

Abstract
Aims
Acute myocardial infarction (MI) is the major cause of chronic heart failure. The activity of blood coagulation factor XIII (FXIIIa) plays an important role in rodents as a healing factor after MI, whereas its role in healing and remodelling processes in humans remains unclear. We prospectively evaluated the relevance of FXIIIa after acute MI as a potential early prognostic marker for adequate healing.
Methods and results
This monocentric prospective cohort study investigated cardiac remodelling in patients with ST‐elevation MI and followed them up for 1 year. Serum FXIIIa was serially assessed during the first 9 days after MI and after 2, 6, and 12 months. Cardiac magnetic resonance imaging was performed within 4 days after MI (Scan 1), after 7 to 9 days (Scan 2), and after 12 months (Scan 3). The FXIII valine‐to‐leucine (V34L) single‐nucleotide polymorphism rs5985 was genotyped. One hundred forty‐six patients were investigated (mean age 58 ± 11 years, 13% women). Median FXIIIa was 118% (quartiles, 102–132%) and dropped to a trough on the second day after MI: 109% (98–109%; P < 0.001). FXIIIa recovered slowly over time, reaching the baseline level after 2 to 6 months and surpassed baseline levels only after 12 months: 124% (110–142%). The development of FXIIIa after MI was independent of the genotype. FXIIIa on Day 2 was strongly and inversely associated with the relative size of MI in Scan 1 (Spearman's ρ = –0.31; P = 0.01) and Scan 3 (ρ = –0.39; P < 0.01) and positively associated with left ventricular ejection fraction: ρ = 0.32 (P < 0.01) and ρ = 0.24 (P = 0.04), respectively.
Conclusions
FXIII activity after MI is highly dynamic, exhibiting a significant decline in the early healing period, with reconstitution 6 months later. Depressed FXIIIa early after MI predicted a greater size of MI and lower left ventricular ejection fraction after 1 year. The clinical relevance of these findings awaits to be tested in a randomized trial.
Keywords: Blood coagulation factor XIII, ST‐elevation myocardial infarction, Healing and remodelling processes, Cardiac magnetic resonance imaging
Introduction
Myocardial infarction (MI) and the subsequent loss of contractile myocardium is the most frequent cause of chronic heart failure. In humans, larger infarcts induce a process of deleterious cardiac remodelling, which is characterized by changes of both the infarcted and residual non‐infarcted myocardiums. 1 The macroscopic substrate of such remodelling is adverse changes in size, mass, geometry, and function of the heart 1 ; the microscopic process is the replacement of damaged myocardium by scar tissue. 2 , 3 , 4 The size of MI is primarily determined by the infarcted vessel's territory and eventually available collateral flow. Necrosis develops several hours after coronary artery occlusion (infarct extension), if reperfusion is not established. 5 , 6 The infarcted tissue may further expand or contract following reperfusion 7 in the first weeks and months (infarct expansion) even after the extension of necrosis is over; thus, reperfusion becomes a critical determinant of remodelling and prognosis. 8 Microscopically, the extent of collagen damage correlates with the degree of infarct expansion. 9 Reperfusion therapy may reduce MI size and salvage viable myocardium and compensatory mechanisms may enhance healing and mitigate remodelling.
Our group managed to identify blood coagulation factor XIII (FXIII) as an important mediator of myocardial healing after MI in animal models. Functionally, FXIII is a pro‐transglutaminase involved in the last step of the coagulation cascade being activated by thrombin and stabilizing the fibrin clot. 10 , 11 FXIII‐deficient mice suffered from impaired cardiac remodelling after MI and died of left ventricular (LV) rupture but had normal survival rates after FXIII replacement therapy. 12 Reduced FXIIIa was also reported in patients with cardiac rupture after MI. 13 However, the influence of FXIIIa on LV remodelling and infarct expansion in humans remains unknown.
Carriers of certain gene variants, for example, the valine‐to‐leucine (V34L) single‐nucleotide polymorphism (SNP) rs5985 that associates with higher FXIIIa, were reported to have a lower risk of ischaemic heart disease 14 and exhibited an improved survival after MI. 15 V34L polymorphism is located near the thrombin activation site of FXIII and promotes more pronounced and rapid susceptibility of FXIII to thrombin activation. However, FXIIIa specific enzyme activity is not affected by genetic polymorphisms. 10 , 11 , 16 Furthermore, fibrin clots formed in the presence of V34L polymorphism tend to be thinner and less porous. 11 The increased rate of fibrin stabilization brought about by the V34L FXIII seems to be paradoxically associated with a protective effect against pathological thrombosis in ischaemic heart disease, but not in ischaemic stroke. 10 , 16
In our clinical study, we aimed to evaluate the role of FXIIIa on infarct size (IS), infarct expansion, LV morphology, and function after ST‐elevation MI in humans. We hypothesized that FXIIIa might predict cardiac remodelling after MI, whereas low FXIIIa would be associated with adverse remodelling.
Methods
Study design and ethical considerations
The patients included in the following analysis participated in the ‘Etiology, Titre‐Course, and Survival’ (ETiCS) study performed in Würzburg. 17 The ETiCS study is an investigator‐initiated prospective multicentric diagnostic study conducted under the auspices of the Competence Network Heart Failure in Germany. It has an independent Data Monitoring and Endpoint Committee and complies with the Good Clinical Practice standards. The study conformed to the Declaration of Helsinki and was approved by the responsible ethics committees. The sub‐study presented here reports on findings in patients recruited at the study centre in Würzburg. For the additional diagnostic evaluations, an additional positive ethics vote and informed consent were collected.
Patient selection
The details of study protocol have been reported earlier. 17 In brief, patients with ST‐elevation MI or equivalent defined as newly documented left bundle branch block with typical MI symptoms, aged >18 years, without the history of diseases covered by exclusion criteria, 17 , 18 and willing to provide a written informed consent, were included in the study from May 2010 to May 2017. The comprehensive list of eligibility criteria is provided in Table S1 .
Study flow
Study inclusion took place as early as possible after admission to the hospital for acute MI and subsequent revascularization (mean: 1.2 ± 0.5 days after admission). Patients' records were used as the initial screening procedure. Figure 1 details the sequence of study procedures. Baseline data collection comprised patients' medical history, current medication, physical examination, and records from left heart catheterization procedure. Cardiac magnetic resonance imaging (cMRI) tomography was performed within the first 96 h (Scan 1) and on Day 7 to 9 (Scan 2) after MI. Follow‐up visits in 2 and 6 months included cardiological evaluation and echocardiography. The complete cardiological evaluation including cMRI (Scan 3) was repeated 12 months after study inclusion. Blood samples were collected for routine lab and study biosamples at the study inclusion. Creatine kinase‐muscle/brain (CK‐MB) and N‐terminal prohormone B‐type natriuretic peptide (NT‐proBNP) were measured serially on Days 1, 2, and 5, and after 2, 6, and 12 months. Additionally, serial venous blood was sampled to measure FXIIIa on the day of study inclusion (Day 1); Days 2, 3, 5, 7, and 9; and after 2, 6, and 12 months. Further technical information regarding the materials used and details on analysis of blood values is provided in Methods S1.
Figure 1.
