR Ca leak (C). Compared with vehicle, RA608 significantly reduced CaSpF at 1, 3, and 10 μM and to a similar extent as AIP (patients: vehicle n = 82; 1 μM RA608 = 70, 3 μM RA608 = 74, and 10 μM RA608 = 52; and AIP = 73). This also translated into a significantly reduced SR Ca leak at 3 and 10 μM of RA608 and with AIP (patients: vehicle n = 81; 1 μM RA608 = 68, 3 μM RA608 = 68, and 10 μM RA608 = 52; and AIP = 69). (D) Original traces of caffeine‐induced Ca transient and mean ± SEM data for caffeine‐transient amplitude (E) indicate that RA608 (1, 3, and 10 μM) significantly increased SR Ca content compared with vehicle, comparable to AIP (patients: vehicle n = 39; 1 μM RA608 = 39, 3 μM RA608 = 40, and 10 μM RA608 = 31; and AIP = 44). (F) Original recordings of developed tension of isolated human atrial trabeculae before and following a 10 s pause of electrical stimulation. (G) Mean ± SEM data for post‐rest potentiation. Pre‐incubation with either 3 or 10 μM RA608 significantly increased contractile function after pause (10 s), comparable to AIP (patients: vehicle n = 27; 1 μM RA608 = 11, 3 μM RA608 = 8, and 10 μM RA608 = 7; and AIP = 7). (B, C, E, and G left panel) * P < 0.05 for RA608 vs. vehicle by Dunnett's test after a one‐way repeated ANOVA; # P < 0.05 for AIP or KN93 vs. vehicle using one‐way repeated ANOVA. (G right panel) * P < 0.05 for RA608 vs. vehicle by Dunn's test after a Kruskal–Wallis test (after not meeting normality requirements). # P < 0.05 for KN93 vs. vehicle using Mann–Whitney U test.
In accordance with reduction of CaSpF, compared with vehicle, RA608 significantly reduced SR Ca leak at 3 and 10 μM, with a more pronounced inhibition at 3 than 10 μM (Figure 2 C ).
Sarcoplasmic reticulum Ca load is highly depended on SR Ca leak (leak–load relationship 23 ). Therefore, we assessed SR Ca content by rapid application of caffeine in Fluo‐4‐loaded cardiomyocytes of 44 patients. For some patients, these experiments were only possible for a subset of experimental groups (Figure 2 D and 2 E ). In accordance with inhibition of SR Ca leak, compared with vehicle, RA608 significantly increased SR Ca content at 1, 3, and 10 μM (Figure 2 D and 2 E ).
Post‐rest contractions were acquired as a measure of SR Ca leak in atrial trabeculae of 27 patients (Figure 2 F and 2 G ). For some patients, these experiments were only possible for a subset of experimental groups (Figure 2 F and 2 G ). During pauses of 10 s, diastolic SR Ca leak limits the gain in SR Ca content that would normally occur. 24 , 25 Therefore, the ratio of developed forces of the first contraction after the pause and the last contraction before the pause can be used as a measure of Ca leak. Interestingly, exposure to RA608 (3 and 10 μM) significantly increased the post‐rest contraction amplitude, suggesting that SR Ca leak was inhibited (Figure 2 F and 2 G ). Exposure to the CaMKII inhibitor KN93 (5 μmol/L) resulted in a comparable increase in contraction amplitude after pause.
RA608 reduces diastolic tension of human atrial trabeculae
Increased diastolic SR Ca leak may impact contractility by increasing diastolic Ca concentration in cardiomyocytes, which results in diastolic dysfunction. 26 We measured diastolic tension in human atrial trabeculae of 25 patients. These trabeculae were pre‐incubated with either vehicle control or 1, 3, or 10 μM RA608 for 20 min, but for some patients, only a subset of experimental groups could be obtained (Supporting Information, Figure S3A and S3B ). For each patient, diastolic tension was normalized to its vehicle control. RA608 significantly reduced diastolic tension in a dose‐dependent manner, while only a trend to decrease is observed with KN93 (Supporting Information, Figure S3B ). Interestingly, exposure to neither RA608 nor KN93 resulted in significant alterations of relaxation time, which is mainly dependent on the kinetics of Ca elimination by SR Ca ATPase and Na/Ca exchanger (time to 80% of relaxation, Supporting Information, Figure S3C ).
Beside diastolic function, SR Ca load critically determines Ca transient amplitude and systolic contractile function. 27 Thus, we investigated the developed tension in isolated atrial trabeculae of 23 patients. For each patient, developed tension was normalized to its vehicle control. Acute exposure to RA608 did not alter developed tension at 1 Hz at 1 and 3 μM but decreased it at the highest concentration 10 μM, at least in this preparation of atrial tissue (Supporting Information, Figure S4A–S4C ).
