

ABSTRACT
Early dysbiosis of the gut microbiota is associated with the severity of acute pancreatitis (AP), although the underlying mechanism is unclear. Here, we investigated the role of crosstalk between NLRP3 and the gut microbiota in the development of AP utilizing gut microbiota deficient mice, as well as NLRP3 knockout (KO) mouse models. Pancreatic damage and systemic inflammation were improved in antibiotic-treated (Abx) and germ-free (GF) mice, accompanied by weakened activity of the intestinal NLRP3 inflammasome. Interestingly, fecal microbiota transplantation (FMT) reactivated the intestinal NLRP3 inflammasome and exacerbated the disease in Abx and GF mice. Although the gut barrier in GF and Abx mice was disrupted, gut microbiota deficiency ameliorated the severity of AP, probably due to the reduction in bacterial translocation from the gut to the pancreas. The composition of the gut microbiota was significantly different between NLRP3 KO mice and wild-type (WT) mice at baseline, and there were alterations in response to the induction of AP. While a dramatic shift in the gut microbiota with overgrowth of Escherichia-Shigella was observed in WT mice suffering from AP, there was no significant change in NLRP3 KO mice with or without AP, suggesting that NLRP3 deficiency counteracts AP-induced microbial disturbance. With a strengthened gut barrier and decreased systemic inflammation, NLRP3 KO mice showed less severe AP, as revealed by reduced pancreatic neutrophilic infiltration and necrosis. Taken together, these results identified the bidirectional modulation between the gut microbiota and NLRP3 in the progression of AP, which suggests the interplay of the host and microbiome during AP.
KEYWORDS: Acute pancreatitis, gut microbiota, NLRP3, gut barrier, inflammation
Introduction
Acute pancreatitis (AP) is one of the most common acute clinical abdominal diseases and has an increasing incidence. While most attacks of AP are mild and self-resolving, patients with severe AP (SAP) experience high mortality and morbidity due to early multiple organ failures and later development of infectious complications.1 It is believed that the gut is involved in the development of distant organ failure and gut barrier dysfunction is present in three of five patients with AP.2 There is sufficient evidence to confirm that gut failure plays an important role in determining the severity of the disease, as the translocation of bacteria derived from the gut is related to infectious complications, including infectious pancreatic necrosis (IPN) and the risk of death.3 The occurrence of gut-derived infection could exacerbate primary systemic inflammatory response syndrome (SIRS), as well as multiple organ dysfunction syndrome (MODS), which is often known as the “second hit” for AP.4 The mechanisms underlying gut dysfunction include disturbances in the microcirculation (ischemia and reperfusion injury), impaired immune defenses (release of cytokines and mediators) and motility, and changes in the indigenous intestinal microbial ecology, leading to the loss of mucosal integrity, which facilitates the entry of endotoxin and even bacteria into the blood circulation.5
The relationship between the gut microbiota and human health is being increasingly recognized, and dysbiosis results in a range of diseases, including obesity, diabetes, cardiovascular diseases and even carcinoma.6 The normal gut commensals exert specific functions in host nutrient metabolism, maintenance of structural integrity of the intestinal mucosal barrier, immunomodulation, and protection against pathogens.7 Accumulating evidence suggests that gut microbiota dysbiosis is associated with the severity of AP in both human and animal models, although the molecular mechanism is unclear.8-10 Patients with AP harbor a characteristic gut microbiota phenotype with reduced diversity and increased abundance of pathogenic bacteria, which is consistent with injury to the gut barrier.8 Furthermore, antibiotic treatment protects mice from caerulein-induced pancreatic injury and systemic inflammation, indicating the role of the gut microbiota in the development of AP.11,12
Nod-like receptors (NLRs) are intracellular pattern recognition molecules that detect microbial- and danger-associated molecular patterns. NLRP3, one of the NLR proteins, plays an important role in forming inflammasomes, which mediate caspase-1 activation and the secretion of the proinflammatory cytokine IL-1β in response to microbial infection and cellular damage.13 Previous studies have shown that the NLRP3 inflammasome is activated not only in the pancreas but also in the gut of mice with AP, and activation of intestinal NLRP3 is associated with the severity of AP.12,14 The literature on other diseases suggests that modulation of the gut microbiota affects activation of the NLRP3 inflammasome, and the expression of NLRP3 also shapes the composition of the gut microbiota, but a detailed understanding of their interactions in AP is lacking.15,16 The aim of this study was to evaluate the effect of intestinal dysbiosis on the worsening of AP and the crosstalk between the gut microbiota and NLRP3 in this process. In the current study, we used germ-free (GF) mice and antibiotic-treated (Abx) mice with or without fecal microbiota transplantation (FMT) to evaluate the effect of remodeling the gut microbiota on the activation of intestinal NLRP3 after AP induction. In addition, alterations in the gut microbiota were analyzed in NLRP3 knockout (KO) mice with or without caerulein stimulation to determine the impact of NLRP3 on the gut microbiome in the course of AP. This work has clinical significance as it demonstrates the destructive role and underlying mechanisms of intestinal dysbiosis in AP, and suggests the importance of early maintenance of gut homeostasis in the treatment of AP.
Results
AP-induced gut disorder is gradually restored with recovery from the disease
First of all, the animal model of AP was established by hourly intraperitoneal injections of caerulein for 11 times followed by one dose of lipopolysaccharide. Then, the mice were sacrificed at different time points (36 h and 7 days) to observe the pathological changes of not only pancreas but also gut and lung during the development of AP. Histological examination at 36 h after the first injection showed pancreatic injury characterized by marked edema, inflammatory cell infiltration and a large number of necrotic acinar cells, which were reconstructed and recovered to the normal architecture after 7 days (Figure 1(a)). The pancreatic histopathology score and the serum levels of amylase and lipase in the 7-day group were downregulated and similar to the control levels compared to those of 36 h group (Figure 1(a,b)). The marked increase in neutrophil sequestration in tissues including the pancreas and lung at 36 h, as measured by myeloperoxidase (MPO) activity, was also gradually downregulated toward the level of the control group at 7 days (Figure 1(c) and Supplement figure 1a). The serum levels of inflammatory cytokines, such as IL-10 and IL-6, were also decreased in the 7-day group compared to the 36 h group, which indicates the recovery from systemic inflammation caused by AP (Supplement figure 1b).
Figure 1.
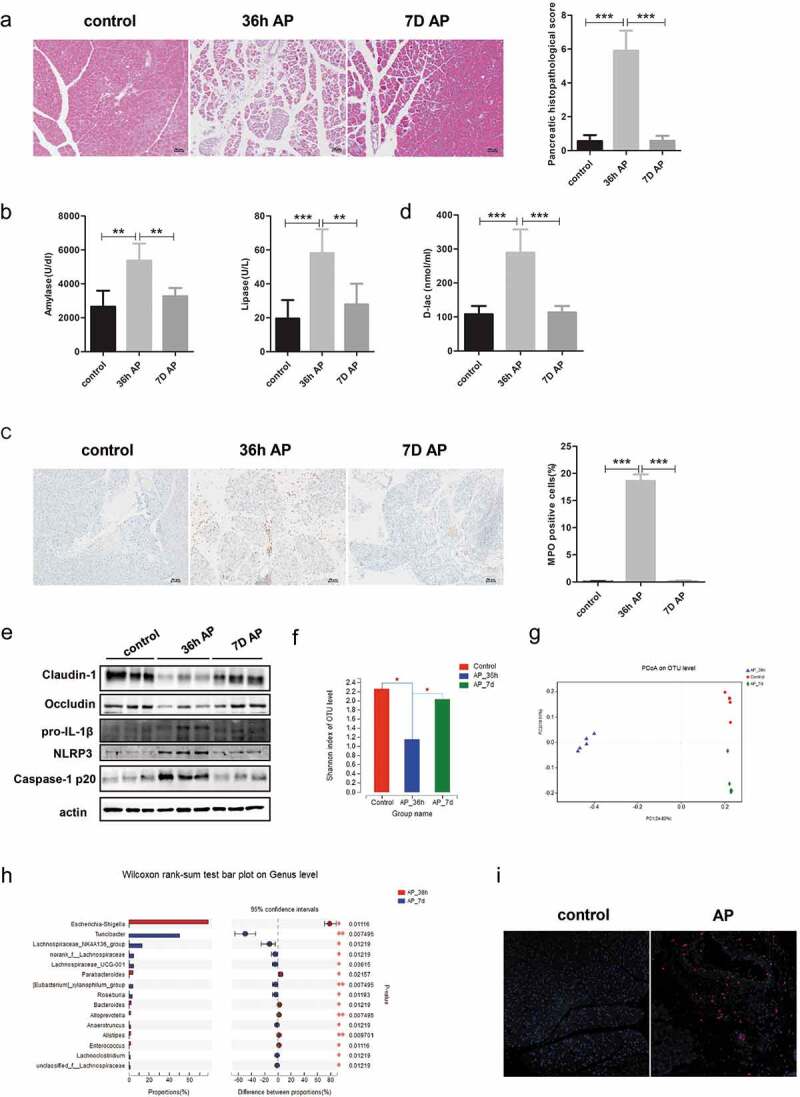
Gut dysfunction was restored with recovery from AP. Mice were sacrificed at 36 h and 7 days after the first injection of cerulein. (a) Representative H&E staining of pancreatic tissue from each group showing pancreatic injury at 36 h that was repaired at 7 days. Scale bar: 50 μm. Histopathological scoring of pancreatic damage. (b) Serum amylase and lipase activity. (c) Representative immunohistochemical staining of MPO which reflects pancreatic inflammation. Scale bar: 50 μm. (d) Serum concentration of the gut barrier index D-lac. (e) Immunoblots of intestinal epithelial tight junction proteins Claudin-1, Occludin and NLRP3 inflammasome proteins NLRP3, IL-1β, Caspase-1 p20. (f) The α diversity of gut microbiota, as determined by the Shannon index, in the 7-day group was increased to a level similar to that of the controls compared to 36 h group. *p < .05. (g) Comparison of β diversity among the different groups. (h) Alterations in the top 15 most differentially abundant genera between 36 h and 7-day group. *p < .05, **p < .01, ***p < .001. (i) Pancreatic staining of E. coli protein (red staining) and DAPI (blue) to visualize nuclei (×100). n = 6 per group, *p < .05, **p < .01, ***p < .001.
