

ABSTRACT
The symptoms of infectious diarrheal disease are mediated by a combination of a pathogen’s virulence factors and the host immune system. Campylobacter jejuni is the leading bacterial cause of diarrhea worldwide due to its near-ubiquitous zoonotic association with poultry. One of the outstanding questions is to what extent the bacteria are responsible for the diarrheal symptoms via intestinal cell necrosis versus immune cell initiated tissue damage. To determine the stepwise process of inflammation that leads to diarrhea, we used a piglet ligated intestinal loop model to study the intestinal response to C. jejuni. Pigs were chosen due to the anatomical similarity between the porcine and the human intestine. We found that the abundance of neutrophil related proteins increased in the intestinal lumen during C. jejuni infection, including proteins related to neutrophil migration (neutrophil elastase and MMP9), actin reorganization (Arp2/3), and antimicrobial proteins (lipocalin-2, myeloperoxidase, S100A8, and S100A9). The appearance of neutrophil proteins also corresponded with increases of the inflammatory cytokines IL-8 and TNF-α. Compared to infection with the C. jejuni wild-type strain, infection with the noninvasive C. jejuni ∆ciaD mutant resulted in a blunted inflammatory response, with less inflammatory cytokines and neutrophil markers. These findings indicate that intestinal inflammation is driven by C. jejuni virulence and that neutrophils are the predominant cell type responding to C. jejuni infection. We propose that this model can be used as a platform to study the early immune events during infection with intestinal pathogens.
KEYWORDS: Disease model, innate immunity, proteomics, intestinal disease, pathogenesis
Introduction
The initiation of diarrheal disease is a multifactorial process involving the host immune system and the invading pathogen. Understanding the interplay between the host and pathogen is necessary for mitigating the impact of intestinal pathogens on human health. Due to widely varying virulence strategies, some bacterial pathogens cause minimal damage to the cells lining the intestinal tract, whereas other invasive pathogens cause excessive intestinal epithelial cell (IEC) death and severe gut pathology. In either case, an infection may result in an inflammatory response and loss of intestinal barrier function.1 The degradation of the intestinal barrier can facilitate the proliferation of the pathogen and potentiate more severe disease symptoms. It is not only the intestinal immune cells that mediate inflammatory responses, but the IECs are also actively involved in intestinal mucosal immunity by producing IL-8 and antimicrobial peptides.2-4 Therefore, IECs can be key players in the severity of diarrheal disease.5 Campylobacter jejuni is a common cause of intestinal illness; however, it is not known what specific host factors respond to the infection, nor is it known how quickly these host responses occur. Our present understanding of disease relies on the appearance of clinical symptoms, which takes several days. In this study, a new methodology has been developed to investigate both bacterial and host responses in an animal model.
C. jejuni is a Gram-negative, microaerophilic, curved-rod shaped bacterium that belongs to the epsilon division of the Proteobacteria. This highly motile organism is one of the most common bacterial causes of infectious gastroenteritis in humans. C. jejuni-mediated enteritis (campylobacteriosis) is characterized by acute bloody diarrhea, intense abdominal pain/cramps, and fever.6 Furthermore, due to molecular mimicry by specific serotypes of sialylated lipooligosaccharide, C. jejuni is the most common initiating event of paralyzing autoimmune disorders, including Guillain-Barré syndrome and Miller Fisher syndrome.7-9 C. jejuni is a zoonotic pathogen because it primarily resides in animals that directly or indirectly transmit the bacterium to humans. C. jejuni is particularly effective at colonizing the intestines of chickens, where it can reach bacterial loads in excess of 108 colony forming units (CFU)/gram of cecal contents without causing apparent disease.6,10 Consumption of foods contaminated by raw chicken is considered the single most common source of C. jejuni infections in humans; however, raw milk, contaminated water, and contaminated produce are also significant sources of infection.11
Human infections with C. jejuni are often associated with intense inflammatory responses in the intestine. In human infection studies, stools become C. jejuni culture-positive 48 hours after bacterial ingestion, and there is, on average, 88.5 hours between ingestion and the onset of diarrhea.12 A study that followed a single patient before and after C. jejuni infection found that there was a strong induction of IL-1β, IL-8, IL-6, IFN-γ, and C-reactive protein in the serum at 3 to 10 days after onset of symptoms.13 C. jejuni infection also leads to increased levels of Campylobacter-reactive IgA, IgG, and IgM in the blood, and the amount of antibody is correlated with the severity of the infection.12 Furthermore, histopathology of intestinal biopsies from infected individuals indicates that there is neutrophil infiltration in the intestine and mucin depletion from the epithelium.14 However, it is not known what initiates the inflammatory process. It is possible that either the resident intestinal cells sense the presence of C. jejuni, or that the process of cell invasion by C. jejuni triggers inflammatory cytokine production. Noteworthy is that C. jejuni is not a potent activator of innate immune receptors. For example, the C. jejuni flagellum is unique in that it does not activate immune sensing by TLR5. Indeed, the flagellar structure appears to cause the production of the anti-inflammatory IL-10 as a result of its glycosylation.15 Furthermore, the C. jejuni capsule reduces TLR2 and TLR4 signaling, presumably by masking the lipooligosaccharide.16 Due to the invasive nature of C. jejuni, we hypothesized that the epithelial cells targeted by C. jejuni initiate intestinal inflammation.
The goal of this study was to understand the events that initiate inflammation during C. jejuni infection. Specifically, we wanted to dissect the differences in the initiation of inflammation by a virulent C. jejuni wild-type strain and a ∆ciaD mutant that is significantly reduced in epithelial cell invasion. The ciaD gene encodes a secreted effector protein that promotes C. jejuni uptake and stimulates IL-8 secretion from cultured epithelial cells.17 In previous studies, pigs have proven useful in investigating the pathogenicity of Campylobacter due to the anatomical similarity to humans and because pigs develop C. jejuni-mediated disease that recapitulates human symptoms. Taylor and Olubunmi18 observed inflammatory lesions and shortening of the villous epithelium in the small and large intestines of weaned piglets that resulted from a natural infection with Campylobacter fetus subspecies coli. Histopathology results from this study were similar to those lesions described in human biopsies from Campylobacter-induced enteritis.14,19 Babakhani et al.20 also observed diarrhea with blood and mucus following infection of colostrum-deprived newborn piglets with C. jejuni. Histological examination of intestinal biopsies revealed rounding of the mucosal epithelial cells and cell necrosis, as evident from exfoliated cells in the lumen.20 These studies illustrate the usefulness of the piglet model in defining the pathogenesis of campylobacteriosis in an immunocompetent animal.