RA608 does not prolong action potential duration
To explore the drug safety, we measured cardiac APs from isolated Purkinje fibres (New Zealand male rabbits) by microelectrode technique. Compared with vehicle, increasing concentration of RA608 (from 0.3 to 10 μmol/L) was devoid of biologically relevant effect on AP duration or maximal rate of depolarization (V max, Figure 3 A, n = 3). At 30 μmol/L, however, RA608 slightly decreased AP duration. APD50 decreased by −12 ± 5%, −14 ± 5%, and −18 ± 5% at 3, 1, and 0.25 Hz, respectively. However, APD90 remained unchanged at 3 Hz and slightly decreased by −4 ± 2% and −10 ± 4% at 1 and 0.25 Hz, respectively. AP amplitude slightly decreased by −5 ± 1%, −2 ± 0%, and −4 ± 2% at 3, 1, and 0.25 Hz, respectively.
Figure 3.
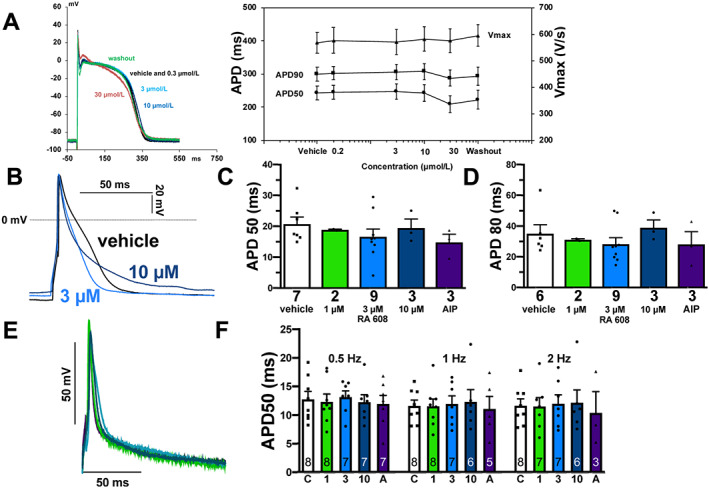
RA608 did not affect action potential characteristics. (A) Left panel: original registrations of APs elicited in isolated Purkinje fibres (n = 3 rabbits) at 1 Hz. There was no notable effect of RA608 exposure on AP shape and characteristics. Right panel: mean ± SEM for AP duration at 50% or 90% repolarization (APD50 and APD90), or maximal AP upstroke velocity (V max, n = 3 rabbits, each). (B) Original recordings of APs elicited in isolated human atrial cardiomyocytes. Mean ± SEM data for APD50 (C, patients: vehicle n = 7; 1 μM RA608 = 2, 3 μM RA608 = 9, and 10 μM RA608 = 3; and AIP = 3) or APD80 (D, patients: vehicle n = 6; 1 μM RA608 = 2, 3 μM RA608 = 9, and 10 μM RA608 = 3; and AIP = 3) for vehicle, 1, 3, and 10 μM RA608, and AIP indicated that there was no significant effect of RA608 or AIP on AP duration. (C, D) Dunnett's test for RA608 vs. vehicle following a one‐way ANOVA; AIP vs. vehicle using Student's t‐test. (E) Original recordings of APs elicited in murine ventricular cardiomyocytes. (F) Mean ± SEM data for APD50 (n in the bars = cells, five mice) upon control (C), RA608 (1 = 1 μM, 3 = 3 μM, and 10 = 10 μM), or 2 μmol/L myristoylated AIP (A) show that there was no significant effect of RA608 or AIP on AP duration (two‐way ANOVA followed by Tukey's post‐test).
Cardiac APs were also measured in human atrial cardiomyocytes of seven patients by whole‐cell patch clamp (Figure 3 B – 3 D ). In accordance with the animal data, compared with vehicle, exposure to RA608 did not significantly alter AP duration (Figure 3 B – 3 D ). AP amplitude, maximal rate of depolarization (V max), and resting membrane potential were also not significantly altered by RA608 nor AIP (Supporting Information, Table S3 ). AP duration was also unchanged by RA608 in murine ventricular cardiomyocytes (Figure 3 E and 3 F ), thus showing an encouraging safety profile.
RA608 prevents heart failure progression in vivo
To test if RA608 improves cardiac function in vivo, we investigated RA608 in the murine HF model induced by TAC. In these mice, left ventricular ejection fraction (LVEF) was assessed at baseline, 2 weeks after TAC, and after an additional week of oral treatment with RA608 (30 mg/kg BW/day) or vehicle control. As published previously, TAC surgery resulted in HF development. 28 Two weeks after TAC and before the oral treatment was started, LVEF was reduced compared with baseline from 62.6 ± 2.9% to 47.0 ± 2.9% and from 59.0 ± 3.0% to 47.2 ± 3.3% in the mice allocated to vehicle or RA608 treatment groups, respectively (Figure 4 A and 4 B ). However, compared with vehicle, 7 days of oral RA608 treatment of TAC mice significantly prevented deterioration of LVEF. At 3 weeks after TAC, vehicle‐treated mice showed further deterioration of LVEF to 34.9 ± 2.6%, but the RA608‐treated mice showed an LVEF of 46.1 ± 3.7% (Figure 4 A and 4 B ). In accordance, at 3 weeks after TAC, left ventricular end‐diastolic dimension tended to be increased more in the vehicle group compared with the RA608 group (Supporting Information, Figure S5B ). Furthermore, the increased lung weight (normalized to tibia length), which can be observed after 3 weeks after TAC as a marker of pulmonary oedema, was decreased after RA608 treatment, suggesting reduced congestion (Supporting Information, Figure S5A ).