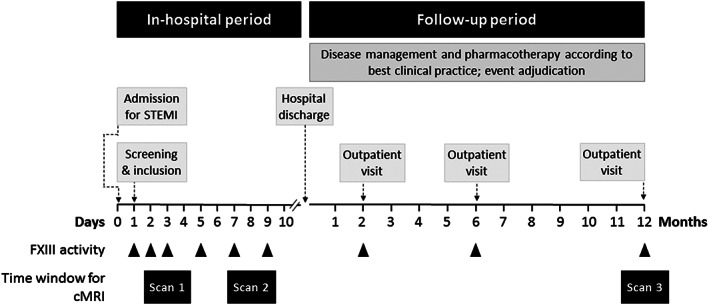
Study flow. cMRI, cardiac magnetic resonance imaging; FXIII, factor XIII.
Measurement of factor XIII activity
FXIIIa was measured by photometry on BCS‐XP analyser (Siemens Healthcare GmbH, Erlangen, Germany) using Berichrom FXIII (OWSU11) with reagents and protocols from the manufacturer according to the following principle. FXIII of the sample is activated to FXIIIa through thrombin of the assay. FXIIIa cross‐links a small‐molecular‐weight substrate to a glutamine‐containing oligopeptide, and ammonia is released in this transglutaminase reaction. Ammonia release is measured by glutamate dehydrogenase mediated NADPH‐dependent indicator reaction (ammonia release assay). The results for FXIIIa are given in % of normal. The reference interval for FXIIIa (70–140% of the norm; values 70–140 U/dL or 0.70–1.4 U/mL) is determined by Standard Human Plasma as a calibrator (Siemens Healthcare Diagnostics Products GmbH, Marburg, Germany).
Cardiac magnetic resonance imaging
Image acquisition
A 3.0‐T MR scanner (MAGNETOM Trio and MAGNETOM Prisma, Siemens Healthcare, Erlangen, Germany) was used. The protocol included three steady‐state free precession cine images in long axis and a full stack of short‐axis cine images from the mitral valve plane to the apex. Typical image parameters were repetition time (TR) 40 ms, echo time (TE) 1.5 ms, flip angle 50°, slice thickness 8 mm without interslice gap, field of view (FOV) 320–380 mm, and matrix size 256 × 216. For late gadolinium enhancement (LGE), the imaging of inversion recovery gradient echo sequence in long and short axes was acquired 15 min after administration of 0.2 mL/kg of gadobenate dimeglumine (MultiHance®, Bracco Imaging, Konstanz, Germany). The stack of short‐axis images ranged from base to apex with typical image parameters TR 750 ms, TE 1.5 ms, flip angle 40°, slice thickness 8 mm without interslice gap, FOV 320–380 mm, and matrix 256 × 256. The inversion time was individually adapted to null normal myocardium with typical values ranging from 250 to 350 ms.
Image analysis
The images were analysed by a single observer with >5 years of experience in cMRI (T. G.) using commercially available certified cMRI evaluation software (cmr42, Circle Cardiovascular Imaging, Calgary, Canada). The reader was blinded to all further data. Endocardial and epicardial contours were manually delineated on all short‐axis slices. LV end‐diastolic (LVEDV), end‐systolic (LVESV) and stroke volume (LVSV), LV ejection fraction (LVEF), and LV mass (LVM) were calculated from the short‐axis cine images. To obtain the myocardial mass, the volume was multiplied by the standard for myocardial density (1.05 g/mL). 19 IS expressed in grams was assessed from LGE short‐axis images. Semi‐automated threshold detection was used to identify infarcted myocardium defined as signal intensity > 5 standard deviations over a region of interest placed in the remote myocardium. Hypo‐intense areas representing microvascular obstruction in the LGE images were manually included in the infarct volume. Relative IS (rIS) was defined as the percentage of IS by systolic LVM. With regard to early healing processes, the term infarct expansion has been coined, describing the disproportionate thinning and dilatation of the infarcted segment, which probably begins within hours after MI and usually reaches a peak in 7 to 14 days. 20 To assess the initial period of infarct expansion, the differences between rIS on Days 7–9 vs. Days 3–4 were applied.
Genomic DNA extraction and genotyping
The blood was taken fresh using S‐Monovette® 7.5 mL K3E (REF 01.1605.001, SARSTEDT AG & Co., Nürnbrecht, Germany) and directly provided for further processing to the institute of pharmacology responsible for gathering and storage of samples until analysis. The blood for DNA analysis was immediately frozen and stored at −80°C until DNA extraction. Following manufacturer's instructions, genomic DNA was isolated from 200 μL of EDTA anticoagulated whole blood using the ‘QIAamp DNA Blood Mini Kit’ (Cat No. 51106; Qiagen, Hilden, Germany). DNA concentration was measured with a NanoDrop Spectrophotometer (Thermo Scientific, Waltham, USA), and samples were diluted to a concentration of 8 ng/μL. Five microlitres of DNA was used for genotyping by Kompetitive Allele Specific PCR (KASPTM, LGC Genomics, Teddington, UK). KASP assay mix (two SNP‐discrimination primers and one common primer) was purchased from LGC Genomics using ‘KASP by design’ (Catalogue No. KBS‐2100‐100). The SNP under investigation (rs5985) and a 50‐base‐pair surrounding sequence were submitted to LGC Genomics (ATAACTCTAATGCAGCGGAAGATGACCTGCCCACAGTGGAGCTTCAGGGC[G/T]TGGTGCCCCGGGGCGTCAACCTGCAAGGTATGAGCATACCCCCCTTCCCC). In silico primer design was done by LGC Genomics using a software developed by the company (‘Kraken™’, LGC, Biosearch Technologies, England, UK). The samples were analysed in 96‐well plates (Bio‐Rad, Hercules, USA; Catalogue No. MLL‐9601). Each 96‐well plate included four negative controls and all three genotypes (G/G, G/T, and T/T; control genotypes were confirmed by Sanger sequencing). PCR was carried out by a CFX96TM Real‐Time PCR System (Bio‐Rad, Hercules, USA). The thermal cycling program starts with an activation incubation at 94°C for 15 min. PCR cycles were two‐step cycles [denature at 94°C for 20 s, annealing/elongation for 60 s (10 cycles starting at 61°C and dropping 0.6°C each cycle to 55°C, followed by 26 cycles at 55°C)]. Allelic discrimination data were displayed in a relative fluorescence unit graph. Successful genotyping resulted in a three‐cluster pattern.
Data analysis
Data are expressed as mean (standard deviation), median (quartiles, range), or n (%), as appropriate. Group‐wise comparisons were performed using Fisher's exact test, χ 2 test, Mann–Whitney U‐test, or Kruskal–Wallis test, as appropriate. Correlation analysis included the lowest FXIII values after MI with the above‐mentioned cMRI outcome parameters. Spearman's correlation coefficient (rho) is given as well as the respective P‐values. Owing to missing values of FXIIIa at different time points, FXIIIa values of Days 3–4, 5–6, and 7–9 were aggregated to enable meaningful statistical evaluations. Consequently, the reported numbers per investigated time point in tables and figures vary slightly.
