

ABSTRACT
The swine gut microbiome has received remarkable attention in recent years given that pigs serve not only as important sources for animal-derived food but also as excellent biomedical models for human health. However, despite recent advances in the understanding of the swine gut microbiome, many important biological and ecological questions are still largely unanswered. In a recent study, we characterized the life-long dynamics of the swine gut microbiome from birth to market. We showed distinct shifts in gut microbiome structure along different growth stages mainly driven by diet. Here, we summarize these discoveries and provide additional data related to the core swine gut microbiome, probiotics development in the swine industry, and foodborne pathogens in the pork supply chain.
KEYWORDS: Swine gut microbiome, probiotics, core microbiome, food safety
Introduction
The human microbiome has been extensively studied in the last decade and substantial progress has been made in that field regarding their membership, structure, dynamics, and correlations with different diseases.1-5 Pigs serve as important protein sources for human beings. With the increase in global population and preference for animal protein sources, it is critical to maintain sustainable and efficient swine production. Given the importance of pigs as both livestock species and as medical models for human diseases, the swine gut microbiome has been characterized by many groups throughout the world. It has been reported that the swine gut microbiome correlated with feed efficiency, fat deposition, and growth performance.6–10 Despite this progress toward understanding the swine gut microbiome, many important ecological questions are yet to be answered.
The swine industry faces enormous challenges. The emergence and spread of antimicrobial resistance (AMR) from the swine industry due to antibiotic application for disease treatment, prevention, and growth promotion have drawn pressing public health concerns,11 necessitating the use of alternatives to antibiotics, such as probiotics, in the swine industry. Furthermore, live pigs on farm serve as one of the major sources of foodborne pathogens in the pork supply chain and the environment. Given the critical roles that the gut microbiome plays in swine health and production, it is crucial to better understand the mechanisms underlying the assembly and succession of the microbial communities in pigs to identify beneficial bacteria that might serve as probiotics. It is also important to track the dynamics of the pathogens to establish an effective management strategy to reduce, if not prevent product contamination. Recently, we investigated the life-long dynamics of the swine gut microbiome in a longitudinal study at pre-harvest and addressed many important ecological questions.12 Here, we build on the discoveries from that study, particularly on the core microbiome and stage-specific bacteria and discuss their implications in the context of probiotics development and detection of foodborne pathogens.
Life-long dynamics of the swine gut microbiome
In a test animal trial, we followed 18 pigs from birth to market and found that their gut microbiome structures significantly shifted during different growth stages: lactation, nursery, growing, and finishing. Dramatic changes in gut microbiome structure occurred during weaning, when the pigs were separated from the sows and provided solid food rather than sow milk. Gradual but significant changes in swine gut microbiome structures were also observed during the different growth stages when the pigs were on a solid diet. Different signature bacterial taxa for these growth stages that drive the shifts in swine gut microbiome structure are listed in Figure 1.
Figure 1.
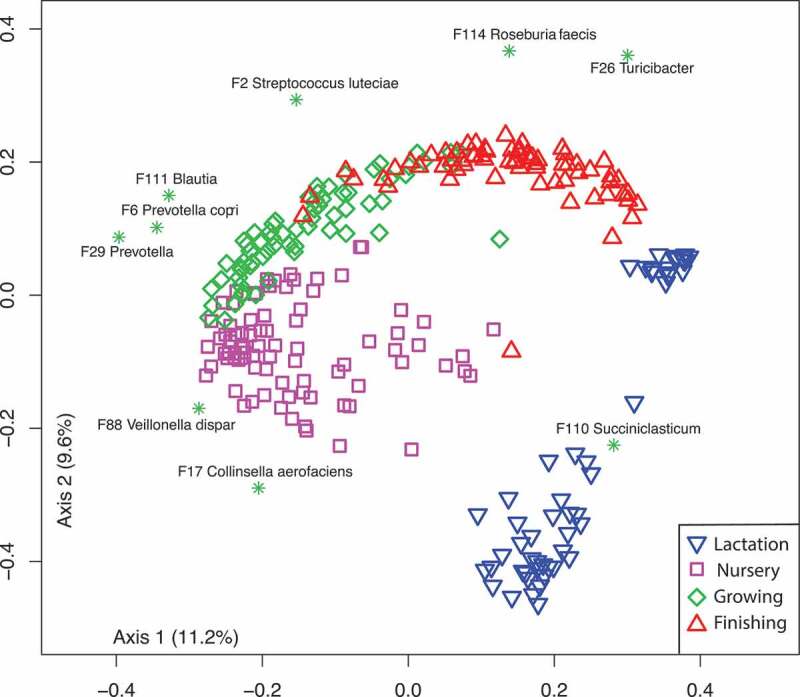
Bi-plot showing the life-long dynamics of the swine gut microbiome and the stage associated Prevotella.
