

ABSTRACT
Up to 10% of women use selective serotonin reuptake inhibitor (SSRI) antidepressants during pregnancy and postpartum. Recent evidence suggests that SSRIs are capable of altering the gut microbiota. However, the interaction between maternal depression and SSRI use on bacterial community composition and the availability of microbiota-derived metabolites during pregnancy and lactation is not clear.
We studied this using a rat model relevant to depression, where adult females with a genetic vulnerability and stressed as pups show depressive-like behaviors. Throughout pregnancy and lactation, females received the SSRI fluoxetine or vehicle. High-resolution 16S ribosomal RNA marker gene sequencing and targeted metabolomic analysis were used to assess the fecal microbiome and metabolite availability, respectively.
Not surprisingly, we found that pregnancy and lactation segregate in terms of fecal microbiome diversity and composition, accompanied by changes in metabolite availability. However, we also showed that fluoxetine treatment altered important features of this transition from pregnancy to lactation most clearly in previously stressed dams, with lower fecal amino acid concentrations. Amino acid concentrations, in turn, correlated negatively with the relative abundance of bacterial taxa such as Prevotella and Bacteroides.
Our study demonstrates an important relationship between antidepressant use during the perinatal period and maternal fecal metabolite availability in a rat model relevant to depression, possibly through parallel changes in the gut microbiome. Since microbial metabolites contribute to homeostasis and development, insults to the maternal microbiome by SSRIs might have health consequences for mother and offspring.
KEYWORDS: Fecal microbiome, fecal metabolome, pregnancy, lactation, depression, SSRI antidepressants, fluoxetine, rat, serotonin transporter, 16S rRNA
Introduction
Dysregulation of the neurotransmitter serotonin (also referred to as 5-hydroxytryptamine or 5-HT) in the brain is a widely recognized hallmark of major depressive disorder.1 Selective serotonin reuptake inhibitor (SSRI) antidepressants target the serotonin transporter (SERT), thereby influencing the serotonergic tone.2 Serotonin and SERT are particularly well-known for playing crucial regulatory roles in the brain, but they also act in many peripheral tissues.3 Nearly 95% of the body’s serotonin resides within the gastrointestinal tract, where it is mainly produced by enterochromaffin cells.4 Interestingly, germ-free and antibiotic-treated mice show lower levels of colonic and serum serotonin. This is reversible by microbial colonization, suggesting an important role for members of the gut microbiome in modulating serotonin availability.5 Indeed, the gut microbiota synthesizes tryptophan, the precursor of serotonin, and promotes serotonin biosynthesis in the host organism.5 Conversely, host serotonin signaling impacts the gut microbiota composition as we have shown in rats,6 and others in mice,7 with reduced or no SERT gene expression. Correspondingly, in vitro studies have shown that serotonin affects the growth of microbes such as Escherichia coli and Rhodospirillum rubrum,8 and activates bacterial quorum sensing.9 Several studies have found alterations in the gut microbiota composition of patients with major depression10-16 and rodents with depressive-like symptoms.17-21 Moreover, recent human population-based studies,16,22,23 as well as rodent experiments,24-29 showed that SSRIs modulate the gut microbiota composition, an effect that may be related to their in vitro antimicrobial properties.24,30,31
The gut microbiota synthesizes a variety of metabolites that reach the systemic circulation,32,33 which is of special relevance during pregnancy and lactation when there is a high metabolic demand.34 Indeed, pregnancy is characterized by hormonal, immunological and metabolic changes, paralleled by remodeling of the maternal gut microbiome.35 Environmental and pharmacological insults disrupt the compositional and functional states of the maternal gut microbiota, as demonstrated by several animal studies focusing on stress,36 diet,37 and antibiotic use during pregnancy.38 Despite clear evidence that the microbiome interacts with serotonin homeostasis,39 it is currently unknown how alterations in serotonin signaling affect the maternal gut microbiome during the perinatal period. An estimated 7 to 13% of women suffer from a major depressive disorder in the perinatal period.40 It was recently shown that there are associations between anxiety and the fecal microbial composition during pregnancy.41 Between 1 and 10% of pregnant women take SSRI antidepressants,40 further highlighting the relevance of a potential effect of SSRI medication during this period on the gut microbiota and its metabolic output.
We investigated the hypothesis that a depressive-like phenotype, SSRI antidepressant treatment, and their combination affect the microbial community composition and function during pregnancy and the postpartum period. To this end, we used a genetic rat model of maternal vulnerability (MV) in combination with early life stress (sMV) or control handling (cMV). In adulthood, sMV females show a depressive-like phenotype, reflected in a lower sucrose preference and lower neural growth factor gene expression than cMV females.42 We treated sMV and cMV rats dams daily with fluoxetine (FLX), a commonly used SSRI, or vehicle (Veh) throughout pregnancy and lactation. Weekly fecal samples were collected for 16S rRNA gene sequencing. In addition, targeted metabolomic analysis of amino acids, short-chain fatty acids and bile acids was done on a subset of samples. We examined the hypothesis that depressive-like symptoms and antidepressant treatment during pregnancy and lactation affect the maternal fecal microbiome and its functional capacity. We expected the combination of sMV and FLX to have the strongest effect.
Results
We examined the hypothesis that depressive-like symptoms and antidepressant treatment with FLX during pregnancy and lactation affect the maternal fecal microbiome composition and function. Maternal vulnerability (MV) rats were used, since early life stress produces depressive-like symptoms and shifts in fecal microbiome community composition in these animals (Figure 1(a)).6,42 In line with this, we observed pre-conception differences between the stressed (sMV) and the control (cMV) groups, with the sMV group showing higher alpha diversity than the cMV group (Supplementary Figure 1a, t30 = −2.0709, p< .05). This coincided with differences in Operational Taxonomic Unit (OTU) relative abundance (Supplementary Figure 1b). Then, throughout gestation and the postpartum period, sMV and cMV females were treated with FLX or Veh daily (Figure 1(b)). Weekly fecal pellets were collected for 16S rRNA sequencing and a subset was used for targeted metabolomic analysis in addition to sequencing (Figure 1(b)).
Figure 1.
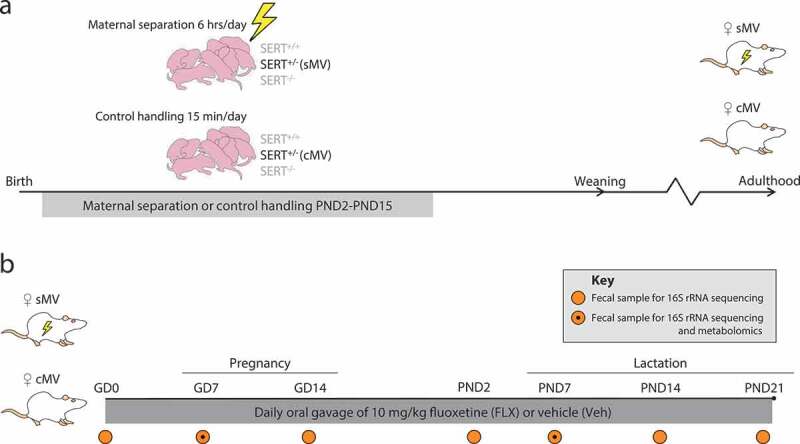
Overview of study design and sampling schedule. (a) Early life stress protocol. SERT+/- females were crossed with SERT+/- males, yielding nests with offspring genotypes SERT+/+, SERT+/-, and SERT−/-. The SERT+/- females are our model of maternal vulnerability (MV). From postnatal day (PND) 2 to PND15, pups were either maternally separated for 6 hours per day (early life stress – sMV) or control handled (cMV) for 15 minutes per day. Pups were weaned at PND21. sMV- and cMV females were group housed (same treatment) until adulthood. (b) Fluoxetine treatment during pregnancy and lactation, and fecal sampling schedule. Adult sMV and cMV females (N = 14–18/group) were crossed with wildtype males. Throughout pregnancy and lactation, from gestational day (GD)1 until PND21, females received a daily oral injection of either 10 mg/kg fluoxetine (FLX) or methylcellulose (Veh). Thus, there were 4 groups of females: cMV-Veh, sMV-Veh, cMV-FLX, and sMV-FLX (N = 6–11/group). Fecal pellets for 16S rRNA gene sequencing were freshly collected before conception at GD0, during pregnancy at GD7 and GD14, at PND2, and during lactation at PND7, PND14 and PND21 (N = 192 in total). Selected fecal samples from GD7 and PND7 were also used for metabolomic analysis (N = 4-5/group per time point, N = 36 in total).
Pregnancy and lactation have distinct fecal microbial signatures
To examine whether pregnancy and lactation have distinct fecal microbial signatures, we first analyzed the 16S rRNA sequencing data in terms of microbial alpha diversity and community structure over all treatment groups. Alpha diversity was higher during pregnancy than during lactation, as expressed in the Shannon diversity index (Figure 2(a), t158 = 7.5845, p< .0001). The differences in phylogenetic structure between samples were explored by assessing weighted UniFrac distances. A separation between samples obtained during pregnancy versus lactation was revealed by Principle Coordinates Analysis (PCoA) (Figure 2(b), PERMANOVA p= .001).
Figure 2.