LVEDV, LVESV, LVSV, and LVM were indexed to body surface and labelled LVEDVi, LVESVi, LVSVi, and LVMi, respectively. LVEF, rIS, and infarct expansion were included in prognostic analyses as cardiac MRI outcome variables. Three different time points of cMRI scans were used to address early healing processes and long‐term remodelling. To assess the role of FXIIIa on long‐term cardiac remodelling, the differences between LVEDVi, LVEF, and rIS measured at Month 12 vs. Days 3–4 were used. Analysis of covariance was performed to analyse the influence of FXIII V34L SNP (rs5985) genotype distribution on the outcome cMRI values. All data analyses were performed on a complete case basis. Missing values were not imputed; thus, the number of observations in each analysis depends on the availability of data of all variables and covariables in a model. Potential confounders of trough levels of FXIIIa were analysed using multiple linear regression including maximal CK‐MB measured within the first 5 days after MI and pain‐to‐balloon time. The estimated regression coefficients are given as well as the standard errors and P‐values. All tests are given two‐tailed and should be considered exploratory. Hence, no correction for multiple testing was introduced. A probability of P < 0.05 was considered statistically significant. Statistical analysis was performed using R3.1.1. 21
Results
Baseline characteristics
One hundred fifty patients were initially included in the study; 34 patients left the study prematurely owing to various reasons. Three patients died: one patient on Day 9 owing to pneumonia; one patient on Day 11 owing to LV rupture; and one patient on Day 7 owing to refractory ventricular arrhythmias. Of the remaining 31 patients, 17 patients terminated the study while still in hospital: eight for inability to undergo cMRI and nine for other patient‐specific reasons. The remaining 14 patients withdrew the study consent after discharge from hospital. As 30 of 34 patients (88%) who prematurely terminated the study had undergone some of initial diagnostic procedures, the information was used in analysis as appropriate.
Therefore, information on baseline characteristics is provided on 146 patients aged 58.2 ± 11.4 years (range 25–85 years; 13% female). Their clinical characteristics, presented in Table 1 , are in accordance with published data on cardiovascular risk profile in patients with MI. 25 , 26
Table 1.
Group‐wise comparison of baseline characteristics of study participants according to the median value of factor XIII activity
| All patients | Factor XIII activity (%) | P‐value | ||
|---|---|---|---|---|
| n = 146 | <109%, n = 61 | ≥109%, n = 62 | ||
| Age (years) | 57.7 (11.5) | 57.9 (12.5) | 57.1 (11.0) | 0.711 |
| Female sex | 19 (13.0) | 4 (6.6) | 9 (14.5) | 0.240 |
| Vital signs at admission | ||||
| Systolic BP (mmHg) | 122 (21) | 116 (19 | 126 (20) | 0.009 |
| Diastolic BP (mmHg) | 73 (14) | 71 (14 | 75 (14) | 0.094 |
| Heart rate° (1/min) | 79 (14) | 80.9 (12) | 77 (15) | 0.090 |
| Co‐morbidities | ||||
| Hypertension | 74 (50.7) | 27 (44.3) | 35 (56.5) | 0.209 |
| Diabetes mellitus | 28 (19.2) | 10 (16.4) | 12 (19.4) | 0.815 |
| Dyslipidaemia | 47 (32.4) | 17 (28.3) | 21 (33.9) | 0.561 |
| BMI (kg/m2) | 27.9 (4.5) | 28.0 (4.9) | 27.7 (4.1) | 0.731 |
| Current smoker | 68 (46.6) | 22 (36.1) | 32 (51.6) | 0.103 |
| Previous CAD | 59 (40.4) | 20 (32.8) | 30 (48.4) | 0.099 |
| Previous myocardial infarction | 15 (10.3) | 9 (14.8) | 4 (6.5) | 0.154 |
| Previous heart failure | 14 (9.6) | 5 (8.2) | 6 (9.7) | 1.000 |
| Atrial fibrillation | 5 (4.1) | 1 (2.1) | 3 (5.4) | 0.622 |
| Peripheral vascular disease | 7 (20.6) | 2 (13.3) | 5 (35.7) | 0.215 |
| Cerebrovascular disease | 2 (5.7) | 0 (0) | 1 (7.1) | 0.467 |
| COPD | 5 (12.5) | 3 (17.6) | 1 (5.6) | 0.338 |
| Anaemia | 23 (16) | 13 (21.3) | 7 (11.3) | 0.150 |
| Renal dysfunction | 11 (7.6) | 7 (11.5) | 2 (3.2) | 0.095 |
| Uncured malignancy | 3 (7.5) | 1 (5.9) | 2 (11.1) | 1.000 |
| Depression | 12 (30) | 6 (35.3) | 3 (16.7) | 0.264 |
| Pre‐procedural medication | ||||
| Unfractionated heparin | 124 (84.9) | 49 (80.3) | 55 (88.7) | 0.222 |
| LMW heparin | 7 (4.8) | 5 (8.2) | 1 (1.6) | 0.114 |
| Aspirin | 131 (91.6) | 54 (91.5) | 57 (93.4) | 0.741 |
| Thienopyridine | 19 (13.4) | 7 (11.9) | 8 (13.3) | 1.000 |
| Procedure | ||||
| Pain‐to‐balloon time (min) | 202 (120–510) | 280 (150–594) | 168 (95–298) | 0.007 |
| Culprit vessel | ||||
| LAD | 66 (53.2) | 32 (60.4) | 27 (50.0) | 0.333 |
| CX | 22 (27.5) | 8 (27.6) | 8 (22.9) | 0.774 |
| RCA | 58 (39.7) | 21 (34.4) | 27 (43.5) | 0.357 |
| Successful intervention | 140 (95.9) | 58 (95.1) | 60 (96.8) | 0.680 |
| Stent implantation | 140 (95.9) | 59 (96.7) | 59 (95.2) | 1.000 |
| Classification of CAD | ||||
| 1‐vessel | 76 (52.1) | 31 (50.8) | 34 (54.8) | 0.719 |
| 2‐vessel | 44 (30.1) | 22 (36.1) | 16 (25.8) | 0.246 |
| 3‐vessel | 26 (17.8) | 8 (13.1) | 12 (19.4) | 0.465 |
| Peri‐procedural medication | ||||
| Unfractionated heparin | 143 (97.9) | 60 (98.4) | 60 (96.8) | >0.99 |
| Fondaparinux | 1 (0.7) | 0 (0.0) | 1 (1.6) | >0.99 |
| Aspirin | 4 (2.7) | 2 (3.3) | 1 (1.6) | 0.619 |
| Thienopyridine | 105 (71.9) | 45 (73.8) | 44 (71) | 0.841 |
| GP‐IIbIIIa inhibitor | 42 (28.8) | 25 (41) | 13 (21) | 0.020 |
| Laboratory parameters | ||||
| Haemoglobin | 14.1 (13.1–14.9) | 14.1 (13.1–14.9) | 14.1 (13.2–14.9) | 0.595 |
| Creatinine | 0.9 (0.8–1.0) | 1.0 (0.8–1.0) | 0.9 (0.8–1.0) | 0.156 |
| eGFR (MDRD) | 89 (77–99) | 88 (76–99) | 92 (78–98) | 0.326 |
| CK‐MB Day 1 | 108 (57–180) | 126 (79–208) | 94 (54–177) | 0.172 |
| CK‐MB peak value | 137 (72–256) | 158 (77–291) | 11 (55–202) | 0.136 |
| Leukocytes | 10.9 (9.2–12.9) | 11.1 (8.9–13.7) | 10.6 (9.1–11.7) | 0.246 |
| C‐reactive protein | 0.76 (0.34–1.93) | 0.84 (0.36–3.11) | 0.52 (0.25–1.25) | 0.035 |
BMI, body mass index: weight in kilograms divided by the square of height in meters; BP, blood pressure; CAD, coronary artery disease; CK‐MB, creatine kinase MB isoenzyme; COPD, chronic obstructive lung disease; CX, circumflex artery; eGFR, estimated glomerular filtration rate; LAD, left anterior descending artery; LMW, low molecular weight; RCA, right coronary artery.