Prevotella is the most abundant genus in the swine gut microbiome. Eleven of the top 30 features belong to this genus. Although three of these Prevotella features belong to the “core” gut microbiome (Table 1), especially Feature 9, which showed the highest abundance at lactation, most of these Prevotella-associated features are stage-specific (Figures 1 and 2). One of them (F29) started appearing at the grower stage whereas eight of these Prevotella features emerged only after weaning when the pigs were introduced to solid food (Figure 2). Prevotella is one of the most abundant genera in humans and its role in human health has been controversial.13 Prevotella species are associated with dietary carbohydrates in humans,14 and produce acetate, an energy source for some butyrate producers such as Ruminococcus, Clostridium, and Blautia. In fact, members of these genera such as Blautia also emerged together with Prevotella at the beginning of nursery stage (Figure 2), supporting the co-occurence and possible cross-feeding between these bacteria in pigs.
Table 1.
The swine core gut microbiome.
| Feature# | Feature ID | Phylum | Order | Family | Genus | Species |
|---|---|---|---|---|---|---|
| F1 | 77560703da191 f21e7d250845229fe06 | Firmicutes | Clostridiales | Veillonellaceae | Megasphaera | |
| F2 | e11db671d9c36b550f08a6ee36ba2cef | Firmicutes | Lactobacillales | Streptococcaceae | Streptococcus | luteciae |
| F3 | 1919b6828724477c2ab08fd9efe3bcd9 | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | copri |
| F5 | 002109bc8b4bcf7a97a7794f4cffda2b | Firmicutes | Lactobacillales | Lactobacillaceae | Lactobacillus | |
| F6 | 9c4260f79a8007b4d15a1e6fe1129ce1 | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | copri |
| F7 | 15caa2d41590f4361bd4ef0b6453fe1 c | Proteobacteria | Enterobacteriales | Enterobacteriaceae | Escherichia | coli |
| F8 | 230f858e6622e1a686ad91373adc20b9 | Firmicutes | Clostridiales | Veillonellaceae | Phascolarctobacterium | |
| F9 | 38ec373490dc98851951b38c50961207 | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | |
| F10 | 21709f541225b3d1e30e195bced4322b | Bacteroidetes | Bacteroidales | [Paraprevotellaceae] | YRC22 | |
| F12 | 8f194030281f1b235fa5874aa0426bb0 | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | |
| F13 | 48ea88e5e788894c86b04d558c7ea12d | Firmicutes | Clostridiales | Veillonellaceae | Anaerovibrio | |
| F17 | 7ef3b00cc85b6d0730685b757440e392 | Actinobacteria | Coriobacteriales | Coriobacteriaceae | Collinsella | aerofaciens |
| F20 | 20a99a2686ac6a717b01361cac773046 | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | stercorea |
| F21 | f0e22244687ff7f51926d7b54adaec4b | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | |
| F23 | 522ba9d695541c9ea478db7cafeb4355 | Bacteroidetes | Bacteroidales | S24-7 | ||
| F28 | fa9cbb7e45355cb861070b68d9a5653b | Firmicutes | Clostridiales | Ruminococcaceae | Oscillospira | |
| F46 | b88939309e235a04fdcff68d166139ab | Bacteroidetes | Bacteroidales | [Paraprevotellaceae] | [Prevotella] | |
| F48 | 27a8a06bc2b03e2ee032a7aeef16dbe3 | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | stercorea |
| F50 | 36585850033948d7966edf6717266cc0 | Firmicutes | Clostridiales | [Mogibacteriaceae] | Mogibacterium | |
| F52 | f324a15262fda881e9e0f6b59a20b21d | Bacteroidetes | Bacteroidales | [Paraprevotellaceae] | CF231 | |
| F53 | f2c72e7978ae955c780882f71f689e9b | Firmicutes | Clostridiales | Christensenellaceae | ||
| F62 | 2307b48380cd51b78c43a1da261bdde9 | Bacteroidetes | Bacteroidales | |||
| F63 | fdf37b54fbf9f5a82616d90386ade16d | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | |
| F70 | 47ee99688649fa7c2937db076f2039de | Firmicutes | Lactobacillales | Lactobacillaceae | Lactobacillus | reuteri |
| F77 | 55d98aa0513d69baf52b439bb4e4207a | Proteobacteria | Campylobacterales | Campylobacteraceae | Campylobacter | |
| F82 | 1e3a3bda59a3e703d9ebadfb122655d7 | Firmicutes | Clostridiales | Ruminococcaceae | Oscillospira | |
| F86 | 4539eb8d334b124b12ef2f840be6c5d7 | Firmicutes | Clostridiales | Ruminococcaceae | Oscillospira | |
| F87 | fd2a145204502d5d1453ac09704619ad | Firmicutes | Clostridiales | Ruminococcaceae | ||