Additionally, we evaluated gut function during the recovery from AP, since the gut is one of the most frequently affected organs in disease progression. The serum level of D-lactate (D-lac), which represents gut permeability, was significantly increased in the 36 h group compared to the controls and was subsequently reduced at 7 days (Figure 1(d)). Meanwhile, the expression of Claudin-1 and Occludin was decreased in the 36 h group but was restored to the level of the controls at 7 days (Figure 1(e)). The intestinal NLRP3 inflammasome was activated in the 36 h group, as evidenced by increased expression of pro-IL-1β, NLRP3 and Caspase-1 p20, which returned to normal by 7 days (Figure 1(e)).
Our previous studies showed that dysbiosis of the gut microbiota plays a role in the development of SAP, yet the dynamic changes in the gut microbiota during the recovery from SAP were unclear.8 The decreased intraindividual diversity as measured by the Shannon index after 36 h of AP induction was elevated toward the level of the control group at 7 days (Figure 1(f)). Principal coordinate analysis based on the unweighted UniFrac distance showed significant differences in taxonomic composition between the 36 h group and 7-day group, with the latter samples clustered closer to those of the control group (Figure 1(g)). At the phylum level, the 7-day group harbored a higher relative abundance of Firmicutes and a lower abundance of Proteobacteria than the 36 h group (Supplement figure 2). At the genus level, the relative abundance of Escherichia-Shigella, Enterococcus and Alistipes was depleted in the 7-day group, whereas Lachnospiraceae and Roseburia were over-represented in the 7-day group relative to the 36 h group (Figure 1(h)). To determine whether the enriched gut bacteria could translocate to distant tissue through the impaired gut barrier, we analyzed the level of Escherichia coli (E. coli) protein in the pancreas. Interestingly, pancreatic levels of E. coli protein were increased after 36 h of AP induction, as evaluated by immunofluorescent staining of the pancreas (Figure 1(i)).
Gut microbiota deficiency regulated AP-induced gut dysfunction and alleviated disease severity
Our previous study demonstrated that pancreatic injury in Abx mice and GF mice was ameliorated compared to that in the control group after AP induction.8 The present study further showed that the number of MPO-positive cells was lower in not only pancreatic but also pulmonary tissues of Abx and GF mice than in those of the control group (Figure 2(a–d)). Serum levels of inflammatory cytokines, including TNF-α, IL-1β, IL-6 and IL-10, were also markedly decreased in Abx and GF mice compared to the controls after induction of AP (Figure 2(e–f)).
Figure 2.
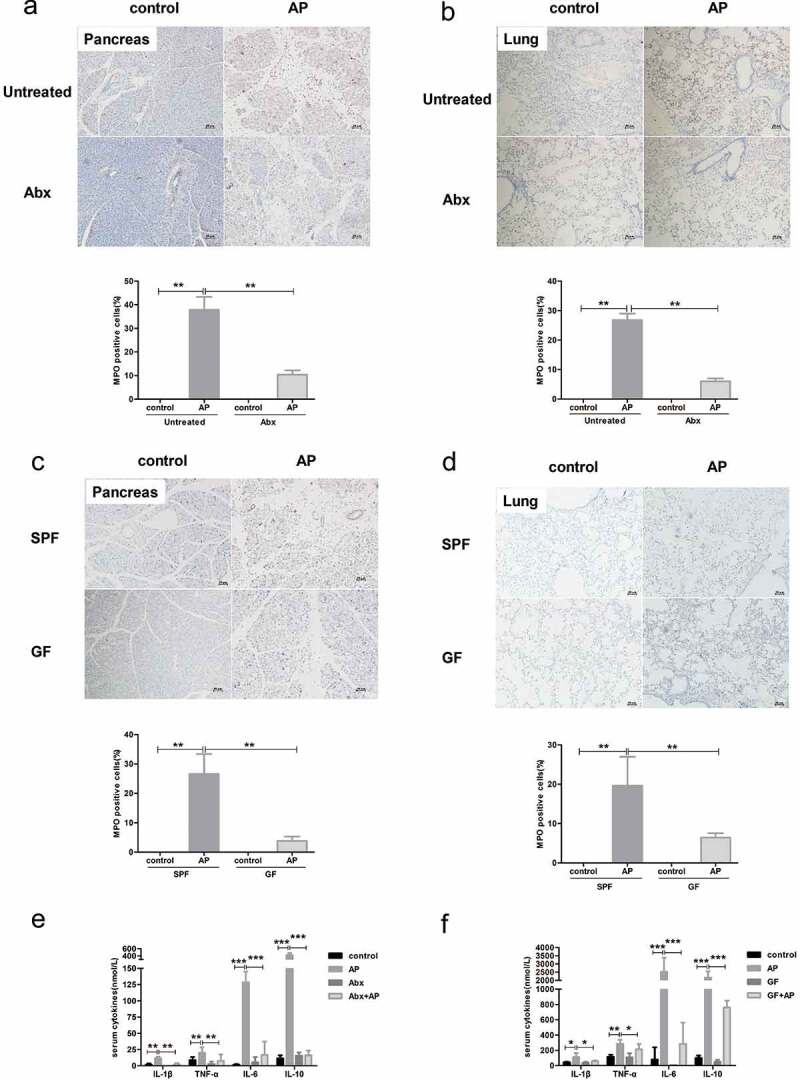
The deficiency of gut microbiota alleviated the systemic inflammatory response induced by AP. Decreased staining of MPO in the pancreas and lung of Abx mice (a, b) and GF mice (c, d) compared to SPF mice after AP induction. Scale bar: 50 μm. Serum levels of cytokines were lower in Abx (e) and GF (f) mice than in SPF mice. n = 6 per group, *p < .05, **p < .01, ***p < .001.
We further examined changes in gut function in Abx and GF mice with AP. Activation of the intestinal inflammasome was strikingly suppressed in both Abx and GF mice with AP compared to specific pathogen free (SPF) mice, as evidenced by the downregulated protein level of NLRP3, pro-IL-1β and caspase-1 p20 (Figure 3(a,b). The serum level of D-lac was increased in both control and AP mice with antibiotic treatment, while GF mice with AP showed lower levels of D-lac than SPF mice (Figure 3(c,d)). Similarly, the protein level of Occludin and Claudin-1 was decreased in Abx and GF mice compared to SPF mice independent of AP induction, which indicates the important role of the gut microbiota in the maintenance of barrier function (Figure 3(e,f)). Due to the deficiency in bacteria in the gut of Abx and GF mice, the level of E. coli protein in the pancreas was significantly reduced after the induction of AP (Figure 3(g)). Thus, we hypothesize that gut microbiota-modulated inflammation, as well as bacterial translocation, plays a more important role than the integrity of the gut barrier in the severity of AP.