We utilized a pig ligated intestinal loop model to investigate the early events during C. jejuni infection and to determine the events leading to intestinal inflammation. This model permits testing of multiple intestinal loops within each animal and provides enough material for analyses of both host and pathogen responses. An additional benefit is that both test and control samples are all within one animal, reducing the confounding effects of animal-to-animal variation. In this study, we investigated the role of C. jejuni intestinal cell invasion in host immune stimulation. More specifically, a C. jejuni ∆ciaD mutant was used because it has identical surface structures as the parental wild-type strain but is lacking a secreted effector protein that potentiates intestinal cell invasion. This study design allowed us to gain a better understanding of why C. jejuni infection with an invasive strain and an attenuated mutant results in different clinical outcomes20 and to identify the source of the initial immune stimulation. We utilized histopathology, immunoassays, microbiome analysis, and proteomics to evaluate the host response, and we used transcriptomics to analyze the mRNA profile of the C. jejuni within the pig intestinal loops.
We found that inoculation of pig intestinal loops with C. jejuni results in an inflammatory response as early as 12 hours post-infection, and this was coincident with the appearance of chemotactic and antimicrobial proteins in the intestinal lumen. Based on the identity of the proteins in the intestine at 30 hours after infection, neutrophils were identified as key players during C. jejuni pathogenesis. Intestinal permeability was also increased by 30 hours, as evidenced by an increase in the concentration of IL-17A. The appearance of neutrophil-related proteins and histopathological lesions were greater after inoculation with the fully virulent C. jejuni wild-type strain than the ∆ciaD mutant. This data suggests that the CiaD-mediated process of cellular invasion by C. jejuni enhances cytokine production and activated neutrophil influx. Furthermore, detailed proteomic analysis of the intestinal lumen during inoculation with C. jejuni revealed numerous antimicrobial proteins that are indicators of intestinal infection and revealed signaling pathways that participate in the development of the disease.
Results
Establishment of pig ligated intestinal loops
Minnesota Mini Pigs were obtained from the National Swine Research and Resource Center located at the University of Missouri-Columbia and confirmed to be Campylobacter culture negative by plating fecal suspensions on Campy-Cefex agar. Intestinal loops were constructed by double ligating 10-cm sections of the intestine beginning at the ileocecal junction and creating loops toward the jejunum. The ileum, which is the distal portion of the small intestine, was chosen because it has been shown to have the highest microbial load of any location in the small intestine, while still retaining the ability to initiate an innate immune response.21 The loops were inoculated in an alternating pattern, where C. jejuni-infected loops were separated by a control loop. Two loops in each animal were inoculated with the C. jejuni wild-type strain, and two loops were inoculated with a C. jejuni ∆ciaD mutant.17 The intestine was returned to the abdominal cavity, and the pig was maintained under general anesthesia between 3 and 30 hours. Three animals were sampled at 3, 6, 12, and 30 hours after infection. A schematic representation of the ligated loops and a representative image of the intestinal tissue after removal from the animal is shown in Figure 1. Gross pathological differences between infected and control loops were not apparent in the intestinal tissues at any time point. The pigs remained stable over the course of the 30 hours under anesthesia; however, it was necessary to provide a 50% oxygen/50% air mix, as opposed to the more commonly used 100% oxygen, to prevent injury to the lungs. Beginning 4 to 6 hours after anesthesia, it was also necessary to supplement the saline drip with glucose to prevent hypoglycemia. None of the animals developed a fever.
Figure 1.
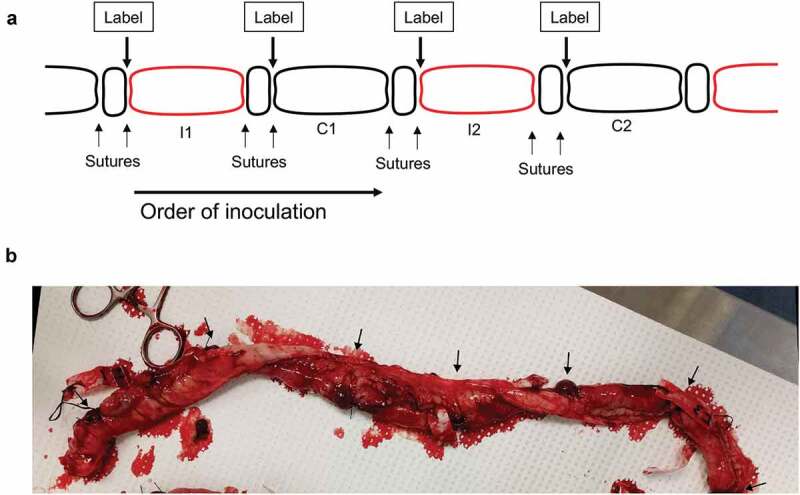
Schematic of the ligated loop creation and representative image of tissue removed from the pig. Panels a: Schematic representation of the intestinal pig loops, beginning at the ileocecal junction at the left, and ascending into the small intestine to the right. Loops in red were inoculated with C. jejuni, and loops in black are control loops. ‘I’ indicates an infected loop, and ‘C’ indicates a control loop. b: Representative tissue removed from the pig at 30 hours post-infection, oriented in the same direction as in Panel A. Arrows indicate the location of the double ligation between each individual intestinal loop.