Data are mean (SD), median [quartiles], or n (%).
Definitions applied: hypertension, history of hypertension; atrial fibrillation, diagnosed from the electrocardiogram or electrocardiogram; COPD, history of this condition requiring bronchiolytic treatment, or newly diagnosed according to GOLD 2016 criteria 22 ; anaemia, haemoglobin < 12 g/dL in women and <13 g/dL in men (WHO criteria) 23 ; renal dysfunction, estimated glomerular filtration rate < 60 mL/min/1.7 3m2. 24
Heart rate according to electrocardiogram.
Changes in factor XIII activity after myocardial infarction
Median FXIIIa dropped from baseline (118%, 102–132%) and reached a trough on the second day after MI (109%, 98–109%; P < 0.001). FXIIIa recovered slowly over time and reached levels similar to those measured at baseline after a period of 2 to 6 months. Only at the time point 12 months after MI had FXIIIa surpassed baseline values (Figure 2 and Table S2 ).
Figure 2.
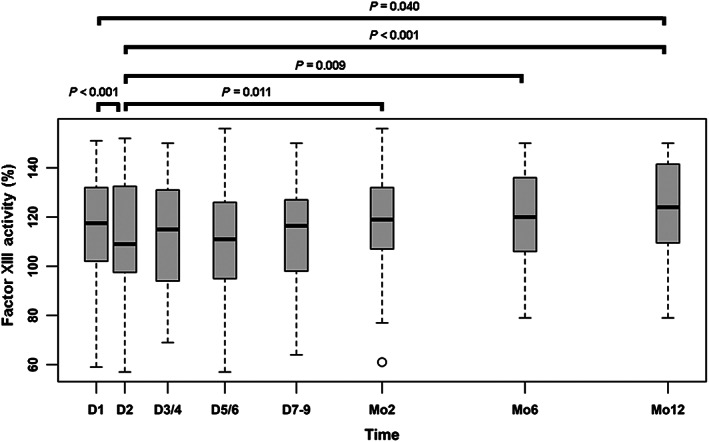
Short‐term and long‐term changes of FXIII activity levels after myocardial infarction. FXIIIa significantly declined in the course of the first days after myocardial infarction and recovered within 2 months. After 12 months, the levels of FXIIIa were significantly higher compared with those on the first post‐MI measurement on Day 2. D, day; FXIII, factor XIII; Mo, month.
The lowest FXIIIa after MI was observed on Day 2 (median FXIIIa 109%). We therefore compared patient groups with FXIIIa above and below 109% with regard to their clinical characteristics that revealed some differences between the groups (Table 1). Patients with higher FXIIIa had higher systolic blood pressure (P = 0.009), experienced chest pain prior to hospitalization for a shorter duration (P = 0.007), required treatment with glycoprotein‐IIb/IIIa inhibitors (P = 0.020) slightly less often, and exhibited lower C‐reactive protein values (P = 0.035). Gender appeared to have no relevant impact (P = 0.240) on FXIIIa values in our cohort.
Correlation of factor XIIIa with other cardiac markers
Data of 116 patients who completed the 12 month examination were used for the long‐term analysis of remodelling processes after MI. The lowest individual level of FXIIIa was inversely associated with the highest CK‐MB values (ρ = –0.201; P = 0.035) and the highest NT‐proBNP values (ρ = –0.235; P = 0.009) after MI.
Relation of factor XIIIa with cardiac magnetic resonance imaging parameters
A positive correlation of FXIIIa trough levels with LVEF was observed both at baseline cMRI Scan 1 (ρ = 0.32; P < 0.01) and after 12 months (cMRI Scan 3; ρ = 0.24; P = 0.04). Additionally, LVEF at cMRI Scan 1 was associated with all the measurements of FXIIIa during the first 9 days after MI. The respective association of LVEF at cMRI Scan 2 was less consistent (Figure 3A ; Table S4 ; Figures S2 and S3). However, no association was found between trough FXIIIa and short‐term (ρ = –0.13; P = 0.31) and long‐term (ρ = –0.05; P = 0.69) change in LVEF (calculated as the respective difference of Scan 2 minus Scan 1, and Scan 3 minus Scan 1; Table S4 ).
Figure 3.
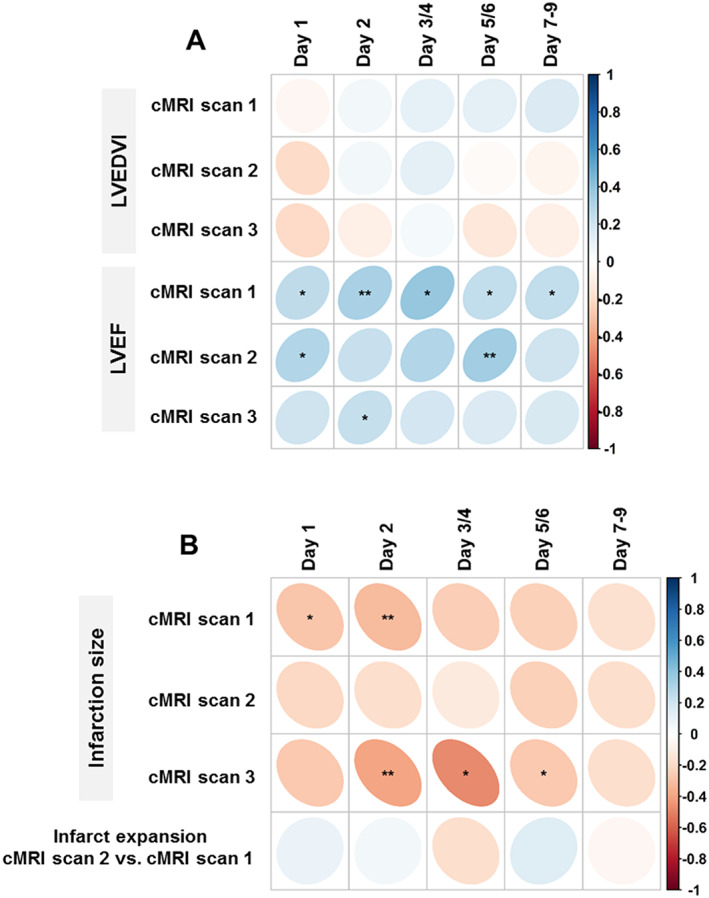
Relation of FXIIIa levels with indices of cardiac remodelling. Spearman correlation between FXIIIa levels measured within the first 7 to 9 days after myocardial infarction and repetitively measured cardiac MRI parameters. (A) Strength of correlation with LVEDVi and LVEF at baseline (i.e. within the first 96 h after MI; cMRI Scan 1), after 7–9 days (Scan 2), and after 12 months (Scan 3). (B) Strength of correlation with rIS at baseline (i.e. within the first 96 h after MI; cMRI Scan 1), after 7–9 days (Scan 2), and after 12 months (Scan 3); and with infarct expansion represented by the difference in rIS between Scan 2 and Scan 1. The colour represents the correlation coefficient, with bluish colours for positive and reddish colours for negative correlations. The area of the individual dot represents the number of patients in each separate analysis. LVEDVi, left ventricular end‐diastolic volume indexed to body surface area; LVEF, left ventricular ejection fraction; rIS, relative infarct size. * indicates P < 0.05, and ** P < 0.01.
