

ABSTRACT
In this review, we highlight the variations of gut resistome studies, which may preclude comparisons and translational interpretations. Of 22 included studies, a range of 12 to 2000 antibiotic resistance (AR) genes were profiled. Overall, studies defined a healthy gut resistome as subjects who had not taken antibiotics in the last three to 12 months prior to sampling. In studies with de novo assembly, AR genes were identified based on variable nucleotide or amino acid sequence similarities. Different marker genes were used for defining resistance to a given antibiotic class. Validation of phenotypic resistance in the laboratory is frequently lacking. Cryptic resistance, collateral sensitivity and the interaction with repressors or promotors were not investigated. International consensus is needed for selecting marker genes to define resistance to a given antibiotic class in addition to uniformity in phenotypic validation and bioinformatics pipelines.
KEYWORDS: Resistome, antibiotic resistance, gut microbiota, meta-genomics, fecal microbiota transplantation
Introduction
Antibiotic resistance is a global public health concern and thus recognition of its reservoirs could facilitate the control of its dissemination. Traditionally, studies of antimicrobial resistance (AR) genes in bacteria started from massive screening of antibiotic resistance phenotypes using macro- or micro-broth dilution methods.1–3 For bacterial isolates displaying resistance phenotypes, total genomic DNA is extracted and probed for candidate AR genes using polymerase chain reaction (PCR)-based method or comparative whole-genome sequencing with reference to sensitive type strains.2 If a candidate resistance gene could not be identified, genomic DNA is cloned into expression vectors and then transformed into a heterologous sensitive bacterial host for molecular and phenotypic characterization.3 In addition to being time-consuming, this technique is limited to bacteria that can be cultivated.
In recent years, the advent of affordable high-throughput sequencing and analysis applied to online antibiotic resistance gene databases enables the avoidance of bacterial culture, facilitating massive resistome-wide studies of potential reservoirs of antibiotic resistance genes.4–8 The ubiquity of resistance genes was exemplified by their unanticipated isolation from various environmental habitats waste water treatment plant and soil to food production chain and wild animals.9–11
The term resistome is defined as the complete collective assemblage of antibiotic, antiseptic and heavy metal resistance genes in a microbial ecosystem.12 Studies have applied this definition variably with considerable heterogeneity in candidate genes, methodology and bioinformatics pipelines, precluding direct comparison across studies.5–7,13,14 Several reports employed high-throughput microfluidic PCR or customized microarrays by designing probes with reference to existing AR gene databases and targeted a selection of resistance genes,15–17 with the resulting number of AR genes varying from less than 507,14,18 to thousands.15,19 Across studies, disparate genes were selected to represent resistance to a given antibiotic class.7,14,16,20 Another issue is in comparing gene and/or protein sequence similarities and defining functional conservation. In sequence-based meta-genomic studies, de novo assembled contigs were compared to the existing AR gene databases. Where the assembled contigs displayed sequence similarity beyond a defined threshold, phenotypic resistance was assumed. The degree of similarity was arbitrary, ranging from 80% of amino acid sequence identity6 to 95% nucleotide sequence identity.21,22 The function of the detected resistance genes was only occasionally validated by expressing in competent bacterial hosts.8,19,20,23 It is well-established that a single amino acid substitution could sufficiently change the susceptibility level of a strain by altering the binding affinity of the drug target site to a corresponding antibiotic.24 In cases where up to 20% difference in the amino acid sequence was observed, the resistance phenotype of the bacterial strains concerned would therefore be questionable. In sequence-based meta-genomic studies, the genetic context of the resistant genes, or the flanking regions containing promoter- and repressor-sequences that define the bacterial host origin, was not consistently reported. This should provide important information on possible bacterial hosts, gene expression, and horizontal transferability. The possible bacterial hosts for a given set of antibiotic resistance genes were occasionally predicted using network analysis lacking downstream resistance phenotype validation.25
We reviewed the literature of the human gut resistome determined in healthy populations from various geographical locations. We highlight the characteristics of these studies; the methodologies used in their analyses, and the range of the magnitude of the resistome. We also identify variables that require careful definition and the adoption of a consensus to enable comparisons to be made across studies.
Methods
Bibliographic search and study selection
Search strategy was developed using the MEDLINE Ovid platform (Table S1), which was then translated for use in electronic databases EMBASE, MEDLINE (PUBMED), Web of Science, and Scopus. Briefly, relevant articles were identified using Medical Subject Heading (MeSH) or Title/Abstract keywords from inception to May 2019. All citations were imported into Endnote X8 software by which duplicates were removed. Two researchers (JH and NB) performed article screening and data verification, respectively, according to predefined inclusion and exclusion criteria. Disagreement on study selection was resolved through discussion. This study was reported according to the Preferred Reporting Items for Systematic Reviews and Meta-analysis (PRISMA) statement.26
Inclusion and exclusion criteria
All primary studies with human fecal specimens collected from healthy population or secondary analyses of its kind were included. Studies on population with high risk for carrying antibiotic resistance genes, such as the critically ill, were excluded. Conference abstracts, editorials, systematic reviews, and meta-analyses were excluded. Other exclusion criteria were set in terms of study population, publication type, and study type. The pre-defined inclusion and exclusion criteria are summarized in Table S2.
Data extraction and quality assessment
Full text of the eligible studies were reviewed and the following data extracted: First author, year of publication, study design, number of fecal specimens, specific antibiotic resistance genes, laboratory methods, and bioinformatic pipelines. Whenever appropriate, the corresponding authors would be contacted by e-mail for clarification. The quality of the included studies was rated by sub-items of Newcastle–Ottawa scale for non-randomized observational studies, a tool with established content validity and inter-rater reliability.27 The representativeness of the study cohort and the ascertainment of antibiotic exposure were determined. Two independent reviewers (JH and NB) rated the studies according to the scale. To ensure reliability of the review process, 10% of the articles in the final data set were reviewed by another reviewer (MI). Any discrepancies were resolved by discussion.