Fluoxetine treatment alters the microbiome during pregnancy and lactation in rats with a depressive-like phenotype. (a) Microbial alpha diversity. A t-test was performed to assess the overall difference between pregnancy and lactation. Within each period, a two-way ANOVA was performed. (b) Structure of the microbial communities during pregnancy and lactation. Communities were clustered using PCoA of the weighted UniFrac distance matrix. Each point corresponds to the microbial community of one sample. The percentage of variation explained by the PC is indicated on the axes. Colors correspond to period; all samples were grouped within pregnancy and lactation. (c) The effect of FLX on the structure of the sMV microbiome during pregnancy and lactation. Each point corresponds to the microbial community of one sample that was collected during pregnancy (upper graph) or lactation (lower graph). Colors correspond to group; sMV-Veh in white and sMV-FLX in gray. (d) Heatmap of relative abundances of Random Forests-identified OTUs distinguishing between pregnancy and lactation, ordered by unsupervised clustering. (e) Relative abundance of selected Random Forests-identified OTUs. Two-way ANOVAs within pregnancy and lactation were used to analyze OTU relative abundance. Note: this analysis was performed on centered log-ratio transformed data (see Methods). N = 12–22 samples/group from pregnancy (N = 64 in total), N = 18–33/group from lactation (N = 96 in total).
In order to identify the bacterial signatures that underlie the observed shift in community structure between pregnancy and lactation, a machine-learning approach called Random Forests was used to analyze the data of the cMV-Veh group. This generated a model containing 28 OTUs at the genus-level, discriminating between pregnancy and lactation. A large reorganization of the microbiome composition from gestation to lactation was revealed, consistent with structural remodeling. Particularly within the Bacteroidales and Clostridiales orders, OTUs changed in their relative abundance from pregnancy to lactation (Supplementary Figure 2a).
Fluoxetine treatment alters the maternal fecal microbial signatures of pregnancy and lactation in a rat model for depressive-like behavior
To test whether early life stress, FLX, and their combination alter the observed fecal microbial signatures of pregnancy and lactation, we first compared the groups with respect to alpha- and beta diversity measures. During pregnancy, we found a main effect of early life stress on microbial alpha diversity, with the sMV groups showing a higher Shannon diversity index than the cMV groups (Figure 2(a), F1,60 = 4.690, p< .05). Post hoc analysis revealed no significant differences between groups. During lactation, there was a significant FLX * early life stress interaction effect (Figure 2(a), F1,92 = 5.256, p< .05) and a trend toward a main effect of stress (F1,92 = 2.784, p= .0986). Post hoc analysis revealed that the Shannon diversity index was significantly higher in the sMV- than in the cMV group during lactation (p< .05). In terms of microbial community structure, the treatment groups dissociated based on PCoA of the weighted UniFrac distances (Supplementary Figure 2b, PERMANOVA p= .001). In particular, microbial community structure in the sMV-FLX group differed from the structure in the sMV-Veh group, both during pregnancy and lactation (Figure 2(c), pregnancy sMV-Veh vs sMV-FLX PERMANOVA p< .05, lactation sMV-Veh vs sMV-FLX PERMANOVA p= .001). This suggests that, in addition to the dynamic restructuring of the fecal microbiome across pregnancy and lactation, fluoxetine treatment modulated community structure of the fecal microbiota in the sMV females during these periods.
Next, the relative abundance of the 28 OTUs constituting the Random Forests-generated model discriminating between pregnancy and lactation in the cMV-Veh group was examined for the treatment groups. Globally, the pattern of OTUs distinguishing between pregnancy and lactation in the treatment groups was similar to that of the cMV-Veh group (Figure 2(d)). However, specific OTUs showed variations in their relative abundance dependent on treatment as tested by two-way ANOVAs (FLX * early life stress) within pregnancy and lactation (Figure 2(e); Supplementary Figure 2c). For instance, the 2 OTUs with the largest contribution to the accuracy of the Random Forests model, Bacteroides and Prevotella from the Bacteroidales order, were sensitive to FLX treatment. Bacteroides relative abundance was lower in the FLX groups than in the Veh-treated groups during pregnancy (Figure 2(e), main effect of FLX, F1,60 = 13.206, p< .001). Post hoc testing revealed that specifically the cMV-FLX group had a lower relative abundance of Bacteroides than the cMV-Veh group (p< .01). During lactation, a trend for a FLX * early life stress interaction was found for Bacteroides relative abundance (F1,92 = 3.774, p= .0551), with a trend for a lower relative abundance in the sMV-FLX group than in the sMV-Veh group (p= .0764). The opposite effect was found for Prevotella, with a main effect of FLX on Prevotella relative abundance during pregnancy (F1,60 = 16.488, p< .001). Specifically, there was a trend for higher Prevotella relative abundance in the cMV-FLX than in the cMV-Veh group (p= .0944), and a statistically significant higher Prevotella relative abundance in the sMV-FLX group than in the sMV-Veh group (p< .01). During lactation, a FLX * early life stress interaction effect was found (F1,92 = 8.520, p< .01), as well as a main effect of FLX (F1,92 = 4.249, p< .05). Post hoc testing revealed a higher Prevotella relative abundance in the sMV-FLX group than in the sMV-Veh group (p< .01) and the cMV-FLX group (p< .05), reaching almost 25% relative abundance. Similarly, there was a main effect of FLX during pregnancy on Ruminococcus relative abundance (F1,60 = 26.166, p< .0001), where both the cMV-FLX group relative to the cMV-Veh group (p< .05) as well as the sMV-FLX group relative to the sMV-Veh group (p < .001) had a higher relative abundance of Ruminococcus. During lactation, a FLX * early life stress interaction was found (F1,92 = 4.245, p< .05), and a trend for a main effect of FLX (F1,92 = 3.575, p= .0618). Post hoc analysis revealed a higher Ruminococcus relative abundance in the sMV-FLX group relative to the sMV-Veh group (p< .05). Early life stress by itself did not have effects as striking as FLX, although during pregnancy relative abundance of Adlercreutzia and UC Lachnospiraceae were higher and Lactobacillus lower in stressed dams than in controls (Supplementary Figure 2c).
Considering that OTUs within a community display co-occurrence and co-exclusion relationships,43 we performed a correlation analysis to look at these relationships within our samples, regardless of time of sampling or treatment. A correlation matrix between the 28 selected OTUs revealed that Prevotella was negatively correlated with a cluster consisting mainly of Clostridiales and Desulfovibrionales (Supplementary Figure 2d). Ruminoccus was the exception within the Clostridiales, being positively correlated with Prevotella.
Together, these results confirm that pregnancy and lactation are distinct with regard to fecal microbiota diversity and composition. Although early life stress increased alpha diversity during lactation, there were limited effects of early life stress alone on either microbial community structure or on the relative abundance of OTUs resulting from our Random Forests model. However, FLX impacted community structure during pregnancy and lactation, as well as the relative abundance of a number of OTUs selected for their ability to distinguish between pregnancy and lactation. FLX in combination with early life stress had the largest effects on the maternal microbiome.
Pregnancy and lactation have distinct fecal metabolic signatures
Bacterial communities metabolize and synthesize a number of metabolites that are essential for both dam and offspring. Shifts in bacterial community composition and predictive function may suggest alterations in the capacity of these communities to synthesize or metabolize these metabolites. We first performed PICRUSt predictive analysis on the OTU data to explore the hypothesis that the functional potential of pregnancy and lactation is different, since they have different microbial signatures. To this end, Random Forests analysis was performed on PICRUSt-generated data from the cMV-Veh group. Nine KEGG pathways were significantly changed across pregnancy and lactation, including the pathway of lysine biosynthesis; the pathway of alanine, aspartate, and glutamate metabolism; and the pathway of arginine and proline metabolism (Figure 3(a); Supplementary Figure 3a). Overall, pathways related to amino acid synthesis and metabolism were among the metabolic pathways best able to characterize the difference in the predicted function of the maternal bacterial community during pregnancy vs lactation.
Figure 3.

Fluoxetine alters fecal metabolite availability during pregnancy and lactation in rats with a depressive-like phenotype. (a) Heatmap of Random Forests-identified PICRUSt-generated KEGG pathways distinguishing between pregnancy and lactation, ordered by unsupervised clustering. (b) Structure of the metabolomic composition during pregnancy and lactation. Samples were clustered using PCA of the metabolomics concentration table. Each point corresponds to the metabolic capacity of one sample. The percentage of variation explained by the PC is indicated on the axes. Colors correspond to period in the upper graph, and to group in the lower graph. (c) Heatmap of Random Forests-identified metabolite concentrations distinguishing between pregnancy and lactation. (d) Concentrations of selected amino acids, analyzed with two-way ANOVAs within pregnancy and lactation. (e) Metabolite Set Enrichment Analysis of pregnancy vs lactation per group. Only the pathways that were significantly altered between pregnancy and lactation are shown here; the full list of metabolic pathways is shown in Supplementary Figure 3e. For a: N = 12–22 samples/group from pregnancy (N = 64 in total), N = 18–33/group from lactation (N = 96 in total). For b-e: N = 4-5/group from pregnancy (N = 18 in total), N = 4-5/group from lactation (N = 18 in total). Abbreviations: biosynth. = biosynthesis; degrad. = degradation; metab. = metabolism.