Figure 3.
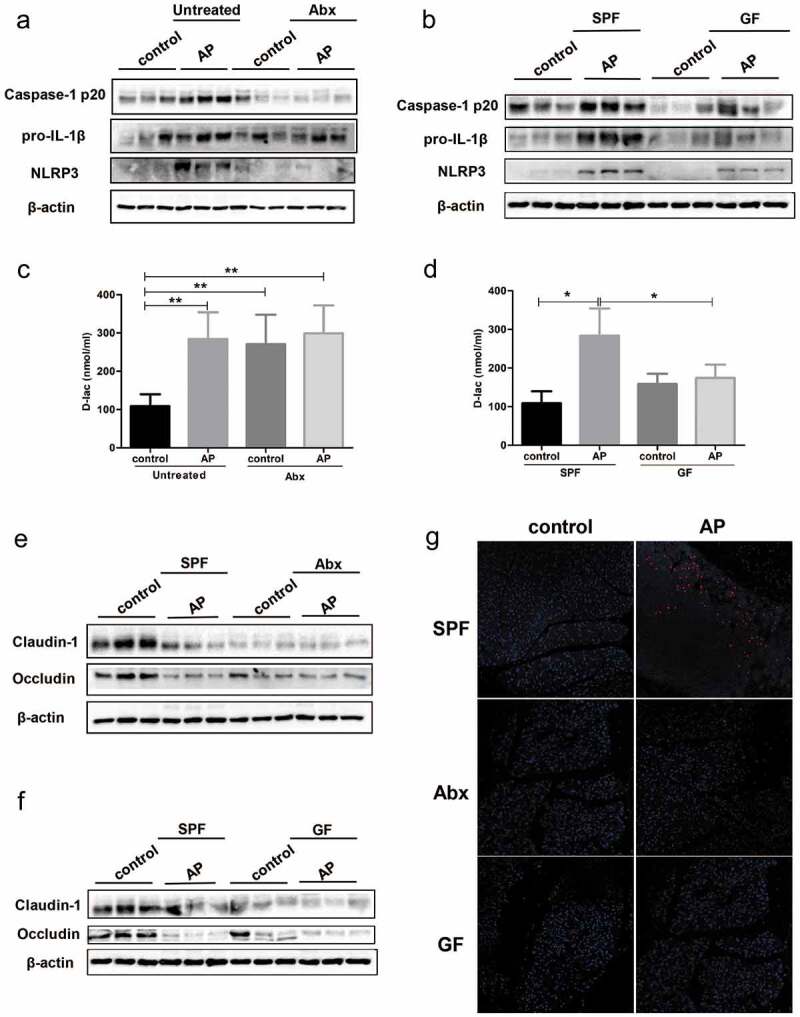
Effect of gut microbiota deprivation on epithelial barrier integrity and activation of the NLRP3 inflammasome. The protein level of NLRP3 inflammasome proteins in the gut of Abx (a) and GF (b) mice was determined by Western blot. The levels of serum D-lac as gut barrier index were measured in Abx (c) and GF mice (d) with or without AP. The level of epithelial tight junction proteins was downregulated in Abx (e) and GF (f) mice compared to SPF mice independent of AP induction. (g) Expression of E. coli protein in the pancreas, as determined by immunofluorescence, was weakened in Abx and GF mice compared to SPF mice. n = 6 per group, *p < .05, **p < .01.
Recolonization of the gut microbiota by FMT exacerbated AP-induced gut disorders and increased the severity of AP
To further demonstrate the role of the gut microbiota in AP, we recolonized Abx and GF mice with bacteria from SPF mice by FMT followed by induction of AP. The percentage of MPO-positive cells was increased in both pancreatic and pulmonary tissues of Abx and GF mice that received FMT (Figure 4(a-d)). The serum levels of TNF-α and IL-1β were also higher in the FMT group than in the control group of Abx mice, whereas after FMT, GF mice had higher concentrations of IL-10 than control mice (Figure 4(e-f).
Figure 4.
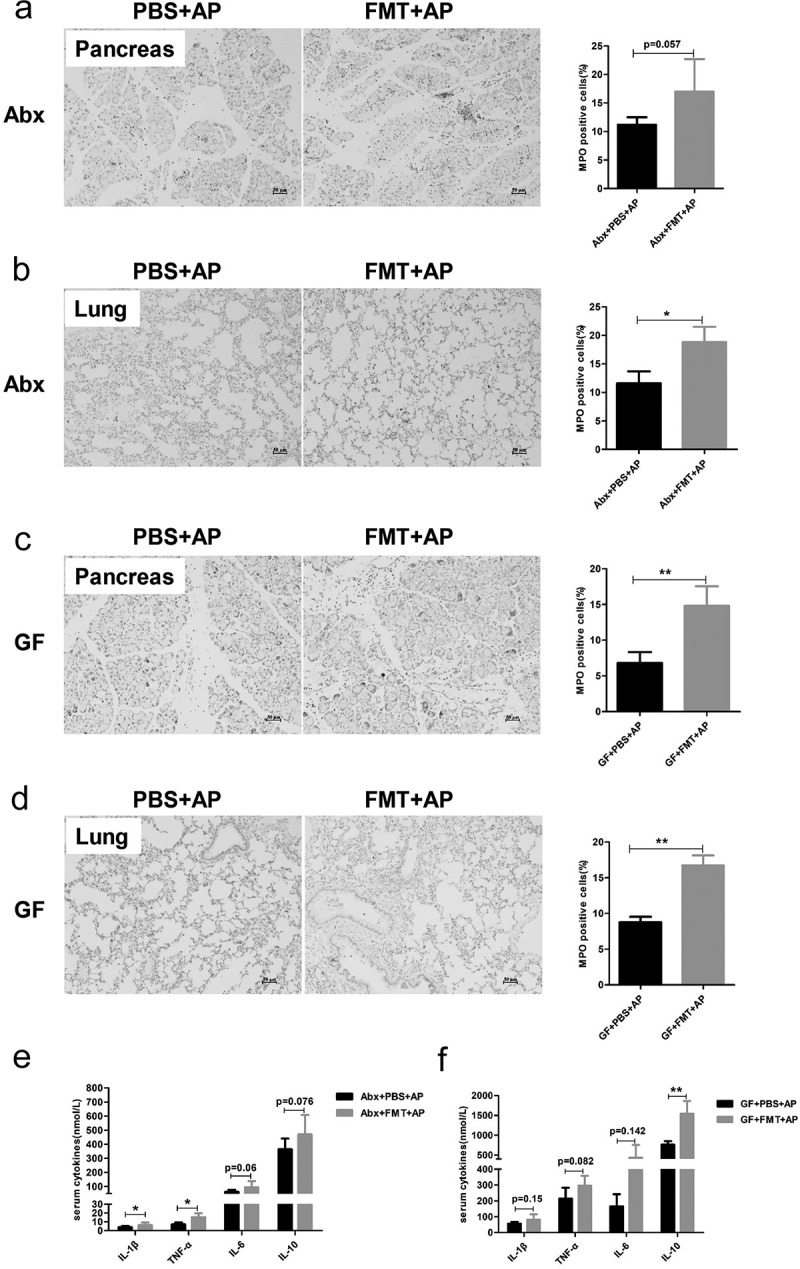
Recolonization with gut microbiota exacerbated local pancreatic damage and systemic inflammation. The fecal microbiota from untreated mice were transplanted to Abx and GF mice before AP induction, and the mice were sacrificed 36 h after the first injection. Representative images and scoring of immunohistological evaluation of MPO in the pancreas and lung tissues of Abx (a, b) and GF mice (c, d) suffering from AP. Scale bar: 50 μm. Comparison of serum cytokine levels between Abx (e) and GF (f) mice with or without FMT. n = 6 per group, *p < .05, **p < .01.
With the recolonization of bacteria in the FMT group, intestinal inflammasome was reactivated, as shown by increased protein level of NLRP3, pro-IL-1β and Caspase-1 p20 (Figure 5(a,b)). The serum level of D-lac, which reflects gut barrier permeability, was significantly increased in both Abx and GF mice with FMT (Figure 5(c,d)). Similarly, FMT downregulated the level of gut epithelial tight junction proteins including Claudin-1 and Occludin in two gut microbiota-deficient mouse models of AP (Figure 5(e,f)). The level of E. coli protein increased dramatically in the pancreas of Abx and GF mice after FMT, which indicates the bacterial translocation in AP due to the impaired gut barrier (Figure 5(g)).