No correlation was found between trough levels of FXIIIa and the LVEDVi at different time points after MI (Figure 3A ; Table S3 ). Trough FXIII levels were also not associated with short‐term (ρ = −0.12; P = 0.36) and long‐term (ρ = –0.09; P = 0.45) development of LVEDVi after MI (calculated as the respective differences of Scan 2 minus Scan 1, and Scan 3 minus Scan 1; Table 2; Table S3 ).
Table 2.
Cardiac MRI outcome variables
| Number of patients | Result | |
|---|---|---|
| LVEDVi (Scan 1), ml/m2 | 97 | 87.44 (14.80) |
| LVEDVi (Scan 2), mL/m2 | 73 | 90.77 (14.95) |
| LVEDVi (Scan 3), mL/m2 | 86 | 86.36 (17.55) |
| Δ LVEDVi (Scan 2 minus Scan 1), mL/m2 | 63 | 1.68 (8.37) |
| Δ LVEDVi (Scan 3 minus Scan 1), mL/m2 | 75 | 0.45 (12.43) |
| Δ LVEDVi (Scan 3 minus Scan 2), mL/m2 | 56 | −3.11 (11.94) |
| LVEF (Scan 1), % | 97 | 48.12 (9.81) |
| LVEF (Scan 2), % | 73 | 49.86 (9.66) |
| LVEF (Scan 3), % | 86 | 53.56 (9.55) |
| Δ LVEF (Scan 2 minus Scan 1), % | 63 | 1.45 (5.96) |
| Δ LVEF (Scan 3 minus Scan 1), % | 75 | 2.79 (6.65) |
| Δ LVEF (Scan 3 minus Scan 2), % | 56 | 0.29 (7.06) |
| Infarct size (Scan 1), % | 85 | 0.20 (0.13) |
| Infarct size (Scan 2), % | 66 | 0.17 (0.10) |
| Infarct size (Scan 3), % | 75 | 0.10 (0.08) |
| Infarct expansion (Scan 2 minus Scan 1), % | 54 | −0.015 (0.045) |
Data are mean (SD). Delta (Δ) values were calculated as the differences of respective cMRI scans, as indicated.
cMRI, cardiac magnetic resonance imaging; LVEDVi, end‐diastolic volume of the left ventricle indexed to body surface area; LVEF, left ventricular ejection fraction.
The rIS at cMRI Scan 1 inversely correlated with FXIIIa measured on Day 1 (ρ = –0.28; P = 0.03) and Day 2 (ρ = –0.31; P = 0.01). Additionally, FXIII activities on Days 2 (ρ = –0.39; P < 0.01), 3–4 (ρ = –0.47; P = 0.03), and 5–6 (ρ = –0.26; P = 0.04) were inversely associated with rIS assessed in cMRI Scan 3 after 12 months. No significant association was found between infarct expansion and FXIIIa (Figure 3B; Table 2 ; Table S3 ; Figure S4).
In search of potential factors confounding the association of trough levels of FXIIIa and above reported markers of cardiac remodelling, multivariable regression modelling was used to adjust for clinical markers of myocardial injury. In a model predicting rIS, maximal CK‐MB measured within the first 5 days after MI and pain‐to‐balloon time emerged as relevant and independent clinical variables. We found that association of FXIIIa trough levels with LVEF was confounded by these factors, whereas FXIIIa remained an independent predictor of rIS, regardless of maximal CK‐MB levels and pain‐to‐balloon time (Table 3 ).
Table 3.
Multiple regression analysis testing the independency of the association of the trough factor XIIIa level with cardiac MRI outcome variables
| Outcome | Variable | B | Standard error | T | P‐value |
|---|---|---|---|---|---|
| LVEF (Scan 1) | CK‐MB maximal | −0.019 | 0.006 | −3.084 | 0.003 |
| Pain‐to‐balloon time | 0.001 | 0.002 | 0.556 | 0.580 | |
| FXIIIa Day 2 | 0.077 | 0.046 | 1.689 | 0.095 | |
| LVEF (Scan 3) | CK‐MB maximal | −0.033 | 0.009 | −3.522 | 0.001 |
| Pain‐to‐balloon time | 0.001 | 0.002 | 0.605 | 0.547 | |
| FXIIIa Day 2 | 0.061 | 0.049 | 1.251 | 0.216 | |
| Infarct size (Scan 1) | CK‐MB maximal | 0.000 | 0.000 | 3.957 | <0.001 |
| Pain‐to‐balloon time | 0.000 | 0.000 | −0.158 | 0.875 | |
| FXIIIa Day 2 | −0.001 | 0.001 | −1.770 | 0.082 | |
| Infarct size (Scan 3) | CK‐MB maximal | 0.000 | 0.000 | 1.165 | 0.249 |
| Pain‐to‐balloon time | 0.000 | 0.000 | −0.551 | 0.584 | |
| FXIIIa Day 2 | −0.001 | 0.000 | −2.103 | 0.040 |
T indicates comparative importance of a variable in the model.
cMRI, cardiac magnetic resonance imaging; LVEF, left ventricular ejection fraction; FXIIIa, factor XIII activity; CK‐MB, creatine kinase MB isoenzyme.
Genetic analyses
Consent for genetic analyses was available for 124 patients; 59% (n = 73) of the patients had G/G, 37% (n = 46) G/T, and only 4% (n = 5) T/T genotype, which was in Hardy–Weinberg equilibrium (χ 2 = 0.46; P = 0.5) 27 as far as genotype frequencies in accordance with the published literature on the general population. 28 The groups did not differ on the time course of FXIIIa dependent on the genetic polymorphism (Figure S1), although the availability of FXIIIa values was strongly underrepresented in the T/T group, most likely by chance. Therefore, the interpretation of these results is limited.
As only three patients receiving cMRI were of T/T genotype, we excluded this genotype group from further analysis. Having compared cMRI outcome in G/G and G/T group, we found that after 12 months LVEDVi in the G/G group was slightly smaller (80.9 vs. 89.7 mL/m2; P = 0.032) and LVEF tended to be slightly higher (56.1% vs. 51.9%; P = 0.059) (Table S4 ).