To determine whether this altered predictive metabolic capacity of the microbiome was associated with actual metabolite availability, we measured the concentration of amino acids, bile acids, and short-chain fatty acids in a subset (N = 4–5 per group in pregnancy and lactation) of fecal pellets previously used for 16S rRNA sequencing (Supplementary Table 1). Corresponding to the microbiome results, principle component analysis (PCA) revealed a clear distinction in metabolic profile between samples obtained during pregnancy versus lactation (Figure 3(b), upper graph).
To reveal the metabolic signatures of pregnancy and lactation, Random Forests analysis was performed on the metabolite data from the cMV-Veh group. Fourteen metabolites were included in the resulting model to distinguish between pregnancy and lactation (Supplementary Figure 3b). Most of them were amino acids, and 11 out of 14 metabolites were higher in concentration during pregnancy than during lactation (Figure 3(c)). For instance, the amino acids isoleucine and methionine had higher concentrations during pregnancy than during lactation, while proline concentrations were higher during lactation than during pregnancy (Supplementary Figure 3c). The bile acids deoxycholic acid and taurochenodeoxycholic acid were both higher in concentration during pregnancy than during lactation. Of the short-chain fatty acids included in the Random Forests model, isocaproic acid concentrations were very low during pregnancy, whereas isovaleric acid concentrations were low during lactation. Overall, the metabolomic results were consistent with the microbiome findings and the predictive functional analysis in discriminating between pregnancy and lactation and pointing toward amino acids as being central to this shift.
Fluoxetine treatment alters maternal fecal metabolite availability during pregnancy and lactation in a rat model for depressive-like behavior
To investigate whether early life stress, FLX and their combination affect the abovementioned fecal metabolic signatures of pregnancy and lactation as observed in the cMV-Veh group, we compared these groups with respect to predicted- and measured metabolic data. The effects of early life stress and FLX on the abundance of predictive pathways were mixed, with the sMV-FLX group having generally the lowest predicted abundance of these metabolic pathways (Figure 3(a)). Visual inspection of the PCA graph based on the metabolite concentration table revealed no clear effect of early life stress or FLX treatment (Figure 3(b), lower graph).
Next, the concentrations of the 14 metabolites included in the Random Forests-generated model discriminating between pregnancy and lactation in the cMV-Veh group were examined for the treatment groups. Similar to the microbiome results, the overall pattern of metabolite availability changes between pregnancy and lactation was similar in all groups (Figure 3(c)). However, fecal amino acid concentrations were affected by FLX treatment (Figure 3(d), Supplementary Figure 3c). There was a trend toward a main effects of FLX on aspartic acid concentrations during pregnancy (F1,16 = 4.090, p= .0602) and a significant main effect during lactation (F1,16 = 9.689, p< .01). There was also a trend toward a main effect of early life stress on aspartic acid concentration during lactation (F1,16 = 3.868, p= .0668). Between-group post hoc analysis revealed a significantly lower concentration of aspartic acid in the sMV-FLX group relative to the sMV-Veh group (p< .05). For serine concentrations, we found a FLX * early life stress interaction effect during pregnancy (F1,16 = 5.868, p< .05), as well as a main effect of FLX (F1,16 = 25.314, p< .001). In particular, there was a trend for lower serine concentrations in the cMV-Veh group than in the sMV-Veh group (p= .0565), and significantly lower concentrations of serine in the sMV-FLX group than in the sMV-Veh group (p< .001). For threonine concentrations, we found a trend for a main effect of FLX during pregnancy (F1,16 = 3.598, p= .076), where the FLX groups show decreased threonine levels.
Since, by restricting ourselves to Random Forests-identified metabolites, we might miss interesting effects of our treatments on metabolites that do not differ between pregnancy and lactation, we decided to also analyze those metabolites as well with two-way ANOVAs within pregnancy and lactation (Supplementary Figure 3d). We found that FLX was associated with lower concentrations of the amino acid glutamic acid during lactation (F1,16 = 8.200, p< .05), and with lower levels of the bile acids betamuricholic acid (F1,16 = 5.543, p< .05) and omegamuricholic acid (F1,16 = 7.002, p< .05) during pregnancy. Interestingly, within the short-chain fatty acids, isobutyric acid concentrations were decreased by FLX during pregnancy (trend, F1,16 = 3.952, p= .0642) and during lactation (F1,16 = 5.902, p< .05), while butyric acid was increased by FLX during lactation (F1,16 = 5.019, p< .05).
Next, we used Metabolite Set Enrichment Analysis (MSEA) to identify the fecal metabolite sets that were enriched during either pregnancy or lactation. In the cMV-Veh group, there were several pathways that significantly differed between these two periods, again containing amino acid-related pathways such as glycine and serine metabolism (Figure 3(e); Supplementary Figure 3e). Then, we performed MSEA on the sMV-Veh, cMV-FLX and sMV-FLX data to determine which metabolites involved in specific metabolic processes were enriched in either pregnancy or lactation. The sMV-Veh group, like the cMV-Veh group, contained a number of metabolite sets that distinguished between pregnancy and lactation, such as methionine metabolism, and glycine and serine metabolism. In contrast, in the cMV-FLX and sMV-FLX groups no metabolite sets were identified that differed significantly between pregnancy and lactation. Thus, fluoxetine treatment lowered the fecal concentrations of several key amino acids during the dynamic periods of pregnancy and lactation in our model for maternal depressive-like symptoms. In addition, fluoxetine treatment abolished the differences in MSEA-identified pathways that distinguished the fecal metabolome during pregnancy from lactation as observed in the Veh-treated groups.
The relative abundances of fecal bacterial taxa correlate with metabolite concentrations
Finally, we generated a correlation matrix to further explore the link between the composition of the microbiome and the availability of metabolites (Figure 4). We collapsed all treatment groups in this analysis to capture the full range of variation in both OTU abundance and metabolite concentrations. A cluster consisting of OTUs from the Clostridiales (including UC Peptococcaceae, Clostridia; other and Oscillospira) and Desulfovibrionales (Desulfovibrio and UC Desulfovibrionaceae) orders was positively correlated with amino acid concentrations. Showing almost the reverse pattern, the relative abundance of Prevotella was negatively correlated with availability of isobutyric acid and a collection of amino acids; alanine, isoleucine, glycine, methionine, and serine. Associating to a different set of metabolites, the relative abundance of Ruminococcus was inversely correlated with concentrations of aspartic acid and some bile acids; betamuricholic acid, omegamuricholic acid, and alphamuricholic acid. Overall, the OTUs that were most affected in relative abundance by FLX treatment (Bacteroides, Prevotella and Ruminococcus) were correlated with amino acid availability.
Figure 4.

Correlation matrix of metabolites and OTUs throughout pregnancy and lactation. Spearman’s rank correlations were determined between the 14 Random Forests-identified metabolites and the 28 Random Forests-identified OTUs. The circles represent the correlation between each metabolite and OTU. The size of the circle increases with decreasing p-value-, and the color of the circle corresponds to the ρ-value of the correlation (the strength of the correlation). Only significant (p < .05) correlations are plotted, and only the OTUs with at least 1 significant correlation are shown here. The matrix is ordered by hierarchical clustering. N = 4-5/group from pregnancy (N = 18 in total), N = 4-5/group from lactation (N = 18 in total).
Discussion
Our study shows that antidepressant treatment with fluoxetine (FLX) during pregnancy and lactation disrupts metabolite availability in a rat model for maternal depressive-like symptoms, possibly through alterations in bacterial community composition and function. In short, maternal vulnerability (MV) female rats were exposed to early life stress (sMV). In adulthood, sMV and control (cMV) females were treated throughout gestation and lactation with the SSRI FLX or vehicle (Veh). Fecal samples were collected for 16S rRNA sequencing and targeted metabolomics.
We first explored the distinct fecal microbial signatures of pregnancy and lactation in cMV-Veh females. Human studies showed a remodeling of the microbiome from the first- to the third trimester, with changes in diversity and composition.44 Similarly, the maternal microbiome in mice changes from early to late pregnancy.36 Paradoxically, here we observed stability of the microbiome over pregnancy, potentially related to the fact that we had no samples during the first week of gestation, only starting at GD7. Likewise, a human study in which fecal sampling did not cover the first few weeks post-fertilization reported stability of the microbiome over the course of pregnancy.45 In addition, stability of the human microbiome over lactation has been observed.46 We complement these studies by instead defining distinct microbial signatures associated with pregnancy versus lactation, periods with dynamic hormonal, immunological and metabolic changes.35 Specifically, we showed that the transition from pregnancy to lactation in rats was characterized by a decrease in alpha diversity and a large reorganization of the structure of the fecal microbial community, with an increase in Prevotella at the cost of Bacteroides within the Bacteroidales, and an increase in Ruminococcus at the cost of other Clostridiales OTUs.
We then hypothesized that early life stress and FLX alter the observed fecal microbial signatures of pregnancy and lactation. It has been shown repeatedly that early life stress by maternal separation in rodents produces long-term effects on the gut and its microbiome.47-52 We showed here that adult sMV females had a higher fecal alpha diversity than cMV females pre-conception, during pregnancy and during lactation. We have previously found this tendency in young rats,6 suggesting that our stress protocol induces an immediate and life-long increase in microbial alpha diversity. This is surprising, since other studies of early life stress by maternal separation in rodents have reported a decrease in alpha diversity,21,51 or no effect.52 In fact, a more diverse microbiome is usually positively associated with health.53 However, it likely depends on the identity of the bacteria that make a more diverse community, whether health outcomes are positive or negative. Stressed females had a reduced relative abundance of Lactobacillus during pregnancy, a microbe known for its beneficial health effects.14 Early life stress did not affect the overall composition of the maternal microbiome like it did pre-conception, possibly due to a masking effect by pregnancy and lactation.