Effect sizes and publication bias
Despite multiple reports of the prevalence of antibiotic resistance genes on the same population were identified, the small sample size and inconsistent use of reference genes precluded such an effect size from pooling. In terms of publication bias, a traditional funnel plot using logarithm of effect size against standard error does not apply to proportion data sets due to its inherent nature of asymmetry for extreme proportions. Neither classic fail-safe N nor Orwin’s fail-safe N would be applicable for prevalence data because statistical significance or trivial levels do not exist for proportion estimates.
Results and discussion
Study selection, countries of origin, cohort representativeness, and exposure ascertainment
The initial search returned 107 articles. After screening titles and abstracts, 86 full-texts were reviewed. Nineteen of these met the inclusion criteria but were not exempted from the exclusion criteria. Three additional studies were identified by manually checking reference lists of these 19 studies (Figure 1). Of the 22 studies included in this review (Table S3), 15 were primary studies involving subject recruitment and specimen collection.4,6,7,14–20,22,23,28–30 These covered populations originated from 18 countries, namely Afghanistan, Canada, China, Denmark, Eritrea, Finland, France, Germany, Iraq, Italy, Norway, Peru, Spain, Syria, Tanzania, The Netherlands, United Kingdom, and the United States. No studies were conducted in 90% of the countries as defined by the United Nations which included more than 193 countries. The remaining seven studies were re-analysis of publicly available datasets.5,8,13,21,25,31,32 With reference to the number of population by countries reported by the United Nations,33 the number of recruited subjects ranged from 0.72 in China to 765 in the Netherlands per 10 million population (median = 19.07, interquartile range 4.56–37.10) (Figure 2). Except for a Dutch study for which a general population-based cohort involving 1,135 individuals derived from 167,000 subjects across three provinces,18 the remaining studies were either based on convenient samples or did not describe how the cohorts were derived. Antibiotic intake is one of the key factors that may alter the collection of antibiotic resistance genes or resistome.16 In this connection, ascertainment of antibiotic exposure prior to fecal specimen collection is important. Nonetheless, the period free from antibiotic exposure ranged from 3 months,3 6 months,16,22 to 12 months.29,30 Notably, the remaining primary studies did not describe antibiotic exposure in their population. However, they did not report higher number of resistant genes. The details of the included studies are presented in Table 1.
Figure 1.

Study selection.
Figure 2.
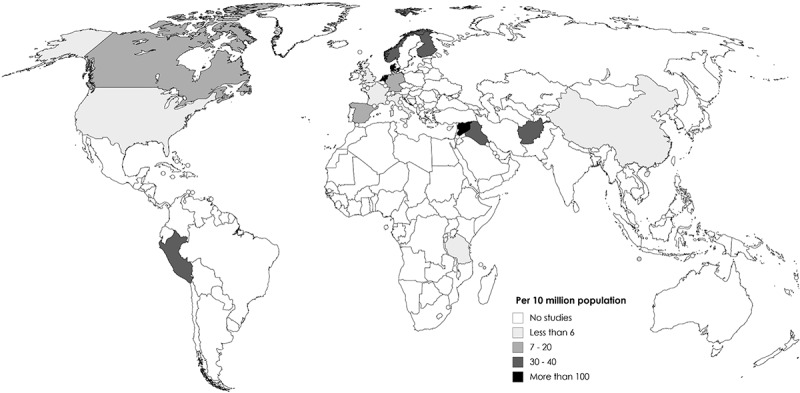
Geographical distribution of studies included in this review. The color intensity indicates the number of participants sampled per 10 million population by countries according to data in the World Data Bank, the United Nations.
Table 1.
Summary of studies included in this review.
| Ref. | Country(ies) | Sample size | Method(s) | ARG (N)a | Pipeline(s) | Reference Database(s) | Key findings | Limitations | |
|---|---|---|---|---|---|---|---|---|---|
| 4 | Canada | 24 | SeqMeta | 13,218 | RAY Meta 2.2.1 | MERGEM | Culture-enrichment in prior to sequencing allows identification of core and accessory ARGs in rare gut microbial species. | Enrichment step has eliminated otherwise prevalent Akkermansia and Prevotella. | |
| 5 | Demark, Spain | 663 | SeqMeta | 6,095 | Prodigal, CD-HIT v4.5.7 | Resfinder, ARG-ANNOT, Lahey Clinic, RED-DB | The predicted ARGs had 29.8% amino acid identity to known ARGs, supporting ARG transfer between microbes is rare. | ARGs in the subdominant microbial species and that non-homologous to the known ARG classes may be missed. | |
| 6 | Tanzania, Italy | 38 | SeqMeta | 2,570 | TBLASTN | ARDB; ResFams | The gut resistome of the Hadza hunter-gatherer resembles the soil, with less syntenic mobile elements than that of the Italian. | Survey of ARGs was limited to known genes. Functional validation was not performed. | |
| 7 | Syria, Iraq, Iran, Eritrea, Afghanistan, Germany | 606 | Mf-PCR | 42 | NA | NA | Refugees had more than five ARGs, with high prevalence of blaCTX-M, blaSHV, blaoxa-1, vanC1, and qnrB. German had three or less ARGs. | Mobility of the ARGs were not investigated and thus their transferability was unclear. | |
| 8 | China, Denmark, Spain | 162 | SeqMeta; FunMeta | 1,093 | BLASTx | ARDB | Chinese had the highest abundance of ARGs, which are phylogenetically distant from others. | The limited samples may not reflect population-wide profile. | |
| 13 | USA, China, Spain, Denmark | 1,267 | SeqMeta | 112 | BLASTx, SOAP | ARDB, ARG-ANNOT | ARG composition was similar among all population studied. Chinese had the highest ARG abundance than others. | Insufficient epidemiologic data that supported gut ARG being affected by antibiotic policy. | |
| 14 | The Netherlands | 122 | PCR | 10 | NA | NCBI | The prevalence of blaCTX-M, qnrB and qnrS increased from 6.6% to 9% before travel to 33.6% to 55.7% after international travel. | Small study population size precluded risk analysis. | |
| 15 | China | 77 | Microarray | 2,915 | NA | ARDB | ARG diversity is related to age. | False-positive or -negative hybridization signals may exist. | |
| 16 | The Netherlands | 10 | Mf-PCR | 81 | NA | ResFinder, NCBI | The catA and tetW genes were more abundant in healthy subjects than the critically ill. | The findings may not apply to countries where resistant bacteria is prevalent. | |