Discussion
This study provides new and important insight into the dynamics of myocardial healing and the role of coagulation factor XIII activity in early healing processes and cardiac remodelling after acute MI. FXIIIa was depressed following MI, reaching a trough on Day 2 and exhibited very prolonged recovery. Higher levels of FXIIIa at trough were associated with better LVEF and reduced IS both at baseline and after 12 months. Our findings point towards an important role of FXIIIa during infarct healing, which should be systematically explored as a potential target influencing cardiac remodelling processes in patients after MI.
Development of factor XIII activity after myocardial infarction in the context of published data
As of yet, the data on the role and development of FXIIIa after MI are very limited. In the studies performed at our institution, FXIII deficiency is associated with deleterious influence on early healing processes after experimental MI. 12 Additionally, in a case series of nine patients with cardiac rupture after myocardial infarction, FXIIIa was strongly depressed. 13 Consistently with these findings, FXIIIa experienced a decline within the first 2 days but returned to normal values in several months after MI in our cohort.
In the prospective study comprising 350 patients with MI, decrease of FXIIIa was associated with the development of heart failure and increased mortality risk. 14 , 15 , 29 Compared with our data, the patients in the study of Gemmati et al. had lower baseline FXIIIa (95.0 ± 10.1%) and experienced stronger decline in FXIIIa within the first 3 to 4 days after MI (54.4 ± 6.5%); that is, relative decline of 40% in FXIII values from Day 0 to Day 4. The decline of FXIIIa was considerably less pronounced in our cohort, that is, from 118% to 109% (relative decline of 7.6%). This may be explained either by the first measurement of FXIII only a day after MI or by a less serious condition of our MI patient group. However, it remains unclear on which day FXIIIa reached its trough in the cohort of Gemmati et al.
Factor XIII activity and clinical outcome
Compared with published mortality data for patients with MI, the mortality in our sample over the study duration of 1 year was low. The low rate is likely to be influenced by selection bias: only stable patients who were able to undergo a sophisticated cMRI protocol of around 120 min were included in the study. It is worth noting that all three patients who died within the 12 month follow‐up of our study exhibited considerably diminished FXIII activity. The clinical characteristics of our patients presented in Table 1 correlate with the published data on cardiovascular risk profile in MI patients. 25 , 26
Factor XIII activity and imaging‐based outcome of patients
Currently, there are no published data with regard to the role of FXIII on cardiac remodelling after MI as measured by sequential cardiac MRI in humans. We have analysed the interaction of trough FXIIIa with predefined MRI outcome parameters and have found intriguing associations of trough FXIIIa after MI with rIS and LVEF both at baseline and after 12 months. Lower FXIIIa was associated with lower LVEF and larger rIS, meaning that FXIIIa as early as 2 days after MI might inform on adverse outcomes after 12 months.
Echocardiography‐based assessment of LVEF was performed by Gemmati et al. only once in their cohort of patients with MI, 29 in contrast to the sequential assessment of cardiac functioning via cMRI in our cohort. Therefore, Gemmati et al. reported no sequential LVEF assessment; and LVEF, in general, was reported only for patients experiencing adverse events. Furthermore, the timing of the LVEF assessment was not defined in previously published data. It is also worth noting that the lowest LVEF values were observed among the patients with heart failure, and those who died during the study had the lowest FXIIIa as postulated by Gemmati et al. Higher FXIIIa and higher LVEF were found in the subgroup with major adverse cardiac event of this previously published cohort. 29
Genetic factor XIII polymorphism and cardiac magnetic resonance imaging‐based outcome of patients
The current study is first to report on the association of the most common FXIII SNP rs5985 with MRI‐based outcome after MI. Gene variants (V34L) increasing FXIIIa were associated with protective inherited predisposition to ischaemic disease 14 and improved survival after MI. 15 The development of FXIII after MI in our cohort was independent of the genotype, although the T/T genotype (corresponding to LL34 homozygotes) that possibly conveys the highest FXIII activity 15 , 30 was strongly underrepresented in the subgroup with available MRI data. Surprisingly, patients with G/G genotype (corresponding to VV34 homozygotes) and assumed lowest FXIII activity, according to the literature, had smaller ventricles and higher LVEF 12 months after MI, than had the G/T group.
Mechanistic considerations
FXIII is an enzyme of blood coagulation system that cross‐links the fibrin mesh to a stable fibrin clot. This final step of the coagulation cascade is common for both intrinsic and extrinsic pathways of haemostasis. The role of FXIII deficiency is addressed in several publications where several genetic polymorphisms of FXIII subdomains were characterized. 31 Furthermore, the predictive role of FXIII deficiency for cardiovascular or bleeding events has also been analysed. Nevertheless, the literature seems to be inconsistent with the effect of low FXIII activity; it is associated with bleeding, thrombotic, and embolic diseases. 32 Besides its role in coagulation, FXIII has a role in extracellular matrix biology as it acts as a transglutaminase that aids collagen cross‐linking.
Accordingly, the consistent clinical data observed here and by others 14 , 15 , 29 as well as the experimental data 12 , 13 , 33 strongly suggest that FXIII potentially plays a critical role in remodelling processes after MI. For example, all mice lacking FXIII died from LV rupture after MI, and low FXIII values are associated with unfavourable outcome in human cohorts.
We observed the initial decline of FXIIIa after MI, its recovery 2–3 months after MI and, finally, the surpassed baseline values only after 6–12 months, irrespective of the genotype. Thus, FXIII may undergo heavy consumption during early healing and subsequent remodelling processes after MI. The severity of FXIII consumption is likely to depend on the size of the MI, whereby larger MI size seems to be associated with stronger FXIII consumption, as represented by MRI data of our patients. We did not confirm our initial hypothesis that FXIIIa might be associated with infarct expansion; this probably happened because of the time points we chose for the first and second MRIs. These time points seemed to be unable to reveal the relevant association of FXIII with infarct expansion because the expansion started within the first hours, and we analysed a time frame of 4 to 7 days after MI and probably missed the initial phase of infarct expansion.
Limitations
We acknowledge several limitations of our study. Venous blood sampling for FXIIIa was not feasible on the day of admission for ST‐elevation MI (i.e. Day 0). Hence, we might have underestimated the drop occurring in the context of MI, which might explain the slightly deviating findings compared with the report of Gemmati et al. 29 The study size was modest and not large enough to comprehensively mirror the complex phenotypes emerging from MI and fully account for important factors including patient age, co‐morbidities, previous infarcts, size of infarcted area, time to and success of intervention, and post‐intervention complications. Sampling at the multiple time points mandated by the protocol was not 100% complete and resulted in missing values. Hence, diverging numbers of subjects were available for analysis per time point (i.e. matching pairs of FXIIIa and cMRI). This might have induced a bias that could work in either direction. Because imputation strategies have their limitations, too, particularly in the context of pathophysiologically driven research questions, we decided to employ a complete case approach. Finally, the reported polymorphism rs5985 (Val34Leu) is only one among several others whose role awaits clarification.