Treatment with the SSRI fluoxetine, however, was associated with major alterations in the maternal fecal microbiome. Human population-based studies also show that SSRI use associates with distinct characteristics of the microbiome.16,22,23 Moreover, recent experiments showed that FLX alters the gut microbiota in rats,24,27 and in mice.25,26,28,29 Interestingly, the specific bacteria underlying this difference do not match between studies (Table 1). Whereas we found significant increases in Prevotella and Ruminococcus relative abundance during pregnancy and lactation in FLX-treated rats, Cussotto et al. report a complete depletion of some Prevotella OTUs in FLX-treated male rats,24 and Lukić et al. found many Ruminococcaceae OTUs to be lower in FLX- and escitalopram-treated mice relative to controls.26 Whether inconsistencies like these are due to differences in life stage and sex – none of the other studies was done in females in the perinatal period – or other factors such as genetic background remains to be investigated. In addition, it is not clear how SSRIs might achieve their effect on the microbiota. In vitro studies suggest that SSRIs possess antimicrobial properties,24,30,31 possibly by inhibiting efflux pumps or by acting on bacterial homologs of the SERT or biogenic amine receptors.31,54,55 Alternatively, it is possible that SSRIs alter the intestinal habitat through their known impact on host SERT functioning and gut homeostasis.3 Our results here confirm that SSRIs have the potential to modulate the fecal microbiome during pregnancy and lactation in rats.
Table 1.
Overview of changes in the gut microbiome after FLX treatment in rodent studies.
| Study | Study design | Highlighted findings FLX-treated vs vehicle-treated animals |
Corresponds to current results? |
|---|---|---|---|
| Cussotto et al. 201924 | Male rats, 28 days of 10 mg/kg/day, in drinking water | Alpha diversity: no significant difference | Yes |
| OTU abundance: depletion of Prevotella 7 and 9 | No; we found increases in Prevotella | ||
| Lyte et al. 201925 | Male mice, 29 days of 20 mg/kg/day, oral gavage | Alpha diversity: no significant difference | Yes |
| OTU abundance: decrease in S24–7 (Bacteroidales) | Potentially; we found decreases in several Bacteroidales OTUs | ||
| Lukić et al. 201926 | Male mice, 21 days of 10 mg/kg/day, IP injections | Alpha diversity: decrease | No |
| OTU abundance: decrease in Ruminococcus | No | ||
| Zhu et al. 201927 | Male rats, 21 days of 2 mg/kg/day, oral gavage, daily stress | Alpha diversity: no significant difference | Yes |
| OTU abundance: Prevotellaceae Ga6A1 decreased, and Ruminoclostridium 6 and Ruminococcaceae NK4A214 increased | Potentially; we found increases in Ruminococcus | ||
| Fung et al. 201928 | Male and female mice, 7 days of 10 mg/kg/day, oral gavage | OTU abundance: decrease in Turicibacter | No (data not shown) |
| Sun et al. 201929 | Male mice, 21 days of 12 mg/kg/day, oral gavage, daily stress | Alpha diversity: no significant difference | Yes |
| OTU abundance: decrease in Bacteroides | Yes |
Since we have previously shown that early life stress interacts with a reduction in serotonin transporter availability to inflate its effect on the gut microbiome composition, we expected our sMV-FLX animals to show the largest effects.6 Indeed, FLX significantly affected overall bacterial community structure only in sMV females. It might be that the sMV microbiome, belonging to a depressive-like host, is more unstable and sensitive to disruptions than the cMV microbiome, especially during the dynamic perinatal period. In this proposed double-hit model, it is the combination of early life stress and FLX that shifts the balance in the gut microbial community. Analogously, another study using early life stress by maternal separation reported a change in the gut microbiome only after a second stressor in adulthood,56 strengthening this double-hit hypothesis.
Next, we tested the hypothesis that the effects of FLX on the sMV maternal microbiome during pregnancy and lactation would be reflected in an altered functional output of the microbial community. It was shown recently that fluoxetine treatment in rats is associated with several altered metabolic pathways in feces, including neurotransmitter synthesis, fatty acid metabolism, and bile acid metabolism.57 It is also known that gut microbes greatly affect host plasma and fecal metabolite availability.32,33 Microbes modify and produce amino acids, bile acids and short-chain fatty acids that become available to the host.58 Thus, we measured the concentrations of these metabolites in a subset of fecal samples that were used for microbiome analysis. Most metabolites were higher in concentration during pregnancy than during lactation, reflecting either greater metabolite intake or production during pregnancy, or more breakdown or transfer to the offspring during lactation. sMV-FLX animals had lower levels of serine during pregnancy, and lower levels of aspartic acid during lactation than sMV-Veh animals. Interestingly, amino acids that were relatively low in FLX-treated animals, such as threonine, can serve as precursors of microbial short-chain fatty acid synthesis.59 We found a corresponding decrease in isobutyric acid concentration as a result of FLX treatment. Cussotto et al. also found a lower mean isobutyric acid concentration in the feces of FLX-treated animals than in controls, although this was not a statistically significant effect.24 Overall, FLX treatment was associated with lower fecal availability of amino acids, most strongly in sMV females, pointing to a potential role for FLX in altering bacterial nitrogen regulation.60
Finally, to link microbes to metabolites, we generated correlation matrices both within the bacterial taxa that changed from pregnancy to lactation, as well as between the relative abundance of these taxa and metabolite concentrations that changed from pregnancy to lactation. We found that the relative abundance of Prevotella, enhanced in the sMV-FLX group, was negatively correlated with a cluster of Clostridiales and Desulfovibrionales genera. Interestingly, these same Clostridiales and Desulfovibrionales OTUs were positively associated with availability of amino acids (with the exception of aspartic acid). Previous literature describes that some of the most prevalent amino acid fermenting microbes indeed belong to the Clostridiales and Proteobacteria taxa.61 The second highly abundant OTU in the sMV-FLX group, Ruminococcus, was negatively correlated to the primary (beta-muricholic acid) and secondary (omega-muricholic acid) bile acids that were in pregnancy in the feces of FLX treated females, potentially mediated by a decrease in Lactobacillus relative abundance. Bile acids have been reported to induce serotonin release from enterochromaffin cells,62 hinting to a potential negative feedback mechanism. The associations we report here yield a working hypothesis that FLX during the perinatal period – at least in an animal model for depressive-like behavior – tips the balance toward higher abundance of Prevotella and Ruminococcus and lower abundance of amino acid-fermenting and bile acid-associated OTUs, leading to lower fecal availability of these metabolites. Since Prevotella and Ruminococcus are known mucin-degraders,63 and breakdown of the protective mucin layer of the gastrointestinal tract could have health effects for the host, it would be interesting to study whether mucin-degrading enzymes are enriched in FLX-treated animals in the future.
This study has several limitations. Using the 16S rRNA marker gene sequencing approach, our analysis was constrained to the genus level. Species within the same genus, however, might differ in their capacity to produce metabolites,64 which creates a limitation in finding and interpreting associations between OTUs and metabolites in this study. In addition, we are aware that the microbiome and its functional capacity varies across locations in the gastrointestinal tract,65 whereas we only used fecal samples. In fact, the effect of FLX on the microbiome likely varies along the gastrointestinal tract, since fluoxetine is absorbed almost completely.66 However, FLX can still be recovered from feces as was shown in humans,67 and in this way could have an effect even on distal portions of the gut. Lastly, since we only measured metabolite concentrations in feces, it is still unclear whether and how the effect of FLX on the gut microbiome is associated with their systemic availability.
The benefits of performing microbiome research on laboratory rodents as opposed to humans deserve mentioning here. For example, diet composition could be kept stable throughout the study, whereas diet composition changes considerably from pre-conception to pregnancy in humans and even short-term changes in diet are known to affect the gut microbiome.68,69 In addition, we are able to study the independent effect of SSRI use in rodents, as well as its interactions with a depressive-like phenotype.40 At the moment, there is a paucity of studies into the effect of SSRI on the microbiome from clinical studies on depression or animal studies mimicking this condition. For instance, it is unclear whether any effects SSRIs might have on the microbiome are part of their therapeutic potential or should rather be seen as side effects.70 However, the biggest asset of the current study is our ability to link changes to the microbiome and its predicted functional capacity to changes in metabolite availability.