Figure 4.
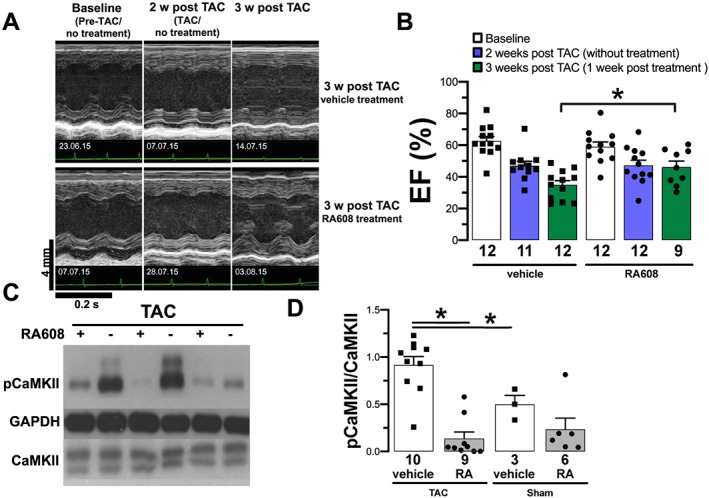
RA608 attenuates heart failure in TAC. After 2 weeks of TAC, mice were treated with either vehicle or oral 30 mg/kg BW RA608 q.d. for 7 days. Original echocardiography recordings (A, time stamp of acquisition date in the lower left corner; all acquisitions took place in the morning hours between 9 a.m. and 11 a.m.) and mean ± SEM (B, n = 12 mice per group) show that TAC mice develop a reduction in LVEF (%) 2 weeks after TAC compared with baseline. After a week of treatment with RA608, TAC mice show a significantly greater ejection fraction compared with the vehicle‐treated TAC mice (significant difference indicated by ‘*’). The level of autophosphorylated CaMKII relative to total CaMKII (p‐CaMKII/CaMKII) was analysed after oral treatment [original Western blots (C) and mean ± SEM (D)]. Compared with vehicle, TAC mice treated with RA608 had significantly reduced p‐CaMKII/CaMKII levels comparable to sham (mice: vehicle + TAC n = 10, RA608 + TAC n = 9, vehicle + sham n = 3, and RA608 + sham n = 6). * P < 0.05 two‐way ANOVA with factors ‘surgery’ and ‘treatment’ was performed. Then two contrast analyses were carried out: one to compare surgeries for vehicle groups and one to compare the RA608 group to the vehicle group for TAC surgery. For LVEF, the statistical analyses were performed 3 weeks after TAC (1 week after treatment).
In contrast to contractile function, treatment with RA608 did not prevent development of left ventricular hypertrophy. At 3 weeks after TAC, in both vehicle‐treated and RA608‐treated mice, heart weight to BW ratio and septum thickness were significantly increased (compared to sham) with no difference between vehicle and RA608 treatment (Supporting Information, Figure S5C and S5D ). Of note, sham‐operated mice did not show any significant difference in ejection fraction for vehicle vs. RA608 treatment (Supporting Information, Table S4 ).
To assess CaMKII activity, the hearts were explanted after the treatment period and subjected to Western blotting. CaMKII autophosphorylation at threonine 287 as a marker of CaMKII activity 6 , 29 was significantly increased in TAC vehicle mice compared with sham vehicle mice (Figure 4 C and 4 D ). Treatment of TAC mice with RA608 resulted in a significant reduction in CaMKII autophosphorylation (Figure 4 C and 4 D ).
RA608 prevents atrial and ventricular arrhythmias in vivo
Ca/calmodulin‐dependent kinase II dysfunction is known to be highly pro‐arrhythmogenic. 15 , 16 Therefore, we measured arrhythmias in a murine model of cardiomyocyte‐specific CaMKIIδ overexpression (CaMKIIδc TG). We chose this model, because CaMKIIδc TG mice are known to develop HF and are especially susceptible to arrhythmias upon exposure to ISO. 11 , 12 To test the antiarrhythmic effect of RA608, 12‐week old male CaMKIIδc TG mice were treated orally with a single dose of either vehicle control or RA608 (30 mg/kg BW) <4 h prior to the experiment. The inducibility of atrial and ventricular arrhythmias was then assessed by programmed electrical stimulation (burst stimulation) at the level of the right atrium or right ventricle (before and after i.p. injection of ISO, Figure 5 ). Developing arrhythmias were deemed relevant when they persisted for at least 1 s after the end of burst stimulation.
Figure 5.
