

Abstract
Intervertebral disc degeneration is the most significant, and least understood, cause of chronic back pain, affecting almost one in seven individuals at some point of time. Each intervertebral disc has three components; central nucleus pulposus (NP), concentric layers of annulus fibrosus (AF), and a pair of end plate (EP) that connects the disc to the vertebral bodies. Understanding the molecular and cellular basis of intervertebral disc growth, health, and aging will generate significant information for developing therapeutic approaches. Rapid and efficient preparations of homogeneous and pure cells are crucial for meaningful and rigorous downstream analysis at the cellular, molecular, and biochemical level. Cross‐sample contamination may influence the interpretation of the results. In addition to altering gene expression, slow or delayed isolation procedures will also cause the degradation of cells and biomolecules that create a bias in the outcomes of the study. The mouse model system is extensively used to understand the intervertebral disc biology. Here we describe two protocols: (a) for efficient isolation of pure NP, AF, and EP cells from mouse lumbar intervertebral disc. We validated the purity of the NP and AF cells using Shh Cre/+; R26 mT/mG/+ dual‐fluorescent reporter mice where all NP cells are GPF+ve, and by the sensitive approach of qPCR analysis using TaqMan probes for Shh, and Brachyury as NP‐specific markers, Tenomodulin as AF‐specific marker, and Osteocalcin as bone‐specific marker. (b) For isolation of high‐quality intact RNA with RIN of 9.3 to 10 from disc cells. These methods will be useful for the rigorous analysis of NP and AF cells, and improve our understanding of intervertebral disc biology.
Keywords: development, genetics, Preclinical models, regenerative medicine
This methods article describes the optimized and step‐by‐step protocols for rapid and efficient isolation of all the three components of the mouse intervertebral disc: NP, AF, and EP cells, and isolation of high‐quality, intact RNA. Quick and efficient pure sample preparation may be useful for molecular and biochemical analysis of each component of the disc during period of healthy growth, differentiation, and aging, and also under different experimental conditions.

1. INTRODUCTION
High throughput molecular and biochemical analysis has rapidly evolved, providing sophisticated approaches of ‐omics to determine the molecular and biochemical signature of specific cells in a spatial and temporal pattern and under different stages of development, health, and disease (Figure 1). However, the purity of samples concerning specific cell type of interest is an onging challenge faced in several areas of biomedical research. A homogenous cell population is a prerequisite, and crucial for precise molecular and biochemical analysis of specific cell types, which aims to understand its molecular signature during development and disease. Moreover, pure cells are also necessary for molecular and biochemical analysis using approaches like NanoString, 1 RT‐qPCR, 2 Western blot, 3 metabolomic profiling, 4 , 5 , 6 immunoprecipitation, 7 chromatin immunoprecipitation, 8 RNP immunoprecipitation, 9 and electrophoretic mobility shift assays. 10 , 11
FIGURE 1.
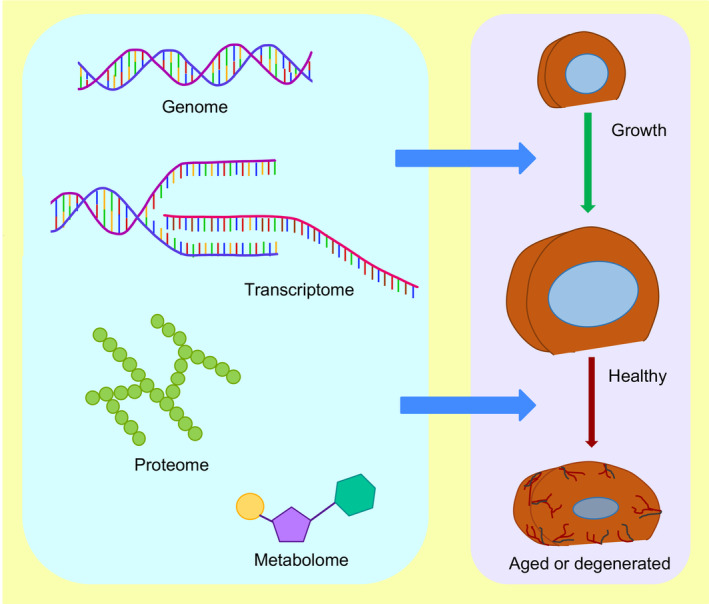
Profiling the genome, transcriptome, proteome, and metabolome of each cell type of the intervertebral disc will provide crucial information about the molecular and biochemical basis of the intervertebral disc growth, maturation, aging, and degeneration, and identify potential therapeutic targets
The application of high throughput molecular and biochemical analysis to understand the changes in the intervertebral disc at genomic, transcriptomic, proteomic, or metabolomic levels will significantly expand our understanding of its growth and progression toward aging and degeneration (Figure 1). Intervertebral discs are the largest avascular structure in the body, forming a joint between two adjacent vertebral bodies. Degeneration of the intervertebral disc, with aging as a major risk factor, is a significant cause of chronic neck and back pain. 12 Due to the extensive use of the mouse model system to understand the cellular, molecular, and biochemical basis of the intervertebral disc, this method paper will focus on the murine model system. The mouse vertebral column consists of 59 to 61 vertebrae 13 distributed into five anatomical regions shown by the radiographs captured in the sagittal and dorsal plane of an adult mouse (Figure 2A,B). Each mouse has seven cervical vertebrae (C1‐C7), 13 thoracic vertebrae (T1‐T13), five to six lumbar vertebrae (L1‐L5/6, depending on genetic background, 14 ), four sacral vertebrae (S1‐S4) and about 29 to 31 coccygeal vertebrae (Co1‐Co29/31, 13 ). The intervertebral disc is a heterogeneous tissue with three different cellular compartments that have distinct developmental origins (reviewed by Reference 15). The core of the disc is formed by nucleus pulposus (NP; Figure 2C,D) cells that are derived from the embryonic notochord. 16 , 17 , 18 , 19 , 20 The proteoglycan‐rich NP cells are surrounded by orthogonal layers of collagen‐rich annulus fibrosus (AF, Figure 2C,D) which are derived from the syndetome compartment 21 of the sclerotome. Together NP and AF are sandwiched between cartilaginous end plates (EPs), derived from sclerotome, connecting the disc to the growth plate of the vertebral bodies (Figure 2C,D). While the intervertebral disc itself is heterogeneous, its three cell types have a distinct spatial location within each disc and throughout the mouse spine.
FIGURE 2.
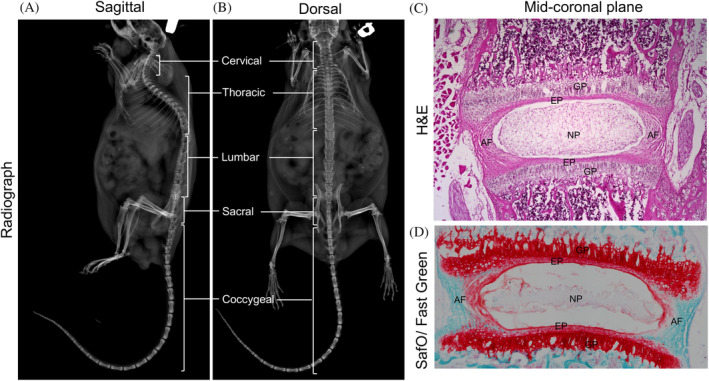
Representative radiograph images in the sagittal, A and dorsal, B plane showing the cervical, thoracic, lumbar, sacral, and coccygeal vertebrae in the spine of a 15‐month‐old male mouse. Representative mid‐coronal section of the lumbar intervertebral disc from a three‐week‐old mouse stained with H&E, C, shows the major components of the disc; SafO/Fast Green, D, staining shows the relative distribution of proteoglycan (stained orange‐red) and collagen (stained fast green). AF, annulus fibrosus; EP, endplate; GP, growth plate; NP, nucleus pulposus
Several research groups studying disc biology use the mouse model system to elucidate changes in gene expression of NP cells. 22 , 23 , 24 , 25 , 26 , 27 , 28 , 29 , 30 , 31 A few recent studies have also analyzed the gene expression of the mouse AF cells. 24 , 28 However, the details on method for isolation of the purified NP or AF cells, as well as validation of their purity, are not elaborated. Moreover, the method for isolation of EP cells is not described in the current literature. We have developed a protocol to isolate the NP, AF, and EP cells by microdissection of the mouse intervertebral disc which can be quickly adapted by researchers studying the molecular and cellular processes in these cells. The following protocol may be used to microdissect and isolate each component of the disc from E16.5 to all ages during the postnatal stage in mice. The purity of the NP and AF samples was validated by analyzing the expression of specific molecular markers using Brachyury (Bra, Tbxt, or T, 32 ) and sonic hedgehog (Shh, 33 ) for NP cells, and tenomodulin (Tnmd, 34 ) for AF cells. While this methods paper describes cell isolation from the lumbar disc, as degeneration and chronic low back pain is highly prevalent at this level of spine in mice, 26 , 35 we currently use the same methodology for microdissection and isolation of equally pure populations of NP and AF cells from the cervical, thoracic, and caudal discs of postnatal mice of all ages.