In conclusion, we showed here that fluoxetine treatment modulated key aspects of maternal microbial community dynamics and metabolite output, mainly in sMV females. Our results add to the growing body of research linking serotonin signaling to the gut microbiome, as well as to the emerging awareness that the maternal microbiome is plastic and vulnerable to environmental and pharmacological insults. Maternal depression and SSRI use during pregnancy are both associated with detrimental developmental outcomes in offspring, such as lower birth weight, delayed motor development, and increased anxiety.71 Future studies will assess whether and to what extent the maternal gut microbiome and metabolome might mediate these effects. Amino acids play essential roles in both homeostatic physiology in adults as well as in development and survival of the fetus.34 Microbial molecules have been shown to transfer from mother to offspring to support the development of the brain and the immune system, emphasizing their importance.72,73 If the dampening effect of SSRI treatment on fecal amino acid concentrations is replicable, and if these concentrations correlate with plasma levels and with maternal and offspring health outcomes, modification of the maternal microbiome (by prebiotics or probiotics) or dietary supplementation of amino acids (i.e. postbiotics) alongside SSRI treatment might be explored as interventions.
Materials and methods
Experimental animals
The animals came from our colony of serotonin transporter knockout (SERT−/−, Slc6a41Hubr) Wistar rats at the University of Groningen (Groningen, the Netherlands), and were derived from outcrossing SERT+/− rats.74 Animals were supplied ad libitum with standard lab chow (RMH-B, AB Diets; Woerden, the Netherlands) and water and were kept on a 12:12 h light-dark cycle (lights off at 11:00 h), with an ambient temperature of 21 ± 2°C and humidity of 50 ± 5%. Cages were cleaned weekly, and animals were provided with a wooden stick for gnawing (10x2x2cm) and nesting material (Enviro-dri™, Shepherd Specialty Papers, Richland, MI, USA). During pregnancy, females were housed individually in type III Makrolon cages (38.2x22.0x15.0cm) and stayed with their pups until postnatal day (PND)21. Animals were weaned at PND21 and group housed in same-sex cages of 3–5 animals in type IV (55.6x33.4x19.5cm) Makrolon cages. Genotyping of the animals was performed as described previously.6 All experimental procedures were approved by the Institutional Animal Care and Use Committee of The University of Groningen and were conducted in agreement with the Law on Animal Experiments of The Netherlands.
Early life stress in a rat model of maternal vulnerability
Our rat model of maternal vulnerability (MV) consists of heterozygous SERT knockout (SERT+/−) female rats. The early life stress protocol was conducted as previously described.6 In short, SERT+/− females were mated with SERT+/− males (F0). Females were left undisturbed until delivery, which was defined as PND0. From PND2 to PND15, pups were either separated from the dam for 6 hours per day – early life stress, or handled 15 minutes per day to control for litter disturbances – control handling (Figure 1(a)). Randomization was performed by alternating assignment of stress or control condition to litters placed in the same room. Both groups were handled by the same experimenters. The SERT+/− female pups from these nests (F1) then matured to become the sMV- and cMV females.
Breeding and fluoxetine treatment
sMV and cMV females were mated with wildtype males. Females were between the age of 3 and 6 months, and mated when in estrus (checked with a disinfected impedance meter, model MK-11, Muromachi, Tokyo, Japan). This was termed gestational day (GD)0. The males were removed after 24 hours, and the females stayed isolated in a type III cage. Throughout pregnancy and lactation, from GD1 until PND21, the dams were weighed and received an oral gavage of either a clinically relevant dose of 10 mg/kg fluoxetine (FLX, Fluoxetine 20 PCH, Pharmachemie BV, the Netherlands),75 or vehicle (Veh) daily at 11:00AM (Figure 1(b)). Methylcellulose (Sigma Aldrich Chemie BV, Zwijndrecht, the Netherlands) was used for Veh injections since it is the constituent of the fluoxetine capsule. FLX (5 mg/mL) and MC (1%) solutions were prepared with autoclaved water. The gavage volume ranged from 0.9 mL to 2.0 mL (depending on body weight). Animals were treated orally by gently picking up the animal without restraint, and using flexible PVC feeding tubes (40 cm length, Vygon, Valkenswaard, the Netherlands) in order to minimize discomfort and stress in the animals. It should be noted that our lab has been confronted with thus far unexplained mortality in about one fourth of MV females as a result of fluoxetine treatment, a phenomenon we have described elsewhere.76 This may be the result of the SERT+/− genotype of the MV females. In future studies, wildtype SERT+/+ rats will have to be included in order to establish whether this is indeed a genotype * drug effect. None of the females included in the current study showed any signs of toxicity as a result of FLX. In total, 4 groups of MV females were sampled for this study: cMV-Veh (N = 20), sMV-Veh (N = 13), cMV-FLX (N = 34) and sMV-FLX (N = 25). The group sizes were uneven because we were dependent on the number of animals included in ongoing experiments. Based on the availability of fecal samples from all time points, the final number of animals selected for 16S rRNA gene sequencing for this study were 11 cMV-Veh, 8 sMV-Veh, 8 cMV-FLX and 9 sMV-FLX. Because littermates can share microbiotas, no females from the same litter were used within each group for the analysis. To this end, we randomly excluded 4 animals that had a litter mate in the same group (IDs 997, 1076, 1113, 1727), leaving us with final sample sizes of 11 cMV-Veh, 8 sMV-Veh, 7 cMV-FLX and 6 sMV-FLX.
Fecal sample collection
Fecal samples to be sequenced were collected at GD0 (before conception), GD7, GD14, PND2, PND7, PND14 and PND21 (Figure 1(b)). We chose not to collect samples closer to the day of giving birth to avoid unnecessary stress. The samples were freshly collected directly from the animal between 10:00AM and 11:00AM, and precautions were taken to minimize sample contamination, such as the use of gloves and disinfection of work surfaces. The samples were placed in clean 2.0 mL Safe-Lock Eppendorf tubes (Nijmegen, The Netherlands), snap frozen in liquid nitrogen, and stored at −80°C.
16S rRNA gene sequencing and analysis
Genomic DNA was isolated from fecal pellets using the PSP® Spin Stool DNA Kit (STRATEC Molecular GmbH, Berlin, Germany), according to the manufacturer’s instructions for difficult to lyse bacteria. About 100–200 mg fecal matter per sample was used for the DNA isolation; the remainder of the sample was stored for metabolite measurements. A dual-index sequencing strategy targeting the V4 region of the 16S rRNA gene was employed, using barcoded primers for the Illumina platform, as described previously.77 Library concentration was quantified using Qubit and an additional primer dimer clean-up step was conducted using the AMPure XP beads according to the manufacturer’s protocol (Beckman Coulter, Brea, CA, USA). Sequencing was executed on a MiSeq instrument (Illumina, San Diego, CA) using 250 base paired-end chemistry at the University of Pennsylvania Next Generation Sequencing Core. For quality control purposes, a sample of the Human Microbiome Project Mock Community was included as a positive control, and water as a negative control. Quality filtering and chimera checking yielded 7,867,454 quality-filtered sequences with a mean ± SD depth of 35,860 ± 2560 reads per sample. Paired-end reads were assembled and quality filtered to include sequences with a Q score ≥ 30.
16S rRNA gene analysis was performed as described before,36 using mothur v. 1.39 and QIIME version 1.9.1 (Quantitative Insights Into Microbial Ecology).78,79 Sequences < 248 bp and > 255 bp in length and sequences with homopolymers > 10 bp in length were removed. Operational Taxonomic Units (OTUs) at genus level were defined with 97% sequence similarity using clustering method CD-HIT. A representative sequence from every OTU containing ≥ 10 sequences was selected for downstream analyses (based on the most abundant sequence). Chimeric sequences were removed using ChimeraSlayer. Representative sequences were assigned to genera using the Ribosomal Database Project (RDP) classifier v 2.2, multiple sequence alignment was performed using PyNAST, and a phylogeny was built with FastTree. The samples were rarified to 1,000 sequences per sample before calculating diversity metrics. The Shannon diversity index was used to calculate alpha diversity, and weighted UniFrac distances were used to calculate beta diversity.
For quality control purposes, water samples and genomic DNA from abovementioned mock communities were sequenced and analyzed through the bioinformatics pipeline and the expected sequences were compared to the obtained sequences. Taxa identified as cyanobacteria or ‘unclassified’ at the phylum level were removed.
Analysis of microbiome data
The samples from GD7 and GD14 were grouped as pregnancy and the samples from PND7, PND14 and PND21 as lactation, since we observed no significant differences within these periods in terms of alpha diversity and beta diversity of the microbiome (Supplementary Figure 4). Conversely, the samples from PND2 were left out of the analysis, since they did not fit either category (Supplementary Figure 4). To identify the fecal microbial OTUs that differentiate between pregnancy and lactation, the machine-based learning algorithm Random Forests was used in R version 3.4.1 as previously described.36,80 OTU importance was ranked by the percent increase in prediction error of the model as a result of removal of that particular OTU from the model. The resulting model, derived from cMV-Veh samples, was then applied to OTU tables from the other groups, to see how early life stress, FLX and their combination alter the microbial signature of pregnancy and lactation. The R package “gplots” was used to plot OTU relative abundances as heat maps. To predict the metabolic capacity of the gut microbiome during pregnancy and lactation, a computational approach known as Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) was used.81 This algorithm uses the 16S rRNA data to predict the total metagenomic content, and these metagenomes are then mapped onto functional Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways in order to predict the full functional capacity of the microbial community, as previously described.36 Random Forests was applied to the dataset of predicted metabolic pathways to identify those that shift from pregnancy to lactation in the cMV-Veh group. Then, the impact of early life stress, FLX, and their combination on the proportional representation of these predicted microbial metabolic pathways was plotted in a heat map.