Figure 5.
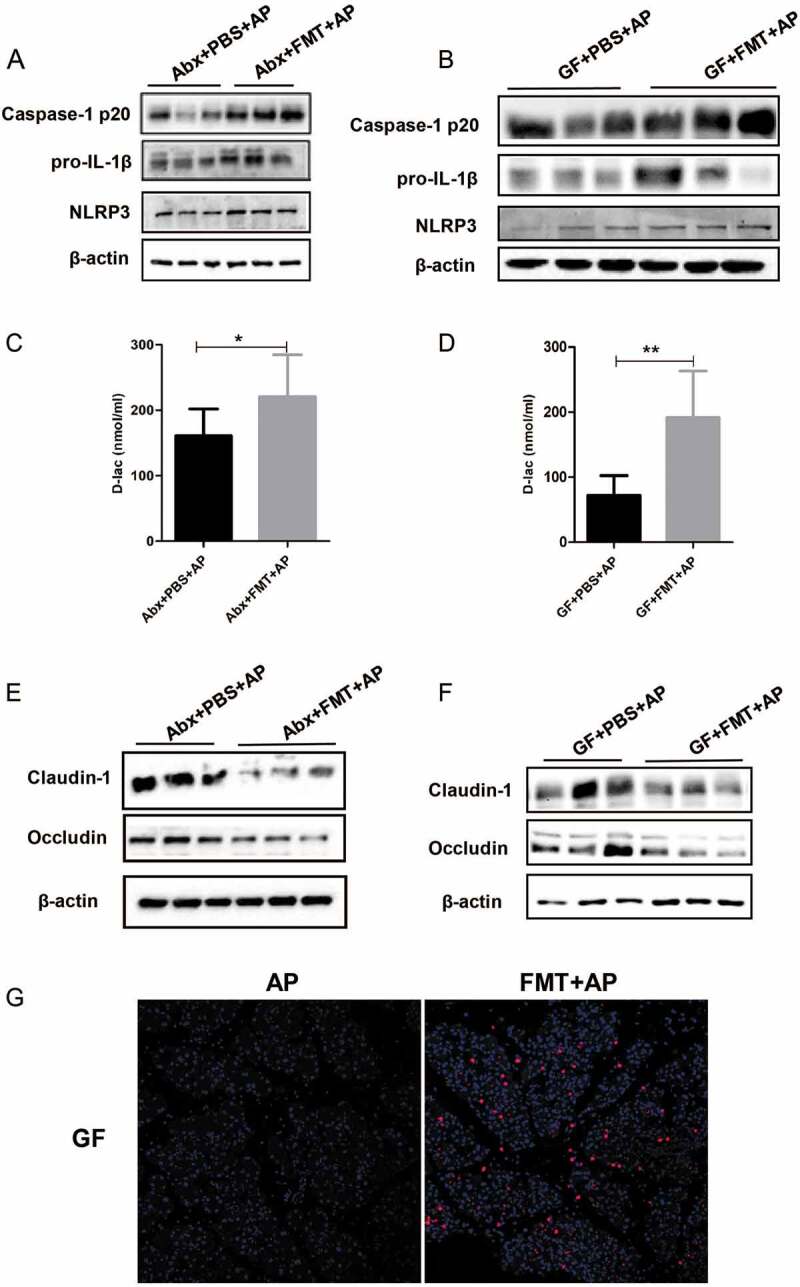
Leaky gut and NLRP3 inflammasome reactivation after FMT in AP- challenged mice. The expression of intestinal NLRP3 inflammasome pathway proteins was analyzed in Abx (a) and GF (b) AP mice with or without FMT. The serum concentrations of D-lac were also compared between these two groups (c, d). Expression of intestinal epithelial tight junction proteins was downregulated in Abx (e) and GF (f) AP mice after FMT. (g) Immunofluorescence showed increased intensity of E. coli expression in the pancreas of GF mice with AP after FMT. n = 6 per group, *p < .05, **p < .01.
Knockout of NLRP3 reshaped the gut microbiota and protected the mice from AP-induced gut injury
Activation of the NLRP3 inflammasome regulates the inflammatory and anti-inflammatory response in AP, which determines the severity of disease.17 Recent studies have reported that NLRP3-mediated modulation of gut microbiota composition and the overgrowth of pathogenic bacteria are associated with gut dysbiosis, which increases the susceptibility to several diseases.16,18 Morphologically, H&E staining showed that edema, inflammatory infiltration and pancreatic acinar cell necrosis were mitigated in NLRP3 KO mice compared to wild-type (WT) mice after AP induction (Figure 6(a)). Lipase activity was significantly lower in NLRP3 KO mice than in WT mice with AP, whereas amylase activity showed a decreasing trend, although the difference was not significant (Figure 6(b)). MPO staining showed less neutrophil infiltration in pancreatic and pulmonary tissues in the NLRP3 KO group than in the WT group (Figure 6(c,d)), suggesting a weaker acute inflammatory response in NLRP3-deficient mice. Systemic inflammation was also attenuated in NLRP3 KO mice with AP, as shown by the reduced serum level of IL-6 (Figure 6(e)).
Figure 6.
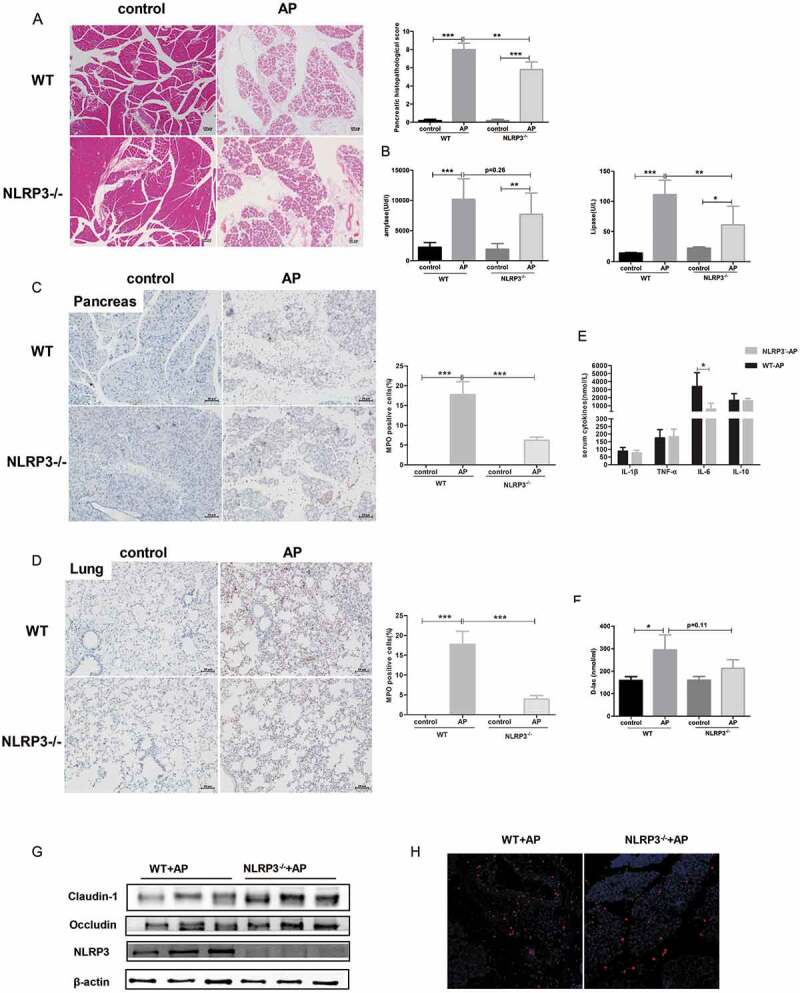
Knockout of NLRP3 ameliorated the severity of AP and improved AP-induced gut barrier impairment. Representative photomicrographs of H&E-stained pancreas sections of WT and NLRP3 KO mice with or without AP induction (a) and their pathological scoring. Scale bar: 50 μm. (b) Serum amylase and lipase activity in each group. Immunohistochemical analysis revealed lower pancreatic (c) and pulmonary (d) expression of MPO in the NLRP3 KO AP group than in the WT AP group. (e) Serum concentrations of cytokines were determined using ELISA. (f) D-lac as a gut barrier index was measured in the serum of mice. (g) Western blot analysis showed the expression of intestinal epithelial tight junction proteins in WT and NLRP3 KO mice during AP. (h) Pancreatic expression of E. coli in WT and NLRP3 KO groups suffering from AP.