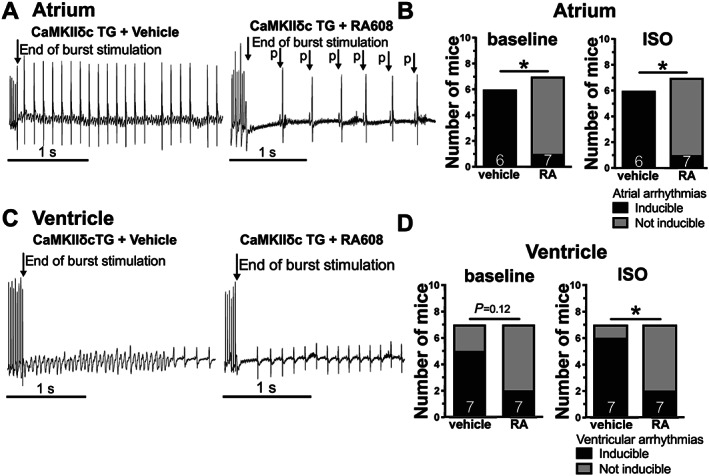
RA608 attenuates atrial and ventricular arrhythmias in vivo. CaMKIIδc TG mice were treated with either vehicle or a single oral dose of the RA608 (30 mg/kg BW) <4 h prior to programmed electrical burst stimulation in vivo. Original surface ECG recordings (A) show atrial burst stimulation followed by atrial fibrillation upon vehicle treatment but sinus rhythm upon RA608 treatment. Frequency of atrial arrhythmias (B) indicated that RA608 significantly reduced the susceptibility to induced atrial arrhythmias at baseline and upon administration of ISO (mice: vehicle n = 6 and RA608 n = 7). Original surface recordings (C) show ventricular burst stimulation followed by ventricular tachycardia upon vehicle treatment but sinus rhythm upon RA608 treatment. Frequency of ventricular arrhythmias upon administration of ISO (D) was significantly reduced upon treatment with RA608 (but only by trend at baseline, mice: vehicle n = 7 and RA608 n = 7). (B, D) * P < 0.05, n − 1 – χ 2, as recommended by Campbell.30
Interestingly, pretreatment with RA608 significantly reduced both the incidence of atrial fibrillation and flutter at baseline and upon administration of ISO (Figure 5 A and 5 B ) and ventricular tachyarrhythmias upon ISO (Figure 5 C and 5 D ). We also performed experiments investigating ventricular arrhythmias in the CaMKIIδc TG mice using an S1S2 stimulation protocol 31 , 32 : RA608 also prevented induction of arrhythmias using this methodology (Supporting Information, Figure S6A and S6B ). In accordance with unaltered APs, heart rate‐corrected QT interval was not different at the level of the surface ECG (Supporting Information, Figure 7B ) and also at intracardiac ECG (data not shown). There were also no significant differences in other ECG parameters such as QRS interval, PR interval, or heart rate (Supporting Information, Figure S7C–S7E ).
Discussion
Here, we report human cardiac tissue and in vivo animal data about the novel orally administrable, ATP‐competitive CaMKII inhibitor RA608. This compound was derived from a series of thienopyrimidines, representing a new class of CaMKII inhibitors with several mechanistic advantages over other compounds like KN93. The RA608 dual inhibition of CaMKIIδ and CaMKIIɣ (geometric mean of IC50 at 22 and 51 nM, respectively), exerting redundant pathological effects at the cardiac level, is expected to be beneficial in the context of HF. Based on its ATP‐competitive mechanism of action, RA608 should inhibit CaMKII whatever its activation status, offering a significant mechanistic advantage over KN93—an allosteric inhibitor that cannot inhibit CaMKII when already activated. Its pharmacokinetic profile combining long terminal half‐life in mice (around 7 h) and high absolute oral bioavailability (around 100%) is another attractive point of differentiation from previous tool inhibitors such as AIP, which suffered from low tissue permeability and insufficient bioavailability. This study is the first to investigate the efficacy of RA608 in one of the largest human cardiac tissue data sets that has ever been used for such a purpose. Moreover, we evaluated the oral efficacy of RA608 in two different models of HF in vivo.
Significant outcomes of the present animal study were attenuated HF progression and a reduced susceptibility to atrial and ventricular arrhythmias upon treatment with RA608. We propose a reduction of CaMKII‐dependent SR Ca leak by RA608 as underlying mechanism. This was confirmed by measurement of SR Ca leak in atrial cardiomyocytes of 119 patients undergoing CABG. In these myocytes, RA608 significantly reduced SR Ca leak and increased SR Ca load, most likely by reduced CaMKII‐dependent phosphorylation of serine 2814 of the cardiac ryanodine receptor, 15 which has been shown numerous times before. As a potential consequence, diastolic cytosolic Ca concentration should be reduced, which would improve diastolic function. Indeed, we found a significant reduction in diastolic stiffness of human atrial trabeculae. Since increased diastolic stiffness is also a hallmark of diastolic HF, CaMKII inhibition with RA608 may also be a possible translational treatment option for this pathology.
Interestingly, while the SR Ca content in isolated cardiomyocytes was significantly increased (and thus the amount of Ca available for a systolic release and contraction), this did not translate into increased steady‐state contractility in isolated atrial trabeculae. However, the human cardiac tissue data were acquired after acute exposure to the drug. The atrial trabeculae were isolated from patients with multiple long‐lasting clinical pathologies that involve atrial structural and electrical remodelling. It seems obvious that these alterations cannot be reversed by CaMKII inhibition of such short duration. Alternatively, CaMKII has also been implicated in the regulation of myofilament Ca sensitivity via titin phosphorylation, 33 as such RA608 could also affect the myofilament Ca response.