| F93 | 1f75f87fb55993516a97af66507a0f08 | Bacteroidetes | Bacteroidales | [Paraprevotellaceae] | [Prevotella] | |
| F112 | 04e3a222e2655f35b175436f9cb29844 | Bacteroidetes | Bacteroidales | |||
| F116 | c0e706eff2fbc0db84755141796f11af | Bacteroidetes | Bacteroidales | |||
| F120 | fd4bd6f441389fd712ee14af66eaf217 | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | |
| F121 | 52f6ab8a13bead00126a83f61d959ab6 | Firmicutes | Lactobacillales | Lactobacillaceae | Lactobacillus | delbrueckii |
| F123 | f212b75172ddc5a1ef357a425ea33482 | Firmicutes | Clostridiales | Christensenellaceae | ||
| F131 | c3a1361961c649a26927a4e6b6eaec04 | Firmicutes | Lactobacillales | Lactobacillaceae | Lactobacillus | mucosae |
| F132 | 4b922c405bf97b57647a7f6323092ce1 | Actinobacteria | Coriobacteriales | Coriobacteriaceae | ||
| F136 | d3ea744c5bbb301b99658befe3e1c162 | Firmicutes | Lactobacillales | Lactobacillaceae | Lactobacillus | |
| F144 | 562f555227af77f2f0d0cc3e6462c8d8 | Firmicutes | Clostridiales | Ruminococcaceae | ||
| F149 | 795b1744a93f0c030c7cac5ed1573 c20 | Firmicutes | Clostridiales | Ruminococcaceae | ||
| F153 | 76f995a642c756e4baef9ba3588be99a | Bacteroidetes | Bacteroidales | Prevotellaceae | Prevotella | |
| F156 | c26fdaf9bfb7235e29a45dad8554c06 c | Bacteroidetes | Bacteroidales | |||
| F157 | f2f55ee5a197eaf6b65a1c22bedc016d | Firmicutes | Erysipelotrichales | Erysipelotrichaceae | [Eubacterium] | biforme |
| F169 | b5dcb92af75a84ef7f76c534e644b4dc | Firmicutes | Clostridiales | Christensenellaceae | ||
| F171 | 6fa1b042db7e367356a0e6b600ce46a0 | Firmicutes | Clostridiales | |||
| F189 | 990ed356cd9dae5b83314355569514e3 | Bacteroidetes | Bacteroidales | |||
| F198 | b05ae994010ddd94626d03a8c4ceafe1 | Firmicutes | Erysipelotrichales | Erysipelotrichaceae | [Eubacterium] | biforme |
| F199 | 179a2e52862950c20141c0e376152624 | Firmicutes | Clostridiales | Ruminococcaceae | ||
| F201 | 27e548475cd61c016aaec05c28e95223 | Firmicutes | Clostridiales | Ruminococcaceae | ||
| F214 | 5503fc4199485012d18108218ba0c589 | Proteobacteria | Desulfovibrionales | Desulfovibrionaceae | Desulfovibrio | |
| F215 | deea65343744187b8e911e87975c8979 | Actinobacteria | Coriobacteriales | Coriobacteriaceae | ||
| F229 | fec7ae85682e4fb767970f4dab35bbb5 | Spirochetes | Spirochaetales | Spirochaetaceae | Treponema | |
| F235 | d102b79fac474290436a96751aaccb59 | Proteobacteria | GMD14H09 | |||
| F241 | 3e2fe19948fdefb2fb179e6f8f1def05 | Firmicutes | Clostridiales | Ruminococcaceae | Oscillospira | |
| F271 | 125a703172cdf3ac7155e51b904d6546 | Firmicutes | Clostridiales | Ruminococcaceae | Oscillospira | |
| F275 | 5947530ad170bbc67f9af778d42f8dc7 | Bacteroidetes | Bacteroidales | Porphyromonadaceae | Parabacteroides | |
| F302 | f361d0070bf833c26d64b95fe512847a | Bacteroidetes | Bacteroidales | Prevotellaceae | ||
| F310 | 413d7371b0955033e463b538bfab0cfc | Firmicutes | Clostridiales | Ruminococcaceae | ||
| F312 | 829d3bd6a7df703802fbb4a076f614b5 | Firmicutes | Clostridiales | Ruminococcaceae | ||
| F319 | fc3d8267a88365a43202affb1b1c0d1f | Firmicutes | Clostridiales | Ruminococcaceae | Oscillospira | |
| F357 | 71903137448067873e3ffbf70dea44eb | Firmicutes | Clostridiales | |||
| F394 | 1c4985419653025d3a4a1be178f4676 c | Firmicutes | Erysipelotrichales | Erysipelotrichaceae | p-75-a5 | |
| F410 | 6d3a987e101712b3302f6e8e5beee7ea | Firmicutes | Clostridiales | |||
| F413 | 02c93fa4666bd94365b522152d56bb3c | Firmicutes | Clostridiales | Lachnospiraceae | ||
| F491 | ebf50bcde4063f1d5e957907a2879929 | Firmicutes | Clostridiales | |||
| F502 | 0007527ae916772430dd7897aeeeb0e8 | Firmicutes | Clostridiales | Ruminococcaceae | ||
| F518 | 547cc82727e9e688a34afaefcedd5c11 | Proteobacteria | Burkholderiales | Alcaligenaceae | Sutterella | |
| F521 | ba2b96b172b40e9f509571929d9a082b | Actinobacteria | Coriobacteriales | Coriobacteriaceae | ||
| F524 | 65732a9995f5007582ebb500e7df4ea5 | Firmicutes | Clostridiales | Ruminococcaceae | Oscillospira | |
| F561 | d76796a7df0d1f21a46610474a17f2e4 | Firmicutes | Clostridiales | Ruminococcaceae | Ruminococcus |
Figure 2.
