

Abstract
Purpose & Experimental Design:
Anaplastic thyroid cancer (ATC) comprises ~2% of all thyroid cancers and its median survival rate remains poor. It is responsible for more than one-third of thyroid cancer-related deaths. ATC is frequently resistant to conventional therapy and NFκB-signaling has been proposed to be a feature of the disease. We aimed to assess the activity of the anti-malaria drug quinacrine known to target NFκB-signaling in combination with the clinically relevant kinase inhibitor sorafenib in ATC cells. The presence of NFκB-p65/RelA and its target Mcl-1 was demonstrated in ATC by meta-data gene set enrichment analysis and immunohistochemistry (IHC). We assessed the responses of a panel of human ATC cell lines to quinacrine and sorafenib in vitro and in vivo.
Results:
We detected increased expression of NFκB-p65/RelA and Mcl-1 in the nucleus of a subset of ATC compared to non-neoplastic thyroid. ATC-cells were found to respond with additive/synergistic tumor cell-killing to the combination of sorafenib plus quinacrine in vitro and the drug combination improves survival of immunodeficient mice injected orthotopically with ATC-cells as compared to mice administered either compound alone or doxorubicin. We also demonstrate that the combination of sorafenib and quinacrine is well tolerated in mice. At the molecular level, quinacrine and sorafenib inhibited expression of the pro-survival gene Mcl-1, pStat3 and dampened NFκB signaling.
Conclusion:
The combination of quinacrine and sorafenib targets emerging molecular hallmarks of ATC and shows promising results in clinically relevant models for the disease. Further testing of sorafenib plus quinacrine can be conducted in ATC patients.
Keywords: Thyroid Cancer, Sorafenib, Quinacrine, Mcl-1
One Sentence Summary:
The drug combination of sorafenib and quinacrine targets Mcl-1 and triggers synergistic responses in difficult to treat anaplastic thyroid cancer cells thereby potentially offering a novel therapy combination for further testing in the clinic.
Introduction
Anaplastic thyroid cancer (ATC) is a rare but highly aggressive form of thyroid cancer with a median survival of 3–5 months that remains unchanged despite increased understanding of the etiology of the disease (1). Unfortunately, refractoriness to chemo- and radio-therapy is a common feature and development of novel therapeutics is essential to improve the prognosis of ATC patients. Recent insight into genetic lesions that drive the progression of ATC may help shape concepts of novel strategies of targeted therapeutic approaches. Currently, the proto-oncogenes RET, RAS and BRAF are some of the best described targets in thyroid cancer (2, 3) and specifically activating BRAF V600E mutations have been reported to occur in approximately 25% of ATCs (4, 5).
Given the frequency of activating mutations of the BRAF oncogene in ATC it is perhaps not surprising that the multi-kinase inhibitor Sorafenib (Nexavar®), an approved drug for the treatment of advanced renal carcinoma (6), unresectable hepatocellular carcinoma (7) and progressive radioactive iodine-refractory differentiated thyroid carcinoma (8), has sparked clinical interest in ATC. Sorafenib targets BRAF and CRAF, in addition to several other tyrosine kinases, suggesting that at least a subpopulation of ATC patients might respond to sorafenib. However, sorafenib has shown limited activity in the reported clinical trials of ATC to date (9, 10). One phase-II study of sorafenib in patients with advanced ATC indicated activity but at low frequency in a similar manner as fosbretabulin, a vascular disrupting agent (10). It is becoming increasingly clear that sorafenib may trigger toxicities in thyroid cancer patients that frequently result in dose reduction (11). Thus, treatment with sorafenib alone may be insufficient to evoke a strong anti-tumor response in ATC patients and incorporation of additional targeted therapeutics that exhibit low-toxicity into sorafenib-protocols may be required to improve outcome.
Additional molecular changes occur in ATC cells that may contribute to disease aggressiveness include aberrant activation of NFκB signaling. Imbalanced activation of NFκB may possibly contribute to the treatment refractory pro-inflammatory and metastatic phenotype of ATC. Indeed, the expression of RelA/p65 was found to be increased in ATC tissues compared to that of normal thyroid (12). Several inhibitors of NFκB-signaling such as dehydroxymethylepoxyquinomicin (DHMEQ), triptolide, imatinib and bortezomib have shown promising results in pre-clinical experiments with ATC cells (13–16).
The acridine derivative Quinacrine, used historically for malaria treatment, is a potent inhibitor of NFκB-signaling (17), and is currently being evaluated in phase-II cancer clinical trials (18). Its extensive use during the Second World War by over three (3) million soldiers makes it a well-studied drug with a safety profile based on extensive epidemiological data. Moreover since quinacrine is currently used for the treatment of giardiasis or lupus and is very affordable (~$30 USD/month of therapy), it is a good candidate compound for repositioning to target malignancies with oncogenic activation of NFκB-signaling. We recently reported the effectiveness of quinacrine with other standard-of-care therapies in liver and colon cancer (19, 20). Quinacrine was found to effectively target NFκB and inhibit Mcl-1 expression in colorectal cancer cells. In addition, we have previously shown that sorafenib inhibits both JAK/STAT3- and NFκB-signaling that also results in the downregulation of Mcl-1 (21, 22).
Herein, we show that quinacrine combines favorably with sorafenib in an additive to synergistic manner and generates a strong anti-tumor response in an orthotopic mice model of ATC without significant toxicity. Treating ATC cells with the sorafenib/quinacrine drug combination dramatically reduced the levels of anti-apoptotic Mcl-1 and triggered Mcl-1-dependent cell death. Mcl-1 protein is overexpressed in a subset of ATC patient specimens compared to non-neoplastic thyroid. Furthermore gene set enrichment analysis of meta-data indicates hyperactivation of NFκB-signaling in ATCs. These findings provide a rationale for future clinical trials of the drug combination quinacrine/sorafenib in aggressive thyroid cancers.
Material and Methods
Detailed Materials and Methods are provided as Supplementary Information
Cell lines and reagents
These were as described previously (21).
Immunohistochemistry of clinical normal and anaplastic thyroid cancer (ATC)
Twelve ATC’s and ten ‘normal’ (non-neoplastic) thyroid patient formalin-fixed paraffin embedded (FFPE) tissue specimens were obtained from the Department of Pathology’s tissue bank/Penn State Hershey Cancer Institute. The following antibodies were used for immunohistochemical detection anti-phosphotyrosine-Stat3 (Y705), anti-NFκB-p65 and anti-Mcl-1. Slides were scored and the expression parameters were correlated with clinical outcome.
Meta-analysis of NFκB-dependent gene expression in clinical ATC’s
Gene expression data were collected through Oncomine™/Gene Expression Omnibus (GEO) and previously published reports containing complete curated expression profiles of resected clinical specimens of indicated tissues. The gene expression profiles were computationally analyzed for statistically significant differences with respect to the expression of four (4) gene sets of putative NFκB-target genes using the Gene Set Enrichment Analysis (GSEA) (http://www.broadinstitute.org/gsea/index.jsp) (23) .
Quantitative RT-PCR
Total RNA was extracted using the RNeasy Mini Kit and converted to cDNA using the First Strand Synthesis kit. For qPCR analysis the SYBR select master mix was used together with the primers for Mcl-1, Stat 3, NFkappaB-p65/RelA.
siRNA knockdown experiments and dose response for sorafenib and quinacrine
The siGENOME Non-Targeting siRNA #1 and Mcl-1 were transfected and cells were collected for dose response CellTiter-Glo™ assay. The same batch of cells was plated for measuring knockdown efficiency of Mcl-1.
Immunoblotting
These are as described previously (21).
Sub-G1 and cell survival analysis
These are as described previously (21)
Mouse orthotopic xenograft model of ATC
A midline cervical incision was made and the underlying submandibular glands were retracted laterally. Gentle retraction of the midline strap muscles was performed to reveal the thyroid glands adjacent to the trachea and visible underneath a deep layer of semitransparent strap muscles. Direct injection of the thyroid was done, the submandibular glands were returned to the original location and the skin closed in a single layer (24). Mice were treated with sorafenib, Doxorubicin and Quinacrine as previously described (21); and followed for tumor burden.
Immunohistochemistry of mouse tissues
Mouse FFPE sections were analyzed by IHC as described for clinical specimens above. Antibodies were used for the detection of VEGF, CD34 and Ki67 are listed in supplementary information. The detection of apoptotic cells were done by using the TUNEL-kit.
Mouse dose-escalation toxicity study
C57BL6/J mice were injected with doses of quinacine (2 times) x 1 dose oral gavage, sorafenib (5 times), whereas doxorubicin (2.5 times) of that used in the orthotopic model. At day seven (7) mice were sacrificed, whole blood was isolated by cardiac puncture for hematology and serum chemistry, and tissues were isolated for histological assessment.
Statistical analysis
All comparisons were made relative to untreated controls, and statistically significant differences are indicated as *p<0.05 and **p<0.005. We used Calcusyn software to determine synergy. Overall survival was determined using Kaplan-Meier method. Cox multi-regression analysis was performed on patient parameters relating to the ATC specimens stained by IHC for pStat3, NFκB-p65 and Mcl-1.
Results
A subset of ATC patients overexpress Mcl-1 mRNA and protein
Targeted therapy acts against specific molecules that are preferentially present in malignant cells and required for cancer cell survival and growth. We have previously shown that both sorafenib and quinacrine can target cell death signaling through modulation of NFκB- and JAK-STAT-signaling in cancer cells (19–22). Although these pathways are frequently activated in cancer (25, 26), it has until recently been largely unknown to what extent these pathways are also aberrantly activated in ATC. To address target availability in ATC we compared the expression of NFκB-p65 (RelA), STAT3 and Mcl-1 mRNA in a panel of human cancer cell lines and resected ATC specimens to that in ‘normal’ (non-neoplastic) thyroid by qRT-PCR (Supplemental Figure 1). The genomic profile of the ATC cell lines used in our study have been established and verified previously (27–29).