2. MATERIALS
2.1. Animals
Mice of both sexes, postnatal day four (P4) to 12 months of age, and in FVB and C57BL/6 background were used for the isolation of NP, AF, and EP cells from the intervertebral discs and isolation of high‐quality, intact total RNA from NP, AF and entire disc. Shh Cre/+ 36 mice were crossed with R26 mT/mG/+ dual‐fluorescent reporter 37 to generate Shh Cre/+; R26 mT/mG/+ line to validate the NP, AF, and EP cell isolation method. Mice were maintained following the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and all experiments were carried out per institutional guidelines and approval by Institutional Animal Care and Use Committee (IACUC). Mice were housed under 12 hours of light/ dark cycle in the WCMC animal facility with food and water ad libitum.
2.2. Tools and reagents required for microdissection and isolation of NP, AF, and EP cells
70% ethanol in spray bottle
Glass bead sterilizer (31178, Simon Keller, Switzerland)
Serrated standard pattern forceps (11002‐13 or 91100‐12, F.S.T., USA)
Sharp end tweezers (11251‐30 or 11200‐33, Dumoxel Biology, FST by Dumont, Switzerland)
Curved fine scissors (14061‐10, F.S.T., USA)
Blunt straight scissors (14074‐11, F.S.T., USA)
Fine straight scissors (14060‐10, F.S.T., USA)
Extra Fine Bonn scissors (14084‐08, F.S.T., USA)
Scalpel handle #3 (10003‐12, F.S.T., USA)
Feather surgical blade no.11 (2976#11, Feather Sterile Surgical blade, Graham‐Field INC.)
Falcon 60 × 15 mm style sterile TC‐treated Petri Dish (353002, Corning, USA)
Falcon 100 × 15 mm style sterile Not TC‐treated Petri Dish (351 029, Corning, USA)
Falcon 50 mL sterile conical centrifuge tube (352098, Corning, USA)
Stereomicroscope (SMZ800, Nikon, Japan; Wild Mz8, Leica, USA)
PIPETMAN Classic 4‐Pipette Kit (F167380, F81024, Gilson, USA)
RNase, DNase, and pyrogen free SHARP Precision Barrier Tips (1158U34, 1159M43, 1159M40, 1159M42, Thomas Scientific, USA)
Ice bucket filled with ice
-
Phosphate‐Buffered Saline (PBS), pH 7.4 ± 0.1.
Prepare 1× PBS by adding 100 mL of 10× PBS (Corning 10X PBS, pH 7.4 ± 0.1, Liquid without calcium and magnesium, RNase−/ DNase‐ and protease‐free, 46‐013‐CM, Corning, USA) to 900 mL of nanopure water. Mix well, adjust pH to ~7.4 and prechill at 4°C. Keep PBS in ice bucket and handy during dissections.
RNAlater Stabilization Solution (AM7021, ThermoFisher Scientific, USA)
2.3. Tools and reagents required for RNA isolation
Alcohol, Ethyl for DNA and RNA (AB00515‐00500, americaBIO, USA)
UltraPure DNase/ RNase‐Free Distilled Water (10977015, Invitrogen, ThermoFisher Scientific, USA)
RNaseZap RNase Decontamination Wipes (AM9786, Invitrogen, USA)
Molecular biology grade chloroform (C2432, Sigma‐Aldrich, USA)
TRI Reagent (T9424, Sigma‐Aldrich, USA)
Polytron Omni Tissue Homogenizer (LR60902, Omni International, USA)
Isotemp heating block (11‐718, Fisher Scientific, USA)
RNase, DNase and pyrogen free Posi‐Click 1.7 mL microcentrifuge tube (1158R19, Thomas Scientific, USA)
RNase, DNase and pyrogen free SHARP Precision Barrier Tips (1158U34, 1159M43, 1159M40, 1159M42, Thomas Scientific, USA)
Bench top Refrigerated Centrifuge (5430R, Eppendorf, USA)
Bench top Room Temperature Centrifuge (5430, Eppendorf, USA)
RNeasy Micro kit (74 004, Qiagen, Germany)
RNeasy Fibrous Tissue Mini Kit (74704, Qiagen, Germany)
NanoDrop One Microvolume UV‐Vis Spectrophotometer (ThermoFisher Scientific, USA)
Kim Wipes (S‐8115, Kimtech Science Brand, USA)
3. METHODS
3.1. A step‐by‐step protocol for isolation of NP, AF, and EP cells from the mouse intervertebral disc
3.1.1. Dissection of the mouse spine
Sterilize all dissecting tools using 70% ethanol, followed by heating for 30 seconds in a glass bead sterilizer set at ≥200°C. Allow the tools to cool before use.
Following euthanasia (per approved IACUC protocol), place the mouse prone with its ventral side on the paper towels covered dissecting table (Figure 3A,B′).
Spray 70% ethanol to avoid fur contamination of the samples.
Holding the skin right above the sacral region, use the serrated standard pattern forceps to make a small lateral incision using the surgical scissors (Figure 3B).
Next, while holding the skin with serrated forceps at the point of incision, use surgical scissors to make a straight vertical incision from caudal (posterior) end along the midline over the spine toward the rostral (anterior) end to expose the dorsal side of the mouse spine (Figure 3C,C″).
To dissect the spine, make a deep incision on the right side of the sacral spine through the pelvic bone. Dissect in a straight line from caudal end cutting through the ribs toward the rostral end. Similarly, start with dissecting through the muscles and pelvic bone on the left side and cut through the ribs toward the rostral end (Figure 3D,D′).
Hold the caudal end of the spine and gently lift and detach the abdominal tissues from caudal to rostral end using surgical scissors (Figure 3E,E′).
Following dissection, immediately immerse the spine in ice‐cold PBS filled 50 mL Falcon tube and place it in an ice bucket.
FIGURE 3.
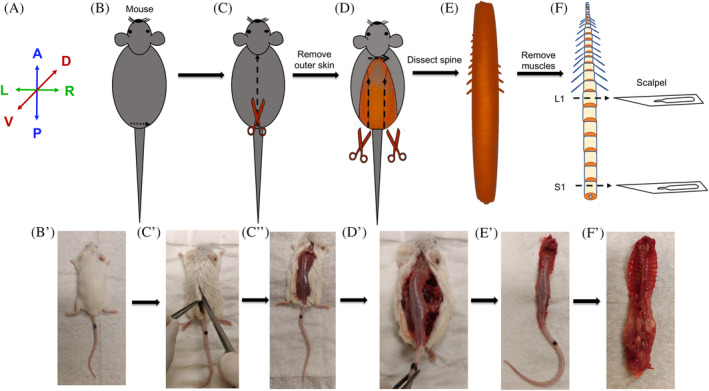
Method for dissection and collection of the mouse spine. A, Anatomical positions for the images presented in B‐F. B and B′, Mouse is placed prone with the ventral side on the dissection table and its back or dorsal side facing the experimenter and doused with 70% ethanol to avoid fur contamination. C, C″, Lift the fur right above the pelvic girdle and create a transverse incision and dissect through the fur from caudal to rostral direction. D and D′, Using a pair of scissors cut from hip joint moving from caudal to rostral end along the lateral side of the spine to dissect the spine. E and E′, Detach and excise the spine from the body. F, Clean the spine by dissecting out the attached muscles. Separate the caudal, lumbar, and thoracic regions of the spine using a scalpel blade or using a pair of fine‐tip and sharp scissors. F′, The ventral view of an excised spine. Images of the mouse were photographed using a cell‐phone camera and are not to scale. A, anterior. P, posterior. R, right. L, left. V, ventral. D, dorsal. L1, first lumbar vertebra. S1, first sacral vertebra
3.1.2. Cleaning of the spine to expose the intervertebral discs
Transfer the spine from the Falcon tube into a 100 mm Petri Dish containing ice‐cold PBS sufficient to immerse the spine completely. Position the spine with the ventral side facing toward the experimenter (Figure 3F′). (Note: Make sure the spine is always submerged in ice‐cold PBS during dissections).
