

ABSTRACT
The bacterial pathogen Shigella flexneri causes more than 250 million cases of bacillary dysentery (blood in stool) every year across the world. This human-specific disease is characterized by profuse bloody diarrhea, dramatic ulceration of the colonic epithelium and immune cell infiltration of the colonic tissue. A major challenge in understanding the mechanisms supporting bacillary dysentery is the reliance on animal models that do not fully recapitulate the symptoms observed in humans, including bloody diarrhea. Here we outline advances provided by a recently developed infant rabbit model of bacillary dysentery. The infant rabbit model defines bacillary dysentery as a critical combination of massive vascular lesions and dramatic epithelial fenestration due to intracellular infection and cell-to-cell spread, respectively. The infant rabbit model provides an unprecedented framework for understanding how the cell biology of Shigella flexneri infection relates to pathogenesis.
KEYWORDS: Shigella flexneri, bacillary dysentery, T3SS, cell-to-cell spread, rabbit, epithelial fenestration, vascular lesion
Shigella flexneri is an important human pathogen
Shigella flexneri is the causative agent of bacillary dysentery (blood in stool) in humans.1 There are more than 270 million cases of shigellosis annually, resulting in more than 200,000 deaths.2 This inflammatory disease is characterized by a dramatic ulceration of the colonic mucosa,3,4 herein referred to as epithelial fenestration. S. flexneri is transmitted via the fecal-oral route and is extremely contagious. Studies in human volunteers showed that the attack rate is above 90% with an infectious dose as low as 100–1000 bacteria per individual.5 Infected patients are usually treated with fluid replacement and antibiotics. The lack of an effective vaccine and the emergence of multiple antimicrobial-resistant (AMR) strains are a major health concern worldwide.6
The cell biology of Shigella flexneri infection
Seminal studies conducted in non-human primates have revealed that S. flexneri is an intracellular pathogen that resides in epithelial cells in the colon.7 Tissue culture systems have been developed very early on to model S. flexneri intracellular invasion.8 The development of in vitro tissue culture systems was instrumental in our understanding of the molecular determinants supporting S. flexneri intracellular invasion. This led to the discovery that S. flexneri invasion relies on the presence of “the invasion plasmid”9 that harbors the 37kb “entry region”10 encoding the type-3 secretion system (T3SS). Bacterial effector proteins, which are delivered into targeted host cells by the T3SS, manipulate various cellular processes. Modulation of the actin cytoskeleton by effector proteins leads to the uptake of the bacteria by non-phagocytic cells, such as epithelial cells, into primary vacuoles (Figure 1(a)).11 Escape from primary vacuoles grants the pathogen access to the host cell cytosol (Figure 1(a)). Cytosolic bacteria express a virulence factor, IcsA,12,13 involved in the recruitment of the host cell actin assembly machinery at the bacterial pole (Figure 1(a)).14,15 Actin polymerization propels the pathogen throughout the cytosol of infected cells, and mediates the formation of membrane protrusions that project into adjacent cells at cell-cell contacts (Figure 1(a)).16-19 The formed protrusions resolve into secondary vacuoles, from which the pathogen escapes, thereby gaining access to the cytosol of adjacent cells and achieving cell-to-cell spread (Figure 1(a)).20 As bacteria grow exponentially and spread from cell to cell, single invasion events lead to the formation of infection foci harboring numerous-infected cells within a few hours (Figure 1(b), WT). This is in contrast with the spreading-defective ΔicsA mutant that is fully invasive but does not spread from cell to cell and remains confined to primarily infected cells (Figure 1(b), ΔicsA).
Figure 1.