To investigate whether the alterations in the gut microbiota were different between WT and NLRP3 KO littermates after the induction of AP, we employed 16 S rRNA gene sequencing. Alpha diversity analysis showed that the Sobs, Shannon and Chao indexes in NLRP3 KO mice with AP were not different from those in the control group, while the WT mice suffered decreased microbial diversity after the induction of AP (Figure 7(a) and Supplement figure 4a). Principal coordinate analysis based on the unweighted UniFrac distance and Bray-Curtis dissimilarity revealed dramatically different microbial communities in WT and NLRP3 KO mice at baseline. Intriguingly, the samples from WT mice with AP clustered separately from those of the control group, while there were high similarities between the AP and control groups of NLRP3 KO mice (Figure 7(b) and Supplement figure 4b).
Figure 8.
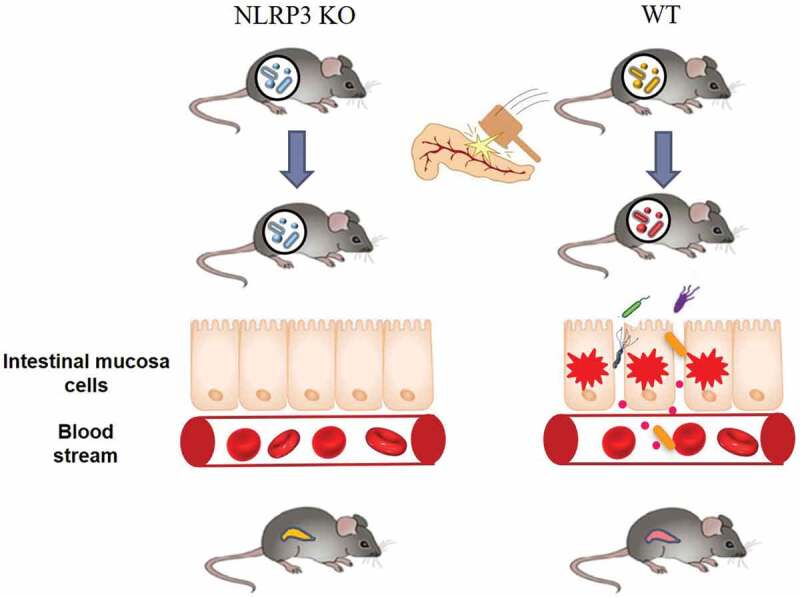
NLRP3 and gut microbiota crosstalk in the exacerbation of AP. The structure of the gut microbiota in NLRP3 deficient mice is distinct from that in WT mice, which might resist the microbial dysbiosis induced by AP and maintain the integrity of the gut barrier. The subsequent prevention of bacterial translocation in NLRP3 KO mice ultimately ameliorated the severity of AP including both pancreatic damage and systemic inflammation.
Figure 7.
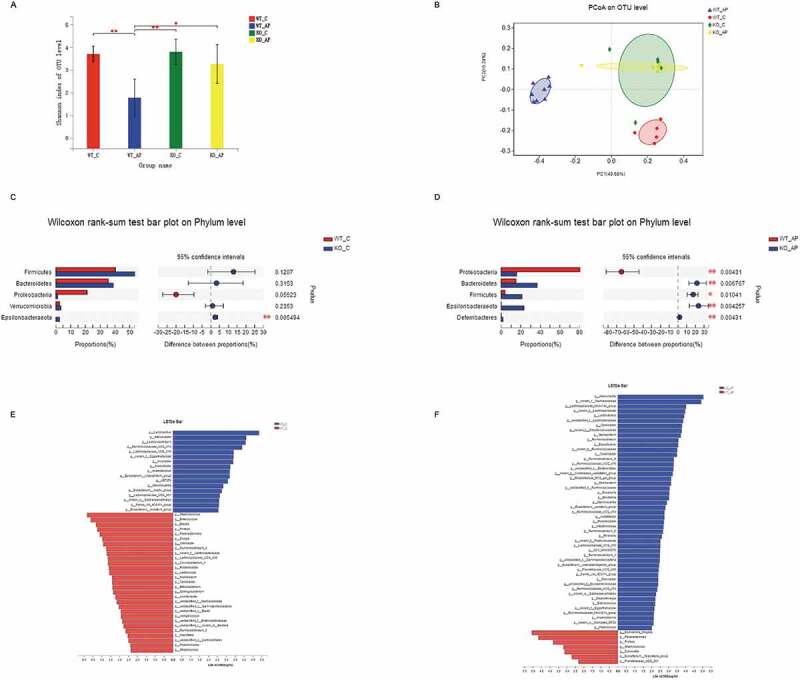
NLRP3 deficiency partially counteracted the gut microbiota dysbiosis induced by AP. (a) Alpha diversity, as revealed by the Shannon index, was analyzed in WT and NLRP3 KO mice with or without AP challenge. *p < .05, **p < .01. (b) Principal coordinate analysis (PCoA) of bacterial beta diversity based on the unweighted UniFrac distance. Samples from the AP group clustered distinctly from those of the control group in WT mice, while samples from NLRP3 KO mice converged together independent of AP induction. (c) Relative abundance of the top 5 phyla in WT and NLRP3 KO mice at baseline. (d) Linear discriminant analysis (LDA) scores were computed for features (at the genus level) that were differentially abundant between the WT and NLRP3 KO groups without treatment. (e) Alterations in the top 5 most abundant phyla between WT and NLRP3 KO mice after AP induction. (f) LEfSe analysis identified the most differentially abundant genera between WT and NLRP3 KO mice with AP.
At the phylum level, the relative abundance of Proteobacteria tended to decrease in the control group of NLRP3 KO mice compared to WT controls, while no significant difference was observed in Firmicutes or Bacteroidetes (Figure 7(c)). After the induction of AP, the phylum Proteobacteria was enriched in the WT group whereas Bacteroidetes and Firmicutes were depleted relative to those of the NLRP3 KO group (Figure 7(d)). To identify differentially abundant taxa, we performed linear discrimination analysis coupled with effect size (LEfSe) analysis on the fecal microbiota of WT and NLRP3 KO mice with or without AP. At the genus level, increased abundance in bacteria including Lactobacillus, Lachnoclostridium, and Ruminococcaceae, and decreased abundance of Staphylococcus, Enterococcus, and Blautia were observed in the NLRP3 KO control group compared to the WT group (Figure 7(e)). The AP-associated genera Escherichia-Shigella, which was demonstrated in our previous study, was more abundant in the WT AP group, while the abundance of Lactobacillus and Roseburia was increased in NLRP3 KO mice (Figure 7(f)).
Additionally, we compared the degree of AP-induced gut injury between WT and NLRP3 KO mice. While increased levels of D-lac were observed in the WT AP group than in the controls, no significant increase in D-lac was found in the NLRP3 KO group with AP (Figure 6(f)). Moreover, the protein level of Claudin-1 and Occludin in the intestinal epithelium, which represents the integrity of the gut barrier, was upregulated in the NLRP3 KO group compared to the WT group with AP (Figure 6(g)). The amount of E. coli protein in the pancreas of NLRP3 KO mice was less than that observed in WT mice after AP induction (Figure 6(h)), which may be due to reconstitution of gut bacteria and strengthening of the gut barrier after NLRP3 knockout.
Discussion
It has long been acknowledged that intestinal dysfunction plays an important role in the severity of AP, and recent accumulating evidence indicates that the bacteria in the gut participate in AP progression, although the mechanism underlying gut-pancreas crosstalk is unclear. Herein, we report the restoration of the gut microbiota during the recovery from AP, accompanied by the attenuation of intestinal NLRP3 activation and reconstruction of the gut barrier. Additionally, activation of intestinal NLRP3 depends on the gut microbiota, which is associated with exacerbation of AP, as demonstrated in Abx and GF mouse models with or without FMT. NLRP3 deficiency reshapes the landscape of the gut microbiota and confers resistance to AP-induced microbial imbalance, which may result in alleviation of the disease.