Detectable expression of Mcl-1, RelA and STAT3 mRNA was found in all five human ATC cell lines tested (Supplemental Figure 1A) tested by qRT-PCR. Expression of Mcl-1 and RelA varied from average to average-high in comparison to the prostate cancer cell lines and the hepatoma cell line HepG2. Expression of STAT3 was variable among the ATC cells, and, interestingly STAT3 mRNA was found underexpressed in the 16T and 21T cell lines. In general, the mRNA expression levels of these oncogenes was lower in ATC cells than the expression found in the untransformed fibroblast cell lines MRC5 and HFF (Supplemental Figure 1A), only the prostate cancer cell line DU145 expressed more Mcl-1, RelA and STAT3 mRNA than the fibroblasts. Thus, a certain level of variability with respect to expression of these genes is present in ATC cells.
In order to better address if expression of these genes may be upregulated by carcinogenesis; we hypothesized that a better comparison would be between resected ATC and non-neoplastic thyroid (Supplemental Figure 1B–D). We observed that the mRNA of Mcl-1 and RelA were expressed in both normal thyroid and ATC tissues, although the sample size was limited due to specimen availability. Interestingly, RelA was underexpressed at the mRNA level in our samples whereas expression data from one ATC sample indicated that potentially a subpopulation of ATC’s may express high levels of Mcl-1 mRNA (Supplemental Figure 1B). However, STAT3 mRNA expression was low or unquantifiable in ATC in comparison to that in normal thyroid tissue (Supplemental Figure 1D).
However, since both NFκB and JAK-STAT signaling can be significantly regulated at the post-transcriptional level; we aimed to address this by assessing the protein expression in the normal and ATC tissues. We performed IHC for NFκB-p65, Mcl-1 and phosphorylated (active) STAT3 (STAT3pY) on resected formalin fixed paraffin embedded (FFPE) patient tissues. Our initial analysis of the stained tissues did not reveal significantly different overall expression (number of positive cells) of NFκB-p65 or STAT3pY between normal thyroid and ATC (Supplemental Figure 2A and B). In fact, some normal thyroid tissues showed higher levels of STAT3pY (Supplemental Figure 2B) consistent with our qPCR data showing lower expression of STAT3 mRNA (Supplemental Figure 1D). STAT3pY staining was found predominantly in vascular endothelial cells and occasionally in thyroid follicular cells (Supplemental Figure 2C). Similar findings with respect to STAT3 have been reported and STAT3 has been proposed to function as a tumor suppressor in thyroid tumorigenesis by suppressing HIF-1alpha-signaling, aerobic glycolysis and tumor growth (30).
We assessed if NFκB-p65 staining (in cytoplasm and nucleus) correlated with overall survival (OS) in ATC patients. Neither ‘low’ (below median) nor ‘high’ (above median) overall expression of NFκB-p65 correlated with OS (P=0.4936) (Supplemental Figure 3A). However, in the case of NFκB-p65 we found that ATC cells would predominantly express NFκB-p65 in the cytoplasm or nucleus and strong nuclear staining was only observed in ATC tissues (Fig. 1A). This finding is consistent with NFκB-p65/RelA being latent in the cytoplasm but translocated into the nucleus to modulate transcription of target genes once activated. Thus, we hypothesized that ATCs with aberrantly activated NFκB-signaling may show primarily nuclear staining of NFκB-p65 as a consequence of activation (12). Four (4) out of eleven (11) ATC patients were classified as having predominately nuclear staining for NFκB-p65 and showed an overall poorer prognosis with median survival of 1.4 months compared to 7.9 months (hazard ratio 7.8; 95% CI: 1.24 – 48.67) for patients with predominantly cytoplasmic staining (Fig. 1B). Nuclear staining for NFκB-p65 was not the only predictor (nor independent) of survival. ATC patients with ‘small’ tumors (greatest dimension <10 cm) and/or who were subjected to surgery as part of their treatment had better OS as compared to patients who had ‘large’ tumors (greatest dimension >10 cm) and who did not receive surgery (Supplemental Figure 3B and C). Although tumor stage (IV-B vs. IV-C) showed a trend towards influencing OS it was not statistically significant (P=0.0690) in the analyzed cohort (Supplemental Figure 3D). A potential explanation for the latter finding may be that our cohort was of modest size even by ATC standards and increased sample size would likely confirm tumor stage to prognosticate survival.
Figure 1: Nuclear NFκB-p65/RelA and Mcl-1 is overexpressed in anaplastic thyroid cancer and may be associated with markers of poor prognosis.
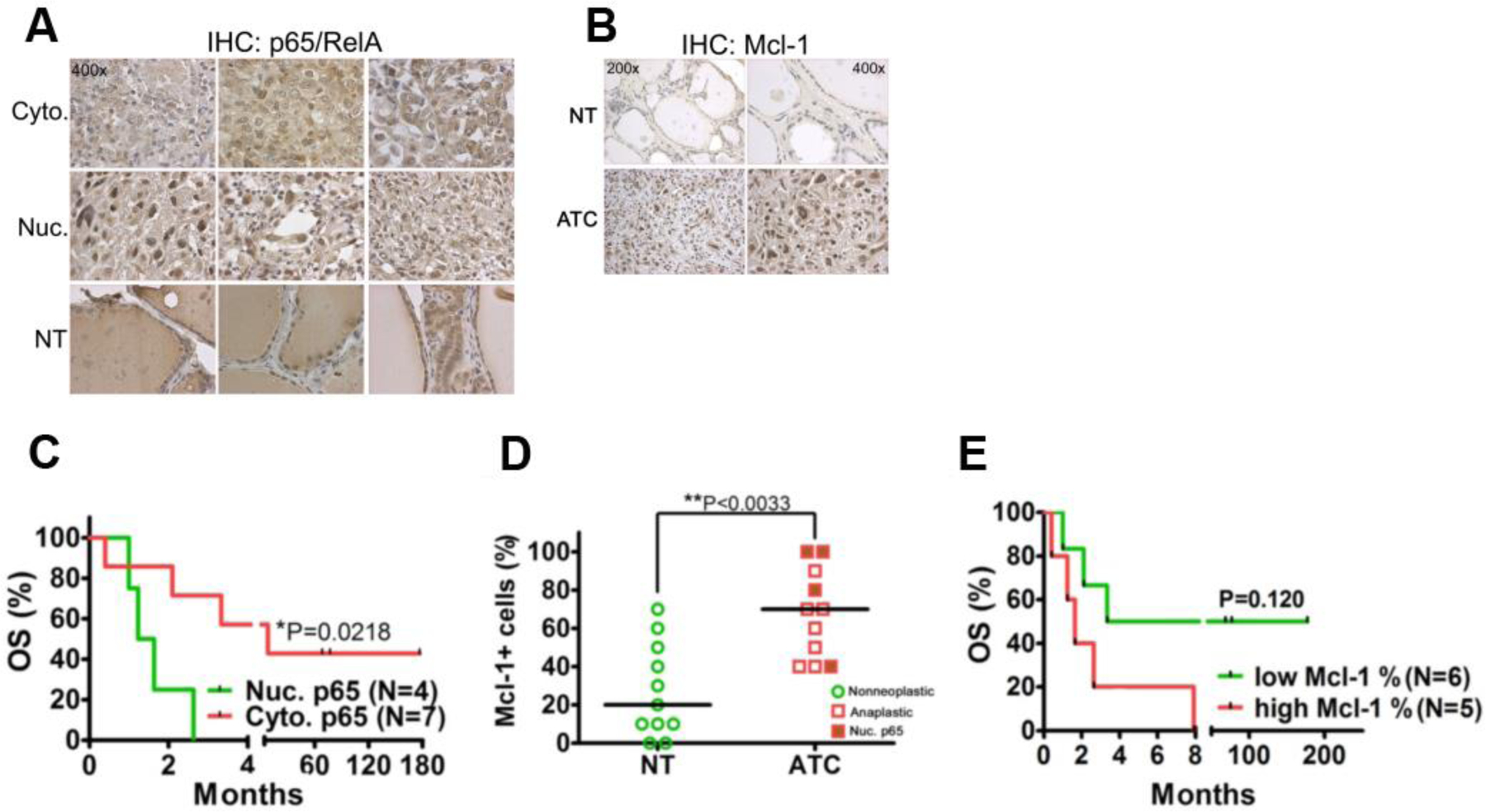
(A) IHC for NFκB-p65/RelA on clinical FFPE non-neoplastic thyroid (NT) and anaplastic thyroid cancer (ATC) specimens. Each panel shows a representative image of a unique individual patient sample with cytoplasmic (Cyto.) and nuclear (Nuc.) expression of NFκB-p65/RelA in ATC or the expression of NFκB-p65/RelA in non-neoplastic thyroid (NT) as detected by IHC. A 400x magnification is shown. (B) Mcl-1 expression in clinical FFPE specimens of NT and ATC. Representative images are shown. (C) Overall survival (OS) of ATC patients with nuclear (Nuc.) and cytoplasmic (Cyto.) expression of NFκB-p65/RelA. The P-value was determined using the Log-rank test. (D) Quantitation of FFPE immunohistochemistry for Mcl-1 in NT (N=11) and ATC tissues (N=11). Medians are shown by black horizontal lines and each data point represents a stained specimen from one patient. Tumors expressing Mcl-1 that had nuclear NFκB-p65/RelA are indicated. The P-value was determined using the Mann-Whitney test. (E) OS of ATC patients in relation to the expression levels of Mcl-1 in their tumors. A “low” and “high” expression level indicates expression (% positive cells in the specimen) below or above the median respectively (as shown in ‘B’). The P-value was determined using the Log-rank test.
ATCs appeared to stain more frequently for the NFκB target Mcl-1 compared to normal thyroid tissue (Fig. 1C and D). Three (3) of four (4) ATCs with nuclear NFκB-p65 also had an expression (% Mcl-1+ cells) above group median (Fig. 1D). Furthermore, by Kaplan-Meier estimates, we observed a trend where patients with ‘high’ (above median) Mcl-1 appeared to have decreased OS compared to patients with ‘low’ (below median) Mcl-1 (Fig. 1E) although this trend was not statistically significant when subjected to multivariate survival analysis using Cox’s regression model (p=0.120).