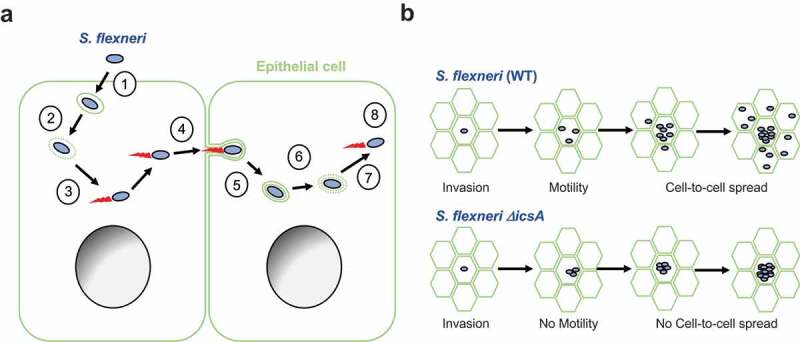
The Cell Biology of S. flexneri. (a) (1) S. flexneri (blue) utilizes its T3SS to trigger its own uptake into epithelial cells (green). (2) S. flexneri escapes primary vacuoles and gains access to the cytosol. (3) S. flexneri hijacks cytoskeleton components and displays actin-based motility. (4) At cell-cell contacts, motile S. flexneri forms membrane protrusions that (5) resolve into secondary (double-membrane) vacuoles. (6) S. flexneri escapes secondary vacuoles and (7) gains access to the cytosol of adjacent cells, where (8) it resumes actin-based motility. (b) Top. Wild type S. flexneri (WT) invades epithelial cells, grows and divides. Concurrently, bacteria acquire actin-based motility and spread from cell to cell, leading to the formation of infection foci. Bottom. The spreading-defective mutant ΔicsA invades epithelial cells, grows and divides, similar to wild type bacteria. The ΔicsA mutant does not acquire actin-based motility and consequently does not spread from cell to cell.
The infant rabbit model of bacillary dysentery
Due to the lack of small animal model of bacillary dysentery, there is a significant gap in knowledge as to how the cell biology of S. flexneri infection relates to pathogenesis. Various small-animal models have been used in the past.21-26 It is noteworthy that these animal models are not relevant to the site of S. flexneri infection in humans, the colon, nor do they recapitulate the hallmark of human shigellosis, bloody diarrhea. Recently, a guinea pig model of colonic infection was shown to display expected innate and adaptive immune responses.27 However, the observed symptoms did not include bloody diarrhea, and critical aspects of S. flexneri infection such as the involvement of the T3SS were not investigated. To address the lack of small animal model of bacillary dysentery, we tested the infant rabbit system that has been successfully used for modeling infections with various human enteric pathogens, including Vibrio cholerae and Enterohemorrhagic Escherichia coli.28,29 We found that infant rabbits infected with S. flexneri developed bloody diarrhea within 24-h post-infection.30 Histological examination revealed numerous red blood cells in the mucosa, indicative of vascular lesions (Figure 2(a), top, RBC). Extensive damage was inflicted on the epithelial structure, resulting in a nearly complete fenestration of the epithelial layer (Figure 2(a), top, EC). Additionally, human shigellosis is characterized by a massive infiltration of polymorphonuclear leucocytes (PMNs), which are referred to as heterophils in rabbits. Similar to the situation observed in infected patients, the mucosa and sub-mucosa appeared densely populated by PMNs and monocytes in infected infant rabbits. As expected, the expression of pro-inflammatory cytokines and chemokines, including IL-6 and IL-8 was dramatically induced in infected colons. Collectively, these results revealed that the infant rabbit model recapitulates all the major symptoms of human shigellosis, including massive epithelial fenestration, immune cell infiltration, and bloody diarrhea.30
Figure 2.