Conclusions
In a medium‐sized cohort of patients with ST‐elevation MI patients, we described for the first time the association between FXIIIa and short‐term and long‐term MRI‐based outcomes. Although no causal role is yet established, our findings are consistent with known pathophysiological mechanisms and, thus, shed a new light on the role of FXIIIa as a potentially healing‐modifying factor after an episode of ST‐elevation MI. An interventional prospective trial studying the substitution of FXIII in patients with depressed FXIIIa after MI is now indicated to clarify if such strategy may improve the outcome of patients after MI. However, such trial has to carefully take into account the not yet quantified potential risk of prothrombotic effects of FXIII substitution. One should take into consideration that in spite of protective effect of V34L polymorphism 16 and association of decreased FXIIIa and negative MRI‐based outcome, elevated FXIII levels seems to be a gender‐specific (female) risk factor of both coronary artery disease and peripheral arterial disease. 16 The trial could have only a proof‐of‐concept character, and one should strongly evaluate the cut‐off value of FXIIIa, causing FXIII substitution and monitor FXIIIa values subsequently to avoid exaggerated substitution. However, therapeutic FXIII substitution is safely performed in patients with congenital severe FXIII deficiency 34 and individuals with severely decreased FXIIIa values under intensive care conditions. 35
Conflict of interest
None declared.
Funding
This work received public funding from the BMBF (Bundesministerium für Bildung und Forschung; MolDiag: 01ES0816, 01ES01901, and 01ES01902; Comprehensive Heart Failure Center: 01EO1004).
Supporting information
Table S1. Inclusion and exclusion criteria for patients in the ETiCS study1.
Table S2. Time course of factor XIII activity (%) in study participants.
Table S3. Correlation of cMRI outcome variables and factor XIII activity within the first 9 days after myocardial infarction.
Table S4. Results for cardiac MRI outcome variables depending on the factor XIII rs5985 genotype distribution.
Figure S1. Time course of factor XIII activity after myocardial infarction by genetic factor XIII polymorphism (rs5985).
Figure S2. Correlation between baseline LVEF (cMRI) and factor XIII activity during hospitalization.
Figure S3. Correlation between LVEF after 7 to 9 days and 12 months (cMRI) and factor XIII activity during hospitalization.
Figure S4. Correlation between relative infarct size at baseline and after 12 months (cMRI) and factor XIII activity during hospitalization.
Acknowledgement
We thank the ETiCS team for their excellent support in the conduct of this comprehensive study.
Frey, A. , Gassenmaier, T. , Hofmann, U. , Schmitt, D. , Fette, G. , Marx, A. , Herterich, S. , Boivin‐Jahns, V. , Ertl, G. , Bley, T. , Frantz, S. , Jahns, R. , and Störk, S. (2020) Coagulation factor XIII activity predicts left ventricular remodelling after acute myocardial infarction. ESC Heart Failure, 7: 2354–2364. 10.1002/ehf2.12774.
References
- 1. Gaudron P, Eilles C, Kugler I, Ertl G. Progressive left ventricular dysfunction and remodeling after myocardial infarction. Potential mechanisms and early predictors. Circulation 1993; 87: 755–763. [DOI] [PubMed] [Google Scholar]
- 2. Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res 2005; 66: 22–32. [DOI] [PubMed] [Google Scholar]
- 3. Ertl G, Frantz S. Wound model of myocardial infarction. Am J Physiol Heart Circ Physiol 2005; 288: H981–H983. [DOI] [PubMed] [Google Scholar]
- 4. Frantz S, Bauersachs J, Ertl G. Post‐infarct remodelling: contribution of wound healing and inflammation. Cardiovasc Res 2009; 81: 474–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jackson BM, Gorman JH, Moainie SL, Guy TS, Narula N, Narula J, John‐Sutton MG, Edmunds LH Jr, Gorman RC. Extension of borderzone myocardium in postinfarction dilated cardiomyopathy. J Am Coll Cardiol 2002; 40: 1160–1167 discussion 1168‐71. [DOI] [PubMed] [Google Scholar]
- 6. Skyschally A, Walter B, Heusch G. Coronary microembolization during early reperfusion: infarct extension, but protection by ischaemic postconditioning. Eur Heart J 2013; 34: 3314–3321. [DOI] [PubMed] [Google Scholar]
- 7. Holmes JW, Yamashita H, Waldman LK, Covell JW. Scar remodeling and transmural deformation after infarction in the pig. Circulation 1994; 90: 411–420. [DOI] [PubMed] [Google Scholar]
- 8. McKay RG, Pfeffer MA, Pasternak RC, Markis JE, Come PC, Nakao S, Alderman JD, Ferguson JJ, Safian RD, Grossman W. Left ventricular remodeling after myocardial infarction: a corollary to infarct expansion. Circulation 1986; 74: 693–702. [DOI] [PubMed] [Google Scholar]
- 9. Whittaker P, Boughner DR, Kloner RA. Role of collagen in acute myocardial infarct expansion. Circulation 1991; 84: 2123–2134. [DOI] [PubMed] [Google Scholar]
- 10. Wartiovaara U, Mikkola H, Szoke G, Haramura G, Karpati L, Balogh I, Lassila R, Muszbek L, Palotie A. Effect of Val34Leu polymorphism on the activation of the coagulation factor XIII‐A. Thromb Haemost 2000; 84: 595–600. [PubMed] [Google Scholar]
- 11. Kobbervig C, Williams E. FXIII polymorphisms, fibrin clot structure and thrombotic risk. Biophys Chem 2004; 112: 223–228. [DOI] [PubMed] [Google Scholar]
- 12. Nahrendorf M, Hu K, Frantz S, Jaffer FA, Tung CH, Hiller KH, Voll S, Nordbeck P, Sosnovik D, Gattenlohner S, Novikov M, Dickneite G, Reed GL, Jakob P, Rosenzweig A, Bauer WR, Weissleder R, Ertl G. Factor XIII deficiency causes cardiac rupture, impairs wound healing, and aggravates cardiac remodeling in mice with myocardial infarction. Circulation 2006; 113: 1196–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nahrendorf M, Aikawa E, Figueiredo JL, Stangenberg L, van den Borne SW, Blankesteijn WM, Sosnovik DE, Jaffer FA, Tung CH, Weissleder R. Transglutaminase activity in acute infarcts predicts healing outcome and left ventricular remodelling: implications for FXIII therapy and antithrombin use in myocardial infarction. Eur Heart J 2008; 29: 445–454. [DOI] [PubMed] [Google Scholar]