Targeted metabolomics
To draw associations between bacterial community composition, predicted gut microbial metabolic capacity, and gut metabolite output, fecal metabolites were quantified in a subset of the same samples that were previously used for 16S rRNA sequencing. Budgetary consideration prevented processing of all samples. GD7 was chosen as a representative time point for pregnancy, and PND7 for lactation. Fecal samples weighing about 300 mg from 5 randomly selected animals per treatment group were used (N = 36 in total). However, before analysis, samples from 2 animals were excluded to avoid having litter mates in the same group (see details above). Targeted metabolomics was performed by the Microbial Culture and Metabolomics Core as part of the PennCHOP Microbiome Program at the University of Pennsylvania via ultra-performance liquid chromatography (Acquity UPLC system, Waters Corporation, Milford, MA). For detailed methods, see Supplementary Table 1. Due to technical challenges, we were unable to acquire measurements of the amino acid tryptophan, the precursor to serotonin.
Analysis of metabolomics data
All metabolites that were detected in at least 40% of samples were included in the analysis. For these metabolites, missing values were replaced by half the minimum positive value found for that metabolite, assuming that missing values were the result of the concentration being below the detection limit. Random Forests was applied as described above to the metabolite concentration table from the cMV-Veh samples in order to reveal which individual metabolites were overrepresented during pregnancy or during lactation. The resulting model was then applied to the data from the treatment groups, and heat maps were generated. For predictive analysis, the open-access online platform MetaboAnalyst 3.0 was used for pathway analysis based on the metabolomics data.82 Metabolite Set Enrichment Analysis, a metabolomic version of Gene Set Enrichment Analysis, was used to investigate which sets of functionally related metabolites differ between pregnancy and lactation per treatment group.83
Additional statistical methods
Microbial alpha diversity was analyzed with a t-test to assess the difference between pregnancy and lactation over all groups using R core package “stats”. Then within each period, a two-way ANOVA was used with FLX and stress as factors. Tukey’s Honest Significant Difference post hoc tests were used for multiple comparisons (cMV-Veh vs sMV-Veh, cMV-Veh vs cMV-FLX, sMV-Veh vs sMV-FLX, cMV-FLX vs sMV-FLX). Permutational multivariate analysis of variance using distance matrices (PERMANOVA) was used to analyze effects of pregnancy vs lactation and treatment group on weighted UniFrac distances.84 PERMANOVA p-values were based on 999 Monte Carlo simulations. Because of the compositional nature of the data, OTU relative abundances were transformed before applying a two-way ANOVA and post hoc tests were as described above. First, zero values were replaced by half the minimum possible value – 0.0005 or 0.05% – and then, using R package “rgr”, relative abundances were transformed using a centered log-ratio transformation.85 Metabolite concentrations were analyzed in the same way but untransformed. Since the OTUs and metabolites analyzed in this way differed between pregnancy and lactation in the cMV-Veh group, following from the Random Forests analysis, further statistics between periods were not conducted. A t-test was used to analyze the GD0 alpha diversity difference between cMV and sMV females. Correlation analysis was done and matrices were made using “Hmisc” and “corrplot”. The statistical significance is indicated as follows: #p< .1, *p< .05; ** p< .01; and *** p< .001, **** p< .0001. Data are presented as mean ± SEM, except OTU relative abundance which is plotted as median ± IQR.
Supplementary Material
Acknowledgments
We thank Judith Swart, Wanda Douwenga and Christa Reitzema-Klein for their assistance with the early life stress procedure, drug administration and sample collection.
Funding Statement
JDAO was supported by the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska Curie Individual Fellowship under Grant 660152-DEPREG; and a NARSAD young investigator grant under Grant 25206. ASR was supported by a scholarship awarded by the Fulbright Center The Netherlands. TLB was supported by the National Institutes of Mental Health under Grant numbers P50-MH099910, MH 104184, MH 091258, MH 087597, MH 073030, and MH 108286. EJ was supported by the National Institutes of Health National Research Service Award F32 under Grant MH 109298.
Availability of data and material
The sequencing dataset supporting the conclusions of this article is available in the European Nucleotide Archive, accession number PRJEB30296 (ERP112729).
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Ethics approval
The animal study was approved by the Institutional Animal Care and Use Committee at the University of Groningen, and was conducted in accordance with the institutional guidelines and the Law on Animal Experiments.
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Coppen A. The biochemistry of affective disorders. Br J Psychiatry. 1967;113:1237–1264. doi: 10.1192/bjp.113.504.1237. [DOI] [PubMed] [Google Scholar]
- 2.Claassen V, Davies JE, Hertting G, Placheta P. Fluvoxamine, a specific 5‐hydroxytryptamine uptake inhibitor. Br J Pharmacol. 1977;60:505–516. doi: 10.1111/j.1476-5381.1977.tb07528.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spohn SN, Mawe GM. Non-conventional features of peripheral serotonin signalling-the gut and beyond. Nat Rev Gastroenterol Hepatol. 2017;14:412–420. doi: 10.1038/nrgastro.2017.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gershon MD, Tack J. The serotonin signaling system: from basic understanding to drug development for functional GI disorders. Gastroenterology. 2007;132:397–414. doi: 10.1053/j.gastro.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 5.Yano JM, Yu K, Donaldson G, Shastri G, Ann P, Ma L, Nagler C, Ismagilov R, Mazmanian S, Hsiao E. Indigenous bacteria from the gut microbiota regulate host serotonin biosynthesis. Cell. 2015;161:264–276. doi: 10.1016/j.cell.2015.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El Aidy S, Ramsteijn, AS, Dini-Andreote, F, van Eijk, R, Houwing, DJ, Salles, JF, Olivier, JD. Serotonin transporter genotype modulates the gut microbiota composition in young rats, an effect augmented by early life stress. Front Cell Neurosci. 2017;11:1–12. doi: 10.3389/fncel.2017.00222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singhal M, Turturice BA, Manzella CR, Ranjan R, Metwally AA, Theorell J, Huang Y, Alrefai WA, Dudeja PK, Finn PW, et al. Serotonin transporter deficiency is associated with dysbiosis and changes in metabolic function of the mouse intestinal microbiome. Sci Rep. 2019;9:1–11. doi: 10.1038/s41598-019-38489-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oleskin AV, Kirovskaia TA, Botvinko IV, Lysak LV. Effect of serotonin (5-hydroxytryptamine) on the growth and differentiation of microorganisms. Mikrobiologiia. 1998;67:305–312. [PubMed] [Google Scholar]
- 9.Knecht LD, O’Connor, G, Mittal R, Liu XZ, Daftarian P, Deo SK, Daunert, S. Serotonin activates bacterial quorum sensing and enhances the virulence of pseudomonas aeruginosa in the host. EBioMedicine. 2016;9:161–169. doi: 10.1016/j.ebiom.2016.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Naseribafrouei A, Hestad, K, Avershina, E, Sekelja, M, Linløkken, A, Wilson, R, Rudi, K. Correlation between the human fecal microbiota and depression. Neurogastroenterol Motil. 2014;26:1155–1162. doi: 10.1111/nmo.12378. [DOI] [PubMed] [Google Scholar]
- 11.Jiang H, Ling Z, Zhang Y, Mao H, Ma Z, Yin Y, Li L.. Altered fecal microbiota composition in patients with major depressive disorder. Brain Behav Immun. 2015;48:186–194. doi: 10.1016/j.bbi.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 12.Zheng P, Zeng B, Zhou C, Liu M, Fang Z., Xu X, Zhang, X. Gut microbiome remodeling induces depressive-like behaviors through a pathway mediated by the host’s metabolism. Mol Psychiatry. 2016;1–11. doi: 10.1038/mp.2016.44. [DOI] [PubMed] [Google Scholar]
- 13.Kelly JR, Borre Y, O’Brien C., Patterson E, El Aidy S, Deane J, Hoban AE. Transferring the blues: depression-associated gut microbiota induces neurobehavioural changes in the rat. J Psychiatr Res. 2016;82:109–118. doi: 10.1016/j.jpsychires.2016.07.019. [DOI] [PubMed] [Google Scholar]
- 14.Aizawa E, Tsuji H, Asahara T, Takahashi T, Teraishi T, Yoshida S, Kunugi H. Possible association of bifidobacterium and lactobacillus in the gut microbiota of patients with major depressive disorder. J Affect Disord. 2016;202:254–257. doi: 10.1016/j.jad.2016.05.038. [DOI] [PubMed] [Google Scholar]