In contrast to the human atrial biopsies, long‐term treatment with RA608 prevented the deterioration of LVEF in TAC mice and attenuated the left ventricular end‐diastolic dimension as a marker of cardiac dilation. Since treatment with RA608 was started 2 weeks after TAC surgery, a time point at which hypertrophy had already developed, it seems conceivable that RA608 was not able to prevent or reduce hypertrophy at least not within 1 week of treatment.
Importantly, RA608 not only was proven to be effective in situ and in vivo for improving contractility but also shows strong in vivo antiarrhythmic properties. Oral RA608 pretreatment prevented the induction of ventricular and atrial arrhythmias in CaMKIIδc TG mice. CaMKII is known to regulate a multitude of cardiac ion channels such as Na, 9 Ca, 34 , 35 and K channels. 8 , 10 Nevertheless, CaMKII inhibition with RA608 did neither significantly alter in vivo ECG parameters nor affected single cell human, rabbit, or murine AP duration, which suggests an encouraging cardiac safety profile of RA608 that could provide the grounds for clinical translation.
Limitations of the study
Potential drawbacks of ATP‐competitive kinase inhibitors are their poor selectivity due to the high level of conservation of ATP binding pockets. It is the reason why RA608 has been tested against a panel of 304 kinases (radioactive assay), showing an acceptable level of selectivity, with only five kinases inhibited at more than 50% by RA608 at 0.1 μM, including CaMKIIδ (99%), CaMKIIɣ (98%), CaMKIIβ (70%), CaMKIδ (61%), and activated CDC42 kinase 1 (68%) (CaMKII⍺ not tested). The big shift of selectivity between CaMKIIδ and CaMKIIβ (22 vs. 1135 nM IC50 in the calliper assay) was achieved despite only one amino acid difference between the ATP binding site of CaMKIIβ and CaMKIIδ isoforms, showing that selectivity is achievable in the case of CaMKII inhibitors.
However, intracellular ATP concentration (mM range) is higher than the one used to assess compound potency in enzymatic CaMKII assays (25–100 μM range). Nevertheless, RA608 displays a cellular potency of 570 nM (IC50) in Cor4U human stem cell‐derived cardiomyocytes (data not shown), which is close to the 190 nM IC50 obtained with the closely analogue RA306 in the same assay. 18 Despite this expected shift of potency between recombinant and cellular assays, both compounds fully inhibit CaMKII at the cardiac level at pharmacological doses. Inhibition of the CaMKII⍺ and CaMKIIβ isoforms at the brain level could be potentially deleterious based on the role of these isoforms in synaptic plasticity and memory as described by several teams. 36 , 37
The previously described RA306 18 has increased potency compared to RA608 (IC50 of 11 nM on CaMKIIδ compared with 22 nM for RA608) and reduced brain/plasma ratio (0.2 vs. 1.5 for RA608). While the reduced brain exposure seems to favour RA306 over RA608 with respect to potential side effects in the nervous system, the IC50 for CaMKIIβ, which is also abundantly expressed in the brain, is much lower for RA306 vs. RA608 (IC50 of 17 vs. 1135 nM). As such, it is still unclear, which inhibitor will offer the best therapeutic value for cardiac CaMKII inhibition in relation to potential neurological side effects.
Furthermore, the role of CaMKII in the immunoresponse, affecting macrophage function and inflammation, also in the context of myocardial infarction, has recently come under new scrutiny and could potentially be affected by RA608. 38 , 39 We also showed high kidney concentrations of RA608, although CaMKII inhibition has also been proposed as protective mechanism in models of kidney injury. 40 , 41 However, as mentioned earlier, there is a big shift of selectivity between CaMKIIδ and CaMKIIβ (22 vs. 1135 nM IC50), showing that deleterious effects could potentially be minimized by dosing. Further testing of this inhibitor on other organs, tissues, and species before progression to human testing will be necessary.
In summary, CaMKII inhibition with RA608 could be a promising strategy in the treatment of systolic and diastolic HF, preventing contractile dysfunction and arrhythmias in vivo. It differs from previously described compounds in that it is orally administrable and thus also an option for long‐term treatment regimens and for translational research in humans. Its ATP competitiveness also renders it more effective than previous substrate‐competitive or CaM‐competitive CaMKII inhibitors, 22 especially when HF co‐morbidities such as diabetes lead to an increased non‐canonical activation of CaMKII. 15 , 42 RA608 may thus be the first‐in‐class substance to be considered for translation into clinical studies.
Conflict of interest
This work is part of a research collaboration between Sanofi R&D and the University of Regensburg and was as such funded in part by Sanofi R&D.