In order to validate our findings with respect to increased NFκB-p65 activation in ATC compared to normal thyroid tissue we performed meta-analysis of previously published gene expression data on ATC. Three studies allowed us access to expression profiles for data mining of a sufficiently large set of genes (31–33). Indeed, GSEA analysis of the expression profiles previously published (33) revealed that normal thyroid and ATC separated in distinct phenotypic expression profiles (Supplemental Figure 4A). We investigated if gene sets with activation of NFκB-signaling were enriched in clinical ATC’s. Subsequently, we matched two gene sets (HALLMARK_INFLAMMATORY_RESPONSES and TNF_SIGNALING_VIA_NFKB) present in the molecular signatures database (MSigDB) with the expression profiles of normal thyroid and ATC. Increased overlap between genes expressed in the ATC expression profile was observed with both of the gene sets (FDR q<0.00001 and nominal P<0.01) (Fig. 2A and B). In addition, we assessed if sets of genes containing the RelA annotation motif GGGAMTTYCC within 2kb of their transcription start site were increasingly expressed in ATC. Such genes were increasingly expressed in clinical ATC specimens compared to normal thyroid (FDR q<0.0001 and nominal P<0.01) (Fig. 2C). We performed GSEA-analysis for gene sets relating to NFκB-activity for ATC-curated gene expression profiles published by Salvatore et al (32) and Hebrant et al (31) (Supplemental Figure 4B and D). Although no enriched expression of NFκB-dependent genes was found with statistical certainty (i.e., FDR q<0.25 and P<0.05) on the curated expression profiles, the ATC expression profiles from both studies contained a larger number of genes linked to NFκB-activity than either normal thyroid tissues or papillary thyroid cancers. We did not find Mcl-1 overexpression in ATC compared to neither normal thyroid tissues nor papillary thyroid cancer (data not shown). By contrast, data from the established expression profile from Hebrant et al (31) indicated reduced mRNA expression of Mcl-1 compared to normal tissues and papillary thyroid cancer (data not shown). Mcl-1 protein expression has been shown to be regulated both at the transcriptional and post translational level (34) and thus they may not always correlate in cancer.
Figure 2: Meta analysis of expression profiles from normal thyroid (NT) tissue and ATC.

(A) Gene set enrichment analysis of clinical meta-data from Giordano et al (Giordano et al 2005). The GSEA analysis was performed for ATC (N=3) and NT (N=3). Gene set enrichment was assessed for the MSigDB gene sets HALLMARK_INFLAMMATORY_RESPONSES (A), HALLMARK_TNF_SIGNALING_VIA_NFKB (B) and N$YKAPPA (genes with RelA binding sites within 2kb of their core promoter) (C).
Combined treatment with sorafenib and quinacrine triggers synergistic anti-tumor responses in ATC cells
We have previously assessed sorafenib (S) and quinacrine (Q)-dependent targeting of the anti-apoptotic protein Mcl-1 to sensitize tumor cells to programmed cell death (apoptosis). Targeting of Mcl-1 by S and Q was previously shown by us to overcome TRAIL- and chemo-resistance in cancer cells (19–22). We hypothesized that Mcl-1 may be a bonafide drug target in ATC.
Q and S displayed single agent anti-tumor activity in human ATC cells. Dose-response relationships for S (Fig. 3A) and Q (Fig. 3B) were established in the ATC cell lines THJ-16T, THJ-21T and THJ-29T (Table I). The THJ-16T cell line appeared to be significantly more resistant to S compared to the THJ-21T and THJ-29T cell lines. By contrast, the same cell lines showed smaller differences with respect to sensitivity to Q (Fig. 3B and Table I). The anti-tumor response appeared to be at least partially explained by apoptosis in the ATC cell population. Increased sub-G1 DNA was observed following short-term treatment with both S (S10) and Q (Q20) alone (Fig. 3C). An additive effect was observed by the combination of S and Q (S10Q20). We further characterized the dose-response characteristics of the combination of S and Q in THJ-29T and THJ-16T cells using the bioluminescent CellTiter-Glo™ cell survival assay (Fig. 3D). A combinatorial effect (reduced fluorescence indicating reduced survival) was observed with S and Q.. We assessed the nature of the S and Q combinatorial drug response in the sorafenib-refractory ATC cell line THJ-16T. We used the theorem of Chou-Talalay (35) to generate combination indices (CI) for the drug combination of S/Q in ATC (Fig. 3E). The best outcome as judged by CI’s generated in our cell survival assay was ‘strong synergism’ indicating that Q may sensitize sorafenib-refractory ATC cells to cell death. Taken together, our data indicate that the drug combination S/Q triggers apoptosis and anti-tumor responses that are synergistic in ATC cells.
Figure 3: Quinacrine and sorafenib synergize in the killing of ATC cells.
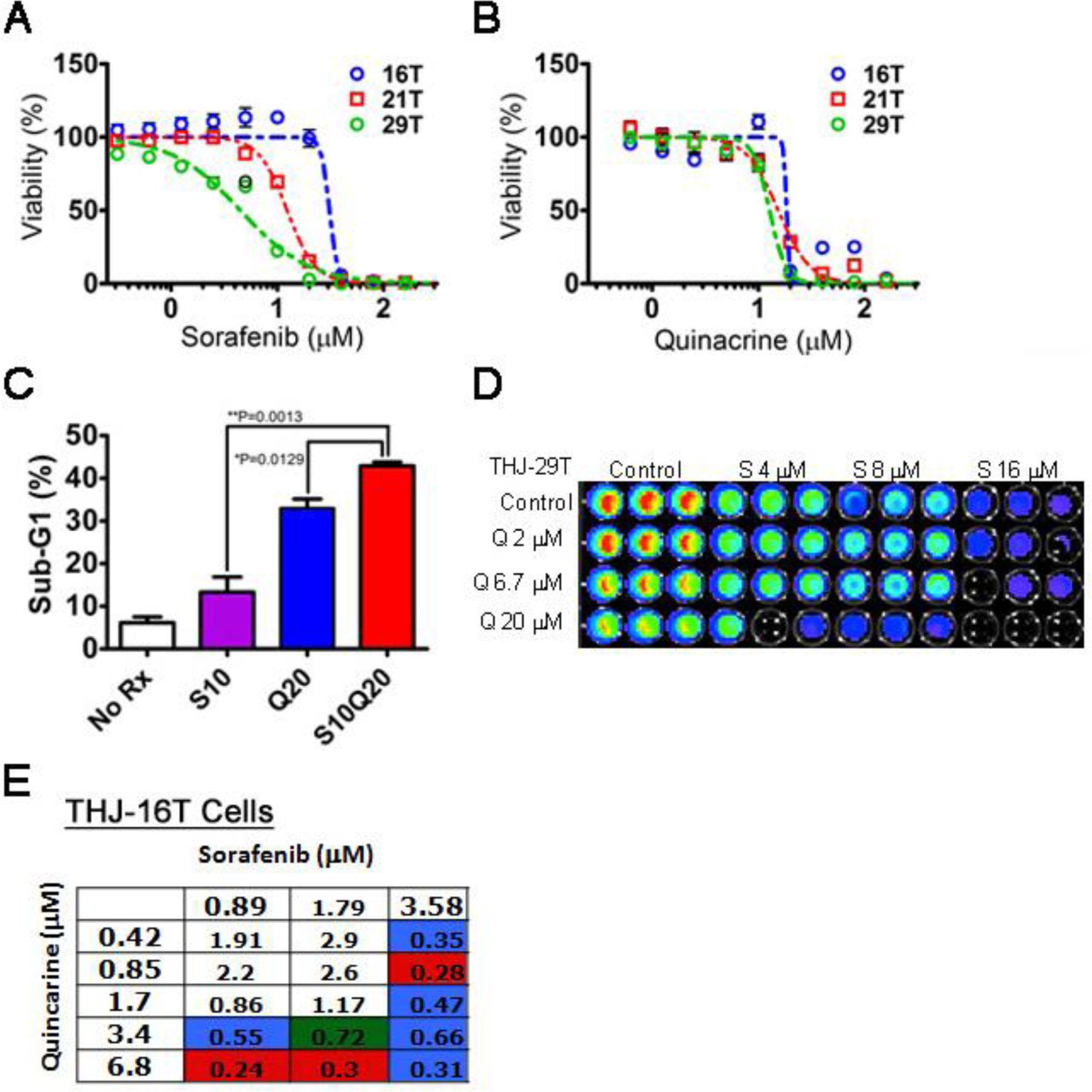
Dose-response curves for the 72-hr survival of ATC cell lines THJ-16T, THJ-21T and THJ-29T following sorafenib (A) and quinacrine (B). The X-axis are shown as log scale. (C) Flow cytometric sub-G1 programmed cell death (apoptosis) assay of the ATC cell line 8505C subjected to treatment with sorafenib (S) and quinacrine (Q) and the combination (S/Q). Averages (N=3) +/− SEM are shown. Statistical analysis was performed using Students t-test where P<0.05 was considered statistically significant. Dose-response modulation of the combinatorial response of Q and S. (D) Cell survival assay (CellTiter-Glo®) of ATC cells treated with different combinatorial concentrations of Q and S. A representative figure of the result of a 72-hr Cell Titer-Glo® assay for the ATC cell line THJ-29T is shown. (C) Combinatorial indices generated using the CalcuSyn 2.0 software employing the method by Chou Talalay for S and Q in the sorafenib-refractory ATC cell line THJ-16T. Red indicates ‘strong drug synergism’, blue – ‘drug synergism’, green – ‘moderate drug synergism’.