| 17 | Canada | 70 | Microarray | 254 | NA | CARD, ResFinder, ARG-ANNOT | Gut resistome in healthy adults showed prompt recovery to baseline in one week after antibiotic perturbation. | The microarray did not include all known ARGs. | |
| 18 | The Netherlands | 1,179 | SeqMeta | 11 | ShortBRED | CARD | Females had higher diversity of ARGs and prevalence of lnu genes. | Small number of male subjects may be underpowered. | |
| 19 | Peru | 263 | SeqMeta | 1,100 | ShortBRED | CARD, Lahey | The resistome from human gut and their associated animals and environment are shared | No functional validation of the ARGs identified. | |
| 20 | China, Europe, USA | 2,768 | SeqMeta | 19,569 | SOAP-denovo | ResFinder | Mobile resistomes are mainly found, in order of frequency, in Proteobacteria (399 genes), Firmicutes (86), Bacteroidetes (46), and Actinobacteria (40). | Nucleotide identity cutoff was set at 95% against ResFinder. | |
| 21 | Caucasian male | 12 | SeqMeta | NA | CARD-RGI | CARD, ResFams, |
Healthy subjects with glycopeptide or aminoglycoside resistance genes were prone to de novo colonization upon antibiotic exposure. | The annotation algorithm was conservative. | |
| 22 | UK, France, Italy, Finland, Norway, Scotland | 22 | Microarray; FunMeta | NA | Chromas, DNAMAN | NCBI | The tet(W) gene and transposons Tn4451 and Tn1549 were highly prevalent in fecal metagenomes. | Only tetracycline and erythromycin resistance genes were investigated. | |
| 23 | Finland, France, Italy, Norway, UK | 100 | PCR; Microarray; FunMeta | 97 | NA | NA | ARGs identified by microarray were not recovered by functional screen. Combining these methods increase the identifiable ARG repertoire. | The cloned ARG in functional screen needs to be expressive in the heterologous host. | |
| 25 | 11 countries | 180 | SeqMeta | 1,209 | ARGs-OAP | SARG | The ermF was the representative macrolide resistant gene in the Chinese population. | Bacterial hosts of ARGs were predicted by network analysis. | |
| 28 | Denmark, Spain, USA | 252 | SeqMeta | 3,496 | MOCAT | ARDB | The most prevalent human gut ARGs were those that encoded resistance to antibiotics used in animals in a country-specific manner. | Only three developed countries were involved. | |
| 29 | USA | 2 | FunMeta | NA | NA | NCBI | Half of the resistance genes recovered from aerobic gut isolates were identical to that in pathogens. | The majority of gut commensals was anaerobic. | |
| 30 | Germany | 2 | SeqMeta | NA | DIAMOND | CARD | The gut resistome in two persons changed differently in response to ciprofloxacin treatment. | Given sequence reads did not span the whole ARG length, its frequency could not be identified. | |
| 31 | USA | 29 | SeqMeta | 21 | Prodigal, BLASTP | DeepARG | Sixty four ARGs were shared amongst the gut resistomes from chickens, pigs and humans. | The genetic contexts of the ARGs were unclear. | |
| 32 | Malawi, USA, Venezuela | 110 | SeqMeta | 70 | BLAST+ | ARDB, ResFams | Antibiotic resistance and virulence genes are correlated and may be co-selected. | Results were based on sequence analysis without downstream laboratory validation. | |
aNumber of antibiotic resistance genes (ARG) surveyed or annotated; FunMeta, functional metagenomics; Marilyn, Marilyn Robert’s website for macrolides and tetracycline resistance genes; MERGEM, Mobile Elements and Resistance Genes Enhanced for Metagenomics; Mf-PCR, microfluidic PCR; PATRIC, Pathosystems Resource Integration Center; Ref, reference; SeqMeta, sequence-based metagenomics; SOAP, short oligonucleotide analysis package.
Ambiguous definition of a healthy human gut resistome
Human gut resistomes are by large defined as the collection of antibiotic resistance genes present in the gut microbiota.14–19 This collection varied from 10 AR genes encoding resistance to five drug classes (β-lactams, tetracyclines, macrolides, aminoglycosides, and quinolones)14 to more than 2,900 AR genes classified into 369 gene types.15 These 369 AR gene types could be broadly classified into resistance to six drug classes including β-lactams and tetracyclines.15 The number of AR genes surveyed for a given antibiotic class also varied considerably. For instance, more than 30 tetracycline resistance genes were tested in one study15 but only tetM and tetQ were investigated in another.14 Nonetheless, they both reported the detection rate of tetracycline resistance in the gut resistome, which was initially limited by the number of genes surveyed. Interestingly, the highly prevalent tetW gene in healthy subjects16 was not investigated in another study targeting healthy people after international travel.14 While the interaction between AR genes per se and their repressors or promoters is well documented, the inclusion of transposons, integrons, and AR gene-associated gene regulators has been variably reported. Several studies did not detect AR gene-associated genetic elements.14,16,20 No study has considered collateral sensitivity and silent AR genes in the healthy gut resistome. In metagenomic studies where de novo assembly was used to detect novel AR determinants, the identity ranged from less than 60% amino acid sequence identity19 to more than 99% nucleotide sequence similarity to define recently horizontal transferred elements.21 A stringent threshold could potentially reduce the number of AR genes identified, and vice versa. Collectively, the difference of the number of AR genes, choice of marker AR gene to represent a functional antibiotic class and the degree of sequence identity to define a putative determinant hinder comparison across studies.
