

ABSTRACT
User-friendly computational tools for 16S ribosomal RNA (rRNA) sequencing analysis enable researchers who are not bioinformaticians to analyze and interpret sequencing data from microbial communities. These tools’ easy-to-use interfaces belie the sophisticated and rapidly-evolving science of their underlying algorithms. When analyzing 16S data from a simple microbiome experiment, we found that superficially unimportant decisions about the bioinformatic pipeline led to results with radically different biological interpretations. We share these results as a cautionary tale whose moral is that, in 16S analysis, the devil is in the details. Wet bench researchers should therefore strongly consider partnering with bioinformaticians or computational biologists when analyzing 16S data.
KEYWORDS: Microbiota, microbiome, bioinformatics, 16s rRNA, intestinal inflammation
Methods
Helicobacter species play a complex role in human health: H. pylori causes peptic ulcers and gastric cancers but reduces the risk of inflammatory bowel disease (IBD), while enterohepatic Helicobacter species confer an increased risk of IBD.1 To explore the effect that Helicobacter species have on other members of the gut microbiota, we colonized one group of mice with a standardized gut community (altered Schaedler flora, ASF) and another group of mice with the ASF community and also Helicobacter bilis.2–4 We performed paired-end 16S sequencing on stool collected from the two groups of mice. We separately denoised forward, reverse, and merged reads with Deblur.5 Finally, we compared closed- and open-reference calling in two popular bioinformatic pipelines, Qiime 1 and Qiime 2.6,7
Results
The different analysis pipelines, run on the different read directions, reported markedly different bacterial community compositions (Figure 1). We point out three examples.
First, when using Qiime 1’s closed-reference calling on the H. bilis-positive samples, the choice of read direction led the H. bilis sequences to be identified three different ways: the reverse reads were identified as Helicobacter, the merged reads were identified as Flexispira, and the forward reads were discarded because they did not match any sequence in the reference database. If we had only used the forward reads, which may have been necessary depending on the sequencing platform and chosen primers, we might have concluded that Helicobacter did not engraft in these mice. We may not have even been aware that a significant portion of sequences were discarded, as it is a common practice for analytical pipelines to silently discard these reads. (The black bars representing discarded sequences in our figure are not present in standard Qiime output files; we added them for emphasis.)
The forward and reverse reads are both derived from the same piece of DNA, so why is one assigned to Helicobacter and the other discarded? The answer is subtle. There is no perfect match for H. bilis in the Greengenes reference database, so the sequence similarity between the sample sequence and the most similar Helicobacter database sequence varies depending on the read region. The forward read is only 96.2% similar to the corresponding region of the database sequence. This identity falls below the default 97% cutoff, and the read is discarded. The reverse sequence, on the other hand, is a perfect match to the corresponding region of the database sequence.
Second, when using Qiime 2’s closed-reference calling on the same sequences, the merged read was classified as Helicobacter, rather than the Flexispira classification made by Qiime 1. Although both Qiime 1 and Qiime 2 implement conceptually identical closed-reference calling, Qiime 1 uses one program (uclust) while Qiime 2 uses another (vsearch), each of which utilizes a slightly different search algorithm, leading to different results.8,9
Third, when using Qiime 1’s closed-reference calling on the H. bilis-negative samples, we again observed that the three read regions identified three different taxonomies for the same sequence: forward reads were classified as Lachnospiraceae, reverse reads as Bacillaceae, and merged reads were discarded. In fact, all three read sections are 100% identical to a database entry for Turicibacter, an experimental contaminant correctly identified by most of the other calling methods.
Why, if the sequences in the sample perfectly match a database entry for Turicibacter, were those sequences identified as anything else? This is because the database search algorithms used by both Qiime 1 and Qiime 2 (uclust and vsearch) are heuristic, which means that they aim to find some database match better than 97% identity, but not necessarily the best match.8,9 (This is also why, in the example above, the merged Helicobacter sequence was identified as Flexispira.) Had we not known this subtlety, we might have erroneously concluded that the experimental contaminant, Turicibacter, was one of the Lachnospiraceae sequences we expected in these mice as part of their defined ASF flora.