Our previous study showed dysbiosis of the gut microbiota with the development of AP in both patients and mouse models, with overgrowth of opportunistic pathogens such as Escherichia-Shigella and reduction in beneficial bacteria such as Bifidobacterium, Blautia and Roseburia.8 In this study, we found that the composition of the gut microbiota in the acute phase in AP mice (36 h after induction) was substantially different from that in the regenerated phase (7 days after induction), which was similar to that of the control group, as evidenced by both alpha and beta diversity analysis. The amount of Escherichia-Shigella was markedly decreased in the 7-day group compared to the 36 h group while Roseburia was increased, which indicates that the gut microbiota is associated with not only AP occurrence but also recovery from AP. Thus, several randomized control trials have investigated the effect of gut microbial modulation, including supplementation with probiotics, prebiotics and synbiotics, on the outcome in patients with SAP. A recent meta-analysis of 13 randomized controlled trials showed a beneficial impact of microbial regulation on SAP patients by reducing the length of hospital stays.19 Other studies in animal models also showed that improvements in the gut microbiota or supplementation with its metabolites ameliorated pancreatic injury by maintaining gut homeostasis.20-23 Here, we observed that depletion of the gut microbiota reduced pancreatic damage and suppressed the systemic inflammatory response, while FMT reversed this process. This finding was consistent with previous studies that showed a protective effect of antibiotics on AP, and this protective effect was weakened after transplanting gut commensal bacteria.11,12 However, the molecular mechanisms underlying the crosstalk between the gut microbiota and pancreas are not well understood.
In this study, we observed synchronous changes in gut microbiota restoration and intestinal NLRP3 inflammasome inactivation during AP recovery. Previous work demonstrated that NLRP3, which mediates assembly of the inflammasome complex in response to stimulation by commensal microbiota or their products, regulates the integrity of the intestinal mucosal barrier and acts as a crucial orchestrator of intestinal homeostasis.24 Consistent with a recent study, induction of the NLRP3 inflammasome in the AP group was weakened in Abx and GF mice, whose gut microbiota was deficient.12 Furthermore, we found that the NLRP3 inflammasome was reactivated in these two mouse models following FMT from control mice, which suggests that activation of the NLRP3 inflammasome in AP depends on the gut microbiota. Two other studies also detected the influence of the gut microbiota on the intestinal NLRP3 inflammasome during AP and demonstrated that pretreatment with probiotic Clostridium butyricum or its metabolite butyrate attenuates colonic injury and the inflammatory response by inhibiting the NLRP3 inflammasome pathway.21,22 Thus, we hypothesize that the disturbance in the gut microbiota in the acute phase of AP stimulates intestinal inflammation via NLRP3 inflammasome activation, which contributes to the compromised epithelial barrier and facilitates the translocation of gut bacteria to distant organs. Therefore, the gut is not only the target organ in the development of AP but also the driver of complications in AP, especially IPN, which is always recognized as a second hit in the course of the disease.3,25
Accumulating evidence suggests that the NLRP3 inflammasome plays a critical role in balancing pro- and anti-inflammation in AP and might be a therapeutic target for patients with AP.17 A study by Fu et al. presented the potential prospect of an NLRP3 inflammasome inhibitor in the treatment of AP, which dramatically reduced the severity of SAP and pancreatitis-associated acute lung injury in the mice model.26 Our study showed that pancreatic and pulmonary inflammation was reduced in NLRP3-deficient mice. This was consistent with previous studies reporting that inhibition of NLRP3 inflammasome activation protects against SAP in mice.26-28 Recent studies have revealed that NLRP3 regulation of the gut microbiota contributes to the progression of some diseases, although their relationship in AP is not well understood. For example, Yao et al. demonstrated that hyperactivation of NLRP3 remodeled the gut microbiota with proliferation of Clostridium XIVa and Lactobacillus murinus, which confer resistance to experimental colitis and colorectal cancer by inducing the regulation of T cells.18 On the other hand, NLRP3 inflammasome deficiency also affects the gut microbiota composition with enrichment of Ruminococcus and depletion of Bacteroides, and this unique microbiota community has been demonstrated to improve chronic unpredictable stress-induced depression-like behavior.16 Notably, we found that NLRP3-deficient mice displayed a different gut microbiota composition compared to WT mice before and after the induction of AP. Interestingly, no significant alterations in gut microbiota were observed in NLRP3-deficient mice following AP induction, while WT mice with AP suffered from microbial dysbiosis. Moreover, a lower abundance of the inflammatory bacteria Escherichia-Shigella and a higher abundance of putative short chain fatty acids (SCFAs)-producing bacteria such as Lactobacillus, and Roseburia were observed in NLRP3 KO mice than in WT mice in the acute phase of AP. SCFAs play an important role in maintaining the integrity of the intestinal epithelial barrier by regulating tight junction proteins.29,30 As expected, we found that NLRP3 deficiency compensates for gut barrier destruction induced by AP, as revealed by increased expression of Claudin-1 and Occludin in NLRP3 KO mice with AP. Collectively, we speculate that the knockout of NLRP3 resists the gut microbial dysbiosis induced by AP, including the enrichment of beneficial bacterial and reduction of pathogenic bacterial, which partly contributes to the alleviation of systematic inflammation (Figure 8). However, there is no doubt that the inactivation of NLRP3 inflammasome per se could downregulate the inflammation in NLRP3 KO mice subjected to AP.
Although our investigations attempted to provide comprehensive insight into the interaction between the gut microbiota and NLRP3 in AP, there are several limitations to be addressed in future studies. First, NLRP3 deficiency was not tissue-specific in the mice, and the alleviation of AP may be due to inhibition of the NLRP3 inflammasome in the pancreas. Gut-specific NLRP3 KO mice are needed to illustrate the impact of intestinal NLRP3 on the severity of AP. Second, our microbial analysis was based on data from 16 S rRNA sequencing. Short-gun sequencing for metagenomics may reveal a more accurate composition, as well as functional information, of the gut microbiome community. Third, we only observed alterations in the gut microbiota in NLRP3-deficient mice, and the role of this specific microbial mixture in the progression of AP needs further investigation by transplanting to Abx or GF mice. However, our study comprehensively revealed the crosstalk between the gut microbiota and NLRP3 during AP, which may influence the severity of the disease. Elimination of the gut microbiota such as antibiotic prophylaxis or NLRP3 inhibition might serve as a potential therapeutic strategy for AP.
Methods
Experimental animal
Six- to eight-week old SPF (male, n = 36) and GF (n = 18 with half male and half female) C57BL/6 mice were purchased from the Shanghai Laboratory Animal Center (SLAC), Chinese Academy of Sciences, Shanghai, China. WT mice and NLRP3 KO mice were purchased from GemPharmatech Company (Nanjing, China). While GF mice were kept in sterile plastic isolators, other animals were housed under controlled environmental conditions. All animal experiments were approved by the Institutional Animal Care and Use Committee of The First Affiliated Hospital of Nanchang University and complied with the national and international guidelines for the Care and Use of Laboratory Animals.
Induction of AP and pretreatment with antibiotics
The induction of AP was performed by administering eleven hourly intraperitoneal injections of caerulein (Sigma-Aldrich, St. Louis, Missouri, USA, 50 μg/kg) immediately followed by one dose of lipopolysaccharide (LPS, Sigma-Aldrich, St. Louis, Missouri, USA, 10 mg/kg). The control group was intraperitoneally injected with saline. The Abx groups were administered broad-spectrum antibiotics (ampicillin 1 g/L, Sigma; neomycin sulfate 1 g/L, Sigma; metronidazole 1 g/L, Sigma and vancomycin 0.5 g/L, Sigma) in drinking water for 4 weeks as previously described.8 Mice were sacrificed 36 h and 7 days after the first injection. Peripheral blood, pancreatic, lung and intestinal tissue were collected.
FMT
Before FMT, the antibiotic-pretreated mice were given water without antibiotics for two days. Fresh fecal pellets (200 mg) from untreated mice were collected and resuspended in 2 ml sterile PBS. After filtering through a sterile 70 μm strainer, the fecal microbial suspension was administered by gavage to Abx and GF at a dose of 200 μl per mouse for 4 consecutive days.31 The groups without FMT were administered sterile PBS as controls.
Measurements of serum amylase, lipase, cytokines and D-lactate (D-lac)
The serum activities of amylase (C016-1) and lipase (A054-2) were measured using commercial kits (Jiancheng Biotech, Nanjing, China) according to the manufacturer’s protocols. Serum cytokine concentrations were quantified using a Bio-Plex multiplex bead array system with the mouse cytokine Th17 panel A (M6000007NY, Bio-Rad). The gut barrier index, as measured by D-lac levels, was determined using a commercial kit (Abcam, Cambridge, UK).