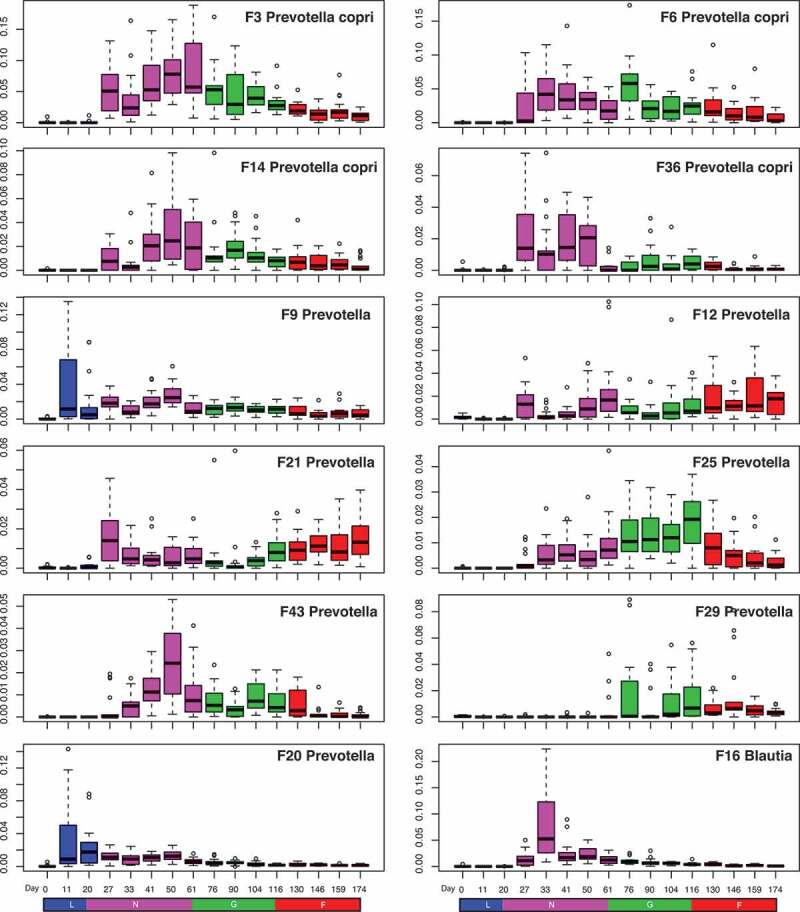
Boxplots showing the dynamics of the top 11 bacterial features associated with Prevotella and one feature associated with Blautia during different growth stages of pigs.
The swine core microbiome
The core microbiome has been well- researched in different species and ecological niches.1,15–18 Identifying a core microbiome is vital to understand its function in the gut to enable manipulation of microbial communities that are beneficial for human health. In general, a core microbiome refers to the common group of bacteria present in all or most (e.g. >90%) of the communities of a special habitat.16,19 However, it has been challenging to define a core microbiome given the many factors that affect the human gut microbiome such as diet, genetics, age, and antibiotics.2,5,19–23
There have also been efforts in defining a core swine microbiome.24,25 In a recent study, Holman and colleagues performed a meta-analysis of 20 data sets and found several shared genera such as Prevotella, Clostridium, Alloprevotella, and Ruminococcus. However, the most striking conclusion from the analysis is that, other than gastrointestinal (GI) tract location, the “Study” factor was also significant in shaping the swine gut microbiota. It is not surprising that different niches along the GI tract harbor distinct gut microbiomes given the unique ecological environments and physiological conditions in each niche. The significant effect of “Study” on the swine gut microbiome underscores the importance of standardizing experimental techniques, e.g. sample collection and storage,26,27 DNA extraction,28–30 hypervariable regions of the 16 S rRNA gene,31 and bioinformatics pipelines,32 to allow cross-study comparisons aiming to identify a core swine gut microbiome.