Start dissecting at 63 to 100× magnification using a bright field stereomicroscope. Carefully dissect the soft tissue over the ventral side of the spine using curved scissors with the convex side of the scissors toward the spine to expose the intervertebral discs and vertebral bodies. Replace the buffer with fresh, ice‐cold PBS.
Next, use curved scissors with the convex side of the scissors toward the spine and the pointed tips away from the spine to dissect the muscles on the lateral sides of the spine. Replace the buffer with fresh, ice‐cold PBS.
Identify the first lumbar vertebra (L1) by its position next to the last thoracic vertebra, which in turn, is determined by the presence of the last pair of ribs at its proximal end (Figure 3F). To dissect the lumbar region of the spine, first, dissect at the center of L1 and next, dissect at the center of the first sacral vertebra (S1), either using straight scissors or using a scalpel blade for younger mice less than 2 weeks of age. Isolating the lumbar spine makes it easy to handle only the lumbar discs during microdissection.
Transfer the lumbar spine to a 60 mm Petri Dish filled with fresh, ice‐cold PBS.
Use Fine Bonn Scissors to laterally cut through the pedicles from L1 toward S1 levels of the spine. Repeat this step on the other lateral side. Separate the posterior element from the anterior body of the spine and to remove the spinal cord. (Note: The spinal cord is removed by gently pulling it with forceps). Replace the buffer with fresh, ice‐cold PBS.
Increase the magnification of the bright field stereomicroscope to about 300 or 350×. Next, clean the spine further by dissecting the surrounding soft tissue, especially around the intervertebral disc, using curved scissors or fine forceps. Gently scrape with a scalpel blade to remove any remaining soft tissue.
3.1.3. Isolation of NP, AF, and EP cells
Move the clean spine to a new 60 mm Petri Dish filled with fresh, ice‐cold PBS and continue dissecting using a bright field stereomicroscope. The ventral side of the spine is curved while the dorsal side is flat. Place the spine with the dorsal side on the surface of Petri Dish and the ventral side facing the experimenter (Figure 4B,B′).
Gently hold L2 vertebra using serrated forceps and make a sharp cut between the AF and EP of the L1 vertebra utilizing a scalpel and Feather surgical blade no.11 (Figure 4C,C′). (Note: Do not use pointed forceps to hold the spine as it may rupture the vertebra and contaminate the buffer with hematopoietic cells).
Now the NP will be exposed from the open end of the disc (Figure 4D). Use the blunt side of the scalpel blade to scoop out the entire NP into the PBS in the Petri Dish (Figure 4E,E′). Else, if interested in collecting entire disc, dissect along with the EP on both ends (Figure 4D′).
Adjust a P200 micropipette to 40 μL volume. Widen the precision tip using a sterile pair of scissors. Gently aspirate the NP cells and use the micropipette to add them in a 1.7 mL Eppendorf tube containing RNAlater (about 200 μL/ tube), or PBS depending upon the downstream application.
Next, use the scalpel blade to cut at the junction of the EP and AF of the L2 vertebra. This step isolates the intact ring of AF that is free of NP and EP cells (Figure 4F,F′). Next, use the scalpel to microdissect EP by cutting adjacent to the soft growth plate cartilage (Figure 4GG′).
Separately collect the AF and EP using fine forceps into Eppendorf tubes containing RNAlater or PBS based on the downstream application.
Repeat steps 17‐21 to collect NP, AF, and EP cells from all the lumbar discs.
FIGURE 4.
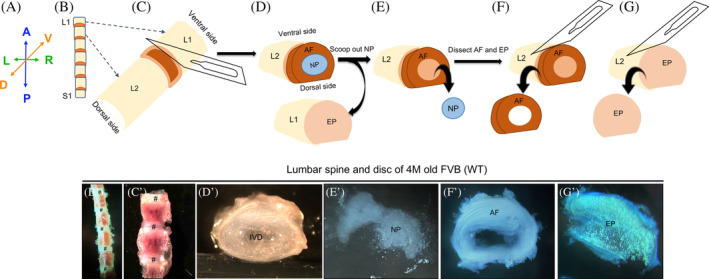
Microdissection procedure for the isolation of the NP, AF, and EP from the intervertebral disc of mouse spine performed using a bright field stereomicroscope. Representative images of the dissected lumbar spine in ventral view, B‐C′, and intervertebral disc in transverse view demonstrating the microdissection procedure, D′,D″, E′, F′ G′, and G″) from a four‐month‐old (4 M) WT FVB mouse captured using an iPhone camera and bright field stereomicroscope. A, Anatomical positions for the images shown in B‐G′. Place the dissected lumbar spine with the dorsal side on the Petri Dish and ventral side facing the experimenter, B and B′. Dissect at the AF and EP junction, C‐C″ to expose gelatinous NP surrounded by AF, D. Transverse view of an intact IVD (D′). Scoop out NP from the AF (E and E′) and collect. Microdissect the AF, F, and F′, and collect. Microdissect the EP, G and G′ and collect. A, anterior. P, posterior. R, right. L, left. V, ventral. D, dorsal. IVD, intervertebral disc. NP, nucleus pulposus. AF, annulus fibrosus. EP, end plate. # represents the disc in B′ and C′
3.1.4. Notes for microdissection procedure
All steps must be performed using ice‐cold PBS. It is essential to replace buffer with fresh and cold PBS every 5 minutes (min) to minimize degradation of the biomolecules.
We have tried several scalpel blades and identified the Feather surgical blade no.11 as the most suitable for precise isolation of all three components of the mouse intervertebral disc. The sharp‐angled tip of the Feather surgical blade no.11 provides a clear view of the cutting area while facilitating precise microdissection.
Do not leave the tissue at room temperature at any time during the procedure. Process and store cells immediately following isolation and collection.
As the EP is also isolated using this method, if required, it may be collected for downstream analysis.
Replace the scalpel blade often, otherwise fine microdissection may not be achieved, which results in the mixing of cell types.
3.2. A step‐by‐step protocol for isolation of high‐quality, intact total RNA from mouse NP, AF, and the whole intervertebral disc
Sterilize the Polytron Omni Tissue Homogenizer with 70% ethanol, followed by incubating in bead sterilizer for 20 minutes. Allow the homogenizer probe to cool before use.
Mount the homogenizer probe on the motor and rinse the probe using three separate aliquots of RNase free water. Then, decontaminate three times with 100% RNase‐free ethanol before one final rinse with chloroform. Allow each rinse cycle to run for about 5 seconds. Following chloroform rinse, dry run the probe for a sec or two to remove any residual chloroform.
-
Add 1 mL of TRI Reagent to the samples (NP, AF, or discs) and vortex to dislodge the tissue from the base of the Eppendorf tube and into the TRI Reagent. Incubate the sample on ice for 5 minutes. Next, disrupt the cells by homogenization using one of the following options:
Immerse the probe into the Eppendorf tube and homogenize the NP cells twice for no more than 15 seconds each. Chill the tubes in ice for a min between each homogenization step.
Homogenize AF (or the whole disc) 3 to 4 times for 20 seconds each by immersing the probe into the Eppendorf tube. Allow the tubes to cool by placing them in ice for a minute between each homogenization step.
Note: Do not homogenize beyond 20 seconds in a given cycle, as this will heat the sample, causing RNA degradation.
Clean the probe between each sample to avoid cross‐sample contamination by rinsing for 5 seconds each, first in RNase‐free water, followed by 100% RNase‐free ethanol and finally in chloroform. Allow the probe to dry before homogenizing the next sample.
After homogenization, centrifuge the samples in a refrigerated centrifuge at 4°C for 12 minutes at 12 000g. Meanwhile, label fresh Eppendorf tubes for the following steps.
-
Following centrifugation, immediately collect the supernatant into a prelabeled new Eppendorf tube and incubate at room temperature (RT) for 5 minutes to facilitate dissociation of the nucleoprotein complexes.