Table I:
Sorafenib and Quinacrine dose-response parameters for three (3) ATC cell lines
| Sorafenib | Quinacrine | |||||
|---|---|---|---|---|---|---|
| Cell line | IC50 (µM) | 95%CI | R2 | IC50 (µM) | 95%CI | R2 |
| THJ-16T | 31.00 | 19.32 to 49.75 | 0.9683 | 18.43 | 14.69 to 23.13 | 0.9732 |
| THJ-21T | 12.38 | 11.79 to 13.00 | 0.9939 | 15.39 | 13.84 to 17.11 | 0.9734 |
| THJ-29T | 4.497 | 3.812 to 5.304 | 0.9636 | 12.74 | 11.65 to 13.93 | 0.9808 |
Quinacrine potentiates sorafenib-dependent eradication of anti-apoptotic Mcl-1 in ATC-cells
We previously showed that treatment of 8505C with increasing concentrations of sorafenib (S) that the drug ameliorates the expression of several pro-survival proteins (21). Among others, phosphorylation of STAT3, MEK and ERK was inhibited. In addition, expression of pro-survival protein Mcl-1, but not Bcl-XL, was inhibited by drug S. Western blot showed that low dose treatment of 8505C cells with Q (1 µM) and S (0.3–3.2 µM) inhibits phosphorylation of STAT3 at the pro-survival phosphorylation site of Y705 (Fig. 4A). At higher doses the drug combination triggers a robust pro-apoptotic response in 8505C cells associated with an essentially complete eradication of Mcl-1 and full-length PARP (Fig. 4B). Expression of mutated p53 was not affected by the drug combination in 8505C cells. Indeed, previous data showed that Q kills cancer cells independent of p53 (20). Q and S/Q inhibited the expression of NFκB-p65 (RelA) in 8505C and THJ-16T cells (Fig. 4C) supporting the notion that the NFκB-pathway is a target in ATC cells. To functionally validate our findings with regards to p65/RelA and Stat3 expressions we used siRNA to target p65/RelA and total Stat3 in 8505C cells prior to treatment with sorafenib and quinacrine. Neither scrambled (Scr) siRNA nor siRNA targeting Stat3 had a significant impact on the cell viability dose-response relationship of sorafenib or quinacrine in 8505C cells (Fig. 4F and G). However, targeting of p65/RelA caused an approximately 2-fold increased sensitization of 8505C cells to sorafenib compared to cells subjected to treatment with Scr siRNA (IC50: 5.15 µM; 95%CI: 4.14–6.41 µM and IC50: 10.49 µM, 95%CI: 7.56–14.5 µM respectively) (Fig. 4F). SiRNA-dependent targeting of p65/RelA did not have any significant impact on the survival dose-response relationship following treatment with quinacrine (Fig. 4G). Thus, our western blot protein dataindicates that the drug combination of S/Q targets several pro-survival pathways and the anti-apoptotic protein Mcl-1 in ATC cells in vitro. However, targeting of NFκB signaling may in particular sensitize ATC cells to sorafenib perhaps indicating that quinacrine may trigger synergism as a result of its capacity to target NFκB-dependent transcription.
Figure 4: Sorafenib (S) and quinacrine (Q) target the anti-apoptotic protein Mcl-1 in ATC cells.
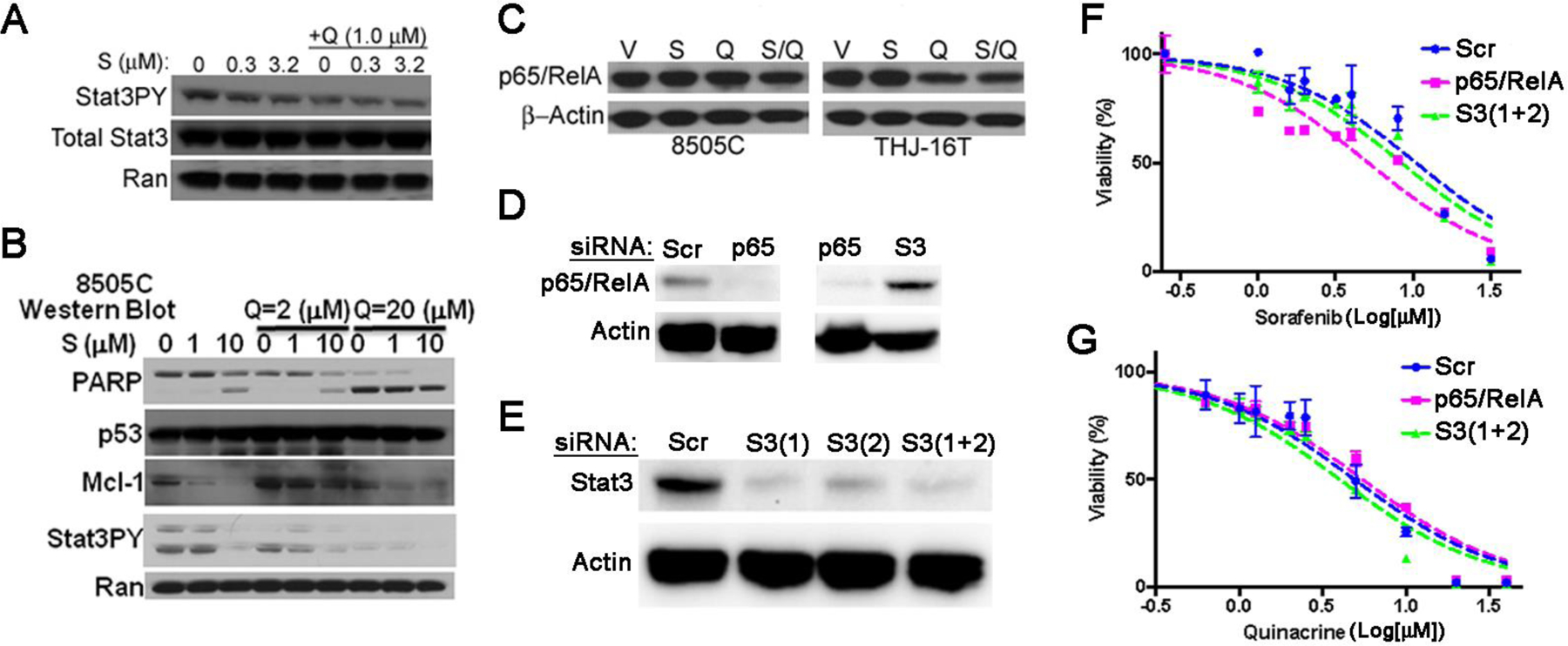
(A) Combination treatment with S and Q triggers inhibition of Stat3 phosphorylation (Stat3PY) in 8505C cells. (B) The effect of S and Q on PARP cleavage, Mcl-1 and Stat3PY expression in 8505C cells. (C) Western blot to analyze impact of S/Q on expression of NFκB-p65/RelA in 8505C and THJ-16T cells. The doses of S and Q used were 10 and 20 µM respectively. (D) Western blot analysis show efficient targeting of p65/RelA in 8505C ATC cells by specific siRNA (p65) (left panel). No effect on p65/RelA is observed by siRNA-mediated targeting of Stat3 (S3 [1+2]) (right panel). Scrambled (Scr) siRNA was used as a control for specific targeting of p65/RelA and Actin was used as a loading control. (E) Western blot analysis show efficient targeting of total Stat3 by two different siRNA (S3[1] and S3[2]) in 8505C cells. A combination of both siRNA (S3[1+2]) was used to achieve efficient knock down of total Stat3. Actin was used as a loading control. Sorafenib (F) and quinacrine (G) dose-response curves following depletion of Stat3 and p65/RelA in 8505C cells using the CellTiter-Glo® assay. The 8505C cells were treated for 72 hrs with the drugs.
Targeting of Mcl-1 impacts the anti-tumor response to sorafenib but not quinacrine in ATC cells
To assess the extent to which Mcl-1 may be required for the therapeutic efficacy of the drug combination S/Q we targeted the expression of Mcl-1 by siRNA gene silencing and assessed how this impacted the dose-response characteristic of S and Q in 8505C cells (Fig. 5A–D). Knockdown of Mcl-1 sensitized 8505C cells to S by 1.4-fold but did not affect the IC50 to Q (Fig. 5C–E). Analyzing the combinatorial effect of S and Q in 8505C cells with and without targeted Mcl-1 expression showed that intact Mcl-1 expression is required to produce a potent combinatorial drug response. Synergism was observed at an ICS80/Q60 combinatorial dose whereas ICS80/Q50 and ICS80/Q30 produced near additive effects in the 8505C cell line (Fig. 5F). By contrast, Mcl-1 knock-down produced an antagonistic relationship between S and Q at the corresponding effective dose levels (Fig. 5G). We also found that Q synergistically sensitized 8505C cells to S at a concentration as low as 125 nM (CI=0.88) and this drug synergism was ameliorated completely by Mcl-1 depletion (CI=1.03) (data not shown). We propose a model where quinacrine-dependent inhibition of Mcl-1 expression contributes to the combinatorial efficacy of S/Q (Supplemental Figure 5). This may favor targeting ATC with high NFκB and Mcl-1.
Figure 5: Mcl-1 expression is required in ATC cells for combinatorial efficacy of the sorafenib (S) and quinacrine (Q).