- 15.Chen Z, Li J, Gui S, Zhou C, Chen J, Yang C, Hu Z, Wang H, Zhong X, Zeng L, et al. Comparative metaproteomics analysis shows altered fecal microbiota signatures in patients with major depressive disorder. Neuroreport. 2018;29:417–425. doi: 10.1097/WNR.0000000000000985. [DOI] [PubMed] [Google Scholar]
- 16.Valles-Colomer M, Falony G, Darzi Y, Tigchelaar EF, Wang J, Tito RY, Schiweck C, Kurilshikov A, Joossens M, Wijmenga C, et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat Microbiol. 2019;4:623–632. doi: 10.1038/s41564-018-0337-x. [DOI] [PubMed] [Google Scholar]
- 17.Park AJ, Collins J, Blennerhassett PA, Ghia JE, Verdu EF, Bercik P, Collins SM. Altered colonic function and microbiota profile in a mouse model of chronic depression. Neurogastroenterol Motil. 2013;25. doi: 10.1111/nmo.12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marin IA, Goertz JE, Ren T, Rich SS, Onengut-Gumuscu S, Farber E, Wu M, Overall CC, Kipnis J, Gaultier A. Microbiota alteration is associated with the development of stress- induced despair behavior. Nat Publ Gr. 2017;1–10. doi: 10.1038/srep43859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golubeva AV, Crampton S, Desbonnet L, Edge D, O’Sullivan O, Lomasney KW, Zhdanov AV, Crispie F, Moloney RD, Borre YE, et al. Prenatal stress-induced alterations in major physiological systems correlate with gut microbiota composition in adulthood. Psychoneuroendocrinology. 2015;60:58–74. doi: 10.1016/j.psyneuen.2015.06.002. [DOI] [PubMed] [Google Scholar]
- 20.Wong M-L, Inserra A, Lewis MD, Mastronardi CA, Leong L, Choo J, Kentish S, Xie P, Morrison M, Wesselingh SL, et al. Inflammasome signaling affects anxiety- and depressive-like behavior and gut microbiome composition. Mol Psychiatry. 2016;21:1–9. doi: 10.1038/mp.2016.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Mahony SM, Marchesi JR, Scully P., Codling C., Ceolho AM., Quigley EM., Dinan TG.. Early life stress alters behavior, immunity, and microbiota in rats: implications for irritable bowel syndrome and psychiatric illnesses. Biol Psychiatry. 2009;65:263–267. doi: 10.1016/j.biopsych.2008.06.026. [DOI] [PubMed] [Google Scholar]
- 22.Jackson MA, Verdi S, Maxan M-E, Shin CM, Zierer J, Bowyer RCE, Martin T, Williams FMK, Menni C, Bell JT, et al. Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nat Commun. 2018;9:1–8. doi: 10.1038/s41467-018-05184-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhernakova A, Kurilshikov A, Bonder MJ, Tigchelaar EF, Schirmer M, Vatanen T, Mujagic Z, Vila AV, Falony G, Vieira-Silva S, et al. Population-based metagenomics analysis reveals markers for gut microbiome composition and diversity. Science. 2016;352:565–569. doi: 10.1126/science.aad3369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cussotto S, Strain CR., Fouhy F., Strain RG.Peterson VL, Clarke, G, Cryan JF. Differential effects of psychotropic drugs on microbiome composition and gastrointestinal function. Psychopharmacology (Berl). 2019;236:1671–1685. doi: 10.1007/s00213-018-5006-5. [DOI] [PubMed] [Google Scholar]
- 25.Lyte M, Daniels KM, Schmitz-Esser S. Fluoxetine-induced alteration of murine gut microbial community structure: evidence for a microbial endocrinology-based mechanism of action responsible for fluoxetine-induced side effects. PeerJ. 2019;7:e6199. doi: 10.7717/peerj.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lukić I, Getselter D, Ziv O, Oron O, Reuveni E., Koren O, Elliott E. Antidepressants affect gut microbiota and ruminococcus flavefaciens is able to abolish their effects on depressive-like behavior. Transl Psychiatry. 2019;9:133. doi: 10.1038/s41398-019-0466-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu H-Z, Liang YD, Ma QY, Hao WZ, Li X. J., Wu MS, Chen JX. Xiaoyaosan improves depressive-like behavior in rats with chronic immobilization stress through modulation of the gut microbiota. Biomed Pharmacother. 2019;112:108621. doi: 10.1016/j.biopha.2019.108621. [DOI] [PubMed] [Google Scholar]
- 28.Fung TC, Vuong HE, Luna CDG, Pronovost GN, Aleksandrova AA, Riley NG, Vavilina A, McGinn J, Rendon T, Forrest LR, et al. Intestinal serotonin and fluoxetine exposure modulate bacterial colonization in the gut. Nat Microbiol. 2019;4:2064–2073. doi: 10.1038/s41564-019-0540-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sun L, Zhang H, Cao Y, Wang C, Zhao C, Wang H, Cui G, Wang M, Pan Y, Shi Y, et al. Fluoxetine ameliorates dysbiosis in a depression model induced by chronic unpredicted mild stress in mice. Int J Med Sci. 2019;16:1260–1270. doi: 10.7150/ijms.37322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muñoz-Bellido JL, Muñoz-Criado S, García-Rodríguez JA. In-vitro activity of psychiatric drugs against corynebacterium urealyticum (corynebacterium group D2). J Antimicrob Chemother. 1996;37:1005–1009. doi: 10.1093/jac/37.5.1005. [DOI] [PubMed] [Google Scholar]
- 31.Bohnert JA, Szymaniak-Vits M, Schuster S, Kern WV. Efflux inhibition by selective serotonin reuptake inhibitors in escherichia coli. J Antimicrob Chemother. 2011;66:2057–2060. doi: 10.1093/jac/dkr258. [DOI] [PubMed] [Google Scholar]
- 32.Wikoff WR, Anfora AT, Liu J, Schultz PG, Lesley SA, Peters EC, Siuzdak G. Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites. Proc Natl Acad Sci U. S. A. 2009;106:3698–3703. doi: 10.1073/pnas.0812874106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marcobal A, Kashyap PC, Nelson TA, Aronov PA, Donia MS, Spormann A, Fischbach MA, Sonnenburg JL. A metabolomic view of how the human gut microbiota impacts the host metabolome using humanized and gnotobiotic mice. Isme J. 2013;7:1933–1943. doi: 10.1038/ismej.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Herrera E. Metabolic adaptations in pregnancy and their implications for the availability of substrates to the fetus. Eur J Clin Nutr. 2000;54:S47–S51. doi: 10.1038/sj.ejcn.1600984. [DOI] [PubMed] [Google Scholar]
- 35.Nuriel-Ohayon M, Neuman H, Koren O. Microbial changes during pregnancy, birth, and infancy. Front Microbiol. 2016;7:1–13. doi: 10.3389/fmicb.2016.01031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jašarević E, Howard CD, Misic AM, Beiting DP, Bale TL. Stress during pregnancy alters temporal and spatial dynamics of the maternal and offspring microbiome in a sex-specific manner. Sci Rep. 2017;7:44182. doi: 10.1038/srep44182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gohir W, Whelan FJ, Surette MG, Moore C, Schertzer JD, Sloboda DM. Pregnancy-related changes in the maternal gut microbiota are dependent upon the mother’s periconceptional diet. Gut Microbes. 2015;0976:310–320. doi: 10.1080/19490976.2015.1086056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan I, Azhar EI, Abbas AT, Kumosani T, Barbour EK, Raoult D, Yasir M. Metagenomic analysis of antibiotic-induced changes in gut microbiota in a pregnant rat model. Front Pharmacol. 2016;7:1–11. doi: 10.3389/fphar.2016.00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Mahony SM, Clarke G, Borre YE, Dinan TG, Cryan JF. Serotonin, tryptophan metabolism and the brain-gut-microbiome axis. Behav Brain Res. 2015;277:32–48. doi: 10.1016/j.bbr.2014.07.027. [DOI] [PubMed] [Google Scholar]
- 40.Olivier JDA, Åkerud H, Sundström Poromaa I. Antenatal depression and antidepressants during pregnancy: unraveling the complex interactions for the offspring. Eur J Pharmacol. 2015;753:257–262. doi: 10.1016/j.ejphar.2014.07.049. [DOI] [PubMed] [Google Scholar]
- 41.Hechler C, Borewicz K, Beijers R, Saccenti E, Riksen-Walraven M, Smidt H, de Weerth C. Association between psychosocial stress and fecal microbiota in pregnant women. Sci Rep. 2019;9:1–10. doi: 10.1038/s41598-019-40434-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Houwing DJ, Ramsteijn AS, Riemersma IW, Olivier JDA. Maternal separation induces anhedonia in female heterozygous serotonin transporter knockout rats. Behav Brain Res. 2019;356:204–207. doi: 10.1016/j.bbr.2018.08.031. [DOI] [PubMed] [Google Scholar]
- 43.Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C. Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol. 2012;8. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Koren O, Goodrich J, Cullender T, Spor A, Laitinen K, Kling Bäckhed H, Gonzalez A, Werner J, Angenent L, Knight R, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell. 2012;150:470–480. doi: 10.1016/j.cell.2012.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DiGiulio DB, Callahan BJ, McMurdie PJ, Costello EK, Lyell DJ, Robaczewska A, Sun CL, Goltsman DSA, Wong RJ, Shaw G, et al. Temporal and spatial variation of the human microbiota during pregnancy. Proc Natl Acad Sci. 2015;112:11060–11065. doi: 10.1073/pnas.1502875112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jost T, Lacroix C, Braegger C, Chassard C. Stability of the maternal gut microbiota during late pregnancy and early lactation. Curr Microbiol. 2014;68:419–427. doi: 10.1007/s00284-013-0491-6. [DOI] [PubMed] [Google Scholar]