Funding
This work is part of a research collaboration between Sanofi R&D and the University of Regensburg and was as such funded in part by Sanofi R&D. This work was supported by Deutsche Forschungsgemeinschaft (DFG) Grants WA 2539/4‐1, WA 2539/5‐1, and WA 2539/7‐1 to S.W. and by Deutsche Stiftung für Herzforschung Grant F/35/15. S.W., S.S., and L.S.M. are funded by DZHK (Deutsches Zentrum für Herz‐Kreislaufforschung—German Centre for Cardiovascular Research). This work was supported by DFG Grants MA 1982/5‐1 and MA 1982/7‐1 to L.S.M. S.W. and L.S.M. are also funded by the DFG SFB 1350 grant (Project Number 387509280, TPA6), are supported by the ReForM C programme of the faculty, and funded by the DZHK (Deutsches Zentrum für Herz‐Kreislauf‐Forschung—German Centre for Cardiovascular Research). J.M. is funded by a grant from the ReForM A programme from the Universität Regensburg and by the Deutsche Gesellschaft für Kardiologie‐Herz und Kreislaufforschung (DGK) Clinician Scientist Program.
Supporting information
Table S1. Eurofins selectivity panel.
Table S2. Human atrial cardiomyocyte spark characteristics.
Table S3. Human atrial cardiomyocyte action potential characteristics.
Table S4. Ejection fraction of sham‐operated mice.
Figure S1. Ion current response to RA608.
Figure S2. Pharmacokinetic profile of RA608 in mice.
Figure S3. RA608 reduces diastolic tension of human atrial trabeculae.
Figure S4. Force development in human atrial trabeculae.
Figure S5. Phenotype and echocardiography parameters of RA608‐treated mice upon TAC.
Figure S6. RA608 prevents ventricular arrhythmias induced by the S1S2 stimulation protocol.
Figure S7. ECG parameters of CaMKIIδc TG mice upon acute RA608‐treatment.
Acknowledgements
We acknowledge the expert technical assistance of Felicia Radtke, Thomas Sowa, Corinne Gomez, Valérie Letang, and Valérie Glenat.
Mustroph, J. , Drzymalski, M. , Baier, M. , Pabel, S. , Biedermann, A. , Memmel, B. , Durczok, M. , Neef, S. , Sag, C. M. , Floerchinger, B. , Rupprecht, L. , Schmid, C. , Zausig, Y. , Bégis, G. , Briand, V. , Ozoux, M.‐L. , Tamarelle, D. , Ballet, V. , Janiak, P. , Beauverger, P. , Maier, L. S. , and Wagner, S. (2020) The oral Ca/calmodulin‐dependent kinase II inhibitor RA608 improves contractile function and prevents arrhythmias in heart failure. ESC Heart Failure, 7: 2871–2883. 10.1002/ehf2.12895.
References
- 1. Bui AL, Horwich TB, Fonarow GC. Epidemiology and risk profile of heart failure. Nat Rev Cardiol 2011; 8: 30–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Mosterd A, Cost B, Hoes AW, de Bruijne MC, Deckers JW, Hofman A, Grobbee DE. The prognosis of heart failure in the general population. The Rotterdam Study. Eur Heart J 2001; 22: 1318–1327. [DOI] [PubMed] [Google Scholar]
- 3. Neef S, Maier LS. Novel aspects of excitation–contraction coupling in heart failure. Basic Res Cardiol 2013; 108: 360. [DOI] [PubMed] [Google Scholar]
- 4. Sag CM, Wadsack DP, Khabbazzadeh S, Abesser M, Grefe C, Neumann K, Opiela M‐K, Backs J, Olson EN, Brown JH, Neef S, Maier SK, Maier LS. Calcium/calmodulin‐dependent protein kinase II contributes to cardiac arrhythmogenesis in heart failure. Circ Heart Fail 2009; 2: 664–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Luo M, Anderson ME. Mechanisms of altered Ca2+ handling in heart failure. Circ Res 2013; 113: 690–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson ME, Brown JH, Bers DM. CaMKII in myocardial hypertrophy and heart failure. J Mol Cell Cardiol 2011; 51: 468–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neef S, Dybkova N, Sossalla S, Ort KR, Fluschnik N, Neumann K, Seipelt R, Schöndube FA, Hasenfuss G, Maier LS. CaMKII‐dependent diastolic SR Ca2+ leak and elevated diastolic Ca2+ levels in right atrial myocardium of patients with atrial fibrillation. Circ Res 2010; 106: 1134–1144. [DOI] [PubMed] [Google Scholar]
- 8. Wagner S, Hacker E, Grandi E, Weber SL, Dybkova N, Sossalla S, Sowa T, Fabritz L, Kirchhof P, Bers DM, Maier LS. Ca/calmodulin kinase II differentially modulates potassium currents. Circ Arrhythm Electrophysiol 2009; 2: 285–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wagner S, Dybkova N, Rasenack ECL, Jacobshagen C, Fabritz L, Kirchhof P, Maier SKG, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca/calmodulin‐dependent protein kinase II regulates cardiac Na channels. J Clin Invest 2006; 116: 3127–3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mustroph J, Maier LS, Wagner S. CaMKII regulation of cardiac K channels. Front Pharmacol 2014; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Maier LS. Transgenic CaMKIIδC overexpression uniquely alters cardiac myocyte Ca2+ handling: reduced SR Ca2+ load and activated SR Ca2+ release. Circ Res 2003; 92: 904–911. [DOI] [PubMed] [Google Scholar]