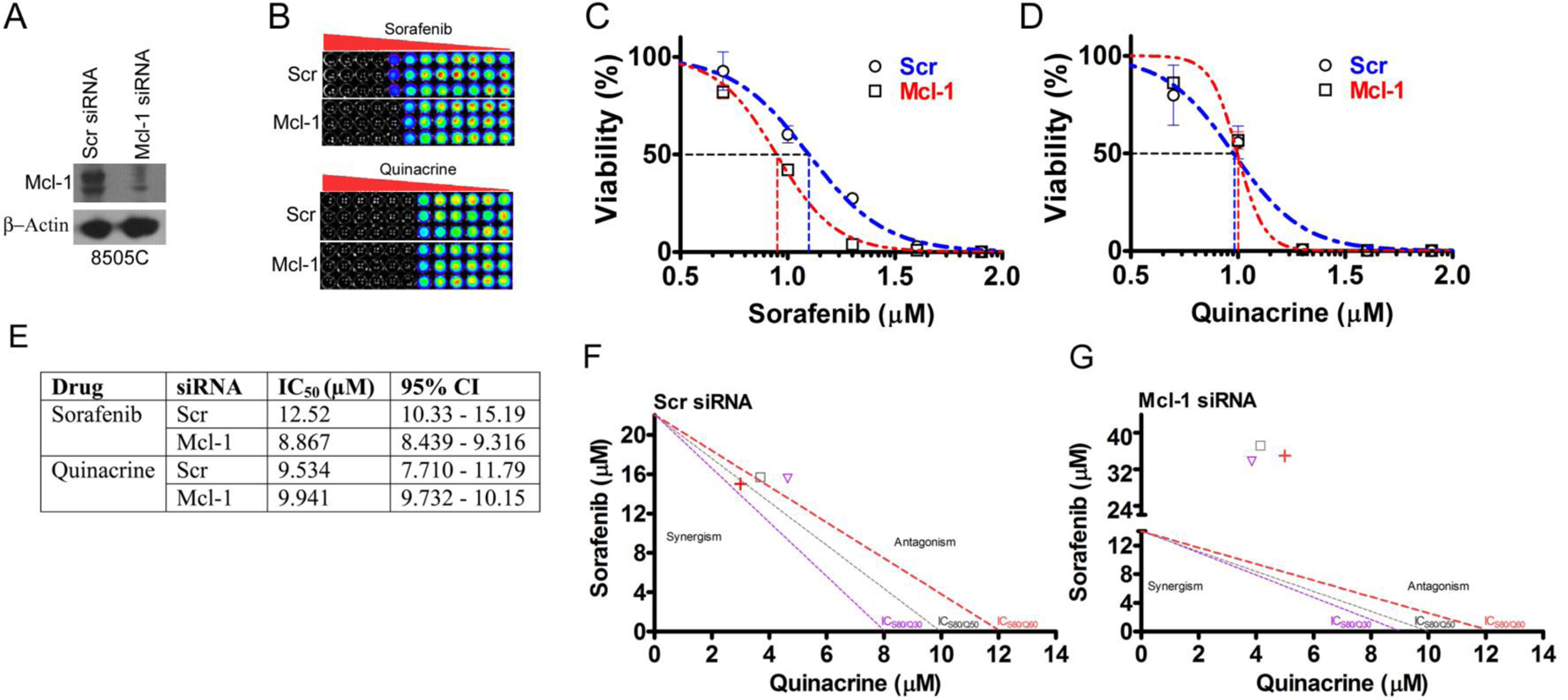
(A) Western blot assessment of the efficacy of small interfering RNA (siRNA)-dependent targeting of Mcl-1 expression in 8505C cells. (B) CellTiter-Glo® cell survival assay. Sorafenib (C) and quinacrine (D) dose-response curves in 8505C cells following siRNA mediated targeting of Mcl-1. The X-axis are shown as log scale. (E) The observed IC50-values following S and Q treatment with and without Mcl-1-targeting. Isobolograms for 8505C cells treated with various effective S and Q concentrations (ICS80/Q30, ICS80/Q50 and ICS80/Q60) in the presence (F) and absence (G) of Mcl-1 knock-down.
Drug combination S/Q improves survival in a mouse orthotopic ATC model
To test the efficacy of drug combination S/Q in vivo we assessed to what extent ATC cells grew as tumors in immunodeficient (Nu/Nu) mice. ATC cells were labeled with fluorescent cell permeable dye CellVue® Maroon and injected orthotopically into recipient mice (Supplemental Figure 6A). At one week following injection mice were subjected to NIR-imaging to visualize fluorescently labeled ATC cells and document the extent of tumor growth. Nu/Nu mice are significantly less sensitive to DNA damage compared to immunodeficient SCID mice that lack the Ku70 subunit of DNA-PK, and thus were the preferred mouse model. Orthotopic models are considered clinically more relevant compared to sub-cutaneous models and therefore we used an orthotopic in vivo model (Supplemental Figure 6B). Injected mice developed tumors in their thyroid gland within seven days and started to show lethargy at day 12 (Fig. 6A). Efforts to assess tumor burden over time in these mice by non-invasive NIR-imaging proved to be difficult as the thyroid is localized behind the salivary gland and so NIR-imaging was used as a post-mortem imaging methodology to verify ATC tumor growth from labeled implanted cells (Fig. 6B, C and Supplemental Figure 6C). IHC data indicated the 8505C tumor xenograft had high expression of Mcl-1 (Supplemental Figure 6D).
Figure 6: The sorafenib (S) and quinacrine (Q) combination improves the survival of mice subjected to a thyroid orthotopic injection of ATC cells.
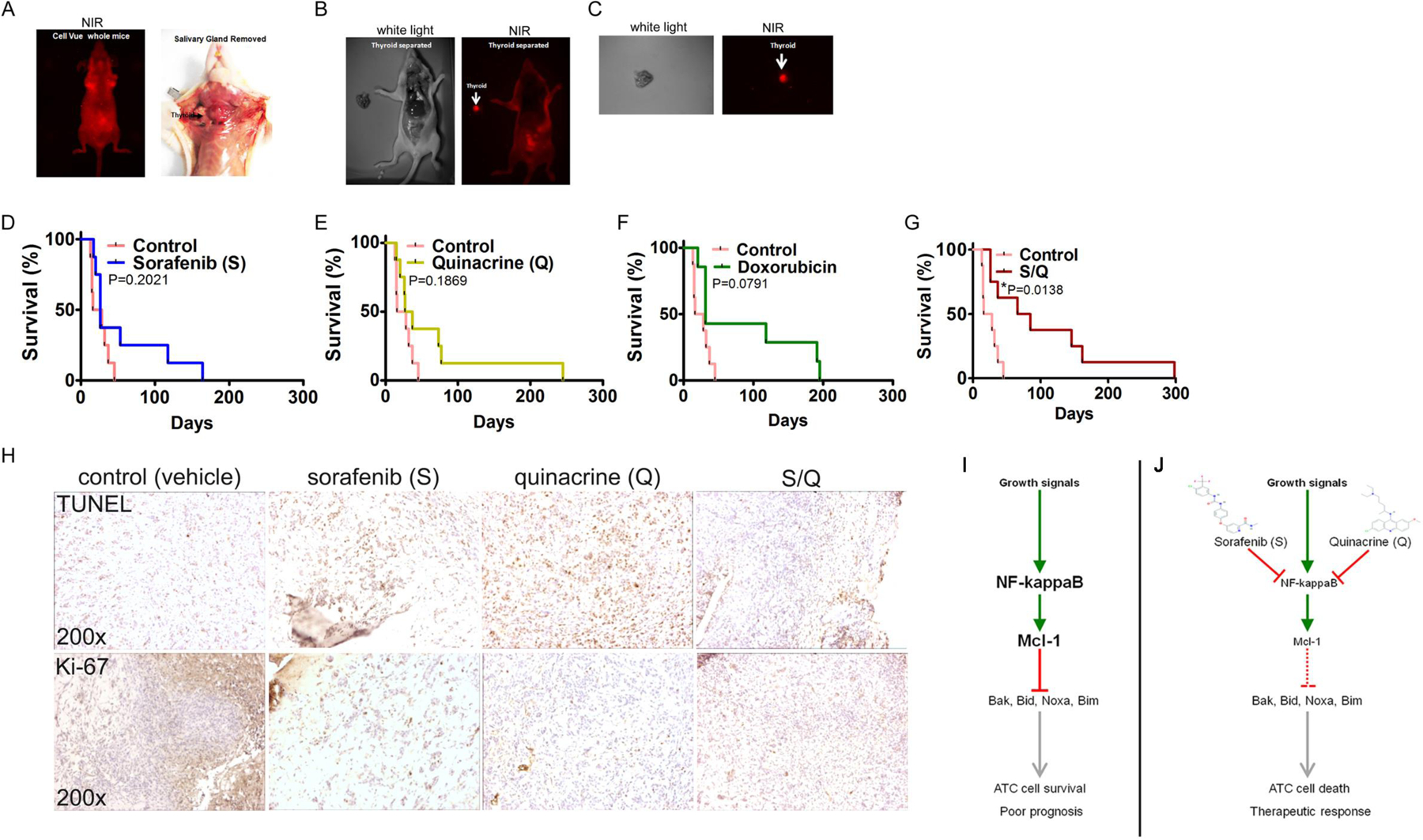
(A) Live whole-body near-infrared imaging (NIR) of whole mice (left panel) and tumor engraftment of 8505C cells in the thyroid of a Nu/Nu mouse (right panel). (B) NIR-imaging of a Nu/Nu mouse subjected to thyroid orthotopic injection of CellVue® NIR815-labeled 8505C cells after the removal of the salivary gland. (C) NIR-imaging of a dissected and isolated tumor ATC-xeno-engrafted mouse (Nu/Nu) thyroid. The injected 8505C cells were labeled with CellVue® NIR815-labeled Survival curves of mice subjected to thyroid orthotopic injection of 8505C cells and subjected to no treatment (Control) and sorafenib (D), quinacrine (E), doxorubicin (F) and the combination of S and Q (G). (H) Immunohistochemistry for TUNEL (apoptosis) and Ki-67 (proliferating cells) on mouse thyroid xenograft tumors of the ATC cell line 8505C. (I) Growth signals trigger constitutively active NFκB signaling and Mcl-1 expression in ATC cells. Mcl-1 inhibits the activity of the pro-apoptotic proteins Bak, Bid, Noxa and Bim. In addition, Mcl-1 may also influence cell cycle progression and proliferation (not depicted). (J) Both S and Q inhibit NFκB activity and ameliorate Mcl-1 expression. Selectivity of the compounds is achieved through increased activity of NFκB in ATC’s. Reduced expression of Mcl-1 allows the pro-apoptotic proteins Bak, Bid, Noxa and Bim to trigger cell death (apoptosis) and a therapeutically relevant anti-tumor response in ATC.
Median survival for untreated mice was twenty-two (22) days (Table II). Although S, Q and doxorubicin did increase median survival (26, 32 and 31 days respectively) these trends did not reach statistical significance when compared to the median survival of untreated (control) mice (Fig. 6D–F and Table II). These data are consistent with the clinical experience where sorafenib and doxorubicin provide a limited impact on OS for ATC patients. By contrast, we found the sorafenib and quinacrine (S/Q) combination significantly increases the survival of mice carrying 8505C xenografts in their thyroid gland compared to untreated mice (median survival 75.5 days, p=0.0138, Gehan-Breslow-Wilcoxon Test) (Fig. 6G and Table II). Analysis of FFPE thyroid tumors indicated increased TUNEL-positivity and reduced expression of the proliferation marker Ki-67 following treatment with the different modalities (Fig. 6H) (19). In summary, although sorafenib, quinacrine and doxorubicin fail to produce a significant survival advantage in this ATC orthotopic mouse model the combination of sorafenib and quinacrine increases the median survival three (3)-fold compared to that of untreated (control) mice without any apparent toxicity.