- 14. Ansani L, Marchesini J, Pestelli G, Luisi GA, Scillitani G, Longo G, Milani D, Serino ML, Tisato V, Gemmati D. F13A1 gene variant (V34L) and residual circulating FXIIIA levels predict short‐ and long‐term mortality in acute myocardial infarction after coronary angioplasty. Int J Mol Sci 2018; 19: 2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gemmati D, Federici F, Campo G, Tognazzo S, Serino ML, De Mattei M, Valgimigli M, Malagutti P, Guardigli G, Ferraresi P, Bernardi F, Ferrari R, Scapoli GL, Catozzi L. Factor XIIIA‐V34L and factor XIIIB‐H95R gene variants: effects on survival in myocardial infarction patients. Mol Med 2007; 13: 112–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bagoly Z, Koncz Z, Harsfalvi J, Muszbek L. Factor XIII, clot structure, thrombosis. Thromb Res 2012; 129: 382–387. [DOI] [PubMed] [Google Scholar]
- 17. Deubner N, Berliner D, Schlipp A, Gelbrich G, Caforio AL, Felix SB, Fu M, Katus H, Angermann CE, Lohse MJ, Ertl G, Stork S, Jahns R, Etiology T‐C, Survival‐Study G . Cardiac beta1‐adrenoceptor autoantibodies in human heart disease: rationale and design of the Etiology, Titre‐Course, and Survival (ETiCS) Study. Eur J Heart Fail 2010; 12: 753–762. [DOI] [PubMed] [Google Scholar]
- 18. Antman EM, Anbe DT, Armstrong PW, Bates ER, Green LA, Hand M, Hochman JS, Krumholz HM, Kushner FG, Lamas GA, Mullany CJ, Ornato JP, Pearle DL, Sloan MA, Smith SC Jr, Alpert JS, Anderson JL, Faxon DP, Fuster V, Gibbons RJ, Gregoratos G, Halperin JL, Hiratzka LF, Hunt SA, Jacobs AK, American College of Cardiology/American Heart Association Task Force on Practice G . ACC/AHA guidelines for the management of patients with ST‐elevation myocardial infarction—executive summary: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to Revise the 1999 Guidelines for the Management of Patients With Acute Myocardial Infarction). Circulation 2004; 110: 588–636. [DOI] [PubMed] [Google Scholar]
- 19. Shimamoto R, Suzuki J, Nishikawa J, Fujimori Y, Nakamura F, Shin WS, Tomaru T, Toyo‐oka T. Measuring the diameter of coronary arteries on MR angiograms using spatial profile curves. AJR Am J Roentgenol 1998; 170: 889–893. [DOI] [PubMed] [Google Scholar]
- 20. Weisman HF, Healy B. Myocardial infarct expansion, infarct extension, and reinfarction: pathophysiologic concepts. Prog Cardiovasc Dis 1987; 30: 73–110. [DOI] [PubMed] [Google Scholar]
- 21. Team RC . R: A language and environment for statistical computing. http://www.R-project.org/
- 22. From the global strategy for the diagnosis, management and prevention of COPD, Global Initiative for Chronic Obstructive Lung Disease (GOLD) 2016. Available from: http://goldcopd.org/ (5 October 2017).
- 23. Adams EB. Nutritional anaemias. Br J Clin Pract 1968; 22: 501–504. [PubMed] [Google Scholar]
- 24. Foundation NK . K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39: S1–S266. [PubMed] [Google Scholar]
- 25. Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, White HD. Executive Group on behalf of the Joint European Society of Cardiology/American College of Cardiology/American Heart Association/World Heart Federation Task Force for the Universal Definition of Myocardial I. Fourth universal definition of myocardial infarction (2018). Circulation 2018; 138: e618–e651. [DOI] [PubMed] [Google Scholar]
- 26. Levine GN, Bates ER, Blankenship JC, Bailey SR, Bittl JA, Cercek B, Chambers CE, Ellis SG, Guyton RA, Hollenberg SM, Khot UN, Lange RA, Mauri L, Mehran R, Moussa ID, Mukherjee D, Ting HH, O'Gara PT, Kushner FG, Ascheim DD, Brindis RG, Casey DE Jr, Chung MK, de Lemos JA, Diercks DB, Fang JC, Franklin BA, Granger CB, Krumholz HM, Linderbaum JA, Morrow DA, Newby LK, Ornato JP, Ou N, Radford MJ, Tamis‐Holland JE, Tommaso CL, Tracy CM, Woo YJ, Zhao DX. 2015 ACC/AHA/SCAI Focused Update on Primary Percutaneous Coronary Intervention for Patients With ST‐Elevation Myocardial Infarction: an update of the 2011 ACCF/AHA/SCAI guideline for percutaneous coronary intervention and the 2013 ACCF/AHA Guideline for the Management of ST‐Elevation Myocardial Infarction. J Am Coll Cardiol 2016; 67: 1235–1250. [DOI] [PubMed] [Google Scholar]
- 27. Stern C. The Hardy–Weinberg law. Science 1943; 97: 137–138. [DOI] [PubMed] [Google Scholar]
- 28. Pruissen DM, Slooter AJ, Rosendaal FR, van der Graaf Y, Algra A. Coagulation factor XIII gene variation, oral contraceptives, and risk of ischemic stroke. Blood 2008; 111: 1282–1286. [DOI] [PubMed] [Google Scholar]
- 29. Gemmati D, Zeri G, Orioli E, Mari R, Moratelli S, Vigliano M, Marchesini J, Grossi ME, Pecoraro A, Cuneo A, Ferrari R, Pinotti M, Serino ML, Ansani L. Factor XIII—a dynamics in acute myocardial infarction: a novel prognostic biomarker? Thromb Haemost 2015; 114: 123–132. [DOI] [PubMed] [Google Scholar]
- 30. Gemmati D, Tognazzo S, Serino ML, Fogato L, Carandina S, De Palma M, Izzo M, De Mattei M, Ongaro A, Scapoli GL, Caruso A, Liboni A, Zamboni P. Factor XIII V34L polymorphism modulates the risk of chronic venous leg ulcer progression and extension. Wound Repair Regen 2004; 12: 512–517. [DOI] [PubMed] [Google Scholar]
- 31. Ariens RA, Lai TS, Weisel JW, Greenberg CS, Grant PJ. Role of factor XIII in fibrin clot formation and effects of genetic polymorphisms. Blood 2002; 100: 743–754. [DOI] [PubMed] [Google Scholar]
- 32. Dorgalaleh A, Rashidpanah J. Blood coagulation factor XIII and factor XIII deficiency. Blood Rev 2016; 30: 461–475. [DOI] [PubMed] [Google Scholar]
- 33. Nahrendorf M, Weissleder R, Ertl G. Does FXIII deficiency impair wound healing after myocardial infarction? PLoS ONE 2006; 1: e48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hsieh L, Nugent D. Factor XIII deficiency. Haemophilia 2008; 14: 1190–1200. [DOI] [PubMed] [Google Scholar]
- 35. Gorlinger K, Fries D, Dirkmann D, Weber CF, Hanke AA, Schochl H. Reduction of fresh frozen plasma requirements by perioperative point‐of‐care coagulation management with early calculated goal‐directed therapy. Transfus Med Hemother 2012; 39: 104–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Inclusion and exclusion criteria for patients in the ETiCS study1.
Table S2. Time course of factor XIII activity (%) in study participants.
Table S3. Correlation of cMRI outcome variables and factor XIII activity within the first 9 days after myocardial infarction.
Table S4. Results for cardiac MRI outcome variables depending on the factor XIII rs5985 genotype distribution.
Figure S1. Time course of factor XIII activity after myocardial infarction by genetic factor XIII polymorphism (rs5985).
Figure S2. Correlation between baseline LVEF (cMRI) and factor XIII activity during hospitalization.
Figure S3. Correlation between LVEF after 7 to 9 days and 12 months (cMRI) and factor XIII activity during hospitalization.
Figure S4. Correlation between relative infarct size at baseline and after 12 months (cMRI) and factor XIII activity during hospitalization.