- 12. Zhang T. The δC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res 2003; 92: 912–919. [DOI] [PubMed] [Google Scholar]
- 13. Voigt N, Li N, Wang Q, Wang W, Trafford AW, Abu‐Taha I, Sun Q, Wieland T, Ravens U, Nattel S, Wehrens XHT, Dobrev D. Enhanced sarcoplasmic reticulum Ca2+ leak and increased Na+–Ca2+ exchanger function underlie delayed afterdepolarizations in patients with chronic atrial fibrillation. Circulation 2012; 125: 2059–2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Fischer TH, Herting J, Tirilomis T, Renner A, Neef S, Toischer K, Ellenberger D, Förster A, Schmitto JD, Gummert J, Schöndube FA, Hasenfuss G, Maier LS, Sossalla S. Ca2+/calmodulin‐dependent protein kinase II and protein kinase A differentially regulate sarcoplasmic reticulum Ca2+ leak in human cardiac pathology. Circulation 2013; 128: 970–981. [DOI] [PubMed] [Google Scholar]
- 15. Mustroph J, Neef S, Maier LS. CaMKII as a target for arrhythmia suppression. Pharmacol Ther 2017; 176: 22–31. [DOI] [PubMed] [Google Scholar]
- 16. Hund TJ, Mohler PJ. Role of CaMKII in cardiac arrhythmias. Trends Cardiovasc Med 2015; 25: 392–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Beauverger P, Begis G, Biscarrat S, Duclos O, McCort G. 5 oxo‐5,8‐dihydropyrido[2,3‐d]pyrimidine derivatives as CaMKII kinase inhibitors for treating cardiovascular diseases. Google Patents; 2012.
- 18. Beauverger P, Ozoux M‐L, Bégis G, Glénat V, Briand V, Philippo M‐C, Daveu C, Tavares G, Roy S, Corbier A, Briand P, Dorchies O, Bauchet A‐L, Nicolai E, Duclos O, Tamarelle D, Pruniaux M‐P, Muslin AJ, Janiak P. Reversion of cardiac dysfunction by a novel orally available calcium/calmodulin‐dependent protein kinase II inhibitor, RA306, in a genetic model of dilated cardiomyopathy. Cardiovasc Res 2019; 116: 329–338. [DOI] [PubMed] [Google Scholar]
- 19. Wagner S, Ruff HM, Weber SL, Bellmann S, Sowa T, Schulte T, Anderson ME, Grandi E, Bers DM, Backs J, Belardinelli L, Maier LS. Reactive oxygen species–activated Ca/calmodulin kinase IIδ is required for late I Na augmentation leading to cellular Na and Ca overload. Circ Res 2011; 108: 555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Toischer K, Hartmann N, Wagner S, Fischer TH, Herting J, Danner BC, Sag CM, Hund TJ, Mohler PJ, Belardinelli L, Hasenfuss G, Maier LS, Sossalla S. Role of late sodium current as a potential arrhythmogenic mechanism in the progression of pressure‐induced heart disease. J Mol Cell Cardiol 2013; 61: 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ratkowsky, DA. , Reedy TJ. Choosing near–linear parameters in the four–parameter logistic model for radioligand and related assays. Biometrics, 1986;42(3):575–582. [PubMed] [Google Scholar]
- 22. Pellicena P, Schulman H. CaMKII inhibitors: from research tools to therapeutic agents. Front Pharmacol 2014; 5: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Shannon TR. Quantitative assessment of the SR Ca2+ leak–load relationship. Circ Res 2002; 91: 594–600. [DOI] [PubMed] [Google Scholar]
- 24. Bers DM, Bassani RA, Bassani JWM, Baudet S, Hryshko LV. Paradoxical twitch potentiation after rest in cardiac muscle: increased fractional release of SR calcium. J Mol Cell Cardiol 1993; 25: 1047–1057. [DOI] [PubMed] [Google Scholar]
- 25. Pieske B, Sütterlin M, Schmidt‐Schweda S, Minami K, Meyer M, Olschewski M, Holubarsch C, Just H, Hasenfuss G. Diminished post‐rest potentiation of contractile force in human dilated cardiomyopathy. Functional evidence for alterations in intracellular Ca2+ handling. J Clin Invest 1996; 98: 764–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Selby DE, Palmer BM, LeWinter MM, Meyer M. Tachycardia‐induced diastolic dysfunction and resting tone in myocardium from patients with normal ejection fraction. J Am Coll Cardiol 2011; 58: 147–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittköpper K, Renner A, Schmitto JD, Gummert J, El‐Armouche A, Hasenfuss G, Maier LS. Inhibition of elevated Ca2+/calmodulin‐dependent protein kinase II improves contractility in human failing myocardium. Circ Res 2010; 107: 1150–1161. [DOI] [PubMed] [Google Scholar]