Note: It is important to gently remove the supernatant with the pipette tip without dislodging the pellet. Please make a note of the volume of supernatant collected, as it is required for the next step.
-
Add 1/5th volume of chloroform to the supernatant (eg, add 200 μL of chloroform for 1 mL of supernatant collected, add 150 μL of chloroform if 750 μL of supernatant was retrieved). Mix well by shaking vigorously for 15 seconds by hand until the solution turns homogeneous and bright pink
Note: Do not vortex as it may fragment the RNA.
Incubate the samples for 10 minutes at RT.
Centrifuge the sample for 15 minutes at 12000g and 4°C. Label fresh Eppendorf tubes for the next step.
Immediately after centrifugation, gently aspirate the upper clear aqueous phase into a fresh prelabeled Eppendorf tube using a 200 μL pipette followed by 20 μL pipette to avoid contamination with the interphase.
Add one volume of 70% ethanol (prepared using RNase, DNase‐free water, and ethanol) to the aqueous phase for precipitation of RNA. (eg, add 450 μL of 70% RNase‐free ethanol if 450 μL of the aqueous phase is collected).
Mix well using a 1 mL pipette and transfer 500 μL of the mix to an RNeasy MiniElute spin column inserted in a 2 mL collection tube, both provided with the Qiagen kit.
Gently close the lid and centrifuge the spin column for 15 seconds at ≥8000g using a benchtop centrifuge maintained at RT.
Note: For isolation of RNA from NP cells, a RNeasy Micro (74004, Qiagen) or RNeasy Mini (74104) kit was used. For isolation of RNA from AF cells and the whole disc, the RNeasy Fibrous Tissue Mini Kit (74704, Qiagen) was used.
-
d.
After centrifugation, discard the flow‐through and add the remaining sample mix (RNA + 70% ethanol mix from step 12) into the spin column. Centrifuge the spin column again for 15 seconds at ≥8000g at RT. Discard the flow‐through.
-
e.
Wash the spin column by adding 350 μL of RW1 buffer (provided with the kit) and centrifuge for 15 seconds at ≥8000g at RT. Discard the flow‐through and collection tube.
-
f.
Place the spin column in a new 2 mL collection tube for on‐column DNase digestion using 80 μL of DNase I incubation mix prepared using 10 μL of DNase I and 70 μL of RDD buffer per column, both provided with the kit. Load the DNase I incubation mix directly on the membrane of each spin column and incubate at RT for 10 minutes.
Note: Prepare a tube with RNase‐free water and incubate in a 75°C dry bath until use. The warm RNase‐free water will be used for efficient elution of RNA in the last step.
-
g.
Next, wash the spin column by adding 350 μL of RW1 buffer and centrifuge for 15 seconds at ≥8000g at RT. Discard the flow‐through.
-
h.
Rewash the spin column with 350 μL of RW1 buffer and centrifuge for 15 seconds at ≥8000g at RT. Discard the flow‐through and collection tube.
-
i.
Place the spin column in a new 2 mL collection tube, add 500 μL buffer RPE. Centrifuge for 15 seconds at ≥8000g at RT. Discard the flow‐through.
-
j.
Add another 500 μL of RPE buffer to the spin column and centrifuge for 30 seconds at ≥8000g at RT. Discard the flow‐through.
-
k.
Add 500 μL of 80% ethanol (prepared in RNase/DNase‐free water and ethanol) to the spin column and centrifuge the spin column for 2 minutes at ≥8000g at RT. Discard the flow‐through and collection tube.
-
l.
Place the spin column in a new 2 mL collection tube and centrifuge the spin column with its cap open for 1 minute at ≥8000g at RT (dry spin). Discard the flow‐through and collection tube.
-
m.
Place the column in a new and labeled 1.7 mL Eppendorf tube. Add 15 μL of the prewarmed (75°C) RNase‐free water directly on the membrane and to the center of the column and leave at RT for 5 minutes.
Note: The kit recommends incubating the column for 1 minute before elution. However, we had significant improvement in the RNA yield, without compromising the quality of RNA (Figure 5), by incubating for 5 minutes.
FIGURE 5.
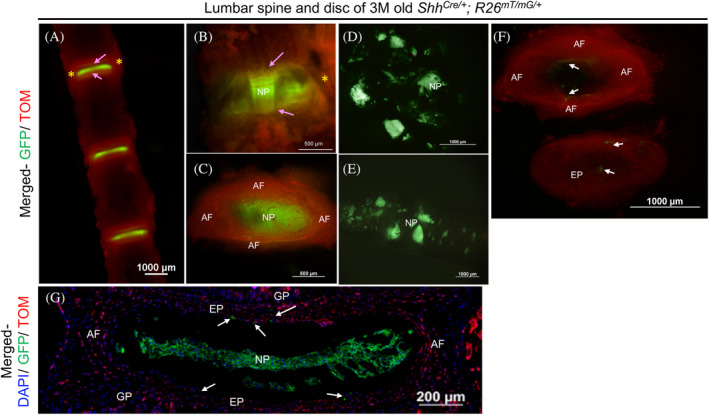
Validation of microdissection procedure using ShhCre/+; R26mT/mG/+ fluorescence reporter line. Representative images of the lumbar spine and intervertebral disc dissected from a three‐month‐old (3 M) ShhCre/+; R26mT/mG/+ mouse where all NP cells appear GFP+ve under an epi‐fluorescence stereomicroscope (A‐G). L1‐L4 spine in ventral view (A), an individual disc at higher magnification in ventral view (B), and a dissected disc in transverse view (C) showing GFP+ve NP cells, TOM+ve AF (yellow asterisk), EP (indicated by pink arrows), and vertebrae from 3 M old ShhCre/+; R26mT/mG/+. NP cells scooped out of the AF (D), collected with a pipette tip (E) are free of any TOM + ve cells. AF and EP are dissected using a fresh scalpel blade (F). C and F are images of the same disc captured in transverse plane. While C shows the intact disc with NP in the center viewed under the thin layer of EP, together surrounded by lamella of AF, F shows the isolated AF and EP following removal of the NP cells. Representative fluorescence image of a mid‐coronal section of a 3 M old ShhCre/+; R26mT/mG/+ with DAPI counterstained nuclei (blue) shows that all notochord‐descendant NP cells are GFP+ve (G). A few notochord‐descendant GFP+ve cells are observed in the AF and EP (white arrows in F and G), which may occur during development as previously reported. NP, nucleus pulposus. AF, annulus fibrosus. EP, end plate. * indicates AF on the lateral side of GFP+ve NP cells in A and B. The pink arrow identifies the EP in A and B
4. TO ELUTE THE RNA, CENTRIFUGE THE SPIN COLUMN FOR 1 MINUTE AT FULL SPEED AT RT
Note: The yield of RNA may further be improved by repeat elution by loading the elute obtained in step 24.
4.1. Multiplex and quantitative real‐time PCR (qPCR) using TaqMan probes
To assess the purity of NP and AF cell populations, we evaluated the expression of molecular markers specific to NP or AF cells. The molecular markers used in the current study, along with details on the respective gene specific Taqman Probes used are listed in Table 1. Soon after elution, the RNA concentration was quantified in duplicates using NanoDrop One Microvolume UV‐Vis Spectrophotometer (ThermoFisher Scientific, USA) and 1.5 μL of RNA was used for sample quality control (QC) using Agilent Bioanalyzer. The remaining RNA was immediately converted into cDNA using SuperScript IV First‐Strand Synthesis System (18 091 050, Invitrogen, ThermoFisher Scientific, USA) and following manufacturer's protocol on Thermal Cycler (C1000 Touch Thermal Cycler, Bio‐Rad Laboratories, USA). Multiplex real‐time PCR (qPCR) was performed using 6 ng of cDNA, iQ Multiplex Powermix (1725849, Bio‐Rad Laboratories, USA), gene‐specific TaqMan probes conjugated to FAM‐MGB, and with Beta‐2‐Microglobulin (B2m) conjugated to VIC‐MGB as an internal control in every PCR reaction (Table 1). No template control (NTC) was included for each experiment to determine nonspecific amplification, which was negative for all TaqMan probes. PCR reactions were run for 40 cycles per manufacturer's recommend program. The qPCR reactions were carried out using CFX96 Touch Real‐Time PCR Detection System (1855195, Bio‐Rad Laboratories, USA). The quantification cycle (Cq) value obtained for each sample was used to calculate dCq by subtracting the Cq of reference gene (B2m) from the Cq of target gene [dCq = Cq (target) − Cq(B2m)]. The relative expression of a particular gene in each sample was expressed as 2^−dCq (Table 2). Data were analyzed using the GraphPad Prism vs 8.3.