Table II:
Survival parameters from the mouse ATC (8505C) orthotopic tumor model.
| Drug | Median Survival (days) | Hazard ratio1 | 95% CI | P value1 |
|---|---|---|---|---|
| Control | 22 | - | - | - |
| Sorafenib (S) | 26 | 0.846 | 0.317–2.25 | 0.202 (NS) |
| Quinacrine (Q) | 32 | 0.687 | 0.258–1.83 | 0.186 (NS) |
| S/Q | 75.5 | 0.2914 | 0.109–0.776 | 0.0138 (*) |
| Doxorubicin | 31 | 0.709 | 0.257–1.95 | 0.0791 (NS) |
Relative Control (untreated) mice
The combination of quinacrine and sorafenib shows limited toxicity in a mouse dose-escalation study as compared to doxorubicin
Although the safety profiles for sorafenib and quinacrine are known in human subjects, the potential for the drug combination to trigger toxicity in an additive/synergistic manner in vivo is largely unknown. We addressed this through a short-term (7 days) dose-escalation study in mice followed by a histopathological assessment and blood correlatives following treatment. No lethality or lethargy was observed in the mice following exposure the super-therapeutic doses of S, Q, doxorubicin and S/Q. However animals subjected to S/Q displayed a transient 15% weight loss nadir at day two (2) following initiation of treatment (data not shown). All of the S/Q-treated animals recovered from this by day five (5). Analysis of serum chemistry and hematology indicated no toxicity of S/Q compared to untreated (control) mice (Supplemental Figure 7 and 8). By contrast, mice subjected to treatment with the first-line chemotherapeutic doxorubicin displayed a reduction in the number of RBC’s and hemoglobin (Hgb) compared to mice treated with S/Q (Supplemental Figure 8B and C). Furthermore, doxorubicin-treated mice had an increased level of platelets in their blood compared to control and S/Q-treated mice (Supplemental Figure 8E). Histopathological evaluation of the bone marrow, gastrointestinal (GI) tract, heart, pancreas, liver and kidney suggested that doxorubicin-treated mice more frequently displayed signs of bone marrow and GI-toxicity than the other treatment groups (Table III). Such signs included prominent congested sinusoids in the bone marrow and villous blunting, crypt dilation, crypt hyperplasia and inflammation of lamina propria in the duodenum. Although doxorubicin is known to cause cumulative-dose cardiotoxicity, we did not find any histopathological evidence of this in our short-term study. Histopathological signs of toxicity from the combination of S/Q were consistent with what was observed with treatment with either compound alone and did not increase in severity following the combination (Table III and data not shown). The only unique histopathological manifestation that we were able to link to S/Q exposure was the appearance of a single small focal chronic cortical infarct with a volume of <0.01% in the kidneys of two out of five (2/5) mice subjected to S/Q (Table III) which was not frequent enough to be statistically significant. Thus, the S/Q combination is well-tolerated even at doses that greatly exceed those used to evoke potent anti-tumor responses in vivo.
Table III:
Histological evidence of toxicity in mice following dose-escalation of sorafenib (S), quinacrine (Q), S/Q and doxorubicin.
| Organ/tissue | Vehicle (V) | Quinacrine (Q) | Sorafenib (S) | Q/S | Doxorubicin |
|---|---|---|---|---|---|
| Bone marrowa, ** | 0/5 (0) | 0/4 (0) | 0/5 (0) | 0/5 (0) | 3/5 (0.60) |
| GIb, ** | 0/5 (0) | 0/4 (0) | 0/5 (0) | 0/5 (0) | 4/5 (0.80) |
| Heartc | 2/5 (0.40) | 1/5 (0.20) | 0/5 (0) | 1/5 (0.20) | 0/5 (0) |
| Pancreas d, ** | 0/5 (0) | 0/4 (0) | 5/5 (1.00) | 4/5 (0.80) | 4/5 (0.80) |
| Liver | |||||
| Vaculation | 2/5 (0.40) | 3/4 (0.75) | 4/5 (0.80) | 4/5 (0.80) | 4/5 (0.80) |
| Glycogene | 2/5 (0.40) | 3/4 (0.75) | 4/5 (0.80) | 2/5 (0.40) | 3/5 (0.60) |
| Hydropic degeneration with basophilic stippling, centrilobular | 0/5 (0) | 2/4 (0.50) | 0/5 (0) | 2/5 (0.40) | 0/5 (0) |
| Lipid, centrilobular | 0/5 (0) | 0/5 (0) | 0/5 (0) | 0/5 (0) | 1/5 (0.20) |
| Kupffer cell hyperplasia, centrilobular | 0/5 (0) | 0/5 (0) | 0/5 (0) | 0/5 (0) | 1/5 (0.20) |
| Hepatocyte apoptosis, centrilobular | 0/5 (0) | 0/5 (0) | 0/5 (0) | 0/5 (0) | 1/5 (0.20) |
| Kidneyf | 0/5 (0) | 0/5 (0) | 0/5 (0) | 2/5 (0.40) | 0/4 (0) |
Characterized by the appearance of prominent congested sinusoids
Characterized by the appearance of villous blunting, crypt dilation, crypt hyperplasia, crypt abscesses and inflammation of lamina propria.
Characterized in (V) by one case of focal segmental medial mineralization in a small mural coronary artery and one case of mineralized necrotic cardiomyocytes affecting <0.1% of the myocardium. In (Q), one case characterized by a nodule of large macrophages and few mast cells between the left ventricle and atrium. In (Q/S), one case displayed several mineralized necrotic cardiomyocytes affecting <0.1% of the myocardium.
Characterized by the appearance of serositis of the pancreatic capsule; subacute or chronic, mild to moderate; necrotizing pancreatitis, subacute, minimal to moderate. This finding may be a result of the intra-peritoneal injection route of drug administration.
Diffuse, centrilobular, midzonal or peri-portal.
Characterized by the appearance of one (1) small focal chronic cortical infarct with a volume of <0.01%.
P<0.05 (Chi-square test).
P<0.01 (Chi-square test).
The drug combination of sorafenib and quinacrine inhibits the expression of angiogenesis markers in mouse 8505C xenografts
Preclinical data indicates that targeting of tumor angiogenesis and VEGF-signaling can trigger anti-tumor responses in models for ATC (36). Preclinical data and two (2) clinical case studies also indicate that receptor tyrosine kinase (RTK) inhibitors that target VEGF-signaling may hold promise for the treatment of ATC (37, 38). In order to address if the combination of sorafenib and quinacrine have potential to modulate tumor angiogenesis we performed IHC for CD34 (a marker for the presence of small vessel endothelium) and VEGF (a pro-angiogenic marker and effector molecule) on FFPE sections from 8505C xenografts from Nu/Nu mice subjected to treatment with S/Q (Fig. S9). The number of CD34-positive vessels was dramatically reduced by S and the combination of S/Q (Fig. S9A and B). Moreover, S alone and in combination with Q exhibited a potent inhibition of VEGF-expression in the tumor xenografts. These data are consistent with sorafenib’s anti-angiogenic properties in addition to BRAF-targeting. Taken together our data indicate that the combination of sorafenib and quinacrine inhibits angiogenesis in addition to the targeting of anti-apoptotic molecules in ATC cells.
Discussion
Despite advances in understanding the molecular pathogenesis of ATC the disease remains associated with a poor prognosis. Increased efforts are being undertaken to define “actionable” genetic changes and develop improved treatment protocols. The multi-kinase inhibitor sorafenib has shown promising results in some solid malignancies but data from recent ATC clinical trials indicate that sorafenib has limited impact on overall survival in ATC (9, 10). Our studies show that sorafenib combines favorably with quinacrine to trigger a combinatorial response in ATC to significantly improve survival of mice implanted orthotopically with ATC cells relative to untreated (control), single agent or doxorubicin-treated mice (Fig. 6). This S/Q drug combination targets NFκB-p65 and Mcl-1 in vitro (Fig. 4 and 5). Both NFκB-p65 and Mcl-1 are overexpressed in a subset of ATC’s (Fig. 1). Furthermore, mouse dose-escalation toxicology data suggests that the S/Q drug combination is safe at the doses tested (Table III and Supplemental Figure 7 and 8). Further assessment of the drug combination in animal models for loco regional toxicities following radiation delivery to the head and neck may be necessary. ATC patients frequently undergo radiotherapy and it is currently unknown if the drug combination of S/Q would have a negative therapeutic index in this context. However, our findings provide a foundation for evaluation of the S/Q drug combination in clinical trials.
Multiple aberrantly activated signal transduction pathways drive ATC pathogenesis
It is somewhat surprising that benefit from sorafenib is limited despite 25% of ATC’s showing BRAF V600E mutations as sorafenib targets multiple pathways with activated tyrosine kinases such as VEGFR and PDGFR-signaling (39, 40). This observation could be due to the fact ATCs also show mutations in beta-catenin that generates hyperactivated Wnt-signaling (41) and that ATC cells undergo EMT and show evidence of increased stemness compared to more differentiated forms of thyroid cancer (42). EMT is associated with refractoriness to therapy and more aggressive behavior in several other malignancies. Moreover, tumor suppressor p53 is frequently lost and mutated in ATC and there is increased inflammatory NFκB-signaling and enhanced activation of matrix metallo-proteinases (MMP-9) which may promote the invasiveness of ATC (43, 44), features supported by our gene set enrichment analysis of meta-data (Fig. 2 and Supplemental figure 4). It is therefore unlikely that targeting a single molecular facet in ATC would yield a significant improvement of patient outcomes.
Interestingly, quinacrine is able to trigger anti-tumor responses independent of mutated p53 through inhibition of NFκB and Mcl-1 (19, 20). Quinacrine can also synergize with the anti-angiogenic drugs such as cediranib that targets VEGF-signaling (45) by blocking autophagy triggered by receptor tyrosine kinase inhibitors suggesting that quinacrine can modulate the activity of anti-angiogenic agents. Sorafenib displays anti-angiogenic activity in our in vivo model and represses VEGF expression including when combined with quinacrine (Supplemental Figure 9), unlike single agent quinacrine (Supplemental figure 9A–C). Thus the combinatorial efficacy, multiple pathway targeting, safety and low expense of quinacrine make the combination of sorafenib plus quinacrine an attractive option to further test in the clinic as ATC treatment.