- 28. Toischer K, Rokita AG, Unsöld B, Zhu W, Kararigas G, Sossalla S, Reuter SP, Becker A, Teucher N, Seidler T, Grebe C, Preuß L, Gupta SN, Schmidt K, Lehnart SE, Krüger M, Linke WA, Backs J, Regitz‐Zagrosek V, Schäfer K, Field LJ, Maier LS, Hasenfuss G. Differential cardiac remodeling in preload versus afterload. Circulation 2010; 122: 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Maier L, Bers D. Role of Ca2+/calmodulin‐dependent protein kinase (CaMK) in excitation–contraction coupling in the heart. Cardiovasc Res 2007; 73: 631–640. [DOI] [PubMed] [Google Scholar]
- 30. Campbell I. Chi‐squared and Fisher–Irwin tests of two‐by‐two tables with small sample recommendations. Stat Med 2007; 26: 3661–3675. [DOI] [PubMed] [Google Scholar]
- 31. Li N, Wehrens XHT. Programmed electrical stimulation in mice. J Vis Exp JoVE MyJove Corporation 2010; 39: e1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Clasen L, Eickholt C, Angendohr S, Jungen C, Shin D‐I, Donner B, Fürnkranz A, Kelm M, Klöcker N, Meyer C, Makimoto H. A modified approach for programmed electrical stimulation in mice: inducibility of ventricular arrhythmias. PLOS ONE Public Library of Science 2018; 13: e0201910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hamdani N, Krysiak J, Kreusser MM, Neef S, dos Remedios CG, Maier LS, Krüger M, Backs J, Linke WA. Crucial role for Ca2+/calmodulin‐dependent protein kinase‐II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res 2013; 112: 664–674. [DOI] [PubMed] [Google Scholar]
- 34. Gao L, Blair LAC, Marshall J. CaMKII‐independent effects of KN93 and its inactive analog KN92: reversible inhibition of L‐type calcium channels. Biochem Biophys Res Commun 2006; 345: 1606–1610. [DOI] [PubMed] [Google Scholar]
- 35. Best JM, Kamp TJ. Different subcellular populations of L‐type Ca2+ channels exhibit unique regulation and functional roles in cardiomyocytes. J Mol Cell Cardiol 2012; 52: 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bachstetter AD, Webster SJ, Tu T, Goulding DS, Haiech J, Watterson DM, Van Eldik LJ. Generation and behavior characterization of CaMKIIβ knockout mice. PLOS ONE Public Library of Science 2014; 9: e105191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Silva A, Paylor R, Wehner J, Tonegawa S. Impaired spatial learning in alpha‐calcium‐calmodulin kinase II mutant mice. Science 1992; 257: 206–211. [DOI] [PubMed] [Google Scholar]
- 38. Doran AC, Ozcan L, Cai B, Zheng Z, Fredman G, Rymond CC, Dorweiler B, Sluimer JC, Hsieh J, Kuriakose G, Tall AR, Tabas I. CAMKIIγ suppresses an efferocytosis pathway in macrophages and promotes atherosclerotic plaque necrosis. J Clin Invest American Society for Clinical Investigation 2017; 127: 4075–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Willeford A, Suetomi T, Nickle A, Hoffman HM, Miyamoto S, Heller Brown J. CaMKIIδ‐mediated inflammatory gene expression and inflammasome activation in cardiomyocytes initiate inflammation and induce fibrosis. JCI Insight The American Society for Clinical Investigation 2018; 3: e97054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Timmins JM, Ozcan L, Seimon TA, Li G, Malagelada C, Backs J, Backs T, Bassel‐Duby R, Olson EN, Anderson ME, Tabas I. Calcium/calmodulin‐dependent protein kinase II links ER stress with Fas and mitochondrial apoptosis pathways. J Clin Invest American Society for Clinical Investigation 2009; 119: 2925–2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bracken C, Beauverger P, Duclos O, Russo RJ, Rogers KA, Husson H, Natoli TA, Ledbetter SR, Janiak P, Ibraghimov‐Beskrovnaya O, Bukanov NO. CaMKII as a pathological mediator of ER stress, oxidative stress, and mitochondrial dysfunction in a murine model of nephronophthisis. Am J Physiol‐Ren Physiol American Physiological Society 2016; 310: F1414–F1422. [DOI] [PubMed] [Google Scholar]
- 42. Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O‐linked glycosylation. Nature 2013; 502: 372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Eurofins selectivity panel.
Table S2. Human atrial cardiomyocyte spark characteristics.
Table S3. Human atrial cardiomyocyte action potential characteristics.
Table S4. Ejection fraction of sham‐operated mice.
Figure S1. Ion current response to RA608.
Figure S2. Pharmacokinetic profile of RA608 in mice.
Figure S3. RA608 reduces diastolic tension of human atrial trabeculae.
Figure S4. Force development in human atrial trabeculae.
Figure S5. Phenotype and echocardiography parameters of RA608‐treated mice upon TAC.
Figure S6. RA608 prevents ventricular arrhythmias induced by the S1S2 stimulation protocol.
Figure S7. ECG parameters of CaMKIIδc TG mice upon acute RA608‐treatment.