C. jejuni is active in the intestinal loops
We initially assessed whether the C. jejuni in the intestinal loop were metabolically active. Because the intestine rapidly absorbed the liquid used for bacterial inoculation, it was necessary to add 5 mL of PBS to the loops to collect the luminal contents. Intestinal contents were serially diluted and plated on selective Campy-Cefex agar to enumerate the number of C. jejuni. Viable C. jejuni was not recovered from any of the control loops (limit of detection ~ 102 CFU/mL). For the loops that were inoculated with either the C. jejuni wild-type strain or the ∆ciaD mutant, the number of C. jejuni recovered was always greater than the inoculum, indicating bacterial replication in the intestine. Comparing bacterial abundance over time, the median bacterial load of the C. jejuni wild-type strain increased 2.6-fold from 3 to 6 hours, remained stable from 6 to 12 hours, and then decreased ~25% from 12 to 30 hours. The C. jejuni ∆ciaD mutant had a similar but delayed trend as compared to the wild-type strain. For the C. jejuni ∆ciaD mutant, the median bacterial load in the loops remained unchanged from 3 to 6 hours, increased 1.4-fold from 6 to 12 hours, and then decreased ~50% from 12 to 30 hours (Figure 2). A caveat of these findings is that this is the number C. jejuni in the lumen of the intestine and does not account for the tissue-associated bacteria. Overall, these data indicate that the C. jejuni were replicating within the intestine. Noteworthy is that the CFU of both the C. jejuni wild-type strain and ∆ciaD mutant decreased at 30 hours post-infection compared to 12 hours; it is possible that the decrease in recoverable bacteria is due to C. jejuni invasion into the tissue, killing by the immune system, or a combination of both.
Figure 2.
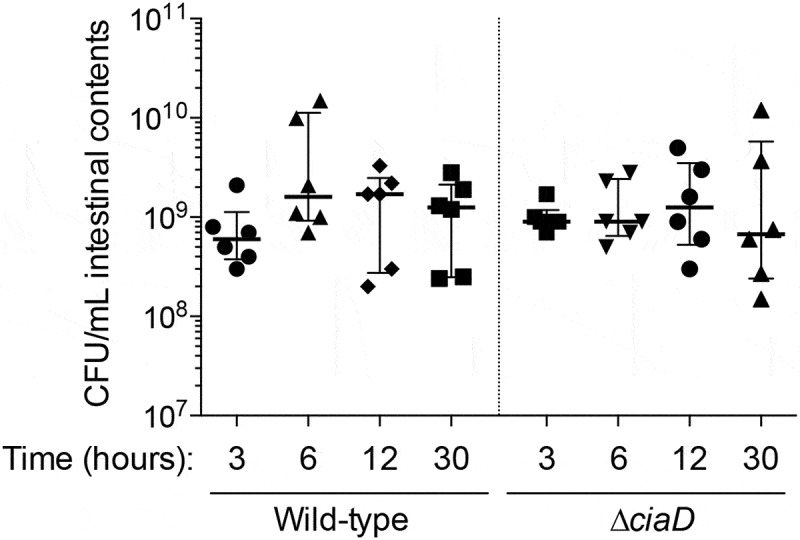
Total colony forming units (CFU) of C. jejuni recovered from the pig loops for each of the three experiments. All loops were inoculated with 10 mL of C. jejuni between 0.8–4 × 109 CFU/mL in phosphate buffered saline. C. jejuni was recovered from the loops at the indicated time points by plating serial dilutions on Campy-Cefex agar. Each dot indicates the CFU/mL from an individual loop. The bars indicate the median ± interquartile range from the indicated samples.
The intestinal environment induces transcriptional changes in C. jejuni
Given the indication that the C. jejuni were metabolically active and multiplying in the intestinal lumen, we then performed RNA-Seq to determine if virulence gene expression specifically occurred in the intestine. The transcriptome of C. jejuni recovered from the intestinal loops was compared with the culture used for inoculation (time zero). While we sequenced samples from every animal, we only acquired sufficient reads (> 10,000) from the 3 and 6 hour samples to determine changes in gene expression. Unfortunately, the 12 and 30 hour samples were not able to be evaluated due to an overabundance of eukaryotic RNA. Nevertheless, to help understand the early signals that C. jejuni responds to in the intestinal environment, the data from the pig intestinal loops was compared to C. jejuni cultured in MH broth with 0.05% sodium deoxycholate (DOC, previously published data)22 and C. jejuni cultured with IPEC-J2 cells (immortalized pig intestinal cells). All comparisons were made against the ‘input’ samples grown in MH broth to provide consistency and highlight the contributions of the specific condition. Noteworthy is the C. jejuni wild-type strain invades IPEC-J2 cells (not shown). These comparisons allowed us to determine if the responses to the intestinal environment were consistent with either exposure to bile or exposure to cells.
Comparing the C. jejuni transcriptome between in vivo and in vitro conditions
Hierarchical clustering was used to evaluate overall trends in the RNA-Seq data (Figure 3) and to help determine what C. jejuni was sensing in the intestine. The analysis resulted in the division of the samples into 5 clusters based on similarity of gene expression (labeled 1, 2, 3, 4A, and 4B). A complete list of the genes in each cluster is in Supplementary Table 1. The most apparent trend in the clustering is that pig and DOC-generated samples are markedly different. Most of the genes with increased expression in DOC had decreased expression in the pig, and most of the genes that had lower expression in DOC had increased expression in the pig; the pig and DOC samples have opposite expression patterns. This result may reflect the fact that in the ileum, where the samples were collected, there is a low concentration of bile, as bile is absorbed for reuse along the length of the small intestine.23
Figure 3.