S/Q targets Mcl-1 an anti-apoptotic protein overexpressed in ATC
We have previously shown that quinacrine targets NFκB/RelA and anti-apoptotic Mcl-1 in cancer cells (19, 20). Mcl-1 is an anti-apoptotic Bcl-2 family member and overexpression of Mcl-1 negatively impacts the responses to some therapeutics in patients with malignancies such as CLL (46). We show Mcl-1 is overexpressed in some ATC’s and expression is inhibited through treatment with the drug combination of sorafenib and quinacrine in vitro (Fig. 1B, 1D, 1E and 4B). Quinacrine has previously been shown to inhibit the transcriptional activity of NFκB/RelA and in addition to reactivate wild-type p53 in renal carcinoma cells (17). Quinacrine can inhibit expression of NFκB-p65 (Fig. 4C). This may be of relevance since our IHC data indicate that nuclear expression of NFκB-p65 is linked to poor prognosis in ATC patients (Fig. 1A and 1C). Three (3) out of four (4) ATC with ‘high’ Mcl-1 expression also showed nuclear expression of NFκB-p65 (Fig. 1D). Mcl-1 is a downstream target gene of NFκB/RelA (47) but we failed to substantiate significantly increased expression of Mcl-1 mRNA in clinical ATC’s through data mining of expression profiles from previously published studies (31). However, several studies indicate that expression of Mcl-1 protein is also regulated post-transcriptionally and its mRNA/protein has a short half-life (34). It is possible that NFκB-p65 contributes to the Mcl-1 mRNA pool and subsequent protein synthesis in ATC’s. Subsequently lesions with nuclear NFκB-p65 may be selectively sensitive to quinacrine treatment. We hypothesize quinacrine disrupts the expression of Mcl-1 through inhibition of NFκB/RelA.
We observed a trend suggesting a link between Mcl-1 overexpression, tumor size and OS (Fig. 1E). Evidence suggests that apoptosis signaling indeed may be impaired in ATC. The inhibitor of apoptosis protein (IAP) Survivin is overexpressed in ATC’s compared to papillary thyroid cancers (31, 33) and Survivin expression has been proposed as a marker for dedifferentiation in thyroid carcinogenesis (48). Interestingly, expression of anti-apoptotic Bcl-2 appears to be lost in ATC compared to well-differentiated papillary and poorly differentiated thyroid cancers (49). . Mcl-1 overexpression has been documented in a number of solid tumors and small molecule compounds targeting Mcl-1 directly are being developed for therapy, but the role of Mcl-1 in ATC remains largely unclear. Mcl-1 shows limited background expression in the follicular epithelium of normal thyroid (50) and Mcl-1 expression is frequently observed in undifferentiated follicular and papillary tumors with low expression of Bax (51). Perhaps not surprisingly indirect targeting of Mcl-1 with sorafenib, may sensitize ATC cells to stimuli that trigger programmed cell death (21). In the absence of additional data it is tempting to speculate that Mcl-1 may be linked to the pathogenesis of ATC and may represent a novel “actionable” scaffold for targeted therapeutic intervention in a subset of ATC patients. Thus Mcl-1 may be a bonafide target in ATC beyond the application of S/Q-treatment.
Expression of STAT3pY is downregulated in ATC
In contrast to immunohistochemical data on prostate cancer (25), overexpression of STAT3pY was not apparent in ATC-tissue (Supplemental figure 2B and C). At lease in some cases more frequent staining for STAT3pY was seen in the non-neoplastic thyroid compared to ATC. ATC’s may potentially also express lower levels of STAT3 mRNA compared to non-neoplastic thyroid as evident from our qRT-PCR analysis (Supplemental figure 1D). As we had limited specimens future studies should repeat this analysis with a bigger sample size.
STAT3 has been suggested to be a tumor suppressor in the thyroid since papillary thyroid tumors that emerge in mice as a result of BRAFV600E mutation are significantly smaller when they retained the STAT3 gene (30). The same study demonstrated an inverse correlation between STAT3pY-expression, tumor size and metastasis. STAT3pY was frequently detected in endothelial cells of papillary thyroid cancer which is consistent with what we observe in non-neoplastic thyroid and ATC (Supplemental figure 2C and data not shown). It seems unlikely that STAT3pY is a rate limiting therapeutic target in ATC. However, it may be plausible that the S/Q drug combination targeting STAT3-signaling in the tumor vasculature of ATC’s may contribute to the overall improved response observed in our orthotopic ATC model (Fig. 6). Additional studies will have to address this therapeutic possibility more directly.
In summary, we show that the combination of sorafenib and quinacrine can trigger synergistic Mcl-1-dependent drug responses in ATC cells in vitro. This combination appears to have limited toxicity in mice and triggers potent anti-tumor responses in an orthotopic model for ATC. Based on our data we propose that sorafenib and quinacrine should be explored as a therapeutic modality for aggressive thyroid cancers in future clinical trials.
Supplementary Material
Translational Relevance.
Anaplastic thyroid carcinoma (ATC) is a fatal cancer with a median survival of 3–5 months after diagnosis. The FDA approval of multi-kinase inhibitor sorafenib for late stage metastatic differentiated thyroid cancer has opened newer avenues for targeting ATC BRAF mutations occur in a quarter of ATCs. Quinacrine, an inexpensive drug with a well-known safety profile, inhibits NFκB signaling and shows anti-cancer activity in preclinical studies. We demonstrate that quinacrine acts additively/synergistically on ATC cells in combination with sorafenib in vitro and confers improved survival in an orthotopic mouse model. The drug combination inhibits NFκB and the expression of the anti-apoptotic molecule Mcl-1, both of which we show are overexpressed in clinical ATC specimens. The drug combination show minimal toxicity compared to current standard of care treatment. Our data indicates sorafenib and quinacrine combination may have potential clinical implication in ATC management and should be further tested in clinical trials.
Acknowledgments:
The authors would like to thank Dr. Henry Crist, Trey Bruggeman and Dr. Nicole C. Williams in identifying suitable patient FFPE specimens for analysis and critiquing IHC staining of said specimens. This work was supported by grants to W.S.E-D; a grant from Thyroid Cancer Survivors Inc. (ThyCa) and the American Thyroid Association (ATA) to N.K.F. (Grant# 143317). W.S.E-D. is an American Cancer Society Research Professor.
Abbreviations:
- ATC
Anaplastic Thyroid Cancer
- ATCC
American Type Culture Collection
- CI
combination index
- FDA
United States Food and Drug Administration
- FFPE
Formalin Fixed Paraffin Embedded
- GI
gastrointestinal
- IC50
concentration that causes 50% cell growth inhibition
- ip
intraperitoneal
- GSEA
Gene set enrichment analysis
- Mcl-1
myeloid cell leukemia sequence 1
- NFκB
nuclear factor kappa B
- NIR
near-infrared
- NT
non-neoplastic thyroid
- OS
overall survival
- Q
quinacrine
- S
sorafenib
- S/Q
sorafenib/quinacrine
- TI
therapeutic index
- STAT
Signal Transducer and Activator of Transcription
- VEGF
vascular endothelial growth factor
Footnotes
Disclosure of Potential Conflicts of Interest: No potential conflicts of interest are disclosed by the authors.