Discussion
Here, we show that implementing different 16S analysis algorithms to profile a commonly used standardized microbial community can lead to drastically different biological interpretations. We illustrate these differences using the popular pipelines, Qiime 1 and Qiime 2, but these general concepts hold true with any 16S analysis pipeline. Rather than say that one analysis approach is better, we merely intend to show that decisions about the bioinformatic pipeline, decisions that might appear innocuous to a novice, can have an enormous impact on the biological interpretation of the results. This variability has two important ramifications.
First, researchers must clearly report their analysis protocols in published work so that individual analyses can be reproduced and the results of different studies can be appropriately compared. This should include information about primer trimming, read merging/joining, denoising, OTU picking, database selection, and taxonomy assignment, specifying software version numbers and when default parameters were used or modified.
Second, wet bench scientists and computational biologists should work together to ensure optimal design of microbiome experiments and interpretation of the resulting sequencing data. If different analysis protocols produce results with different biological interpretations, the choice of protocol must be based on a strong understanding of the computational methods and the underlying biology. Only by working together with a strong respect for the complexity of the algorithms that underlie 16S analysis can we hope to defeat the devil in the details.
Figure 1.
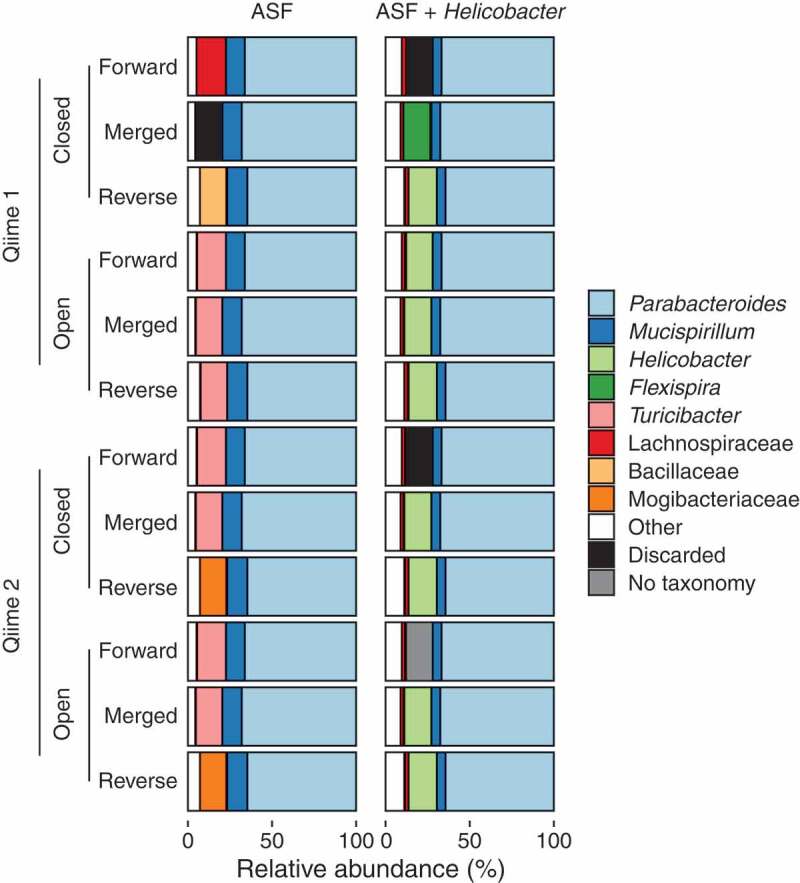
Taxonomic compositions (colors) of two samples (columns) returned by two taxonomy assignment methods (closed- and open-reference) using two different software packages (Qiime 1 and Qiime 2) using the 3 sections of paired-end 16S rRNA sequences (forward, reverse, merged).
Detailed materials and methods
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Boston Children’s Hospital Institutional Animal Care and Use Committee (Protocol Number: 14-04-2677 R).