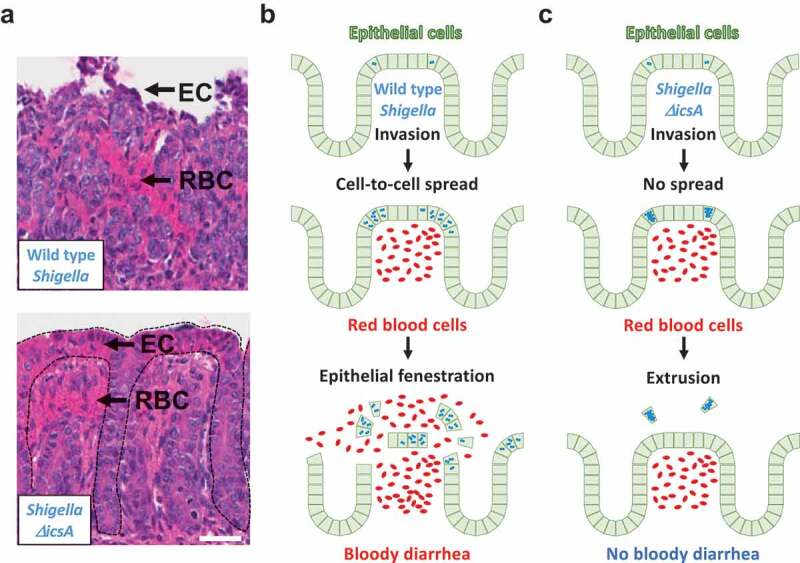
Connecting Cell Biology and pathogenesis. (a) Representative images of hematoxylin- and eosin-stained colonic mucosa of infant rabbit colon infected with wild type S. flexneri (top) and the ΔicsA mutant (bottom). EC, epithelial cell; RBC red blood cell. Dotted lines delineate crypts. Scale bar, 50 μm. (b) Top. Wild type S. flexneri (blue) invades the colonic epithelium (green). Middle. Wild type bacteria spread from cell to cell. Intracellular infection leads to vascular lesions and red blood cell infiltration (red). Bottom. The combination of epithelial fenestration due to cell-to-cell spread and red blood cell infiltration leads to bloody diarrhea. (c) Top. The ΔicsA mutant (blue) invades the colonic epithelium (green). Middle. The ΔicsA mutant does not spread from cell to cell and grows as macro-colonies. Intracellular infection leads to vascular lesions and red blood cell infiltration (red). Bottom. In absence of cell-to-cell spread, cells infected with the ΔicsA mutant are eliminated, presumably through extrusion. The epithelial structure remains intact, and the animals do not experience bloody diarrhea, or any signs of illness.
Connecting the cell biology of S. flexneri infection and pathogenesis
Using the infant rabbit model, we explored in vivo the role of bacterial factors critical to intracellular infection in tissue culture systems, including the T3SS and the dissemination factor IcsA (Figure 1(b), ΔicsA). Fluorescence microscopy revealed that S. flexneri invades epithelial cells in the infant rabbit colon (Figure 2(b), top). As expected, infection with T3SS-defective mutants was asymptomatic due to a failure to invade epithelial cells. Previous studies using the rabbit ileal loop model31 suggested that S. flexneri transit through M cells to invade epithelial cells. Our results however demonstrate that in the colon, S. flexneri directly invade epithelial cells. We next investigated the role of cell-to-cell spread using the ΔicsA bacterial mutant, which is defective in actin-based motility (Figure 1(b), ΔicsA). The ΔicsA mutant was as invasive as wild type (Figure 2(c), top), but it failed to spread from cell to cell, and grew as micro-colonies in primarily infected cells (Figure 2(c), middle). Importantly, animals infected with the ΔicsA mutant did not display any outward signs of disease, including diarrhea. Histological examination revealed that red blood cells accumulated in the colonic mucosa, indicating vascular lesions (Figure 2(a), bottom, RBC). Strikingly, the integrity of the epithelium was almost completely preserved (Figure 2(a), bottom, EC). Animals infected with the ΔicsA mutant did not lose weight and recovered rapidly after infection. These crucial results reveal the critical role of cell-to-cell spread in pathogenesis in infant rabbits. We note that evaluation of S. flexneri vaccine candidate strains suggested that the ΔicsA mutant is attenuated in non-human primates.32 The infant rabbit model, therefore, offers a unique system to investigate the molecular and cellular mechanisms supporting S. flexneri infection in vivo and determine how these mechanisms relate to dysentery, as discussed below.