TABLE 1.
TaqMan Probe used for multiplex qPCR analysis
| Gene name | TaqMan probe | Fluorophore |
|---|---|---|
| Beta‐2‐microglobulin (B2m) | Mm00437762_m1 | VIC‐MGB |
| Brachyury (T, Tbxt, or Bra) | Mm013182496_m1 | FAM‐MGB |
| Sonic hedgehog (Shh) | Mm00436528_m1 | FAM‐MGB |
| Tenomodulin (Tnmd) | Mm00491594_m1 | FAM‐MGB |
| Osteocalcin (Bglap) | Mm03413826_mH | FAM‐MGB |
TABLE 2.
Results of qPCR analysis to validate the purity of isolated NP and AF cells microdissected from 6‐month‐old WT FVB mice using bright field stereomicroscope
| Gene ID | Shh | T (Bra) | Tnmd | Bglap | ||||
|---|---|---|---|---|---|---|---|---|
| Cell type | NP | AF | NP | AF | NP | AF | NP | AF |
| Replicates (n) | 6 | 6 | 3 | 5 | 3 | 3 | 3 | 3 |
| Mean (relative to B2m) | 0.0717 | 0 | 17.18 | 2.00E – 05 | 0 | 1.163 | 0 | 5.00E – 05 |
| Std. deviation | 0.038 | 0 | 0.659 | 5.00E – 05 | 0 | 0.029 | 0 | 9.00E – 05 |
| Std. error of mean | 0.0155 | 0 | 0.381 | 2.00E – 05 | 0 | 0.017 | 0 | 5.00E – 05 |
4.2. Radiography
High‐resolution digital X‐ray Faxitron images of the anesthetized mouse were captured using the UltraFocus high‐resolution digital X‐ray cabinet by Faxitron Bioptics, LLC (Arizona, USA). Before imaging, the imager was calibrated using automatic calibration and automatic exposure for appropriate exposure time and kV settings suitable for imaging the mouse. Mice were anesthetized with 2% isoflurane, placed in the X‐ray chamber and imaged in the dorsal and sagittal plane.
4.3. Microscopy
The lumbar spines were dissected from 4‐month‐old (4 M) wild‐type (WT) FVB mice, collected in ice‐cold PBS, and imaged using bright field stereomicroscope. The lumbar spines of three‐month‐old (3 M) Shh Cre; R26 mTmG mice were used to validate the NP, AF, and EP cell isolation protocol following microdissection. Lumbar spine and intervertebral discs from Shh Cre; R26 mTmG mice were imaged to visualize the endogenous GFP and TOM fluorescence using a Nikon DS‐Qi2 monochromatic digital camera (Nikon, Japan) attached to a Nikon SMZ‐25 Stereo Fluorescence Microscope (Nikon, Japan) and accompanying NIS‐Elements Advanced Research (AR) software (Nikon, Japan). All images captured using GFP and TOM epifluorescence were merged to create a composite image (Figure 5).
For histological preparations, the spine was fixed in freshly prepared 4% buffered paraformaldehyde (PFA) for 6 hours, and decalcified in 0.5 M ethylenediaminetetraacetic acid (EDTA, E9884, Sigma‐Aldrich, USA), pH 7.6 at 8°C on a rocker platform for five days. Following decalcification, the spine was washed three times in cold PBS for 30 minutes each, and cryomolds were prepared in Tissue‐Tek optimum cutting temperature (O.C.T, 102094‐106, VWR, USA) that was immediately snap‐frozen and stored at −80°C until further use. Cryosections in the coronal plane were prepared at 8 μm thickness using a Leica cryostat (CM3050 S, Leica, USA). Slides were stored at −80°C. Midcoronal sections were processed for histological evaluation by hematoxylin and eosin (H&E) staining using a standard protocol, dehydrated, cleared, and mounted in PROTOCOL mounting medium (C.A.S. 23‐245‐691, Fisher Healthcare, USA). For Safranin‐O and Fast Green (SafO/Fast Green) staining, frozen sections were hydrated in distilled water for 5 minutes and then incubated for 12 minutes in aqueous 0.001% Fast Green Solution (C.I. 42053, Sigma‐Aldrich, USA). Fast green stained sections were de‐stained by rinsing with a 1% acetic acid solution and subsequently stained with 0.1% Safranin‐O solution (C.I. 50240, Sigma‐Aldrich, USA) for 20 minutes. The sections were then dehydrated, cleared, and mounted in PROTOCOL mounting medium using a standard protocol. H&E and SafO/Fast Green stained sections were imaged using a DS‐Fi2 digital camera (colored, Nikon, Japan) attached to a Nikon Ni‐E Eclipse Microscope, and accompanying NIS‐Elements Advanced Research (AR) software (Nikon, Japan).
Lumbar spines from Shh Cre/+; R26 mTmG/+ mice were fixed, decalcified, and cryosectioned in the coronal plane. The slides were washed three times in PBS, counterstained in DAPI (D1306, 1:5000, Invitrogen, ThermoFisher Scientific, USA), and mounted in Prolong Diamond (P36962, Invitrogen, ThermoFisher Scientific, USA). Sections were imaged using DAPI, GFP, and TxRd filter cubes, ANDOR Zyla sCMOS monochromatic digital camera (Oxford Instruments, UK) attached to a Nikon Ni‐E Eclipse Fluorescent Microscope, and accompanying NIS Elements AR software (Nikon, Japan).
5. VALIDATION OF THE METHODS
5.1. Purity of isolated NP, AF, and EP cells
Isolation of NP, AF, and EP cells by microdissection was validated using the lumbar disc of a 3 M Shh Cre/+; R26 mT/mG/+ mouse (Figure 5). Before expression of Cre recombinase, all cells in the mouse express membrane‐bound tdTomato (mTOM or mT, 37 ). Following Shh‐mediated recombination, the TOM cassette, along with the stop codon, is excised only in the Shh Cre/+‐expressing cells, hence, permanently turning “ON” the expression of membrane‐bound GFP (mGFP or mG) in these cells as well as their lineages. 15 During mouse embryogenesis, Shh is first expressed by E7.5 in the node, 38 , 39 which gives rise to the notochord. Hence, node, notochord, and their descendant NP cells, even following terminal differentiation into multinucleated lacunae, 26 will be mGFP+ in the Shh Cre/+; R26 mT/mG/+ mice. None of the other cells of the spine express Shh and will continue to be TOM+ve. Both GFP+ve NP cells and non‐NP cells can be visualized with fluorescence microscope (Figure 5A‐G). The NP cells isolated by scooping out from the AF were free of any TOM+ve cells and were all GFP+ve (merged composite of GFP and TOM, Figure 5D,E). The NP cells can easily be collected by aspiration in a wide‐tip pipette (merged composite of GFP and TOM, Figure 5E). The lamella of AF and thin EP can be microdissected and collected from the same disc (Figure 5F). However, a few GFP+ve cells were detected in the AF and EP sample preparations (Figure 5F). These are likely the notochord‐descendant cells that are trapped in the AF and EP during disc formation at the embryonic stage and identified by white arrows in the mid‐coronal section of a 3 M old Shh Cre/+; R26 mT/mG/+ mouse lumbar disc in Figure 5G, and previously reported. 17
5.2. Purity of NP and AF validated by qPCR analysis
As cross‐contamination by a few cells from another compartment of the disc may not be visible under a stereomicroscope, even in the presence of a cell‐specific reporter allele, we further validated the purity of NP and AF cell populations by analyzing the expression of cell‐specific molecular markers for each component using TaqMan probes (Table 1) and qPCR analysis. qPCR is an amplification‐based sensitive technique and can detect even a single copy of a specific transcript. 40 Hence, qPCR analysis using probes for cell‐specific markers offers a robust approach for analyzing cross‐sample contamination in cell isolation procedures. As Shh and Bra are expressed only by the NP cells and not by AF cells, 32 , 33 , 41 these markers were used to identify NP cell contamination in the AF cell preparations. Tnmd is a specific molecular marker of the AF cells and is not expressed by NP cells in the intervertebral disc. 34 Hence, expression analysis of Tnmd was used to validate the absence of AF cells in the NP cell preparations. We also tested for contamination of bone cells in both the NP and AF cell preparations by analyzing the expression of Osteocalcin. Osteocalcin, also known as Bone Gamma‐Carboxyglutamate (Gla) Protein (Bglap), is a molecular marker of osteoblasts of the bone (reviewed by References 42, 43). Multiplex qPCR analysis for these molecular markers was performed using NP and AF cells prepared from 6‐month‐old mice (n ≥ 3), and B2m was used as an internal control in all PCR reactions. As there are no known molecular markers of EP, we did not analyze it further. The amplification results are presented as relative gene expression compared to B2m (Table 2). As expected, expression of Bra and Shh was detected only in the isolated NP cells, and expression of both these NP‐specific markers was undetected (UD) in the AF cells. Amplification for Bra was detected in 39.25 of the 40‐cycle PCR reaction for one replicate of AF and was undetected in the remaining four AF preparations. The expression of Tnmd was detected only in the AF cells and was undetected in the NP cell preparations. Bglap expression was undetectable in all the NP cell preparations, indicating the absence of any bone cell contamination in the isolated NP cells. However, amplification for Bglap was detected at 39.82 cycles of the 40‐cycle PCR reaction in one of the samples of AF cells, while it was undetected in the rest of the AF samples.