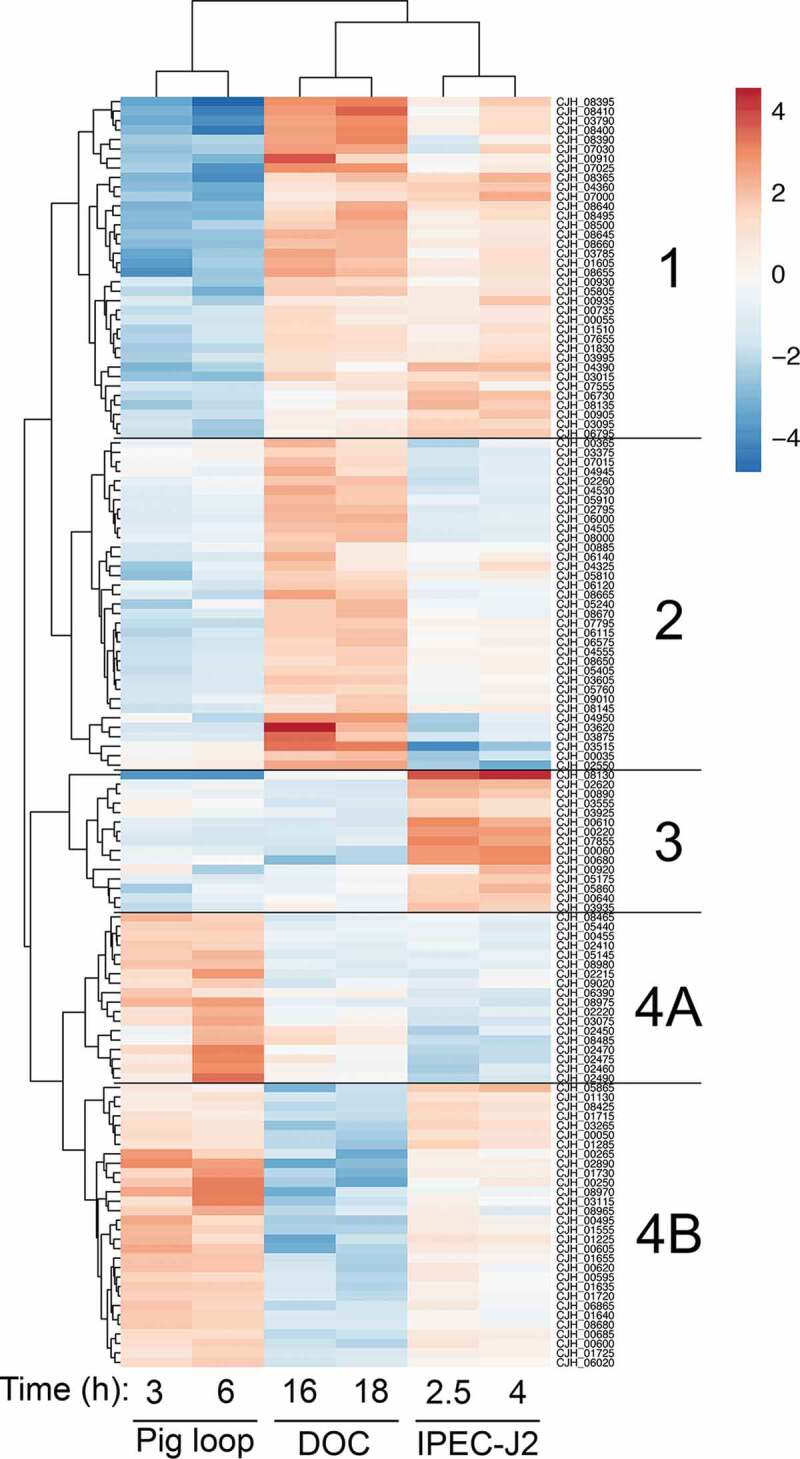
RNA-Seq of the C. jejuni in the pig loops indicates similarities and differences to in vitro culture conditions. The transcriptome of the C. jejuni in the ligated pig loop at 3 and 6 hours post-infection was compared to the transcriptome of C. jejuni cultured in sodium deoxycholate (DOC) for 16 and 18 hours and to C. jejuni co-cultured with IPEC-J2 cells for 2.5 and 4 hours. In each condition, the samples were compared to the respective ‘input’ sample of C. jejuni grown in MH broth. Data was filtered to keep genes that had a Benjamini-Hochberg adjusted p-value (q-value) of ≤ 0.05, and a log2(fold change) of greater than 1.5 (or less than −1.5) in at least four of the six conditions. Hierarchical clustering was applied to the dataset, and this revealed 5 clusters of expression patterns, labeled as 1, 2, 3, 4A, and 4B. In general, the C. jejuni cultured with DOC was the least similar to the C. jejuni recovered from the ligated pig loop. The C. jejuni cultured with the pig intestinal cell line IPEC-J2 had some similarities with both the C. jejuni cultured with DOC and recovered from the pig loop. In the diagram, red indicates genes with increased expression and blue indicates genes with decreased expression. The scale bar indicates log2(fold change).
In contrast to the DOC, C. jejuni gene expression within the intestinal loop was similar to the expression during co-incubation with cultured IPEC-J2 intestinal porcine enterocytes. While there were genes regulated in the opposite direction (clusters 1, 3, and 4A, n = 69 genes), there were two clusters of genes that were regulated similarly in the pig and IPEC-J2 cells (clusters 2 and 4B, n = 65 genes). Cluster 2 has decreased expression in both the pig loop and co-incubation with IPEC-J2 cells and is enriched in the COG category “Inorganic ion transport and metabolism” (enriched 5.08 fold, q-value = 0.016), which is primarily made up of membrane transporters. Cluster 4B has genes that have increased expression in both the pig loop and IPEC-J2 cells and is enriched in the COG category “Amino acid transport and metabolism” (enriched 3.91 fold, q-value = 0.082), which is comprised of the components of the tryptophan biosynthesis pathway. The transcriptome similarity to cultured cells, rather than DOC, indicates that C. jejuni likely penetrates the intestinal mucus, and gains access to the epithelial cells by 3 hours post-infection. This finding indicates that the C. jejuni encounters signals in the IPEC-J2 cell culture system that are also in the intestine. It is important to note that these signals are not limited to the cells themselves, but also include the components of the tissue culture medium and the relative metabolite and oxygen concentrations.
Unique in vivo expression responses induced by the pig
While in vitro tissue culture results in similar expression patterns when compared to the intestinal loop, there were genes in clusters 1 and 4A that had unique expression in the in vivo environment, which contains elements such as mucus and resident microbiota. Genes in cluster 1 had less expression in the pig compared to IPEC-J2 and DOC, while genes in cluster 4A had higher expression in the pig compared to IPEC-J2 and DOC. The unique genes that were in cluster 1 (lower expression in the pig) were enriched in the COG category “Inorganic ion transport and metabolism” (enriched 10.8 fold, q-value = 4.5 × 10−7). This category included several iron acquisition genes (chuA, chuB, chuD, ceuB, cfbpA, cfbpB, cfrA, and cj1658), catalase (katA), and the transcriptional regulator perR. Cluster 4A (higher expression in the pig) is not significantly enriched in any COG category (q-value > 0.6); this category contains several metabolic genes (sdhA, sdhB, leuA, leuB), the flagellar component flgE, and the adhesin flpA. The differential expression of these genes in the pig intestines suggests that the intestinal environment provides cues to the C. jejuni that are not present in the in vitro conditions.