Mechanisms of dysentery: epithelial fenestration
In animals infected with the bacterial mutant ΔicsA, the colon did not undergo the massive epithelial fenestration observed in animals infected with wild type bacteria (compare Figure 2(a) top and bottom, and Figure 2(b,c), bottom). These results point to the essential role of bacterial spread from cell to cell in the fenestration process. In animals infected with the ΔicsA mutant, the epithelium recovered completely within a few days and infected cells had been removed, perhaps through single-cell extrusion, leaving the rest of the epithelial structure intact. In cells infected with wild type S. flexneri, however, bacteria promptly spread from cell to cell, perhaps augmenting the shear magnitude of extrusion events, ultimately leading to total fenestration of the epithelium. In addition, we often observed clusters of cells sloughing off together in animals infected with wild type bacteria. This observation suggests that in addition to single-cell extrusion events, cell-to-cell spread may lead to dramatic exfoliation of the epithelium en masse. We have previously shown that S. flexneri utilizes its T3SS to manipulate tyrosine kinase and phosphoinositide signaling in order to successfully form and resolve membrane protrusions upon cell-to-cell spread.16,17 Moreover, the activity of various T3SS effector proteins interferes with vesicular trafficking,33,34 which may affect the composition of proteins inserted in the plasma membrane and challenge the integrity of cell-cell contacts. Thus, the infant rabbit model offers a new platform to decipher the host/pathogen interface supporting the cell-to-cell spread and the mechanisms leading to epithelial fenestration.
Mechanisms of dysentery: vascular lesions
The presence of blood in stool is a hallmark of bacillary dysentery. Our comparative analysis with bacterial mutants in the infant rabbit model provided critical insight into the mechanisms supporting bloody diarrhea. In animals infected with wild type bacteria, we observed vascular lesions as indicated by massive presence of red blood cells in the intestinal mucosa (Figure 2(a), top, RBC and Figure 2(b), middle). Importantly, we also observed vascular lesions in animals infected with the spreading-defective mutant ΔicsA (Figure 2(a), bottom and Figure 2(c), middle). Since we did not observe signs of vascular lesions in animals challenged with the invasion-defective T3SS mutant, these results indicate that vascular lesions occur in response to intracellular infection of epithelial cells, regardless of the ability of the bacteria to spread from cell to cell. We speculate that in response to the presence of bacteria in the cytosol, epithelial cells produce cytokines that may remotely affect endothelial cell junctions, leading to massive leakage of blood vessels. It is also possible that the production of chemokines leads to infiltration of immune cells, which may also affect the integrity of endothelial cell junctions. The mechanisms supporting S. flexneri cytosolic detection in vivo and leading to cytokine and chemokine expression are poorly understood. In addition to the mechanisms supporting epithelial fenestration, the infant rabbit model offers a new platform to dissect the mechanisms mediating vascular lesions.
Concluding remarks
The infant rabbit model of S. flexneri infection defines bacillary dysentery as a critical combination of massive vascular lesions and dramatic epithelial fenestration (Figure 2(b), bottom). Remarkably, in spite of the early signs of immune cell infiltration and vascular lesions, the animals infected with the spreading-defective mutant ΔicsA did not experience any signs of bloody diarrhea and related weight loss, presumably because the preserved integrity of the epithelial structure as a whole-precluded massive efflux of liquid and blood cells into the lumen (Figure 2(c), bottom). The infant rabbit model therefore highlights the critical role of cell-to-cell spread in bacillary dysentery. It also suggests that targeting cellular mechanisms that support S. flexneri spread from cell to cell may represent a promising medical countermeasure for bacillary dysentery.
Acknowledgments
We thank the members of the Agaisse Lab for stimulating discussions. This work was supported by the National Institutes of Health grant R01AI073904 (H.A).