- 47.Bailey MT, Coe CL. Maternal separation disrupts the integrity of the intestinal microflora in infant rhesus monkeys. Dev Psychobiol. 1999;35:146–155. doi: 10.1002/(ISSN)1098-2302. [DOI] [PubMed] [Google Scholar]
- 48.De Palma G, Blennerhassett P, Lu J, Deng Y, Park AJ, Green W, Denou E, Silva MA, Santacruz A, Sanz Y, et al. Microbiota and host determinants of behavioural phenotype in maternally separated mice. Nat Commun. 2015;6:7735. doi: 10.1038/ncomms8735. [DOI] [PubMed] [Google Scholar]
- 49.Pusceddu MM, El Aidy S, Crispie F, O’Sullivan O, Cotter P, Stanton C, Kelly P, Cryan JF, Dinan TG. N-3 polyunsaturated fatty acids (PUFAs) reverse the impact of early-life stress on the gut microbiota. PLoS One. 2015;10:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O’Mahony SM, Hyland NP, Dinan TG, Cryan JF. Maternal separation as a model of brain-gut axis dysfunction. Psychopharmacology (Berl). 2011;214:71–88. doi: 10.1007/s00213-010-2010-9. [DOI] [PubMed] [Google Scholar]
- 51.Rincel M, Olier M, Minni A, Monchaux de Oliveira C, Matime Y, Gaultier E, Grit I, Helbling J-C, Costa AM, Lépinay A, et al. Pharmacological restoration of gut barrier function in stressed neonates partially reverses long-term alterations associated with maternal separation. Psychopharmacology (Berl). 2019;236:1583–1596. doi: 10.1007/s00213-019-05252-w. [DOI] [PubMed] [Google Scholar]
- 52.Rincel M, Aubert P, Chevalier J, Grohard PA, Basso L, de Oliveira CM, Helbling JC, Lévy É, Chevalier G, Leboyer M, Eberl G, et al. Multi-hit early life adversity affects gut microbiota, brain and behavior in a sex-dependent manner. Brain Behav Immun. 2019;0–1. doi: 10.1016/j.bbi.2019.03.006. [DOI] [PubMed] [Google Scholar]
- 53.Lozupone CA, Stombaugh JI, Gordon JI, Jansson JK, Knight R. Diversity, stability and resilience of the human gut microbiota. Nature. 2012;489:220–230. doi: 10.1038/nature11550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lyte M, Brown DR. Evidence for PMAT- and OCT-like biogenic amine transporters in a probiotic strain of lactobacillus: implications for interkingdom communication within the microbiota-gut-brain axis. PLoS One. 2018;13:e0191037. doi: 10.1371/journal.pone.0191037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou Z, Zhen J, Karpowich NK, Law CJ, Reith ME, Wang DN. Antidepressant specificity of serotonin transporter suggested by three LeuT-SSRI structures. Nat Struct Mol Biol. 2009;16:652–657. doi: 10.1038/nsmb.1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Barouei J, Moussavi M, Hodgson DM. Effect of maternal probiotic intervention on HPA axis, immunity and gut microbiota in a rat model of irritable bowel syndrome. PLoS One. 2012;7:1–13. doi: 10.1371/journal.pone.0046051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhao J, Jung YH, Jin Y, Kang S, Jang CG, Lee J. A comprehensive metabolomics investigation of hippocampus, serum, and feces affected by chronic fluoxetine treatment using the chronic unpredictable mild stress mouse model of depression. Sci Rep. 2019;9:7566. doi: 10.1038/s41598-019-44052-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gill SR, Pop M, Deboy RT, Eckburg PB, Turnbaugh PJ, Samuel BS, Gordon JI, Relman DA, Fraser-Liggett CM, Nelson KE. Metagenomic analysis of the human distal gut microbiome. Science (80-.). 2006;312:1355–1359. doi: 10.1126/science.1124234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lin R, Liu W, Piao M, Zhu H. A review of the relationship between the gut microbiota and amino acid metabolism. Amino Acids. 2017;49:2083–2090. doi: 10.1007/s00726-017-2493-3. [DOI] [PubMed] [Google Scholar]
- 60.Neis EPJG, Dejong CHC, Rensen SS. The role of microbial amino acid metabolism in host metabolism. Nutrients. 2015;7:2930–2946. doi: 10.3390/nu7042930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dai Z-L, Wu G, Zhu W-Y. Amino acid metabolism in intestinal bacteria: links between gut ecology and host health. Front Biosci. 2011;16:1768–1786. doi: 10.2741/3820. [DOI] [PubMed] [Google Scholar]
- 62.Kidd M, Modlin IM, Gustafsson BI, Drozdov I, Hauso O, Pfragner R. Luminal regulation of normal and neoplastic human EC cell serotonin release is mediated by bile salts, amines, tastants, and olfactants. Am J Physiol Liver Physiol. 2008;295:G260–G272. [DOI] [PubMed] [Google Scholar]
- 63.Derrien M, van Passel MW, van de Bovenkamp JH, Schipper R, de Vos W, Dekker J. Mucin-bacterial interactions in the human oral cavity and digestive tract. Gut Microbes. 2010;1:254–268. doi: 10.4161/gmic.1.4.12778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Louis P, Flint HJ. Formation of propionate and butyrate by the human colonic microbiota. Environ Microbiol. 2017;19:29–41. doi: 10.1111/emi.2017.19.issue-1. [DOI] [PubMed] [Google Scholar]
- 65.Li D, Chen H, Mao B, Yang Q, Zhao J, Gu Z, Zhang H, Chen YQ, Chen W. Microbial biogeography and core microbiota of the rat digestive tract. Sci Rep. 2017;8:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hiemke C, Hartter S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol Ther. 2000;85:11–28. doi: 10.1016/S0163-7258(99)00048-0. [DOI] [PubMed] [Google Scholar]
- 67.Lemberger L, Bergstrom RF, Wolen RL, Farid NA, Enas GG, Aronoff GR. Fluoxetine: clinical pharmacology and physiologic disposition. J Clin Psychiatry. 1985;46(3 Pt 2):14–9. [PubMed] [Google Scholar]
- 68.Hillier SE, Olander EK. Women’s dietary changes before and during pregnancy: a systematic review. Midwifery. 2017;49:19–31. doi: 10.1016/j.midw.2017.01.014. [DOI] [PubMed] [Google Scholar]
- 69.David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505:559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cussotto S, Clarke G, Dinan TG, Cryan JF. Psychotropics and the microbiome: a chamber of secrets …. Psychopharmacology (Berl). 2019;236:1411–1432. doi: 10.1007/s00213-019-5185-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Olivier JDA, Åkerud H, Kaihola H, Pawluski JL, Skalkidou A, Högberg U, Sundström Poromaa I. The effects of maternal depression and maternal selective serotonin reuptake inhibitor exposure on offspring. Front Cell Neurosci. 2013;7:73. doi: 10.3389/fncel.2013.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jašarević E, Howard CD, Morrison K, Misic A, Weinkopff T, Scott P, Hunter C, Beiting D, Bale TL. The maternal vaginal microbiome partially mediates the effects of prenatal stress on offspring gut and hypothalamus. Nat Neurosci. 2018. doi: 10.1038/s41593-018-0182-5. [DOI] [PubMed] [Google Scholar]
- 73.Pronovost GN, Hsiao EY. Perinatal interactions between the microbiome, immunity, and neurodevelopment. Immunity. 2019;50:18–36. doi: 10.1016/j.immuni.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Smits BMG, Mudde JB, van de Belt J, Verheul M, Olivier J, Homberg J, Guryev V, Cools AR, Ellenbroek BA, Plasterk RHA, et al. Generation of gene knockouts and mutant models in the laboratory rat by ENU-driven target-selected mutagenesis. Pharmacogenet Genomics. 2006;16:159–169. doi: 10.1097/01.fpc.0000184960.82903.8f. [DOI] [PubMed] [Google Scholar]
- 75.Capello CF, Bourke CH, Ritchie JC, Stowe ZN, Newport DJ, Nemeroff A, Owens MJ. Serotonin transporter occupancy in rats exposed to serotonin reuptake inhibitors in utero or via breast milk. J Pharmacol Exp Ther. 2011;339:275–285. doi: 10.1124/jpet.111.183855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Houwing DJ, Staal L, Swart JM, RamsteijnS, Wohr M, Boer S De, Olivier JD. Subjecting dams to early life stress and perinatal fluoxetine treatment differentially alters social behavior in young and adult rat offspring. Front Neurosci. 2019;13:1–15. doi: 10.3389/fnins.2019.00229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the miseq illumina sequencing platform. Appl Environ Microbiol. 2013;79:5112–5120. doi: 10.1128/AEM.01043-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Liaw A, Wiener M. Classification and regression by randomForest. R News. 2002;2:18–22. [Google Scholar]
- 81.Langille MGI, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013;31:814–821. doi: 10.1038/nbt.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Xia J, Sinelnikov IV, Han B, Wishart DS. MetaboAnalyst 3.0-making metabolomics more meaningful. Nucleic Acids Res. 2015;43:W251–W257. doi: 10.1093/nar/gkv380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Anderson MJ. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 2001;26:32–46. [Google Scholar]
- 85.Gloor GB, Reid G. Compositional analysis: a valid approach to analyze microbiome high-throughput sequencing data. Can J Microbiol. 2016;62:692–703. doi: 10.1139/cjm-2015-0821. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing dataset supporting the conclusions of this article is available in the European Nucleotide Archive, accession number PRJEB30296 (ERP112729).