Histopathology and immunohistochemistry
Pancreatic and pulmonary tissues were fixed in 4% formalin for 24 h, embedded in paraffin and then cut into 4 μm sections for H&E staining. Pancreatic damage was assessed by two pathologists in a blinded manner according to previously described criteria.32 Briefly, evaluation of pancreatic pathology included four categories: edema, inflammatory cell infiltration, necrosis and vacuolization. The infiltration of neutrophils in both pancreatic and pulmonary tissues of mice was evaluated by immunohistochemical staining using anti-myeloperoxidase (MPO) rabbit polyclonal antibody (Abcam, ab9535). After blocking endogenous peroxidase activity with 3% H2O2 for 8 min, the sections were incubated with diluted (1:200) anti-MPO antibody overnight, developed with 3,3-diaminobenzidine (DAB) solution and finally counterstained with hematoxylin followed by dehydration. The MPO-positive cells were calculated by the staining intensity scores (0 (normal), 1 (weak), 2 (medium), 3 (strong)) and staining area (0: 0%, 1: 1%-25%, 2: 26%-50%, 3: 51%-75%, 4: 76%-100%).
Immunofluorescence
Paraffin-fixed sections (4 μm thick) were prepared on slides. The sections were deparaffinized in xylene and rehydrated through a graded ethanol series. Endogenous peroxidase activity was blocked by incubation in a 3% H2O2 solution at room temperature for 8 min. Antigen retrieval was performed using boiling citrate buffer (pH 6.0) in a microwave for 15 min. The membranes were permeabilized by 0.3% triton-X100 for 15 min at 37°C and then blocked with 3% BSA for 60 min at room temperature. The sections were incubated with anti-Occludin and anti-Claudin-1 at 4°C overnight followed by incubation with Alexa Fluor-conjugated secondary antibody (1:500, Invitrogen) at 37ºC for 30 min. DAPI was used to counterstain (Invitrogen) the nuclei. Fluorescence staining was observed under a normal fluorescence microscope.
Western blotting
Ten milligrams of ileum tissue were homogenized in lysis buffer using a homogenate machine. Protein concentrations were measured with a BCA protein quantitative assay kit. An equal amount of protein (20–25 μg) was separated on SDS-PAGE gels and then transferred to a polyvinylidene fluoride (PVDF) membrane. After blocking with 5% nonfat milk in Tris-buffered saline (TBS) containing 0.1% Tween-20 (TBST) at room temperature for 1 h, the membranes were incubated overnight with the primary antibody at 4 °C. Then the membranes were washed and incubated with HRP-conjugated secondary antibody. Antibodies against Claudin-1, Occludin and Caspase-1 p20 were purchased from Santa Cruz (SC-166338, SC-133256 and SC-398715), IL-1β was purchased from Cell Signaling Technology (12507), and NLRP3 was purchased from AdipoGen (AG-20B-0014-C100).
Bacterial translocation
The pancreatic sections were deparaffinized in xylene and rehydrated through a graded ethanol series. The sections were air-dried and covered with hybridization buffer containing 10ng/μl probe, which had been pre-heated to 74.5°C for 10 minutes, and then incubated in a humidified chamber in the dark at 48°C overnight. Hybridized slides were gently rinsed with double distilled water to remove the parafilm and washed in pre-warmed washing bufferⅠ and bufferⅡ in the dark for 15 min at 48°C. Finally, the slides were rinsed in double distilled water and air-dried, and DAPI was used to counterstain (Invitrogen) the nuclei. Fluorescence staining was observed under a normal fluorescence microscope.
Gut microbiome sequencing and analysis
The cecum mucosa samples were collected from different mouse groups, and microbial DNA was extracted from each sample using a QIAGEN DNeasy Kit (QIAGEN, California USA). The V3-V4 region of the bacterial 16 S ribosomal RNA gene was amplified by polymerase chain reaction (PCR) using the following primers: 338 F (5ʹ-ACTCCTACGGGAGGCAGCAG-3ʹ) and 806 R (5ʹ-GGACTACHVGGGTWTCTAAT-3ʹ). The PCR components were as follows: 4 μL of 5 × Fast-Pfu buffer, 0.8 μL of 2.5 mmol/L deoxyribonucleotide triphosphates (dNTPs), 0.8 μL of each primer (5 μmol/L), 0.4 μL of Fast-Pfu polymerase, and 10 ng of template DNA. The PCR conditions were 3 minutes of denaturation at 95 °C, followed by 27 cycles of 30 seconds at 95 °C, 30 seconds annealing at 55 °C, 45 seconds elongation at 72 °C, and finally 10 minutes extension at 72 °C. Amplification products were separated, purified and finally pooled on an Illumina MiSeq platform (Illumina Inc., USA) for sequencing.
The raw data were processed to obtain clean reads by eliminating adaptor pollution and low-quality sequences using Trimmomatic software, and then the reads were truncated at any site with an average quality score < 20 over a 50 bp sliding window. After trimming, Fast Length Adjustment of Short reads (FLASH, v 1.2.11) was used to combine tags with high-quality paired-end reads with an average read length of 252 bp. The effective reads were clustered as operational taxonomic units (OTUs) with a 97% similarity cutoff by the algorithm in USEARCH (version 7.1) software. The phylogenetic taxa of each 16 S rRNA gene sequence was analyzed by the Ribosomal Database Project (RDP) Classifier (http://rdp.cme.msu.edu/) against the Silva (SSU128) 16 S rRNA database using a confidence threshold of 70%. The alpha diversity was calculated based on the OTU table, and beta diversity was evaluated by computing the unweighted UniFrac distances and Bray-curtis distances and was visualized in principal coordinate analysis (PCoA). The differential abundance of phyla was compared using the Wilcoxon rank sum test. LEfSe was performed to identify the differential taxa between groups at the genus or higher taxonomy levels.33
Statistical analysis
The data were expressed as the mean ± SEM. Statistical analysis was performed using the SPSS 13.0 software. Differences between two groups with normal distributions were assessed by Student’s t test, and one-way analysis of variance was used to compare differences between more than two groups. The least significant difference (LSD) post hoc test was performed when ANOVA indicated significance. The value of p < .05 was regarded as the cutoff for statistical significance.
Supplementary Material
Funding Statement
This research is supported by the National Natural Science Foundation of China [81760120, 81960128, 81860106], the Key Research and Development Program from the Science and Technology Department of Jiangxi Province [20171BBG70084, 20192ACBL20037, 20192BBG70037].
Availability of data
The raw sequencing data have been uploaded to the NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra) under accession number SRP237476.
Competing financial interest declaration
The authors declare no conflict of interest.