5.3. Isolation of high‐quality, intact RNA from NP, AF, and entire disc
It is well established that high‐quality, intact RNA is critical for high‐throughput deep sequencing transcriptomic analysis including RNAseq. The importance of intact and high‐quality total RNA for qPCR analysis is also emphasized in the MIQE guidelines, 44 as fragmented RNA may affect the interpretations of qPCR results. Thus, a protocol was optimized to isolate high‐quality and intact total RNA with RNA integrity number (RIN) 9.5 to 10 with a 28S:18S rRNA ratio of over 2.0 for the NP cells (Figure 6A‐C), and RIN ≥9.3 for AF cells or the whole disc (Figure 6D,E). However, the 28S:18S rRNA ratio for RNA isolated from AF cells or the disc as a whole was only over 1.3 (Figure 6D,E). The 5S rRNA band was not detected in any of the samples, which indicates that the RNA is intact and not degraded or fragmented (Figure 6). All RNA samples were also free of any genomic DNA contamination, which, if present, can be seen right below the loading well.
FIGURE 6.
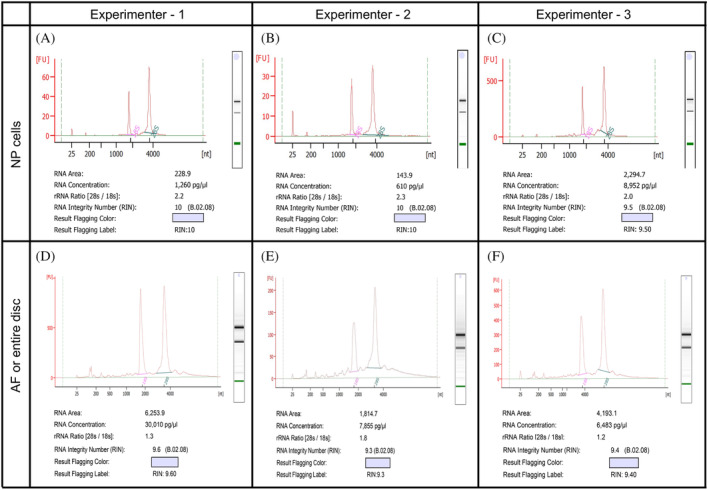
Representative Bioanalyzer electropherogram for quality check analysis of total RNA isolated by three independent experimenters using the NP, A‐C, and AF or whole disc, D‐F, microdissected using the bright field stereomicroscope from the wild‐type FVB mice spine. RNA with a RIN between 9.5 and 10 and free of gDNA contamination was isolated from the NP cells microdissected and collected from one lumbar disc of a year old mouse, A, NP cells from one lumbar disc of postnatal day four mouse, B, and NP cells pooled from six lumbar discs of 6‐month‐old mouse, C. RNA with RIN between 9.3 and 9.6 and free of gDNA contamination was isolated from the whole disc using five intact lumbar discs of 6‐month‐old mouse, D, from the microdissected AF and pooled from two lumbar discs of a month old mouse, E, and from the microdissected and pooled AF from two lumbar discs of a 2‐week‐old mouse (F)
6. DISCUSSION
We are in a molecular era with access to highly sophisticated, sensitive, and high‐throughput ‐omics approaches that may be employed to elucidate the intervertebral disc biology during growth, healthy maturation, and maintenance, and aging (Figure 1). Purity and quality of sample preparation is a prerequisite for successful downstream analysis whether using sophisticated high‐throughput approaches to profile cells at a single‐cell resolution, or conventional methods to determine changes at the molecular and cellular level. The purity of samples is also crucial for reliable and reproducible research findings as intermixing, or cross‐sample contamination can dramatically skew the data as well as the interpretation of results. Intervertebral disc is made up of three different cell types; NP, AF, and EP cells, and it is essential to isolate each component separately to precisely elucidate their molecular and biochemical profile. The mouse model is frequently employed to study the cellular and molecular basis of intervertebral disc formation, development, aging, and degeneration (reviewed by Reference 15). Previously it was demonstrated that all NP cells, even in a middle‐aged mouse disc, are a homogeneous population of notochord‐descendant cells. 17 NP cells from young mice to about 14 to 16 months of age look morphologically similar under a microscope. 26 Still, we do not know whether NP cells are molecularly homogenous or diversify into molecularly heterogenous populations during postnatal stages. Single‐cell RNA sequencing (scRNA seq) offers a robust approach to analyze the transcriptome at single‐cell resolution. 45 One of the significant applications of this approach is to study the molecular heterogeneity within an otherwise homogeneous‐looking cell population using scRNAseq. Due to the sensitivity of scRNA seq, it is necessary to use a pure and very high proportion of viable cells for reliable and reproducible results. Otherwise, the data may be misleading.
To determine whether postnatal NP cells continue to express molecular markers of its notochord precursor, a prior study used laser capture microdissection to obtain a pure population of NP and AF cells from two‐week‐old mouse lumbar discs. The results showed that notochord descendant NP cells continue to express key notochord marker Brachyury while syndetome derived AF does not. 32 Laser capture microdissection was also employed to dissect the newly formed disc and vertebrae from an E13.5 mouse embryo for gene expression analysis. 46 Although laser capture microdissection offers the ability to precisely microdissect specific cell types, even at single‐cell resolution under high magnification, 47 the lengthy process yields low RNA and results in RNA degradation. Moreover, expertise is required for preparation of thin cryosections on membrane slides and requires time to microdissect and collect the cells of interest on specific caps. The high laser power required for microdissecting cells embedded in the extracellular matrix, including AF, burns the tissue and cells, thereby degrading RNA. Lastly, the cost of this approach is also high due to specific membrane coated or metal frame slides, caps, and reagents required for the amplification of RNA. Another process to collect pure population of NP cells is by enzymatic digestion followed by FACS sorting using cell‐specific cell surface markers, or fluorescent reporter alleles. A recent utilized this approach to compare the transcriptomes of E12.5 notochord and NP cells collected from new‐born mouse disc at postnatal day zero (P0). 27 However, brief exposure of NP to normoxia dramatically changes the gene expression of the NP cells, 48 and enzymatic digestion and FACS sorting is a lengthy process for cell isolation. To overcome these caveats, we have developed a protocol for rapid and efficient isolation of all components from the same intervertebral disc to eliminate cross‐sample contamination (Table 2) and ensure preserved samples for downstream analysis, including at a transcriptional level (Figure 6). Since all three components of the disc can be collected from each sample, this method is also ideal for studies aimed at analyzing the cell‐cell crosstalk. However, the purity of EP was not examined in the current study due to lack of specific markers, and needs further validation. In one of the isolated AF samples, we did detect amplification of Bglap, a molecular marker of bone cells, 42 , 43 in the cycle 39.82, and Bra in the cycle 39.25 of the 40‐cycle PCR reaction. As the amplifications of these genes was detected in the last cycle of the PCR reaction, it is likely that a low copy of Bglap or Bra transcripts, from a few bone or NP cells, respectively, were present in these AF cell preparations. Therefore, it is essential to validate the purity of cells for each experiment.