In our recent report, we identified a “core” microbiome of 69 bacterial features that were present in all the growth stages and shared by three groups of pigs in the test and validation trial (Table 1). Consistent with the findings of Holman et al,25 most of these features are associated with the order Clostridiales (n = 28), Bacteroidiales (n = 22), and Lactobacillales (n = 6). At the family level, the top three families are Prevotellaceae (n = 11), Ruminococcaceae (n = 16), and Lactobacillaceae (n = 5). A total of 10 features were not classified to the family level. Although some of the top features such as Megasphaera (F1) and Prevotella (F3) were present during all the growth stages, others such as F4 (unclassified Clostridiaceae) and F10 (Bacteroidetes YRC22), rarely noticeable at lactation and nursery stages, emerged rapidly and became the dominant taxa at the growing and finishing stages. The sequence and taxonomy of these features are listed in Table 1.
Our study has several contributions to the effort of identifying a core microbiome. We used ASVs (amplicon sequence variants) or ESVs (exact sequence variants) to define bacterial features that differed with a single nucleotide. This approach enables cross-study comparisons to determine whether the same bacterial features are shared among different studies.33 For core microbiome studies, our data show that it is important to take into account the stage-specific bacterial taxa. For example, the finishing-stage bacteria (e.g. F4) might be missed in the “core” gut microbiome if only nursery-stage samples were used for comparison.
Swine probiotics development: does one size fit all?
Due to pressures from a public health standpoint, many countries have banned the use of antibiotics for growth promotion in animals. Alternatives to antibiotics are critical to improve animal health and growth performance in the swine industry. Probiotics serve as an important alternative to sub-therapeutic antibiotics.34 As a first step in developing probiotics, we performed a regression-based random forest analysis to identify potentially beneficial bacteria that correlate with growth performance at each growth stage and at the end of the test trials. Apparently, different sets of bacteria associated with growth performance were observed from each stage. In a validation trial, we inoculated 12 post-weaning pigs with fecal samples from a healthy pig at growing stage. Fecal microbiota transplantation (FMT) improved the growth performance of the recipient pigs. Although not statistically significant likely due to the small sample size (n = 12), FMT did increase the body weight of recipient pigs by 4.9 kg on average compared to their litter mates at the end of the validation trial. The fecal sample from the donor is representative of the mature pigs from growing stages regarding the microbiome composition and structure.12 FMT didn’t change the overall gut microbiome structure of the recipients, however, it did enrich several groups of bacterial taxa. Random forest identified these stage-associated bacteria that were enriched in the FMT group and correlated with growth performance in the validation trial. A comparison of the test and validation trial identified shared bacterial features that might serve as potentially beneficial bacteria promoting animal growth performance (Table 2).
Table 2.
Stage-specific potentially beneficial bacteria shared by the two animal trials.
| Lactation | Nursery | Growing | Finishing | Overall |
|---|---|---|---|---|
| F77 | F222 | F100 | F26 | F55 |
| F363 | F604 | F73 | F19 | F4 |
| F182 | F7 | F40 | F27 | F27 |
| F876 | F233 | F333 | F75 | |
| F162 | F339 | F100 | F19 | |
| F1 | F336 | F61 | F26 | |
| F166 | F301 | F394 | F234 | |
| F502 | F454 | F134 | F18 | |
| F21 | F336 | |||
| F53 | F165 | |||
| F247 | F127 | |||
| F103 | ||||
| F377 | ||||
| F196 | ||||
| F433 | ||||
| F231 | ||||
| F376 | ||||
| F100 | ||||
| F17 | ||||
| F330 | ||||
| F307 |
Figure 3 shows the relative abundance of these bacteria in the pigs of the control and the FMT group in the validation trial. Bacterial features associated with Bulleidia (F336) and Lacobacillus mucosae (F454) were more abundant in the FMT group with greater growth performance at the nursery stage, whereas features affiliated with Acidaminococcus (F100) and Prevotella (F73) were over represented in the FMT group at the late nursery and growing stages. Members of Turicibacter (F26), more abundant in the FMT group, didn’t emerge until the finishing stage. Of note, features associated with growth performance, calculated based on the final body weight, were mainly late colonizers of the swine gut. For example, F4 and F18, which were more abundant in the FMT group, started appearing in the gut at the growing stages. Our data show different sets of potentially beneficial bacteria associated with superior phenotypes with a stage-specific pattern. We propose that a mix of probiotics tailored to growth stages of pigs, rather than to a single bacterial strain, should be developed to optimize their beneficial effects on swine health and production.
Figure 3.