Epidemiological factors shaping the healthy gut resistome
At an individual level, the gut resistome landscape is largely shaped by age,15 sex,18 co-morbidities,16 gut microbiota composition,25 living conditions such as household environment,19 and daily life exposure.6 For instance, the Hadza hunter-gatherer had a gut resistome profile similar to that in soil.6 While antibiotic exposure was anticipated to alter the human gut resistome in people with existing co-morbidities,16 the gut resistome appears to be highly resilient in healthy subjects following short-term experimental exposure to antibiotics.17,22 At the population-wide level, a single ecological study suggested that the gut resistotype was associated with the country-level consumption of antibiotics.28 In the 21st century, the mobility of humans across countries also accounts for the variation of gut resistome. Despite an initially low prevalence of blaCTX-M (9%) and qnrB (6.6%) before departure from the Netherlands, a country with low prevalence of antibiotic resistance, as many as 33.6% and 36.9% of the Dutch travelers acquired blaCTX-M and qnrB genes, respectively, following travel to Southeast Asian and the Indian subcontinents.14 Cross-country migrations of refugees and asylum seekers also potentially altered the gut resistome amongst the local population due to higher prevalence of resistance genes in such developing countries as Afghanistan, Iraq, and Syria.7 A recent study in the UK has shown that the two major risk factors for carriage of CTX-M in the healthy community are travel to an area of high occurrence and birth-origin in such countries which is associated with frequent travel to those areas of the world.34,35
Contemporary approaches to investigate human intestinal resistome
Essentially, three approaches of studying the gut resistome have been identified. Classically (and historically), a selected pool of AR genes was detected by either PCR or customized microarrays.7,14–17s Different marker AR genes were chosen to represent resistance phenotype to a given antibiotic class.14–17 These methods were also limited to the small number of AR genes possibly be detected, reducing the representativeness of the AR gene repertoire. The second approach employed total bulk DNA directly isolated from samples, for which only known AR or related genes with similar sequence identity were detected. In this method, DNA was extracted from fecal specimens and sequenced with a depth of 1.2Gb to 12.6Gb per sample.4,19 Contigs were then assembled based on user-defined settings, the sequence of which was compared against existing antibiotic resistance gene databases such as Comprehensive Antibiotic Resistance Database (CARD).,18,19,22,24 Lahey beta-lactamase database,19 Antibiotic Resistance Gene-ANNOTation (ARG-ANNOT),13 Antibiotic Resistance Database (ARDB),13,25,28 and Resfinder.21 A combination of these databases could also be included in a study to increase the detection rate of AR genes.13,19,30 Based on local AR gene distribution, additional augmentation could also be possible.28 The third approach entails direct fragmentation of DNA extracted from fecal samples and cloned into heterologous hosts such as E. coli using shuttle vectors including plasmids,19 fosmids8 or bacterial artificial chromosomes, depending on the insert size.20,23 This method has the advantage of identifying novel AR genes and confirming the role of the AR genes albeit its expression may differ in the heterologous host. Collectively, targeted AR gene detection will be inexpensive, allowing surveys be conducted in resource-limited settings such as low- and middle-income countries. In places where affordability is not a concern, an initial sequence-based metagenomic survey can be conducted to identify putative AR genes, followed by confirming its biological role in an expressive bacterium such as E. coli. All in all, while metagenomic shotgun sequencing can sidestep cultivability of gut commensals in which anaerobic bacteria predominate, its small size of contig assembly renders the characterization of antibiotic-resistant gene harboring transposons or other mobile elements difficult.21 This can be overcome by constructing and screening of fosmid libraries using longer DNA fragments isolated from pulsed-field gel electrophoresis assuming that the gene encodes the same phenotype in both the heterologous host and the native bacterium.8 The bioinformatic pipelines and reference databases for gut resistomes are summarized in Tables 2 and 3, respectively.
Table 2.
Common bioinformatic pipelines for gut resistome analysis.
| Pipeline | Comments | Latest Update | Website | Ref. |
|---|---|---|---|---|
| fARGene | ARGs can be identified and reconstructed directly from metagenomic data, suitable for unknown resistomes. | May 2019 | https://github.com/fannyhb/fargene | 36 |
| ResFinder | Allows the identification of acquired AMRs and chromosomal mutations. | Apr 2019 | https://cge.cbs.dtu.dk/services/ResFinder | 37 |
| ARG-OAP | Online pipeline for fast annotation of ARGs. | June 2018 | http://smile.hku.hk/SARGs | 38 |
| GROOT | Combination of a variation graph representation of genes with a Forest indexing scheme. | April 2019 | https://github.com/will-rowe/groot | 39 |
| ARIBA | Efficient for identifying acquired AMR genes and variants. | June 2019 | https://github.com/sanger-pathogens/ariba | 40 |
| DeepARG | This is a deep machine learning algorithm for ARGs. | June 2017 | https://bench.cs.vt.edu/deeparg | 41 |
| ShortBRED | Profiling protein families of ARGs from shotgun metagenomics data. | Unclear | http://huttenhower.sph.garvard.edu/shortbred | 42 |
ARDB, Antibiotic Resistance Gene Database; CARD, The Comprehensive Antibiotic Resistance Database; MERGEM, Mobile Elements and Resistance Genes database Enhanced for Metagenomics; RED-DB, Resistance Determinants Database; SARG, Structured Antibiotic Resistance Genes.
Table 3.
Reference databases for antibiotic resistance genes.