In summary, we concluded that the C. jejuni are not experiencing stress in the initial stages of intestinal colonization at 3 and 6 hours when injected into the intestinal loops. This is based on the fact that iron acquisition systems are not upregulated, in fact, some are downregulated, and there is an absence of reactive oxygen species (ROS) responsive genes. Indeed, a key regulator of ROS stress responses, perR, has reduced expression in the pig intestinal loop.24 Instead, the C. jejuni are increasing expression of genes involved in amino acid synthesis and tissue adherence through the FlpA adhesin during the early stages of intestinal colonization. We hypothesize that the expression of other virulence-related proteins is altered later than 6 hours during infection.
Initiating the intestinal inflammatory response
C. jejuni causes an increase in intestinal IL-8 and TNF-α
A critical question to be answered by this study is whether the initiation of intestinal inflammation was dependent on the ability of C. jejuni to invade the IECs. To address this question, luminal contents were evaluated for the presence of acute inflammatory cytokines IL-8 and TNF-α at 3, 6, and 12 hours after infection. Compared to the control loops, we found that the amount of IL-8 in the lumen of the intestine is significantly increased in the loops infected with the C. jejuni wild-type strain at 12 hours post-infection. In contrast, the amount of IL-8 in the loops infected with the C. jejuni ∆ciaD mutant was not significantly increased above the control loops (Figure 4a). The amount of TNF-α in the intestine followed the same trend, where loops infected with the C. jejuni wild-type strain had increased amounts of TNF-α at 12 hours post-infection when compared to the controls (Figure 4b). Although not statistically different from the matched control, the average amount of TNF-α at 12 hours post-infection was roughly equivalent between loops inoculated with the C. jejuni wild-type strain and the ∆ciaD mutant. No correlation was observed with the level of IL-8 or TNF-α and the loop position (i.e., proximal or distal end of the series of loops) (Supplementary Figure 1). Furthermore, no correlation was found between the experimental animal and the level of any of the immune cytokines. These data indicate that the inflammatory cytokine response in the loops is independent of the location of the loop, is reproducible among animals, and is driven by the presence of C. jejuni. Based on these results, we concluded that the innate immune response is initiated by 12 hours with the C. jejuni wild-type strain, while this induction is attenuated by infection with the ∆ciaD mutant. Thus, early intestinal inflammation appears to be related to the ability of C. jejuni to invade cells.
Figure 4.
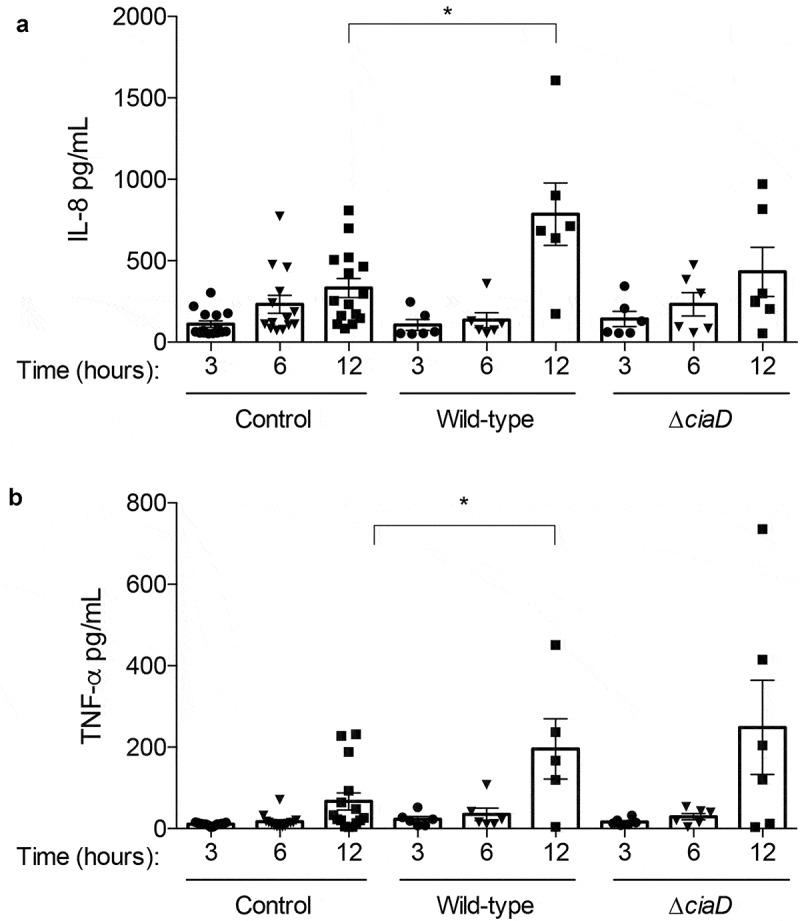
C. jejuni causes an increase of intestinal IL-8 and TNF-α. Cytokines in the lumen of the intestinal loops were evaluated by an ELISA. The concentration of each cytokine is presented for each animal, where a single point represents the concentration in a single loop. Panels a: The luminal concentration of IL-8 is increased in loops inoculated with the C. jejuni wild-type strain at 12 hours post-infection and was not significantly increased by the C. jejuni ∆ciaD mutant compared to the controls at each time point. b: Levels of TNF-α are significantly increased in loops inoculated with the C. jejuni wild-type strain at 12 hours post-infection and was not significantly increased by infection with the ∆ciaD mutant compared to the controls at each time point. Bars indicate mean ± standard error, * indicates p < .05 by one-way ANOVA followed by Sidak’s multiple comparisons test.