Stool samples were collected from mice colonized with ASF with and without H. bilis by holding the mice and allowing them to defecate directly into a sterile microcentrifuge tube. DNA was extracted from stool samples using the PowerSoil DNA Isolation Kit (Mobio). The 16S rRNA V4 region was PCR-amplified using primers adapted from the 515F and 806R primers used by the Earth Microbiome Project and modified to include Illumina paired-end adaptors (forward: CTT TCC CTA CAC GAC GCT CTT CCG ATC TGT GCC AGC MGC CGC GGT AA; reverse GGA GTT CAG ACG TGT GCT CTT CCG ATC TGG ACT ACH VGG GTW TCT AAT).10 Nextera XT indices (Illumina) were attached to the 16S V4 amplicons during a second PCR step. Both PCR steps were performed using 5PRIME HotMasterMix (Quantabio). Cycling conditions for the first step were: 94°C for 3 min; 20 cycles of 94°C for 45 s, 50°C for 60 s, and 72°C for 90 s; then 72°C for 10 min. Cycling conditions for the second step were: 94°C for 3 min; 5 cycles of 94°C for 45 s, 65°C for 60 s, and 72°C for 90 s; then 72°C for 10 min. Amplicons were purified and normalized using the SequalPrep Normalization Plate Kit (Invitrogen). Pooled samples were quantified by quantitative PCR using the KAPA Library Quantification Kit (KAPA Biosystems). Paired-end sequencing was done on the Miseq platform using the 300-cycle v2 kit.
Primer sequences were removed using cutadapt (version 2.7) with default parameters, keeping only trimmed sequences.11 Trimmed forward and reverse reads were merged using fastq-join (version 1.01.759) with default parameters.12 Subsequent analysis was done using Qiime 1 (version 1.9.1) and Qiime 2 (version 2019.10) with default settings.
Funding Statement
Crohn’s and Colitis Foundation (Award #527125), Fred Lovejoy Resident Research and Education Award.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- 1.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Peña AG, Goodrich JK, Gordon JI, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37:852–857. doi: 10.1038/s41587-019-0209-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Piovani D, Danese S, Peyrin-Biroulet L, Nikolopoulos GK, Lytras T, Bonovas S.. Environmental risk factors for inflammatory bowel diseases: an umbrella review of meta-analyses. Gastroenterology. 2019;157:647–659.e4. doi: 10.1053/j.gastro.2019.04.016. [DOI] [PubMed] [Google Scholar]
- 4.Orcutt RP, Gianni FJ, Judge RJ.. Development of an “altered Schaedler flora” for NCI gnotobiotic rodents. Microecl Ther. 1987;17:59. [Google Scholar]
- 5.Wannemuehler MJ, Overstreet A-M, Ward DV, Phillips GJ. Draft genome sequences of the altered schaedler flora, a defined bacterial community from gnotobiotic mice. Genome Announc. 2014;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wymore Brand M, Wannemuehler MJ, Phillips GJ, Proctor A, Overstreet A-M, Jergens AE, Orcutt RP, Fox JG. The altered schaedler flora: continued applications of a defined murine microbial community. ILAR J Natl Res Counc Inst Lab Anim Resour. 2015;56:169–178. doi: 10.1093/ilar/ilv012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Amir A, McDonald D, Navas-Molina JA, Kopylova E, Morton JT, Xu ZZ, Kightley EP, Thompson LR, Hyde ER, Gonzalez A, et al. Deblur rapidly resolves single-nucleotide community sequence patterns. mSystems. 2017;2:e00191–16. doi: 10.1128/mSystems.00191-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinforma Oxf Engl. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 9.Rognes T, Flouri T, Nichols B, Quince C, Mahé F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4:e2584. doi: 10.7717/peerj.2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheiman J, Luber JM, Chavkin TA, MacDonald T, Tung A, Pham L-D, Wibowo MC, Wurth RC, Punthambaker S, Tierney BT, et al. Meta-omics analysis of elite athletes identifies a performance-enhancing microbe that functions via lactate metabolism. Nat Med. 2019;25:1104–1109. doi: 10.1038/s41591-019-0485-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 12.Aronesty E. Comparison of sequencing utility programs. Open Bioinforma J. 2013;7:1–8. doi: 10.2174/1875036201307010001. [DOI] [Google Scholar]