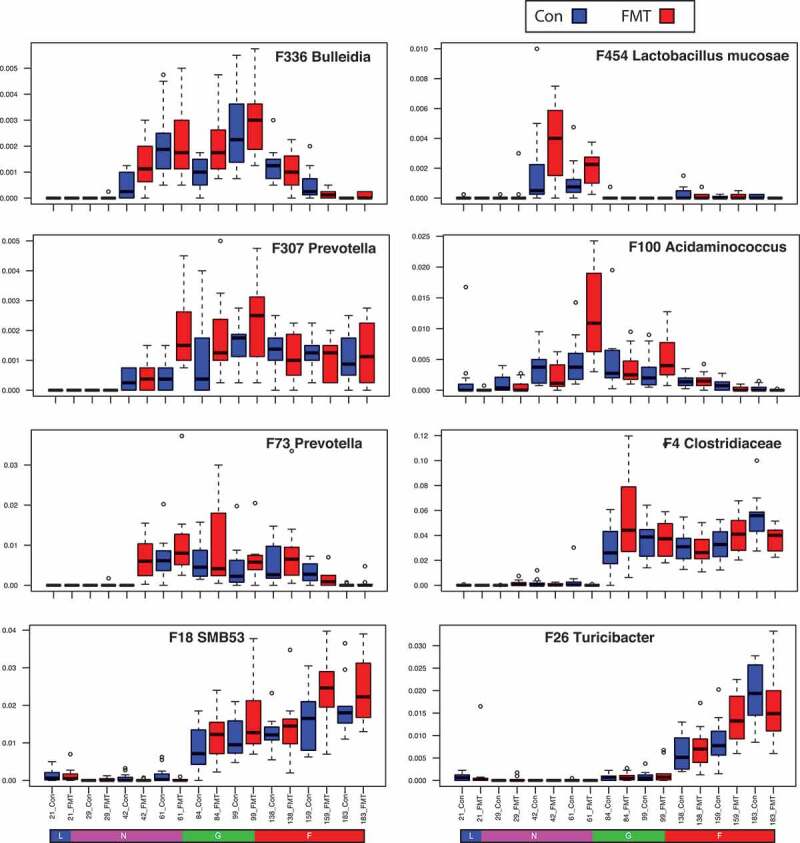
Stage-associated potentially beneficial bacteria in the control and fecal microbiota transplantation (FMT) pigs. FMT remarkably improved swine growth performance. Bacterial features identified by random forest that positively correlate with growth performance were more abundant in the FMT group.
Foodborne pathogens: the best window for intervention
Pork, the most consumed meat worldwide, is a major source of foodborne pathogens, which are a major cause of human morbidity and mortality every year.35 Most of the studies in the food safety area focus on the post-harvest section of the swine industry with very limited information about the live animals on farm during the pre-harvest season. In fact, live pigs are not only the source of foodborne pathogens, which are passed along to the post-harvest supply chain, they also shed these pathogens on the farms, thus exposing farm workers as well.36 Therefore, understanding the dynamics of these pathogens on farm provides information on the best window and strategy to manage these pathogens.
Campylobacter is one of the major foodborne pathogens in the swine industry.35 Our longitudinal study shows the relative abundance and dynamics of 13 bacterial features associated with Campylobacter and one bacterial feature associated with E. coli (Figure 4). E. coli was abundant during the lactation stage and faded out after weaning. Different features of Campylobacter showed different dynamic patterns. Future study is needed to track the dynamics of these pathogens in the whole pork supply chain to determine which features are transmitted from farm to fork so that a treatment window based on this essential information could be determined.
Figure 4.
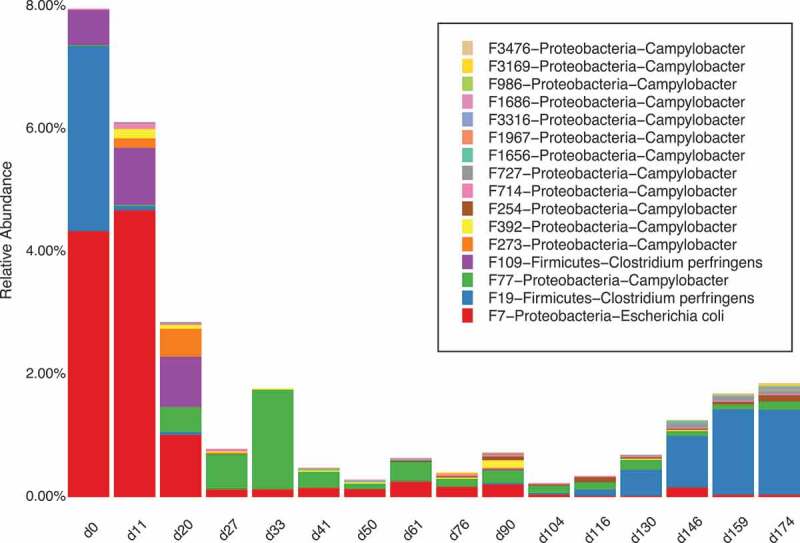
The dynamics of foodborne pathogens (members of Campylobacter and E. coli) in pigs during the pre-harvest section of the pork supply chain.
Conclusions
In conclusion, this longitudinal characterization of the swine gut microbiome provides the foundation for translational research aiming to improve animal health and production. This study not only contributes to our understanding of many key biological and ecological concepts, but also identified potentially beneficial bacteria and pathogens. Characterization of the growth-stage-associated swine gut microbiome emphasizes the importance of optimization of probiotics based on different stages. It also provides insights on the best window to manage foodborne pathogens during the pre-harvest season of the swine industry. We also advocate for standard protocols in swine gut microbiome studies (e.g. sample collection, DNA extraction) to improve reproducibility and cross-study comparability for translational research investigations.