Kinetic changes in neutrophil protein abundance in the intestine
We evaluated the proteome from the supernatant of the intestinal loops to further understand the events occurring in the intestine. The proteome of intestinal supernatants was evaluated at 3 and 12 hours after infection, and the data from infected loops were compared to control loops (Supplementary Table 2). Because the initial analysis suggested that neutrophil markers were abundant in the infected loops, we determined the time frame in which the neutrophil specific proteins appeared. We used CD177 as the primary indicator of neutrophil abundance.25 CD177 was not detectable at 3 hours post-infection but was present at 12 hours post-infection in both the loops inoculated with the C. jejuni wild-type strain and the ∆ciaD mutant. Notably, the amount of CD177 was lower in loops inoculated with the ∆ciaD mutant compared to inoculation with the wild-type strain (Figure 5). Additionally, the abundance of CD177 correlated with other neutrophil markers, including matrix metallopeptidase 9 (MMP9), lipocalin 2 (LCN2), elastase (ELANE), and proteinase 3 (PRTN3). These results are consistent with the finding that the C. jejuni wild-type strain causes accumulation of both TNF-α and IL-8 in the intestinal lumen 12 hours post-infection, while the ∆ciaD mutant induces lower concentrations of IL-8. These data demonstrate that neutrophils are recruited to the site of the infection in a manner that is consistent with the ability of C. jejuni to invade cells.
Figure 5.
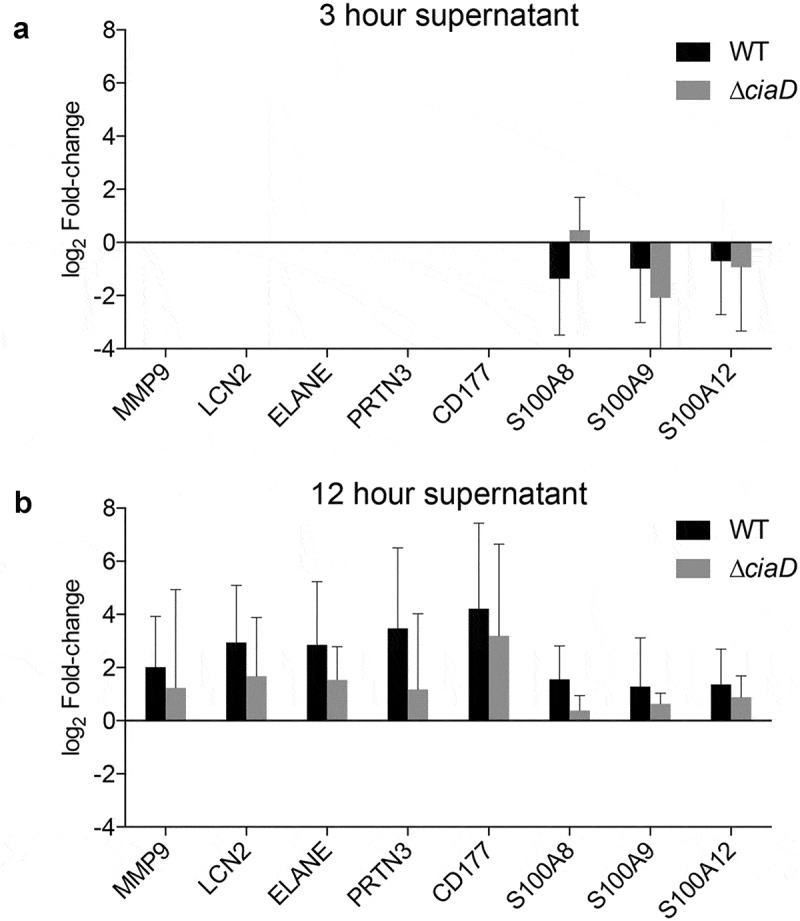
Neutrophil markers increase in the intestinal supernatant in response to C. jejuni infection. The proteome from intestinal loop supernatants at 3 and 12 hours was compared to the matched controls at each time point. Initial analysis indicated enrichment of immune related proteins in the intestinal loops, so the specific abundance compared to control loops log2 (fold change) of the proteins that make up the neutrophil degranulation pathway were plotted. In general, proteins in the neutrophil degranulation pathway were not detected at 3 hours (Panel a) post-infection but were detected at 12 hours (Panel b) post-infection with C. jejuni. Furthermore, the amount of most of the neutrophil markers were higher after a 12 hour incubation period with the C. jejuni wild-type strain than with the C. jejuni ∆ciaD mutant. Error bars indicate standard deviation.
Early signs of disease in the intestine
Histopathological lesions are present in infected loops
Tissues infected with the C. jejuni wild-type strain and non-infected tissues from the three 30-hour experiments were examined for histological changes caused by infection. In the infected loops from two of the three animals, enterocytes at the villous tips were often distorted. Occasional enterocytes were hypereosinophilic and had dense chromatin, indicating necrosis. Associated enterocytes were rounded and dissociated. These markers were not present in the C. jejuni ∆ciaD mutant-infected nor in the non-infected tissues. Overall, the lesions were consistent with Campylobacter infection. These findings support the proposal that damage of the IECs occurs after 30 hours. Representative microscopic sections of the intestinal loops are presented in Figure 6a-c.
Figure 6.