Funding Statement
This work was supported by the National Institute of Allergy and Infectious Diseases [R01 AI073904].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Musher DM, Musher BL.. Contagious acute gastrointestinal infections. N Engl J Med. 2004;351:2417–2427. doi: 10.1056/NEJMra041837. [DOI] [PubMed] [Google Scholar]
- 2.Khalil IA, Troeger C, Blacker BF, Rao PC, Brown A, Atherly DE, Brewer TG, Engmann CM, Houpt ER, Kang G, et al. Morbidity and mortality due to shigella and enterotoxigenic Escherichia coli diarrhoea: the global burden of disease study 1990-2016. Lancet Infect Dis. 2018. doi: 10.1016/S1473-3099(18)30475-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anand BS, Malhotra V, Bhattacharya SK, Datta P, Datta D, Sen D, Bhattacharya MK, Mukherjee PP, Pal SC. Rectal histology in acute bacillary dysentery. Gastroenterology. 1986;90:654–660. doi: 10.1016/0016-5085(86)91120-0. [DOI] [PubMed] [Google Scholar]
- 4.Mathan MM, Mathan VI. Morphology of rectal mucosa of patients with shigellosis. Rev Infect Dis. 1991;13(Suppl 4):S314–8. doi: 10.1093/clinids/13.supplement_4.s314. [DOI] [PubMed] [Google Scholar]
- 5.DuPont HL, Hornick RB, Dawkins AT, Snyder MJ, Formal SB. The response of man to virulent Shigella flexneri 2a. J Infect Dis. 1969;119:296–299. doi: 10.1093/infdis/119.3.296. [DOI] [PubMed] [Google Scholar]
- 6.Puzari M, Sharma M, Chetia P. Emergence of antibiotic resistant Shigella species: a matter of concern. J Infect Public Health. 2018;11(4):451–454. [DOI] [PubMed] [Google Scholar]
- 7.Takeuchi A, Formal SB, Sprinz H. Experimental acute colitis in the Rhesus monkey following peroral infection with Shigella flexneri. An electron microscope study. Am J Pathol. 1968;52:503–529. [PMC free article] [PubMed] [Google Scholar]
- 8.Labrec EH, Schneider H, Magnani TJ, Formal SB. Epithelial cell penetration as an essential step in the pathogenesis of bacillary dysentery. J Bacteriol. 1964;88:1503–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sansonetti PJ, Kopecko DJ, Formal SB. Involvement of a plasmid in the invasive ability of Shigella flexneri. Infect Immun. 1982;35:852–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurelli AT, Baudry B, d’Hauteville H, Hale TL, Sansonetti PJ. Cloning of plasmid DNA sequences involved in invasion of HeLa cells by Shigella flexneri. Infect Immun. 1985;49:164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carayol N, Tran Van Nhieu G. The inside story of Shigella invasion of intestinal epithelial cells. Cold Spring Harb Perspect Med. 2013;3:a016717. doi: 10.1101/cshperspect.a015511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bernardini ML, Mounier J, d’Hauteville H, Coquis-Rondon M, Sansonetti PJ. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci U S A. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Makino S, Sasakawa C, Kamata K, Kurata T, Yoshikawa M. A genetic determinant required for continuous reinfection of adjacent cells on large plasmid in S. flexneri 2a. Cell. 1986;46:551–555. doi: 10.1016/0092-8674(86)90880-9. [DOI] [PubMed] [Google Scholar]
- 14.Suzuki T, Mimuro H, Suetsugu S, Miki H, Takenawa T, Sasakawa C. Neural Wiskott-Aldrich syndrome protein (N-WASP) is the specific ligand for Shigella VirG among the WASP family and determines the host cell type allowing actin-based spreading. Cell Microbiol. 2002;4:223–233. [DOI] [PubMed] [Google Scholar]
- 15.Egile C, Loisel TP, Laurent V, Li R, Pantaloni D, Sansonetti PJ, Carlier MF. Activation of the CDC42 effector N-WASP by the Shigella flexneri IcsA protein promotes actin nucleation by Arp2/3 complex and bacterial actin-based motility. J Cell Biol. 1999;146:1319–1332. doi: 10.1083/jcb.146.6.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dragoi AM, Agaisse H. The serine/threonine kinase STK11 promotes Shigella flexneri dissemination through establishment of cell-cell contacts competent for tyrosine kinase signaling. Infect Immun. 2014;82:4447–4457. doi: 10.1128/IAI.02078-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dragoi AM, Agaisse H. The class II phosphatidylinositol 3-phosphate kinase PIK3C2A promotes Shigella flexneri dissemination through formation of vacuole-like protrusions. Infect Immun. 2015;83:1695–1704. doi: 10.1128/IAI.03138-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gouin E, Gantelet H, Egile C, Lasa I, Ohayon H, Villiers V, Gounon P, Sansonetti PJ, Cossart P. A comparative study of the actin-based motilities of the pathogenic bacteria Listeria monocytogenes, Shigella flexneri and Rickettsia conorii. J Cell Sci. 1999;112:1697–1708. [DOI] [PubMed] [Google Scholar]