| Database | Comments | Latest Update | Information contained | Website | Ref. |
|---|---|---|---|---|---|
| ARDB | No longer being maintained. All information has moved to CARD. | July 2009 |
13,293 genes, 377 types, 257 antibiotics, 632 genomes, 933 species and 124 genera | https://ardb.cbcb.umd.edu/ | 43 |
| ARGO | Data was restricted to β-lactamase, tetracycline, and vancomycin genes. | Unclear | 555 β-lactamases and 115 vancomycin resistance genes reported before 2004. | http://www.argodb.org/ | 44 |
| ARG-ANNOT | Based on a local BLAST program in Bio-Edit software, allowing offline analysis. | Unclear | 1,689 antibiotic resistance genes | No longer available | 45 |
| CARD | This is one of the most extensive AMR sequence databases, covering intrinsic, mutation-driven, and acquired resistance, including mobile genetic elements. | April 2019 | 2570 reference sequences, 1233 SNPs, 7305 plasmids, 5524 chromosomes | https://card.mcmaster.ca/ | 46 |
| Lahey Clinic database | This database provides comprehensive information on β-lactamase genes. | May 2019 | β-lactamase classification and amino acid sequences for TEM, SHV, and OXA genes. | https://www.lahey.org/ | 47 |
| SARG | The redundant sequences were removed and query sequence detection optimized. | June 2018 | 9080 sequence belonged to 189 ARGs subtypes. | http://smile.hku.hk/SARGs | 38 |
| MERGEM | This database contains resistance genes suitable for k-mer filtering. | Unclear | The website is no longer maintained | http://www.mergem.genome.ulaval.ca/ | 48 |
| RED-DB | Both partial and complete coding sequences are available. | May 2019 |
10,742 non-redundant genes covering ten antibiotic classes. | http://www.fibim.unisi.it/REDDB | - |
| ResFams | Database of proteins encoding drug resistance, organized by ontology. | Feb 2018 | 2,454 unique sequences, presenting 54 Resfams protein families. | http://www.dantaslab.org/resfams | 49 |
| AMRFinder | Database of AMR protein and gene nomenclature with hidden Markov models | June 2019 | 4,579 AMR protein sequences and more than 560 hidden Markov models. | https:/www.ncbi.nlm.nih.gov/pathogens/antimicrobial-resistance/AMRFinder/ | 50 |
ARDB, Antibiotic Resistance Gene Database; CARD, The Comprehensive Antibiotic Resistance Database; MERGEM, Mobile Elements and Resistance Genes database Enhanced for Metagenomics; RED-DB, Resistance Determinants Database; SARG, Structured Antibiotic Resistance Genes.
Common antibiotic resistance determinants found in healthy human gut
Of 180 fecal metagenome datasets tested, the AR genes aadE, bacA, acrB, tetM, tetW, vanR, and vanS were present in all people.25 The healthy human gut is expected to carry AR genes which are associated with intestinal anaerobic commensals such as catA and tetW encoding chloramphenicol and tetracycline resistances, respectively.16 Interestingly, functional metagenomic studies have recovered clones with AR genes encoding resistance to β-lactamases (blaTEM), macrolides (ermB, ereA), aminoglycosides (strA, strB), trimethoprim (dfrA14) and sulfonamides (sul2) other than bacitracin and tetracyclines.20 To date, genes conferring resistance to isoniazid, rifampicin, oxazolidinones have not yet been reported in the healthy human gut microbiome.
Quality assessment
Of 22 included studies, seven were re-analysis of publicly available metagenomic datasets whereas 15 were primary studies of which only one was population based.18 The remaining used either convenient samples or less than 10 samples with limited representation of the target population. Only six studies reported prior exposure to antibiotics amongst the participants.5,16,20,22,29,30 The scores for each study as evaluated by Newcastle–Ottawa scale for non-randomized cohort studies are summarized in Table S4.
Discussion
The abundance and diversity of human gut microbiota and its association with physiological functions are well recognized. Nonetheless, their roles as potential carriers of AR genes have received unequal attention. With the increasing prevalence of multiple drug-resistant bacteria worldwide, the potential reservoir of AR genes in the gut microbiota and the possibility of their transfer to human pathogens are of concern. In this article, we systematically reviewed studies on healthy human digestive tract resistome and highlight the methodological heterogeneity across studies which may potentially generate biased results.
The environmental resistome has been extensively studied. It was speculated that AR genes have been selected because of human use of antibiotics. Isolation of AR genes in ancient permafrost and in communities without anthropogenic activity disproved this hypothesis, suggesting that antibiotic resistance genes may pre-date the widespread use of naturally occurring antibiotics.12 The rapid emergence of resistance to sulfonamides in the 1930s demonstrates that a non-natural chemical entity can readily select for genes encoding enzymes with preexisting functionality in bacteria to confer resistance to these synthetic agents. The presence of relevant antibiotic resistance mechanisms in organism such as Streptomyces from which the antibiotics was isolated for clinical use was assumed to be a part of self-protection mechanism for survival.12 This finding led to numerous studies on disparate microbial communities, predominantly in the soil resistome which is reviewed elsewhere.9
Defining an antibiotic resistance gene could be difficult, particularly in cases where sequence-based metagenomic studies have identified a gene with considerable similarity to a known AR gene but without knowledge of its phenotype.31 This is further complicated by using de novo assembly which identifies thousands of potential AR genes with variable degree of similarity to the existing known AR genes.21 The number of candidate AR genes thus identified may actually reflect the cutoff of similarity to define an AR gene per se, without reference to the functional significance of the difference in DNA sequence, such as that based on the active sites that determine resistance. In addition, sequence-based metagenomic approach is limited by the variability of expression of the AR genes in heterologous hosts. Some of the AR genes identified by sequence similarity may be silent or nonfunctional in the native hosts.
In multiple studies, AR genes appear to be exclusively present in gut commensals.5,16,20 Longitudinal analysis of fecal metagenomic assembly from critically ill subjects surprisingly identified that all antibiotic-resistant genes including aph(2”)-Ib and aadE-like gene were carried by mobile genetic elements such as plasmids and insertion sequences in anaerobic gut commensals among Firmicutes, Bacteroidetes, and Actinobacteria.16 The aminoglycoside resistance genes were harbored by butyrate-producing Clostridium cluster IV commensal which are present in healthy humans.16 These genes encoding clinically relevant drug resistance, if transferred to pathogens, will pose a threat to the patients.