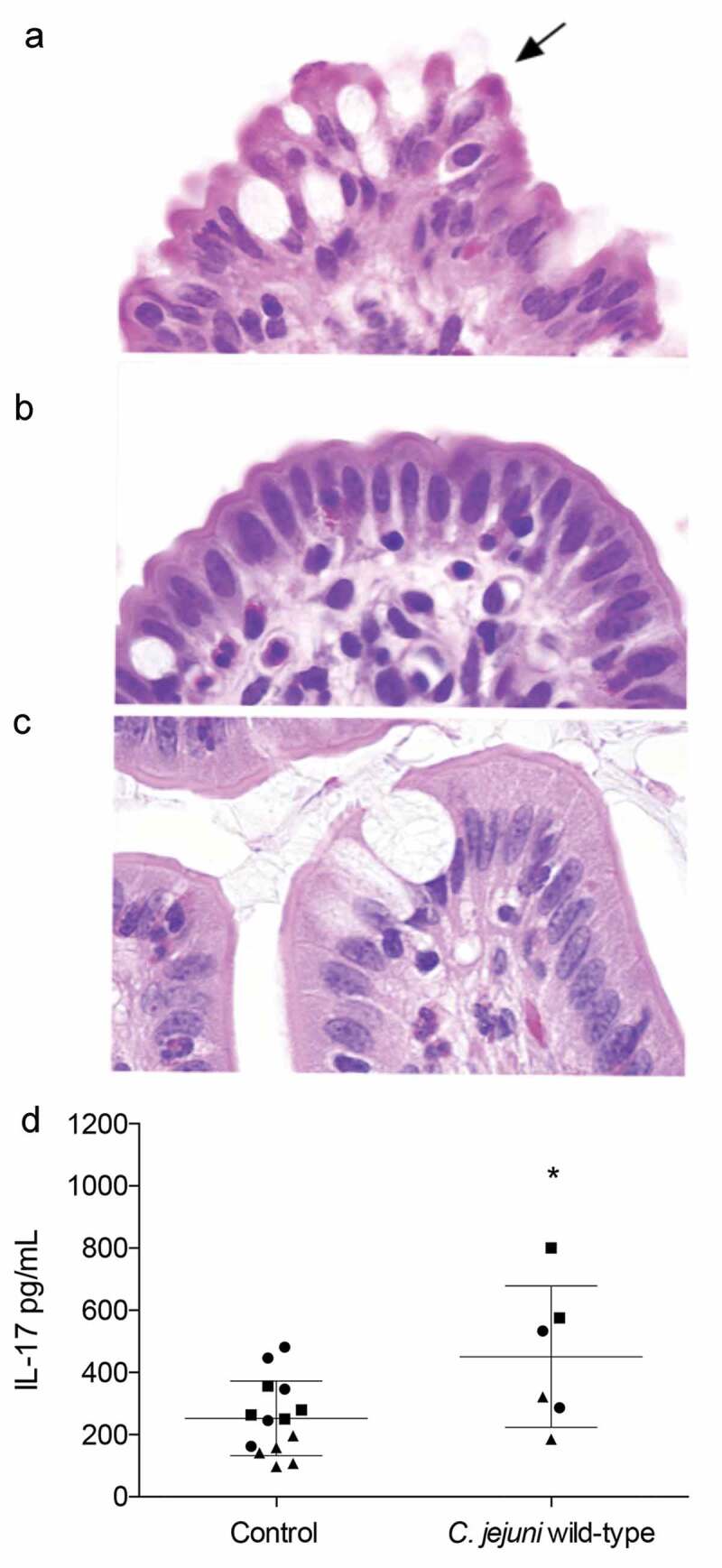
C. jejuni infection induces mild lesions and IL-17A production. Intestinal segments at 30 hours post infection were fixed in neutral buffered formalin, sectioned, and stained with hematoxylin and eosin. All images were taken at 100x magnification. Minimal peracute necrosis was observed in the C. jejuni infected loops. Panels: a: In the loop inoculated with the C. jejuni wild-type strain, enterocytes on the villous tips were mildly distorted. Rarely, cells had hypereosinophilic cytoplasm and a pyknotic nucleus (black arrow), indicating necrosis. b: In the loop inoculated with the C. jejuni ∆ciaD mutant, the villous enterocytes were histologically normal. c: In the control loop, the villous enterocytes were histologically normal. d: Levels of IL-17A were modestly (but statistically significantly) increased in C. jejuni wild-type infected loops. Each animal is represented by a different shape, where the circles represent animal 1, squares represent animal 2, and triangles represent animal 3. Bars indicate mean ± standard deviation, * indicates p ≤ 0.05 by Student’s t-test.
IL-17A is induced by C. jejuni infection
We tested the intestinal lumen for the presence of IL-17A, which is an immune cytokine indicative of compromised intestinal barrier integrity. At 30 hours after infection, IL-17A was detected at a mean level of 450.59 pg/mL in the loops inoculated with the C. jejuni wild-type strain compared to 252.3 pg/mL in the control loops (Figure 6d). The induction of IL-17A in infected loops indicates that there is a specific and localized response to C. jejuni infection. Furthermore, this finding also indicates that there is a significant delay between the appearance of C. jejuni in the intestine and the beginning of intestinal barrier disruption. This is consistent with the finding that humans become culture positive in the stool approximately 40 hours before the onset of diarrheal symptoms.
Intestinal microbiota is consistent among animals
Shifts in intestinal microbiota are frequently associated with disease states, so we determined the total microbiome of the intestinal loops from every time point by 16S rRNA gene sequencing. Approximately 50% of the reads from the infected samples originated from C. jejuni. Because this was a known experimental manipulation, we compared the microbiome from the C. jejuni-infected loops, after the removal of the C. jejuni sequence reads, to that of the non-infected loops. The microbiome findings are summarized in Figure 7. No significant differences were observed in the richness or diversity measures of the microbiota between the C. jejuni-infected and non-infected control loops at any given time point. Primarily, the observable differences reflected the incubation time and not the presence/absence of C. jejuni in the loop. We sampled both the luminal contents and mucosal scrapings for microbiota analysis. While there were small differences in the microbial composition between the mucosal and luminal samples, they largely reflected the same trends in that there were no differences between infected and non-infected loops at a given time point. Importantly, the microbiome data indicated that the microbial communities in the intestine of the three experimental animals at any given time point were consistent. Because these experiments were conducted on different days from individually housed animals, this provides support that the experimental results were not due to varying microbiota among the animals.
Figure 7.
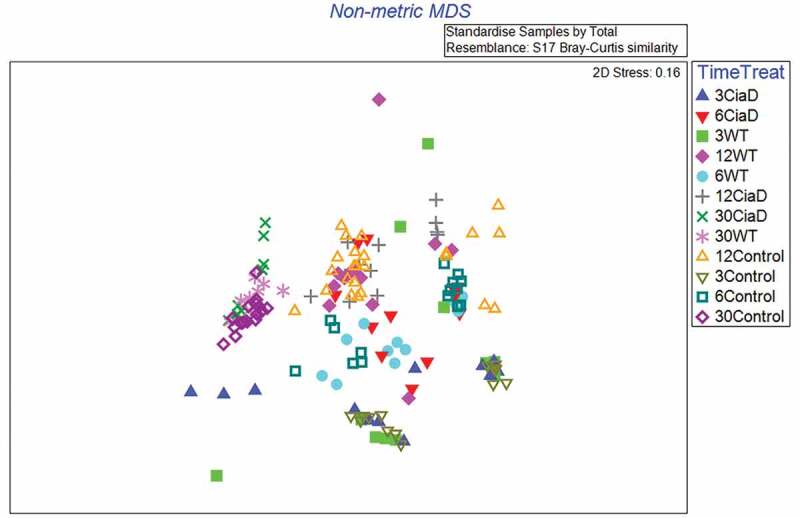
Microbiota in the ligated intestinal loops is insensitive to C. jejuni infection. The microbiota of the luminal contents and mucosal scrapings were analyzed by deep sequencing the 16S rRNA gene. Because the addition of C. jejuni was a known experimental manipulation in the loops, we removed the C. jejuni reads from subsequent analysis so we could observe changes in the resident microbiota. The data below is summarized in a non-metric multidimensional scaling (MDS) plot where data from each individual loop is plotted as a single point. Points are differentiated by both time of incubation (in hours) and infection state. From this data, treatment with C. jejuni does not meaningfully change the microbial composition when compared to the control at the same time point, however, the duration of incubation does alter the microbiome. Because each individual animal was assayed on independent days, the data also indicates that the microbiota among the animals was consistent.