Supplemental material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.van Dijk SM, Hallensleben NDL, van Santvoort HC, Fockens P, van Goor H, Bruno MJ, Besselink MG.. Dutch pancreatitis study G. acute pancreatitis: recent advances through randomised trials. Gut. 2017;66:2024–2032. doi: 10.1136/gutjnl-2016-313595. [DOI] [PubMed] [Google Scholar]
- 2.Wu LM, Sankaran SJ, Plank LD, Windsor JA, Petrov MS. Meta-analysis of gut barrier dysfunction in patients with acute pancreatitis. Br J Surg. 2014;101:1644–1656. doi: 10.1002/bjs.9665. [DOI] [PubMed] [Google Scholar]
- 3.Capurso G, Zerboni G, Signoretti M, Valente R, Stigliano S, Piciucchi M, Delle Fave G. Role of the gut barrier in acute pancreatitis. J Clin Gastroenterol. 2012;46(Suppl):S46–51. doi: 10.1097/MCG.0b013e3182652096. [DOI] [PubMed] [Google Scholar]
- 4.Thomson JE, Nweke EE, Brand M, Nel M, Candy GP, Fonteh PN. Transient expression of interleukin-21 in the second hit of acute pancreatitis may potentiate immune paresis in severe acute pancreatitis. Pancreas. 2019;48:107–112. doi: 10.1097/MPA.0000000000001207. [DOI] [PubMed] [Google Scholar]
- 5.Landahl P, Ansari D, Andersson R. Severe acute pancreatitis: gut barrier failure, systemic inflammatory response, acute lung injury, and the role of the mesenteric lymph. Surg Infect (Larchmt). 2015;16:651–656. doi: 10.1089/sur.2015.034. [DOI] [PubMed] [Google Scholar]
- 6.Sekirov I, Russell SL, Antunes LC, Finlay BB. Gut microbiota in health and disease. Physiol Rev. 2010;90:859–904. doi: 10.1152/physrev.00045.2009. [DOI] [PubMed] [Google Scholar]
- 7.Jandhyala SM, Talukdar R, Subramanyam C, Vuyyuru H, Sasikala M, Nageshwar Reddy D. Role of the normal gut microbiota. World J Gastroenterol. 2015;21:8787–8803. doi: 10.3748/wjg.v21.i29.8787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu Y, He C, Li X, Cai Y, Hu J, Liao Y, Zhao J, Xia L, He W, Liu L, et al. Gut microbiota dysbiosis worsens the severity of acute pancreatitis in patients and mice. J Gastroenterol. 2019;54:347–358. doi: 10.1007/s00535-018-1529-0. [DOI] [PubMed] [Google Scholar]
- 9.Tan C, Ling Z, Huang Y, Cao Y, Liu Q, Cai T, Yuan H, Liu C, Li Y, Xu K. Dysbiosis of intestinal microbiota associated with inflammation involved in the progression of acute pancreatitis. Pancreas. 2015;44:868–875. doi: 10.1097/MPA.0000000000000355. [DOI] [PubMed] [Google Scholar]
- 10.Cen ME, Wang F, Su Y, Zhang WJ, Sun B, Wang G. Gastrointestinal microecology: a crucial and potential target in acute pancreatitis. Apoptosis. 2018;23:377–387. doi: 10.1007/s10495-018-1464-9. [DOI] [PubMed] [Google Scholar]
- 11.Zheng J, Lou L, Fan J, Huang C, Mei Q, Wu J, Guo Y, Lu Y, Wang X, Zeng Y. Commensal escherichia coli aggravates acute necrotizing pancreatitis through targeting of intestinal epithelial cells. Appl Environ Microbiol. 2019;85. doi: 10.1128/AEM.00059-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jia L, Chen H, Yang J, Fang X, Niu W, Zhang M, Li J, Pan X, Ren Z, Sun J, et al. Combinatory antibiotic treatment protects against experimental acute pancreatitis by suppressing gut bacterial translocation to pancreas and inhibiting NLRP3 inflammasome pathway. Innate Immun. 2019;1753425919881502. doi: 10.1177/1753425919881502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: an overview of mechanisms of activation and regulation. Int J Mol Sci. 2019;20. doi: 10.3390/ijms20133328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sendler M, van den Brandt C, Glaubitz J, Wilden A, Golchert J, Weiss FU, Homuth G, De Freitas Chama LL, et al. NLRP3 inflammasome regulates development of systemic inflammatory response and compensatory anti-inflammatory response syndromes in mice with acute pancreatitis. Gastroenterology. 2019. doi: 10.1053/j.gastro.2019.09.040. [DOI] [PubMed] [Google Scholar]
- 15.Wu C, Pan LL, Niu W, Fang X, Liang W, Li J, Li H, Pan X, Chen W, Zhang H, et al. Modulation of gut microbiota by low methoxyl pectin attenuates type 1 diabetes in non-obese diabetic mice. Front Immunol. 2019;10:1733. doi: 10.3389/fimmu.2019.01733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang Y, Huang R, Cheng M, Wang L, Chao J, Li J, Zheng P, Xie P, Zhang Z, Yao H. Gut microbiota from NLRP3-deficient mice ameliorates depressive-like behaviors by regulating astrocyte dysfunction via circHIPK2. Microbiome. 2019;7:116. doi: 10.1186/s40168-019-0733-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sendler M, van den Brandt C, Glaubitz J, Wilden A, Golchert J, Weiss FU, Homuth G, De Freitas Chama LL, Mishra N, Mahajan UM, et al. NLRP3 Inflammasome regulates development of systemic inflammatory respoes in mice with acute pancreatitis. Gastroenterology. 2019. doi: 10.1053/j.gastro.2019.09.040. [DOI] [PubMed] [Google Scholar]
- 18.Yao X, Zhang C, Xing Y, Xue G, Zhang Q, Pan F, Wu G, Hu Y, Guo Q, Lu A, et al. Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat Commun. 2017;8:1896. doi: 10.1038/s41467-017-01917-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tian X, Pi YP, Liu XL, Chen H, Chen WQ. Supplemented use of pre-, pro-, and synbiotics in severe acute pancreatitis: an updated systematic review and meta-analysis of 13 randomized controlled trials. Front Pharmacol. 2018;9:690. doi: 10.3389/fphar.2018.00690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wan YD, Zhu RX, Bian ZZ, Pan XT. Improvement of gut microbiota by inhibition of P38 mitogen-activated protein kinase (MAPK) signaling pathway in rats with severe acute pancreatitis. Med Sci Monit. 2019;25:4609–4616. doi: 10.12659/MSM.914538. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Pan X, Fang X, Wang F, Li H, Niu W, Liang W, Wu C, Li J, Tu X, Pan LL, et al. Butyrate ameliorates caerulein-induced acute pancreatitis and associated intestinal injury by tissue specific mechanisms. Br J Pharmacol. 2019;176(23):4446–4461. doi: 10.1111/bph.14806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pan LL, Niu W, Fang X, Liang W, Li H, Chen W, Zhang H, Bhatia M, Sun J. Clostridium butyricum strains suppress experimental acute pancreatitis by maintaining intestinal homeostasis. Mol Nutr Food Res. 2019;e1801419. doi: 10.1002/mnfr.201801419. [DOI] [PubMed] [Google Scholar]
- 23.He Y, Wu C, Li J, Li H, Sun Z, Zhang H, de Vos P, Pan LL, Sun J. Inulin-type fructans modulates pancreatic-gut innate immune responses and gut barrier integrity during Experimental acute pancreatitis in a chain length-dependent manner. Front Immunol. 2017;8:1209. doi: 10.3389/fimmu.2017.01209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zaki MH, Lamkanfi M, Kanneganti TD. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. 2011;32:171–179. doi: 10.1016/j.it.2011.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McClave SA. Factors that worsen disease severity in acute pancreatitis: implications for more innovative nutrition therapy. Nutr Clin Pract. 2019;34(Suppl 1):S43–S8. doi: 10.1002/ncp.10371. [DOI] [PubMed] [Google Scholar]
- 26.Fu Q, Zhai Z, Wang Y, Xu L, Jia P, Xia P, Liu C, Zhang X, Qin T, Zhang H. NLRP3 deficiency alleviates severe acute pancreatitis and pancreatitis-associated lung injury in a mouse model. Biomed Res Int. 2018;2018:1294951. doi: 10.1155/2018/1294951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou C, Zhu X, Shi C, Peng Y, Huang D, Li Q, Miao Y. Iguratimod (T-614) attenuates severe acute pancreatitis by inhibiting the NLRP3 inflammasome and NF-kappaB pathway. Biomed Pharmacother. 2019;119:109455. doi: 10.1016/j.biopha.2019.109455. [DOI] [PubMed] [Google Scholar]
- 28.Jin HZ, Yang XJ, Zhao KL, Mei FC, Zhou Y, You YD, Wang WX. Apocynin alleviates lung injury by suppressing NLRP3 inflammasome activation and NF-kappaB signaling in acute pancreatitis. Int Immunopharmacol. 2019;75:105821. doi: 10.1016/j.intimp.2019.105821. [DOI] [PubMed] [Google Scholar]
- 29.Morrison DJ, Preston T. Formation of short chain fatty acids by the gut microbiota and their impact on human metabolism. Gut Microbes. 2016;7:189–200. doi: 10.1080/19490976.2015.1134082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Y, Zhang Y, Xu Y, Luo T, Ge Y, Jiang Y, Shi Y, Sun J, Le G. Dietary methionine restriction improves the gut microbiota and reduces intestinal permeability and inflammation in high-fat-fed mice. Food Funct. 2019;10:5952–5968. doi: 10.1039/c9fo00766k. [DOI] [PubMed] [Google Scholar]
- 31.Schuijt TJ, Lankelma JM, Scicluna BP, de Sousa E Melo F, Roelofs JJ, de Boer JD, Hoogendijk AJ, de Beer R, de Vos A, Belzer C, et al. The gut microbiota plays a protective role in the host defence against pneumococcal pneumonia. Gut. 2016;65:575–583. doi: 10.1136/gutjnl-2015-309728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schmidt J, Rattner DW, Lewandrowski K, Compton CC, Mandavilli U, Knoefel WT, Warshaw AL. A better model of acute pancreatitis for evaluating therapy. Ann Surg. 1992;215:44–56. doi: 10.1097/00000658-199201000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing data have been uploaded to the NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra) under accession number SRP237476.