The factors that may affect RNA integrity and purity, all of which must be controlled to optimize the quantity and quality of RNA, include tissue type, postmortem method, and time for tissue processing, storage conditions, and RNA isolation method. Enzymes that degrade cells, tissues, and biomolecules, including nucleic acids, proteins, and carbohydrates, are most efficient at a warm temperature. Tissue degradation initiates soon after euthanasia. Hence, it is crucial to process tissues at lower temperatures using an ice‐cold buffer to slow down the enzymatic and metabolic activity for efficient downstream analysis. Furthermore, nucleic acids are well preserved in RNAlater, thus minimizing the possibilities of RNA degradation. A standard method for quality check of RNA integrity is by Bioanalyzer and determining the RIN value (calculated algorithmically by Agilent Bioanalyzer). The RIN value ranges from 1 to 10, with RIN 1 indicating extremely degraded and RIN 10 representing the highest‐quality intact RNA. Both the quality and purity of RNA affect the gene expression profiles, and in turn, influence the results. 49 Therefore, the quality of RNA can impact the downstream applications, which analyze transcriptional changes in cells under different experimental conditions and include qPCR, micro‐arrays, in‐situ hybridization, bulk RNA seq, scRNA seq, and northern blotting to name a few. We have modified the RNA isolation protocol established by Chomczynski and Sacchi, 50 , 51 and combined it with Qiagen MiniElute spin column‐based kits. Although we used TRI Reagent for cell lysis in our current method, this protocol will work equally well using TRIzol (15596026, ThermoFisher Scientific, USA), and QIAzol (79306, Qiagen, Germany), as all these reagents are based on the Phenol and guanidinium isothiocyanate (GITC) based cell lysis. Moreover, Phenol/ GITC based lysis buffers allow the isolation of DNA, RNA, and protein from the same cells. 50 The collection and evaluation of all biomolecules from each biological sample enables the evaluation of additional outcome measures for rigorous and robust analysis. While standardizing the RNA isolation protocol from the mouse intervertebral disc cells we tested various approaches for tissue lysis including Qiagen TissueLyzer II (11843, Qiagen, Germany) with RIN ~ 5 to 6, Mortar and Pestle with RIN ~ 5 to 6, and syringes only for NP cells with RIN ~ 7. However, these lysis methods yielded low RNA quality and the yield due to inefficient lysis, delay in lysis, or overheating of samples. We found that the use of the polytron homogenizer, in combination with the methods described in this paper, yields the highest quality RNA with RIN of 9.3 to 10. Although we did not test the protocol for isolation of RNA from the microdissected EP cells, the method for RNA isolation entire disc, which also has EP region, should work equally well. We use these methods of isolation of NP and AF and subsequent RNA isolation from mice over 2 years of age and with equal purity and quality. The yield of RNA will vary based on the age of mouse (Figure 6). Younger mice have smaller discs with fewer cells that increases with maturity. 32 However, discs from aged mice are more fibrous and hypocellular, 26 , 30 and the yield of RNA per disc decreases in very aged mice. Also, number of discs used to pool samples for RNA isolation will affect the total yield (Figure 6). However, the RNA quality did not vary with age (Figure 6). Hence, depending on the amount of RNA required for downstream analysis, the researchers may pool samples from adjacent discs.
In summary, this method paper describes the optimized and step‐by‐step protocols for rapid and efficient isolation of all the three components of the mouse intervertebral disc: NP, AF, and EP cells, as well as the isolation of high‐quality, intact RNA from NP, AF, and entire disc. Quick and efficient pure sample preparation may be useful for molecular and biochemical analysis of each component of the disc during healthy growth, differentiation, and aging, and also under different experimental conditions. Additionally, isolating of all three components from the same disc offers the opportunity for their concerted analysis in each biological replicate. Furthermore, high‐quality, and intact RNA preparations are necessary for rigorous transcriptomic approaches, including NextGen sequencing and qPCR analysis. As these protocols are relatively simple and do not require sophisticated tools or reagents, we hope that researchers will adopt them to understand intervertebral disc biology. Lastly, these methods can be modified for isolation of NP, AF, and EP cells from other animal models including, but not limited to rats, guinea pigs, and rabbits.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHOR CONTRIBUTIONS
Chitra Lekha Dahia conceptualized and designed the methods; Vikrant Piprode, Raffaella Bonavita, Sarah Loh, Rajakumar Anbazhagan, Sarthak Mohanty, and Chandan Saini performed and validated the methods; Robert Pinelli and Paul Pricop provided technical assistance; Vikrant Piprode and Chitra Lekha Dahia analyzed the data, prepared figures, and wrote the manuscript. All authors reviewed the manuscript and gave their final approval for submission.
ACKNOWLEDGMENTS
This publication was made possible by Grant Number R01AR065530 from the National Institute of Arthritis and Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health (NIH), and Grant Number S10OD026763 from the NIH both made to CLD. Contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIAMS or NIH. Research endowment supported by Starr Foundation and a research grants from S & L Marx Foundation and OREF Grant Number 11‐228 made to CLD also supported this publication.
Piprode V, Mohanty S, Bonavita R, et al. An optimized step‐by‐step protocol for isolation of nucleus pulposus, annulus fibrosus, and end plate cells from the mouse intervertebral discs and subsequent preparation of high‐quality intact total RNA . JOR Spine. 2020;3:e1108 10.1002/jsp2.1108
Funding information Starr Foundation, S & L Marx Foundation, OREF: 11‐228; National Institutes of Health, Grant/Award Number: S10OD026763; National Institute of Arthritis and Musculoskeletal and Skin Diseases, Grant/Award Number: R01AR065530
REFERENCES
- 1. Geiss GK, Bumgarner RE, Birditt B, et al. Direct multiplexed measurement of gene expression with color‐coded probe pairs. Nat Biotechnol. 2008;26:317‐325. [DOI] [PubMed] [Google Scholar]
- 2. Mullis K, Faloona F, Scharf S, Saiki R, Horn G, Erlich H. Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harbor Symp Quant Biol. 1986;51(Pt 1):263‐273. [DOI] [PubMed] [Google Scholar]
- 3. Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A. 1979;76:4350‐4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kaddurah‐Daouk R, Kristal BS, Weinshilboum RM. Metabolomics: a global biochemical approach to drug response and disease. Annu Rev Pharmacol Toxicol. 2008;48:653‐683. [DOI] [PubMed] [Google Scholar]
- 5. Kristal BS, Shurubor YI, Kaddurah‐Daouk R, Matson WR. High‐performance liquid chromatography separations coupled with coulometric electrode array detectors: a unique approach to metabolomics. Methods Mol Biol. 2007;358:159‐174. [DOI] [PubMed] [Google Scholar]
- 6. Lindon JC, Holmes E, Nicholson JK. Metabonomics in pharmaceutical R&D. FEBS J. 2007;274:1140‐1151. [DOI] [PubMed] [Google Scholar]
- 7. Thurston CF, Henley LF. Direct immunoprecipitation of protein. Methods Mol Biol. 1988;3:149‐158. [DOI] [PubMed] [Google Scholar]
- 8. Jackson V. Studies on histone organization in the nucleosome using formaldehyde as a reversible cross‐linking agent. Cell. 1978;15:945‐954. [DOI] [PubMed] [Google Scholar]
- 9. Keene JD, Komisarow JM, Friedersdorf MB. RIP‐Chip: the isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat Protoc. 2006;1:302‐307. [DOI] [PubMed] [Google Scholar]
- 10. Fried M, Crothers DM. Equilibria and kinetics of lac repressor‐operator interactions by polyacrylamide gel electrophoresis. Nucleic Acids Res. 1981;9:6505‐6525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Garner MM, Revzin A. A gel electrophoresis method for quantifying the binding of proteins to specific DNA regions: application to components of the Escherichia coli lactose operon regulatory system. Nucleic Acids Res. 1981;9:3047‐3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hartvigsen J, Hancock MJ, Kongsted A, et al. What low back pain is and why we need to pay attention. Lancet. 2018;391:2356‐2367. [DOI] [PubMed] [Google Scholar]