References
- 1.Patel KN, Shaha AR. Poorly differentiated and anaplastic thyroid cancer. Cancer control : journal of the Moffitt Cancer Center. 2006;13:119–28. [DOI] [PubMed] [Google Scholar]
- 2.Woyach JA, Shah MH. New therapeutic advances in the management of progressive thyroid cancer. Endocrine-related cancer. 2009;16:715–31. [DOI] [PubMed] [Google Scholar]
- 3.Smallridge RC, Marlow LA, Copland JA. Anaplastic thyroid cancer: molecular pathogenesis and emerging therapies. Endocrine-related cancer. 2009;16:17–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu H, Sun Y, Ye H, Yang S, Lee SL, de Las Morenas A. Anaplastic Thyroid Cancer: Outcome and the Mutation/Expression Profiles of Potential Targets. Pathology oncology research : POR. 2015. [DOI] [PubMed]
- 5.Nagaiah G, Hossain A, Mooney CJ, Parmentier J, Remick SC. Anaplastic thyroid cancer: a review of epidemiology, pathogenesis, and treatment. Journal of oncology. 2011;2011:542358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. The New England journal of medicine. 2007;356:125–34. [DOI] [PubMed] [Google Scholar]
- 7.Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. The New England journal of medicine. 2008;359:378–90. [DOI] [PubMed] [Google Scholar]
- 8.Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014;384:319–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gupta-Abramson V, Troxel AB, Nellore A, Puttaswamy K, Redlinger M, Ransone K, et al. Phase II trial of sorafenib in advanced thyroid cancer. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2008;26:4714–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Savvides P, Nagaiah G, Lavertu P, Fu P, Wright JJ, Chapman R, et al. Phase II trial of sorafenib in patients with advanced anaplastic carcinoma of the thyroid. Thyroid : official journal of the American Thyroid Association. 2013;23:600–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thomas L, Lai SY, Dong W, Feng L, Dadu R, Regone RM, et al. Sorafenib in metastatic thyroid cancer: a systematic review. The oncologist. 2014;19:251–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pacifico F, Mauro C, Barone C, Crescenzi E, Mellone S, Monaco M, et al. Oncogenic and anti-apoptotic activity of NF-kappa B in human thyroid carcinomas. The Journal of biological chemistry. 2004;279:54610–9. [DOI] [PubMed] [Google Scholar]
- 13.Mitsiades CS, McMillin D, Kotoula V, Poulaki V, McMullan C, Negri J, et al. Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. The Journal of clinical endocrinology and metabolism. 2006;91:4013–21. [DOI] [PubMed] [Google Scholar]
- 14.Kim E, Matsuse M, Saenko V, Suzuki K, Ohtsuru A, Mitsutake N, et al. Imatinib enhances docetaxel-induced apoptosis through inhibition of nuclear factor-kappaB activation in anaplastic thyroid carcinoma cells. Thyroid : official journal of the American Thyroid Association. 2012;22:717–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meng Z, Mitsutake N, Nakashima M, Starenki D, Matsuse M, Takakura S, et al. Dehydroxymethylepoxyquinomicin, a novel nuclear Factor-kappaB inhibitor, enhances antitumor activity of taxanes in anaplastic thyroid cancer cells. Endocrinology. 2008;149:5357–65. [DOI] [PubMed] [Google Scholar]
- 16.Zhu W, Ou Y, Li Y, Xiao R, Shu M, Zhou Y, et al. A small-molecule triptolide suppresses angiogenesis and invasion of human anaplastic thyroid carcinoma cells via down-regulation of the nuclear factor-kappa B pathway. Molecular pharmacology. 2009;75:812–9. [DOI] [PubMed] [Google Scholar]
- 17.Gurova KV, Hill JE, Guo C, Prokvolit A, Burdelya LG, Samoylova E, et al. Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:17448–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ehsanian R, Van Waes C, Feller SM. Beyond DNA binding - a review of the potential mechanisms mediating quinacrine’s therapeutic activities in parasitic infections, inflammation, and cancers. Cell communication and signaling : CCS. 2011;9:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gallant JN, Allen JE, Smith CD, Dicker DT, Wang W, Dolloff NG, et al. Quinacrine synergizes with 5-fluorouracil and other therapies in colorectal cancer. Cancer biology & therapy. 2011;12:239–51. [DOI] [PubMed] [Google Scholar]
- 20.Wang W, Gallant JN, Katz SI, Dolloff NG, Smith CD, Abdulghani J, et al. Quinacrine sensitizes hepatocellular carcinoma cells to TRAIL and chemotherapeutic agents. Cancer biology & therapy. 2011;12:229–38. [DOI] [PubMed] [Google Scholar]
- 21.Abdulghani J, Allen JE, Dicker DT, Liu YY, Goldenberg D, Smith CD, et al. Sorafenib sensitizes solid tumors to Apo2L/TRAIL and Apo2L/TRAIL receptor agonist antibodies by the Jak2-Stat3-Mcl1 axis. PloS one. 2013;8:e75414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ricci MS, Kim SH, Ogi K, Plastaras JP, Ling J, Wang W, et al. Reduction of TRAIL-induced Mcl-1 and cIAP2 by c-Myc or sorafenib sensitizes resistant human cancer cells to TRAIL-induced death. Cancer cell. 2007;12:66–80. [DOI] [PubMed] [Google Scholar]
- 23.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim S, Park YW, Schiff BA, Doan DD, Yazici Y, Jasser SA, et al. An orthotopic model of anaplastic thyroid carcinoma in athymic nude mice. Clinical cancer research : an official journal of the American Association for Cancer Research. 2005;11:1713–21. [DOI] [PubMed] [Google Scholar]
- 25.Abdulghani J, Gu L, Dagvadorj A, Lutz J, Leiby B, Bonuccelli G, et al. Stat3 promotes metastatic progression of prostate cancer. The American journal of pathology. 2008;172:1717–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rayet B, Gelinas C. Aberrant rel/nfkb genes and activity in human cancer. Oncogene. 1999;18:6938–47. [DOI] [PubMed] [Google Scholar]
- 27.Schweppe RE, Klopper JP, Korch C, Pugazhenthi U, Benezra M, Knauf JA, et al. Deoxyribonucleic acid profiling analysis of 40 human thyroid cancer cell lines reveals cross-contamination resulting in cell line redundancy and misidentification. The Journal of clinical endocrinology and metabolism. 2008;93:4331–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ito T, Seyama T, Hayashi Y, Hayashi T, Dohi K, Mizuno T, et al. Establishment of 2 human thyroid-carcinoma cell-lines (8305c, 8505c) bearing p53 gene-mutations. International journal of oncology. 1994;4:583–6. [DOI] [PubMed] [Google Scholar]
- 29.Marlow LA, D’Innocenzi J, Zhang Y, Rohl SD, Cooper SJ, Sebo T, et al. Detailed molecular fingerprinting of four new anaplastic thyroid carcinoma cell lines and their use for verification of RhoB as a molecular therapeutic target. The Journal of clinical endocrinology and metabolism. 2010;95:5338–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Couto JP, Daly L, Almeida A, Knauf JA, Fagin JA, Sobrinho-Simoes M, et al. STAT3 negatively regulates thyroid tumorigenesis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E2361–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hebrant A, Dom G, Dewaele M, Andry G, Tresallet C, Leteurtre E, et al. mRNA expression in papillary and anaplastic thyroid carcinoma: molecular anatomy of a killing switch. PloS one. 2012;7:e37807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Salvatore G, Nappi TC, Salerno P, Jiang Y, Garbi C, Ugolini C, et al. A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer research. 2007;67:10148–58. [DOI] [PubMed] [Google Scholar]
- 33.Giordano TJ, Au AY, Kuick R, Thomas DG, Rhodes DR, Wilhelm KG Jr., et al. Delineation, functional validation, and bioinformatic evaluation of gene expression in thyroid follicular carcinomas with the PAX8-PPARG translocation. Clinical cancer research : an official journal of the American Association for Cancer Research. 2006;12:1983–93. [DOI] [PubMed] [Google Scholar]
- 34.Mojsa B, Lassot I, Desagher S. Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells. 2014;3:418–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chou TC. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacological reviews. 2006;58:621–81. [DOI] [PubMed] [Google Scholar]
- 36.Gule MK, Chen Y, Sano D, Frederick MJ, Zhou G, Zhao M, et al. Targeted therapy of VEGFR2 and EGFR significantly inhibits growth of anaplastic thyroid cancer in an orthotopic murine model. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011;17:2281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gomez-Rivera F, Santillan-Gomez AA, Younes MN, Kim S, Fooshee D, Zhao M, et al. The tyrosine kinase inhibitor, AZD2171, inhibits vascular endothelial growth factor receptor signaling and growth of anaplastic thyroid cancer in an orthotopic nude mouse model. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007;13:4519–27. [DOI] [PubMed] [Google Scholar]
- 38.Schoenfeld JD, Odejide OO, Wirth LJ, Chan AW. Survival of a patient with anaplastic thyroid cancer following intensity-modulated radiotherapy and sunitinib--a case report. Anticancer research. 2012;32:1743–6. [PubMed] [Google Scholar]
- 39.Smalley KS, Xiao M, Villanueva J, Nguyen TK, Flaherty KT, Letrero R, et al. CRAF inhibition induces apoptosis in melanoma cells with non-V600E BRAF mutations. Oncogene. 2009;28:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilhelm SM, Adnane L, Newell P, Villanueva A, Llovet JM, Lynch M. Preclinical overview of sorafenib, a multikinase inhibitor that targets both Raf and VEGF and PDGF receptor tyrosine kinase signaling. Molecular cancer therapeutics. 2008;7:3129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kurihara T, Ikeda S, Ishizaki Y, Fujimori M, Tokumoto N, Hirata Y, et al. Immunohistochemical and sequencing analyses of the Wnt signaling components in Japanese anaplastic thyroid cancers. Thyroid : official journal of the American Thyroid Association. 2004;14:1020–9. [DOI] [PubMed] [Google Scholar]
- 42.Liu J, Brown RE. Immunohistochemical detection of epithelialmesenchymal transition associated with stemness phenotype in anaplastic thyroid carcinoma. International journal of clinical and experimental pathology. 2010;3:755–62. [PMC free article] [PubMed] [Google Scholar]
- 43.Liu J, Brown RE. Morphoproteomic confirmation of an activated nuclear factor-κBp65 pathway in follicular thyroid carcinoma. International journal of clinical and experimental pathology. 2012;5:216–23. [PMC free article] [PubMed] [Google Scholar]
- 44.Volpe V, Raia Z, Sanguigno L, Somma D, Mastrovito P, Moscato F, et al. NGAL controls the metastatic potential of anaplastic thyroid carcinoma cells. The Journal of clinical endocrinology and metabolism. 2013;98:228–35. [DOI] [PubMed] [Google Scholar]
- 45.Lobo MR, Green SC, Schabel MC, Gillespie GY, Woltjer RL, Pike MM. Quinacrine synergistically enhances the antivascular and antitumor efficacy of cediranib in intracranial mouse glioma. Neuro-oncology. 2013;15:1673–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Awan FT, Kay NE, Davis ME, Wu W, Geyer SM, Leung N, et al. Mcl-1 expression predicts progression-free survival in chronic lymphocytic leukemia patients treated with pentostatin, cyclophosphamide, and rituximab. Blood. 2009;113:535–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu H, Yang J, Yuan Y, Xia Z, Chen M, Xie L, et al. Regulation of Mcl-1 by constitutive activation of NF-kappaB contributes to cell viability in human esophageal squamous cell carcinoma cells. BMC cancer. 2014;14:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pannone G, Santoro A, Pasquali D, Zamparese R, Mattoni M, Russo G, et al. The role of survivin in thyroid tumors: differences of expression in well-differentiated, non-well-differentiated, and anaplastic thyroid cancers. Thyroid : official journal of the American Thyroid Association. 2014;24:511–9. [DOI] [PubMed] [Google Scholar]
- 49.Saltman B, Singh B, Hedvat CV, Wreesmann VB, Ghossein R. Patterns of expression of cell cycle/apoptosis genes along the spectrum of thyroid carcinoma progression. Surgery. 2006;140:899–905; discussion −6. [DOI] [PubMed] [Google Scholar]
- 50.Krajewski S, Bodrug S, Krajewska M, Shabaik A, Gascoyne R, Berean K, et al. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. The American journal of pathology. 1995;146:1309–19. [PMC free article] [PubMed] [Google Scholar]
- 51.Branet F, Brousset P, Krajewski S, Schlaifer D, Selves J, Reed JC, et al. Expression of the cell death-inducing gene bax in carcinomas developed from the follicular cells of the thyroid gland. The Journal of clinical endocrinology and metabolism. 1996;81:2726–30. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.