In recent decades, the increasing use of disinfectants in the food industry and clinical settings led to research on reduced susceptibility to disinfectants. Surprisingly, studies on gut resistome did not consider the presence of genes encoding reduced susceptibility to biocides such as quaternary ammonium compounds and triclosan. It has been shown that environments with high concentrations of quaternary ammonium biocides select for high carriage rates in bacteria of Class 1 integrons and therefore might be expected to co-select for AR genes.51 Numerous studies employed quantitative polymerase chain reaction and commercially available microarray assays to study gut resistome. These findings would therefore be limited to known resistance genes. Coupled with functional screen of large DNA inserts in appropriate vectors and expression hosts, functional metagenomics appears to be a promising robust molecular tool to identify novel antimicrobial resistance genes and their origins. For instance, functional screens of specimens with known plasmid-borne resistance genes detected in microarray-hybridization did not recover any transformed clones with plasmid-borne resistant determinants but instead detected a clone with resistant gene carried in chromosome.20 This discrepancy may indicate that the genes detected by microarray may be silent and that both determinants may be present in the microbial community.
The use of metagenomic shotgun sequencing circumvents the problem of non-culturable gut commensals.52–54 The results are often limited by the bioinformatics pipelines and ARG database(s) selected for analyses. Recently, the National Center for Biotechnology Information (NCBI) produced a high-quality, curated, AMR gene reference database consisting of up-to-date protein and gene nomenclature. A comparison of the susceptibilities of three common Gram-negative foodborne pathogens against the database gave high consistent predictions of 98.4%. This database is designated as AMRFinder with more than 390,000 entries, which is one of the most comprehensive databases.50 However, the small size of the contigs which are assembled renders the characterization of the resistance-gene harboring transposons or other mobile elements difficult.21 This can be overcome by constructing and screening of fosmid libraries using longer DNA fragments isolated from pulsed-field gel electrophoresis assuming that the gene encodes the same phenotype in both the heterologous host and the native bacterium.8
Strengths and limitations of this review
To our knowledge, this is the first systematic review on studies investigating healthy human gut resistome. Using pre-defined inclusion and exclusion criteria, we comprehensively searched the available databases for relevant studies. Our review highlights the methodological heterogeneity across studies that demands harmonization. However, our review is limited by the small sample size in most of the included studies and that most of them were based on convenient samples or re-analysis of publicly available datasets, a shortcoming which will be addressed as newer technologies are developed and deployed.
Conclusions
In conclusion, there are considerable methodological heterogeneities across studies. The choice of marker genes to represent resistance phenotype to a given antibiotic class varied. Uniformity in phenotypic validation and bioinformatics pipelines will need to be addressed to facilitate inter-study comparisons.
Supplementary Material
Funding Statement
We thank the support of funding from Health and Medical Research Fund (Project number 18170082, PI: MI), Food and Health Bureau of the Hong Kong Special Administrative Region, People’s Republic of China. The study was partially supported by a seed fund for gut microbiota research provided by the Faculty of Medicine, The Chinese University of Hong Kong. The funding bodies did not involve in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Disclosure of potential conflicts of interest
None to declare.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Ip M, Ang I, Liyanapathirana V, Ma H, Lai R.. Genetic analyses of penicillin binding protein determinants in multidrug-resistant Streptococcus pneumoniae serogroup 19 CC320/271 clone with high-level resistance to third-generation cephalosporins. Antimicrob Agents Chemother. 2015;59:4040–12. doi: 10.1128/AAC.00094-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ip M, Chau SS, Chi F, Cheuk ES, Ma H, Lai RW, Chan PK. Longitudinally tracking fluoroquinolone resistance and its determinants in penicillin-susceptible and –nonsusceptible Streptococcus pneumoniae isolates in Hong Kong, 2000–2015. Antimicrob Agents Chemother. 2007;51:2192–2194. doi: 10.1128/AAC.00139-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang Y, Luo M, Zhou H, Li C, Luk A, Zhao G, Fung K, Ip M. Role of two-component system response regulator bceR in the antimicrobial resistance, virulence, biofilm formation, and stress response of group B Streptococcus. Front Microbiol. 2019;10:10. doi: 10.3389/fmicb.2019.00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raymond F, Boissinot M, Quameur AA, Deraspe M, Plante PL, Kpanou SR, Berube E, Huletsky A, Roy PH, Ouellette, et al. Culture-enriched human gut microbiomes reveal core and accessory resistance genes. Microbiome. 2019;7:56. doi: 10.1186/s40168-019-0669-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruppe E, Ghozlane A, Tap J, Pons N, Alvarez AS, Maziers N, Cuesta T, Hernando-Amado S, Clares I, Martinez JL, et al. Prediction of the intestinal resistome by a three-dimensional structure-based method. Nat Microbiol. 2019;4:112–113. doi: 10.1038/s41564-018-0292-6. [DOI] [PubMed] [Google Scholar]
- 6.Rampelli S, Schnorr SL, Consolandi C, Turroni S, Severgnini M, Peano C, Brigidi P, Crittenden A, Henry A, Candela M, et al. Metagenome sequencing of the Hadza hunter-gatherer gut microbiota. Curr Biol. 2015;25:1682–1693. doi: 10.1016/j.cub.2015.04.055. [DOI] [PubMed] [Google Scholar]