Materials and methods
All the animals and sequencing data were from our previous study12. No additional pigs or sequences were included. Sequence process and analysis were performed as described previously12. The biplot in Figure 1 was generated by the corr.axes function (setting: method = spearman; numases = 2) in mothur software package (v.1.40.5).37 Boxplots (Figures 2 and 3) and stacked barchart (Figure 4) were generated by the ggplot2 package of R.
Acknowledgments
We deeply appreciate Dr. Marites A. Sales’ help with proofreading and editing of this manuscript.
Funding Statement
This work was partially supported by the USDA National Institute of Food and Agriculture [2018-67015-27479] to JZ and the National Natural Science Foundation of China [31630074] to JW.
References
- 1.Wang X, Tsai T, Deng F, Wei X, Chai J, Knapp J, Apple J, Maxwell CV, Lee JA, Li Y, et al. Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome. 2019;7(1):109. doi: 10.1186/s40168-019-0721-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhao J, Schloss PD, Kalikin LM, Carmody LA, Foster BK, Petrosino JF, Cavalcoli JD, VanDevanter DR, Murray S, Li JZ, et al. Decade-long bacterial community dynamics in cystic fibrosis airways. Proc Natl Acad Sci U S A. 2012;109(15):5809–5814. doi: 10.1073/pnas.1120577109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. Human gut microbiome viewed across age and geography. Nature. 2012;486(7402):222–227. doi: 10.1038/nature11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clemente JC, Ursell LK, Parfrey LW, Knight R.. The impact of the gut microbiota on human health: an integrative view. Cell. 2012;148(6):1258–1270. doi: 10.1016/j.cell.2012.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kong F, Deng F, Li Y, Zhao J.. Identification of gut microbiome signatures associated with longevity provides a promising modulation target for healthy aging. Gut Microbes. 2018;10(1):1–6. doi: 10.1080/19490976.2018.1455790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsai T, Sales MA, Kim H, et al. Isolated rearing at lactation increases gut microbial diversity and post-weaning performance in pigs. Front Microbiol. 2018;9:2889. Published 2018 Nov 29. doi:10.3389/fmicb.2018.02889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wei X, Tsai T, Knapp J, et al. ZnO modulates swine gut microbiota and improves growth performance of nursery pigs when combined with peptide cocktail. Microorganisms. 2020;8(2):146. Published 2020 Jan 21. doi:10.3390/microorganisms8020146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao YP, Kong FL, Xiang Y, et al. Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs. Sci Rep. 2018;8:5985. doi:10.1038/s41598-018-24289-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H, Xiang Y, Robinson K, et al. Gut microbiota is a major contributor to adiposity in pigs. Front Microbiol. 2018;9:3045. Published 2018 Dec 10. doi:10.3389/fmicb.2018.03045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He B, Bai Y, Jiang L, et al. Effects of oat bran on nutrient digestibility, intestinal microbiota, and inflammatory responses in the hindgut of growing pigs. Int J Mol Sci. 2018;19(8):2407. Published 2018 Aug 15. doi:10.3390/ijms19082407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Looft T, Johnson TA, Allen HK, Bayles DO, Alt DP, Stedtfeld RD, Sul WJ, Stedtfeld TM, Chai B, Cole JR, et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc Natl Acad Sci U S A. 2012;109(5):1691–1696. doi: 10.1073/pnas.1120238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Filippis F, Pasolli E, Tett A, Tarallo S, Naccarati A, De Angelis M, Neviani E, Cocolin L, Gobbetti M, Segata N, et al. Distinct genetic and functional traits of human intestinal prevotella copri strains are associated with different habitual diets. Cell Host Microbe. 2019;25(3):444–53 e3. doi: 10.1016/j.chom.2019.01.004. [DOI] [PubMed] [Google Scholar]
- 14.Wu GD, Chen J, Hoffmann C, Bittinger K, Chen YY, Keilbaugh SA, Bewtra M, Knights D, Walters WA, Knight R, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science. 2011;334(6052):105–108. doi: 10.1126/science.1208344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tschop MH, Hugenholtz P, Karp CL. Getting to the core of the gut microbiome. Nat Biotechnol. 2009;27(4):344–346. doi: 10.1038/nbt0409-344. [DOI] [PubMed] [Google Scholar]
- 16.Shade A, Handelsman J. Beyond the Venn diagram: the hunt for a core microbiome. Environ Microbiol. 2012;14:4–12. doi: 10.1111/j.1462-2920.2011.02585.x. [DOI] [PubMed] [Google Scholar]