- 19.Kadurugamuwa JL, Rohde M, Wehland J, Timmis KN. Intercellular spread of Shigella flexneri through a monolayer mediated by membranous protrusions and associated with reorganization of the cytoskeletal protein vinculin. Infect Immun. 1991;59:3463–3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Agaisse H. Molecular and cellular mechanisms of Shigella flexneri dissemination. Front Cell Infect Microbiol. 2016;6:29. doi: 10.3389/fcimb.2016.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freter R. Experimental enteric Shigella and Vibrio infections in mice and guinea pigs. J Exp Med. 1956;104:411–418. doi: 10.1084/jem.104.3.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sereny B. Experimental keratoconjunctivitis shigellosa. Acta Microbiol Acad Sci Hung. 1957;4:367–376. [PubMed] [Google Scholar]
- 23.Voino-Yasenetsky MV, Voino-Yasenetskaya MK. Experimental pneumonia caused by bacteria of the Shigella group. Acta Morphol Acad Sci Hung. 1962;11:439–454. [PubMed] [Google Scholar]
- 24.Perdomo OJ, Cavaillon JM, Huerre M, Ohayon H, Gounon P, Sansonetti PJ. Acute inflammation causes epithelial invasion and mucosal destruction in experimental shigellosis. J Exp Med. 1994;180:1307–1319. doi: 10.1084/jem.180.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sansonetti PJ, Arondel J, Cavaillon JM, Huerre M. Role of interleukin-1 in the pathogenesis of experimental shigellosis. J Clin Invest. 1995;96:884–892. doi: 10.1172/JCI118135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sansonetti PJ, Arondel J, Huerre M, Harada A, Matsushima K. Interleukin-8 controls bacterial transepithelial translocation at the cost of epithelial destruction in experimental shigellosis. Infect Immun. 1999;67:1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shim DH, Suzuki T, Chang SY, Park SM, Sansonetti PJ, Sasakawa C, Kweon M-N. New animal model of shigellosis in the Guinea pig: its usefulness for protective efficacy studies. J Immunol. 2007;178:2476–2482. doi: 10.4049/jimmunol.178.4.2476. [DOI] [PubMed] [Google Scholar]
- 28.Ritchie JM, Rui H, Bronson RT, Waldor MK. Back to the future: studying cholera pathogenesis using infant rabbits. MBio. 2010;1. doi: 10.1128/mBio.00047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ritchie JM, Waldor MK. The locus of enterocyte effacement-encoded effector proteins all promote enterohemorrhagic Escherichia coli pathogenicity in infant rabbits. Infect Immun. 2005;73:1466–1474. doi: 10.1128/IAI.73.3.1466-1474.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yum LK, Byndloss MX, Feldman SH, Agaisse H. Critical role of bacterial dissemination in an infant rabbit model of bacillary dysentery. Nat Commun. 2019;10:1826. doi: 10.1038/s41467-019-09808-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wassef JS, Keren DF, Mailloux JL. Role of M cells in initial antigen uptake and in ulcer formation in the rabbit intestinal loop model of shigellosis. Infect Immun. 1989;57:858–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sansonetti PJ, Arondel J, Fontaine A, d’Hauteville H, Bernardini ML. OmpB (osmo-regulation) and icsA (cell-to-cell spread) mutants of Shigella flexneri: vaccine candidates and probes to study the pathogenesis of shigellosis. Vaccine. 1991;9:416–422. doi: 10.1016/0264-410x(91)90128-s. [DOI] [PubMed] [Google Scholar]
- 33.Burnaevskiy N, Fox TG, Plymire DA, Ertelt JM, Weigele BA, Selyunin AS, Way SS, Patrie SM, Alto NM. Proteolytic elimination of N-myristoyl modifications by the Shigella virulence factor IpaJ. Nature. 2013;496:106–109. doi: 10.1038/nature12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dong N, Zhu Y, Lu Q, Hu L, Zheng Y, Shao F. Structurally distinct bacterial TBC-like GAPs link Arf GTPase to Rab1 inactivation to counteract host defenses. Cell. 2012;150:1029–1041. doi: 10.1016/j.cell.2012.06.050. [DOI] [PubMed] [Google Scholar]