- 13. Shinohara H. The mouse vertebrae: changes in the morphology of mouse vertebrae exhibit specific patterns over limited numbers of vertebral levels. Okajimas Folia Anat Jpn. 1999;76:17‐31. [DOI] [PubMed] [Google Scholar]
- 14. McLaren A, Michie D. Factors affecting vertebral variation in mice. 4. Experimental proof of the uterine basis of a maternal effect. J Embryol Exp Morphol. 1958;6:645‐659. [PubMed] [Google Scholar]
- 15. Mohanty S, Dahia CL. Defects in intervertebral disc and spine during development, degeneration, and pain: new research directions for disc regeneration and therapy. Wiley Inter Rev Dev Biol. 2019;8:e343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carlier EW. Fate of the notochord and development of the intervertebral disc in the sheep, with observations on the structure of the adult disc in these animals. J Anat Physiol. 1890;24:573‐584. [PMC free article] [PubMed] [Google Scholar]
- 17. Choi KS, Cohn MJ, Harfe BD. Identification of nucleus pulposus precursor cells and notochordal remnants in the mouse: implications for disk degeneration and chordoma formation. Dev Dyn. 2008;237:3953‐3958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Peacock A. Observations on the prenatal development of the intervertebral disc in man. J Anat. 1951;85:260‐274. [PMC free article] [PubMed] [Google Scholar]
- 19. Peacock A. Observations on the postnatal structure of the intervertebral disc in man. J Anat. 1952;86:162‐179. [PMC free article] [PubMed] [Google Scholar]
- 20. Walmsley R. The development and growth of the intervertebral disc. Edinb Med J. 1953;60:341‐364. [PMC free article] [PubMed] [Google Scholar]
- 21. Sugimoto Y, Takimoto A, Hiraki Y, Shukunami C. Generation and characterization of ScxCre transgenic mice. Genesis. 2013;51:275‐283. [DOI] [PubMed] [Google Scholar]
- 22. Alvarez‐Garcia O, Matsuzaki T, Olmer M, et al. FOXO are required for intervertebral disk homeostasis during aging and their deficiency promotes disk degeneration. Aging Cell. 2018;17:e12800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Bratsman A, Couasnay G, Elefteriou F. A step‐by‐step protocol for isolation of murine nucleus pulposus cells. JOR Spine. 2019;2:e1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gorth DJ, Ottone OK, Shapiro IM, Risbud MV. Differential effect of long‐term systemic exposure of TNFalpha on health of the annulus fibrosus and nucleus pulposus of the intervertebral disc. J Bone Mineral Res. 2019;35:725‐737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kushioka J, Kaito T, Chijimatsu R, et al. A novel and efficient method for culturing mouse nucleus pulposus cells. The Spine J. 2019;19:1573‐1583. [DOI] [PubMed] [Google Scholar]
- 26. Mohanty S, Pinelli R, Pricop P, Albert TJ, Dahia CL. Chondrocyte‐like nested cells in the aged intervertebral disc are late‐stage nucleus pulposus cells. Aging Cell. 2019;18:e13006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Peck SH, McKee KK, Tobias JW, Malhotra NR, Harfe BD, Smith LJ. Whole transcriptome analysis of notochord‐derived cells during embryonic formation of the nucleus pulposus. Sci Rep. 2017;7:10504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Veras MA, McCann MR, Tenn NA, Seguin CA. Transcriptional profiling of the murine intervertebral disc and age‐associated changes in the nucleus pulposus. Connect Tissue Res. 2020;61:63‐81. [DOI] [PubMed] [Google Scholar]
- 29. Wei Y, Tower RJ, Tian Z, et al. Spatial distribution of type II collagen gene expression in the mouse intervertebral disc. JOR Spine. 2019;2:e1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Winkler T, Mahoney EJ, Sinner D, Wylie CC, Dahia CL. Wnt signaling activates Shh signaling in early postnatal intervertebral discs, and re‐activates Shh signaling in old discs in the mouse. PloS One. 2014;9:e98444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang Y, Xiong C, Kudelko M, et al. Early onset of disc degeneration in SM/J mice is associated with changes in ion transport systems and fibrotic events. Matrix Biol. 2018;70:123‐139. [DOI] [PubMed] [Google Scholar]
- 32. Dahia CL, Mahoney EJ, Durrani AA, Wylie C. Postnatal growth, differentiation, and aging of the mouse intervertebral disc. Spine. 2009b;34:447‐455. [DOI] [PubMed] [Google Scholar]
- 33. Dahia CL, Mahoney EJ, Durrani AA, Wylie C. Intercellular signaling pathways active during intervertebral disc growth, differentiation, and aging. Spine. 2009a;34:456‐462. [DOI] [PubMed] [Google Scholar]
- 34. Christiani TR, Baroncini E, Stanzione J, Vernengo AJ. In vitro evaluation of 3D printed polycaprolactone scaffolds with angle‐ply architecture for annulus fibrosus tissue engineering. Regenerative Biomater. 2019;6:175‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Vincent K, Mohanty S, Pinelli R, et al. Aging of mouse intervertebral disc and association with back pain. Bone. 2019;123:246‐259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Harfe BD, Scherz PJ, Nissim S, Tian H, McMahon AP, Tabin CJ. Evidence for an expansion‐based temporal Shh gradient in specifying vertebrate digit identities. Cell. 2004;118:517‐528. [DOI] [PubMed] [Google Scholar]
- 37. Muzumdar MD, Tasic B, Miyamichi K, Li L, Luo L. A global double‐fluorescent Cre reporter mouse. Genesis. 2007;45:593‐605. [DOI] [PubMed] [Google Scholar]
- 38. Echelard Y, Epstein DJ, St‐Jacques B, et al. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell. 1993;75:1417‐1430. [DOI] [PubMed] [Google Scholar]
- 39. Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. 1993;75:1401‐1416. [DOI] [PubMed] [Google Scholar]
- 40. Palmer S, Wiegand AP, Maldarelli F, et al. New real‐time reverse transcriptase‐initiated PCR assay with single‐copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J Clin Microbiol. 2003;41:4531‐4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dahia CL, Mahoney E, Wylie C. Shh signaling from the nucleus pulposus is required for the postnatal growth and differentiation of the mouse intervertebral disc. PloS One. 2012;7:e35944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chapurlat RD, Confavreux CB. Novel biological markers of bone: from bone metabolism to bone physiology. Rheumatology. 2016;55:1714‐1725. [DOI] [PubMed] [Google Scholar]
- 43. Zoch ML, Clemens TL, Riddle RC. New insights into the biology of osteocalcin. Bone. 2016;82:42‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bustin SA, Benes V, Garson JA, et al. The MIQE guidelines: minimum information for publication of quantitative real‐time PCR experiments. Clin Chem. 2009;55:611‐622. [DOI] [PubMed] [Google Scholar]
- 45. Tang F, Barbacioru C, Wang Y, et al. mRNA‐Seq whole‐transcriptome analysis of a single cell. Nat Methods. 2009;6:377‐382. [DOI] [PubMed] [Google Scholar]
- 46. Sohn P, Cox M, Chen D, Serra R. Molecular profiling of the developing mouse axial skeleton: a role for Tgfbr2 in the development of the intervertebral disc. BMC Dev Biol. 2010;10:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Emmert‐Buck MR, Bonner RF, Smith PD, et al. Laser capture microdissection. Science. 1996;274:998‐1001. [DOI] [PubMed] [Google Scholar]
- 48. Yang SH, Hu MH, Sun YH, Lin FH. Differential phenotypic behaviors of human degenerative nucleus pulposus cells under normoxic and hypoxic conditions: influence of oxygen concentration during isolation, expansion, and cultivation. The Spine J. 2013;13:1590‐1596. [DOI] [PubMed] [Google Scholar]
- 49. Padhi BK, Singh M, Rosales M, Pelletier G, Cakmak S. A PCR‐based quantitative assay for the evaluation of mRNA integrity in rat samples. Biomol Detect Quant. 2018;15:18‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chomczynski P. A reagent for the single‐step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 1993;15:532‐534. 536–537. [PubMed] [Google Scholar]
- 51. Chomczynski P, Sacchi N. Single‐step method of RNA isolation by acid guanidinium thiocyanate‐phenol‐chloroform extraction. Anal Biochem. 1987;162:156‐159. [DOI] [PubMed] [Google Scholar]