- 7.Hasler R, Kautz C, Rehman A, Podschun R, Gassling V, Brzoska P, Sherlock J, Gräsner J-T, Hoppenstedt G, Schubert S, et al. The antibiotic resistome and microbiota landscape of refugees from Syria, Iraq, and Afghanistan in Germany. Microbiome. 2018;6:37. doi: 10.1186/s40168-018-0414-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N, Pan Y, Li J, Zhu L, Wang X, et al. Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun. 2013;4:2151. doi: 10.1038/ncomms3151. [DOI] [PubMed] [Google Scholar]
- 9.Surette MD, Wright GD. Lessons from the environmental antibiotic resistome. Annu Rev Microbiol. 2017;71:309–329. doi: 10.1146/annurev-micro-090816-093420. [DOI] [PubMed] [Google Scholar]
- 10.Karman A, Do TT, Walsh F, Virta MPJ. Antibiotic-resistance genes in waste water. Trends Microbiol. 2018;26:220–228. doi: 10.1016/j.tim.2017.09.005. [DOI] [PubMed] [Google Scholar]
- 11.McEwen SA, Collignon PJ. Antimicrobial resistance: a one health perspective. Microbiol Spectr. 2018;6:2. doi: 10.1128/microbiolspec.ARBA-0009-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.D’Costa VM, McGrann KM, Hughes DW, Wright GD. Sampling the antibiotic resistome. Science. 2006;311:374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- 13.Yang Z, Guo Z, Qiu C, Li Y, Feng X, Liu Y, Zhang Y, Pang P, Wang P, Zhou Q, et al. Preliminary analysis showed country-specific gut resistome based on 1,267 feces samples. Gene. 2016;581:178–182. doi: 10.1016/j.gene.2016.01.043. [DOI] [PubMed] [Google Scholar]
- 14.von Winstersdorff CJ, Penders J, Stobberingh EE, Oude Lashof AM, Hoebe CJ, Savelkoul PH, Wolffs PFG. High rates of antimicrobial drug resistance gene acquisition after international travel, The Netherlands. Emerg Infect Dis. 2014;20:649–657. doi: 10.3201/eid2004.131718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu N, Hu Y, Zhu L, Yang X, Yin Y, Lei F, Zhu Y, Du Q, Wang X, Meng Z, et al. DNA microarray analysis reveals that antibiotic resistance-gene diversity in human gut microbiota is age related. Sci Rep. 2015;4:4302. doi: 10.1038/srep04302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Buelow E, Gonzalez T, Fuentes S, de Steenhuijsen Piters WAA, Lahti L, Bayjanov JR, Majoor EAM, Braat JC, van Mourik MSM, Oostdijk EAN, et al. Comparative gut microbiota and resistome profiling of intensive care patients receiving selective digestive tract decontamination and healthy subjects. Microbiome. 2017;5:88. doi: 10.1186/s40168-017-0309-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacPherson CW, Mathieu O, Tremblay J, Champagne J, Nantel A, Girard S-A, Tompkins TA.. Gut bacterial microbiota and its resistome rapidly recover to basal state levels after short-term amoxicillin-clavulanic acid treatment in healthy adults. Sci Rep. 2018;8:11192. doi: 10.1038/s41598-018-29229-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sinha T, Vich Vila A, Garmaeva S, Jankipersadsing SA, Imhann F, Collij V, Bonder MJ, Jiang XF, Gurry T, Alm EJ, et al. Analysis of 1135 gut metagenomes identifies sex-specific resistome profiles. Gut Microbes. 2018;29:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pehrsson EC, Tsukayama P, Patel S, Mejia-Bautista M, Sosa-Soto G, Navarrete KM, Calderon M, Cabrera L, Hoyos-Arango W, Bertoli MT, et al. Interconnected microbiomes and resistomes in low-income human habitats. Nature. 2016;533:212–216. doi: 10.1038/nature17672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Card RM, Warburton PJ, MacLaren N, Mullany P, Allan E, Anjum MF, Aziz RK. Application of microarray and functional-based screening methods for the detection of antimicrobial resistance genes in the microbiomes of healthy humans. PLoS One. 2014;9:e86428. doi: 10.1371/journal.pone.0086428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y, Yang X, Li J, Lv N, Liu F, Wu J, Lin IYC, Wu N, Weimer BC, Gao GF, et al. The bacterial mobile resistome transfer network connecting the animal and human microbiomes. Appl Environ Microbiol. 2016;82:6672–6681. doi: 10.1128/AEM.01802-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palleja A, Mikkelsen KH, Forslund SK, Kashani A, Allin KH, Nielsen T, Hansen TH, Liang S, Feng Q, Zhang C, et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat Microbiol. 2018;3:1255–1265. doi: 10.1038/s41564-018-0257-9. [DOI] [PubMed] [Google Scholar]
- 23.Seville LA, Patterson AJ, Scot KP, Mullany P, Quail MA, Parkhill J, Ready D, Wilson M, Spratt D, Roberts AP, et al. Distribution of tetracycline and erythromycin resistance genes among human oral and fecal Metagenomic DNA. Microb Drug Resist. 2009;15:159–166. doi: 10.1089/mdr.2009.0916. [DOI] [PubMed] [Google Scholar]
- 24.Abraham N, Kwon DH. A single amino acid substitution in PmrB is associated with polymyxin B resistance in clinical isolate of Pseudomonas aeruginosa. FEMS Microbiol Lett. 2009;298:249–254. doi: 10.1111/fml.2009.298.issue-2. [DOI] [PubMed] [Google Scholar]
- 25.Feng J, Li B, Jiang X, Yang Y, Wells GF, Zhang T, Li X. Antibiotic resistome in a large-scale healthy human gut microbiota deciphered by metagenomics and network analyses. Environ Microbiol. 2018;20:3855–3868. doi: 10.1111/1462-2920.14009. [DOI] [PubMed] [Google Scholar]
- 26.Moher D, Liberati A, Tetzlaff J, DG A, The PRISMA Group . Preferred reporting items for systematic reviews and meta-analyses: the PRISMA Statement. PLoS Med. 2009;6:e1000097. doi: 10.1371/journal.pmed.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wells GA, Shea B, O’Connell D, Peterson J, Welch V, Losos M, Tugwell. The Newcastle-Ottawa Scale (NOS) for assessing the quality of nonrandomized studies in meta-analyses. Accessed www.ohri.ca/programs/clinical_epidemiology/oxford.asp, 10 Dec 2019. [Google Scholar]
- 28.Foslund K, Sunagawa S, Kultima JR, Mende DR, Arumugam M, Typas A, Bork P. Country-specific antibiotic use practices impact the human gut resistome. Genome Res. 2013;23:1163–1169. doi: 10.1101/gr.155465.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sommer MOA, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science. 2009;325:1128–1131. doi: 10.1126/science.1176950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Willmann M, El-Hadidi M, Huson DH, Schutz M, Weidenmaier C, Autenrieth IB. Antibiotic selection pressure determination through sequence-based metagenomics. Antimicrob Agents Chemother. 2015;59:7335–7345. doi: 10.1128/AAC.01504-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeng J, Pan Y, Yang J, Hou M, Zeng Z, Xiong W. Metagenomic insights into the distribution of antibiotic resistome between the gut-associated environments and the pristine environments. Environ Int. 2019;126:346–354. doi: 10.1016/j.envint.2019.02.052. [DOI] [PubMed] [Google Scholar]