- 17.Zaura E, Keijser BJ, Huse SM, Crielaard W. Defining the healthy “core microbiome” of oral microbial communities. BMC Microbiol. 2009;9:259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lundberg DS, Lebeis SL, Paredes SH, Yourstone S, Gehring J, Malfatti S, Tremblay J, Engelbrektson A, Kunin V, Rio TGD, et al. Defining the core Arabidopsis thaliana root microbiome. Nature. 2012;488(7409):86–90. doi: 10.1038/nature11237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hamady M, Knight R. Microbial community profiling for human microbiome projects: tools, techniques, and challenges. Genome Res. 2009;19(7):1141–1152. doi: 10.1101/gr.085464.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arumugam M, Raes J, Pelletier E, Le Paslier D, Yamada T, Mende DR, Fernandes GR, Tap J, Bruls T, Batto J-M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goodrich JK, Waters JL, Poole AC, Sutter JL, Koren O, Blekhman R, Beaumont M, Van Treuren W, Knight R, Bell J, et al. Human genetics shape the gut microbiome. Cell. 2014;159(4):789–799. doi: 10.1016/j.cell.2014.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng F, Li Y, Zhao J. The gut microbiome of healthy long-living people. Aging (Albany NY). 2019;11(2):289–290. doi: 10.18632/aging.101771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao J, Murray S, Lipuma JJ. Modeling the impact of antibiotic exposure on human microbiota. Sci Rep. 2014;4(1):4345. doi: 10.1038/srep04345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lowe, B.A., Marsh, T.L., Isaacs-Cosgrove, N . et al. Defining the “core microbiome” of the microbial communities in the tonsils of healthy pigs. BMC Microbiol. 2012;12:20. doi: 10.1186/1471-2180-12-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holman DB, Brunelle BW, Trachsel J, Allen HK.Meta-analysis to define a core microbiota in the swine gut. mSyst. 2017: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao J, Li J, Schloss PD, Kalikin LM, Raymond TA, Petrosino JF, Young VB, LiPuma JJ. Effect of sample storage conditions on culture-independent bacterial community measures in cystic fibrosis sputum specimens. J Clin Microbiol. 2011;49(10):3717–3718. doi: 10.1128/JCM.01189-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Song SJ, Amir A, Metcalf JL, Amato KR, Xu ZZ, Humphrey G, et al. Preservation methods differ in fecal microbiome stability, affecting suitability for field studies. mSyst. 2016: 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Costea PI, Zeller G, Sunagawa S, Pelletier E, Alberti A, Levenez F, Tramontano M, Driessen M, Hercog R, Jung F-E, et al. Towards standards for human fecal sample processing in metagenomic studies. Nat Biotechnol. 2017;35(11):1069–1076. doi: 10.1038/nbt.3960. [DOI] [PubMed] [Google Scholar]
- 29.Zhao J, Carmody LA, Kalikin LM, Li J, Petrosino JF, Schloss PD, Young VB, LiPuma JJ. Impact of enhanced Staphylococcus DNA extraction on microbial community measures in cystic fibrosis sputum. PLoS One. 2012;7(3):e33127. doi: 10.1371/journal.pone.0033127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kennedy NA, Walker AW, Berry SH, Duncan SH, Farquarson FM, Louis P, Thomson JM, Satsangi J, Flint HJ, Parkhill J, et al. The impact of different DNA extraction kits and laboratories upon the assessment of human gut microbiota composition by 16S rRNA gene sequencing. PLoS One. 2014;9(2):e88982. doi: 10.1371/journal.pone.0088982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Z, Hui PC, Hui M, Yeoh YK, Wong PY, Chan MCW, et al. Impact of preservation method and 16S rRNA hypervariable region on gut microbiota profiling. mSyst. 2019: 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sinha R, Abu-Ali G, Vogtmann E, Fodor AA, Ren B, Amir A, Schwager E, Crabtree J, Ma S, Abnet CC, et al. Assessment of variation in microbial community amplicon sequencing by the Microbiome Quality Control (MBQC) project consortium. Nat Biotechnol. 2017;35(11):1077–1086. doi: 10.1038/nbt.3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, et al. A communal catalogue reveals Earth’s multiscale microbial diversity. Nature. 2017;551(7681):457–463. doi: 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu WC. Application of complex probiotics in swine nutrition – a review. Ann Anim Sci. 2018;18(2):335-50-2018. doi: 10.2478/aoas-2018-0005. [DOI] [Google Scholar]
- 35.Baer AA, Miller MJ, Dilger AC. Pathogens of interest to the pork industry: A review of research on interventions to assure food safety. Compr Rev Food Sci Food Saf. 2013;12:183–217. [Google Scholar]
- 36.Sun J, Liao XP, D’Souza AW, Boolchandani M, Li SH, Cheng K, Luis Martínez J, Li L, Feng Y-J, Fang L-X, et al. Environmental remodeling of human gut microbiota and antibiotic resistome in livestock farms. Nat Commun. 2020;11(1):1427. doi: 10.1038/s41467-020-15222-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb. 2009;75(23):7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]