- 32.Escudeiro P, Pothier J, Dionisio F. Nogueira. Antibiotic resistance gene diversity and virulence gene diversity are correlated in human gut and environmental microbiomes. mSphere. 2019;4:e00135–19. doi: 10.1128/mSphere.00135-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.United Nations . World population prospects: the 2017 revision. New York (NY): United Nations Department of Economic and Social Affairs, Population Division; 2017. [Google Scholar]
- 34.McBulty CAM, Lecky DM, Xu-McCrae L, Nakiboneka-Ssenabulya D, Chung KT, Nichols T, Thomas HL, Thomas M, Alvarez-Buylla A, Turner K, et al. CTX-M ESBL-producing enterobacteriaceae: estimated prevalence in adults in England in 2014. J Antimicrob Chemother. 2018;73:1368–1388. doi: 10.1093/jac/dky007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bevan ER, McNally A, Thomas CM, Piddock LJV, Hawkey PM. Acquisition and loss of CTX-M producing and non-producing Escherichia coli in the fecal microbiome of travellers to South Asia. MBio. 2018;9:e02408–18. doi: 10.1128/mBio.02408-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berglund F, Osterlund T, Boulund F, Marathe NP, Larsson DGJ, Kristiansson E. Identification and reconstruction of novel antibiotic resistance genes from metagenomes. Microbiome. 2019;7:52. doi: 10.1186/s40168-019-0670-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother. 2012;67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yang Y, Jiang X, Chai B, Ma L, Li B, Zhang A, Cole JR, Tiedje JM, Zhang T. ARGs-OAP: online analysis pipeline for antibiotic resistance genes detection from metagenomic data using an integrated structured ARG-database. Bioinformatics. 2016;32:2346–2351. doi: 10.1093/bioinformatics/btw136. [DOI] [PubMed] [Google Scholar]
- 39.Rowe WPM, Winn MD, Hancock J. Indexed variation graphs for efficient and accurate resistome profiling. Bioinformatics. 2018;34:3601–3608. doi: 10.1093/bioinformatics/bty387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hunt M, Mather AE, Sanchez-Buso L, Page AJ, Parkhill J, Keane JA, Harris SR.. ARIBA: rapid antimicrobial resistance genotyping directly from sequencing reads. Microb Genom. 2017;3:e00131. doi: 10.1099/mgen.0.000131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arango-Argoty G, Garner E, Pruden A, Heath LS, Vikesland P, Zhang L. DeepARG: a deep learning approach for predicting antibiotic resistance genes from metagenomic data. Microbiome. 2018;6:23. doi: 10.1186/s40168-018-0401-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaminski J, Gibson MK, Franzosa EA, Segata N, Dantas G, Huttenhower C. High-specificity targeted functional profiling in microbial communities with ShortBRED. PLoS Comput Biol. 2015;11:e1004557. doi: 10.1371/journal.pcbi.1004557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu B, Pop M. ARDB-antibiotic resistance genes database. Nucleic Acids Res. 2009;37(Database issue):D443–7. doi: 10.1093/nar/gkn656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Scaria J, Chandramouli U, Verma SK. Antibiotic resistance genes online (ARGO): a database on vancomycin and beta-lactam resistance genes. Bioinformation. 2005;1:5–7. doi: 10.6026/97320630001005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gupta SK, Padmanabhan BR, Diene SM, Lopes-Rojas R, Kempf M, Landraud L, Rolain JM. ARG-ANNOT, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob Agents Chemother. 2014;58:212–220. doi: 10.1128/AAC.01310-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira S, Sharma AN, et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017;45(D1):D566–D573. doi: 10.1093/nar/gkw1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Naas T, Oueslati S, Bonnin RA, Dabos ML, Zavala A, Dortet L, Retailleau P, Iorga BI. Beta-lactamase database – structure and function. J Enzyme Inhib Med Chem. 2017;32:917–919. doi: 10.1080/14756366.2017.1344235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Raymond F, Ouameur AA, Deraspe M, Iqbal N, Gingras H, Dridi B, Leprohon P, Plante P-L, Giroux R, Bérubé È, et al. The initial state of the human gut microbiome determines its reshaping by antibiotics. Isme J. 2016;10:707–720. doi: 10.1038/ismej.2015.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gibson MK, Forsberg KJ, Dantas G. Improved annotation of antibiotic resistance functions reveals microbial resistomes cluster by ecology. Isme J. 2014;9:207–216. doi: 10.1038/ismej.2014.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Feldgarden M, Brover V, Haft DH, Prasaad AB, Slotta DJ, Tolstoy I. 2019. Using the NCBI AMRFinder tool to determine antimicrobial resistance genotype-phenotype correlations within a collection of NARMS isolates. Antimicrob Agents Chemother. 2019;63:e00483-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gaze WH, Abdouslam N, Hawkey PM, Wellington EM. Incidence of class 1 integrons in a quaternary ammonium compound-polluted environment. Antimicrob Agents Chemother. 2005;49:1802–1807. doi: 10.1128/AAC.49.5.1802-1807.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chen Z, Yeoh YK, Hui M, Wong PY, Chan MCW, Ip M, Yu J, Burk RD, Chan FKL, Chan PKS. Diversity of macaque microbiota compared to human counterparts. Sci Rep. 2018;8:15573. doi: 10.1038/s41598-018-33950-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yeoh YK, Chen Z, Hui M, Wong MCS, Ho WCS, Chin ML, Ng SC, Chan FKL, Chan PKS. Impact of inter- and intra-individual variation, sample storage, and sampling fraction on human stool microbial community profiles. PeerJ. 2019;7:e6172. doi: 10.7717/peerj.6172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen Z, Hui PC, Hui M, Yeoh K, Wong PY, Chan MCW, Wong MCS, Ng SC, Chan FKL, Chan PKS, et al. Impact of preservation method and 16S rRNA hypervariable region on gut microbiota profiling. mSystems. 2019;4:e00271–e002718. doi: 10.1128/mSystems.00271-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.