Evaluation of the intestinal proteome
Proteomic analysis of the intestinal loops identifies immune markers
We sought to characterize the intestinal proteome in response to the invasive C. jejuni wild-type strain and noninvasive ∆ciaD mutant early in infection. Samples from the lumen of the intestine were evaluated using a label-free quantitative proteomic approach (Supplementary Table 3). In all of the analyses, the C. jejuni-inoculated loops were compared to the control, non-inoculated, intestinal loops at the same time point. Because the intestine is a complex tissue, it was not possible to determine protein abundance changes within a single cell type (i.e., epithelial cells), but the appearance of specific proteins was used to identify specific cell types (i.e., neutrophils) and broad trends in protein abundance.
Immune pathways involved in C. jejuni infection
The entire proteome from the intestinal lumen after 30 hours of infection was analyzed with the Reactome pathway database26 to determine pathway enrichment and guide our analysis. Proteins from both the supernatant and pellet that had a paired p-value of < 0.05 and a log2(fold change) > 0 were combined and analyzed to discover the changes occurring in response to C. jejuni. The analysis was limited to proteins increased in abundance, as we reasoned that these proteins would reveal the host factors that were induced in response to C. jejuni, or the cell types recruited to the areas of infection. From this list of proteins, the top enriched Reactome pathway was ‘Neutrophil Degranulation’ (q-value = 0.0016), which included the CD177 protein, a surface glycoprotein predominantly present on neutrophils. The amount of the S100A8, S100A9, and S100A12 proteins also increased; these three proteins comprise up to 35% of the cytosolic protein content of a neutrophil and are potent neutrophil chemoattractants.27 Further evidence of neutrophil infiltration was the increase in the proteins MMP9, LCN2, ELANE, MPO, and PRTN3 in the lumen of the C. jejuni-inoculated loops compared to the uninoculated loops (Figure 8, Supplementary Figure 2). These proteins are associated with neutrophils and have antimicrobial activities. In total, proteomic analysis has allowed for the development of a model revealing that C. jejuni triggers the influx of neutrophils to the intestine early in infection, and that this process depends on the C. jejuni secreted effector protein CiaD. These findings highlight multiple antimicrobial proteins involved in iron sequestration and bacterial killing in the intestine, including lipocalin-2 and myeloperoxidase.28,29 This data also suggests that once neutrophils are present, further neutrophil recruitment could occur via the S100A8 and S100A9 proteins, which are strong chemoattractants.30
Figure 8.
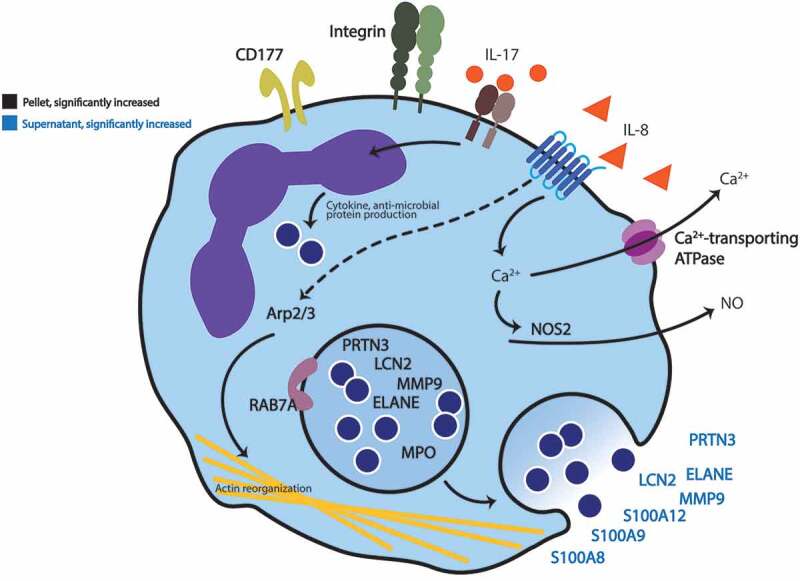
Graphical summary of proteomic analysis. Proteomic analysis of the intestinal loops at 30 hours post infection indicates infiltration of neutrophils. Luminal contents from ligated intestinal loops that were infected with C. jejuni were separated into pellets and supernatants by centrifugation, the proteome of each was determined, and then compared to non-infected loops. Proteins indicated on this model were significantly increased in abundance (p-value of ≤ 0.05 and a log2 fold-change > 0) in intestinal loops infected with C. jejuni compared to non-infected loops. The proteins identified are broadly defined as participating in neutrophil function and include: Integrins (αMβ2), CD177, Arp2/3, RAB7A, and NOS2. Additionally, neutrophil-related secreted proteins were detected: S100A8, S100A9, S100A12, LCN2, MMP9, PRTN3, and neutrophil elastase (ELANE). Bold black text indicates proteins found in the pellets collected from the intestine, and bold blue text indicates proteins found in the intestinal supernatant. Non-bold text indicates that the proteins were identified by immunoassay (IL-8 and IL-17A) or are included for clarity and were not directly identified (Ca2+ and NO).
In addition to immune-related proteins, components of the Arp2/3 complex, which facilitate actin rearrangement, were increased in abundance in the C. jejuni-inoculated loops. One of the key requirements for C. jejuni invasion of epithelial cells is activating the actin rearrangement and adhesive machinery. While the specific cell-type producing these proteins is not known, it is known that C. jejuni relies on actin reorganization to invade epithelial cells.31 However, given the apparent abundance of neutrophil-related proteins, an increase in actin remodeling proteins could also be a result of neutrophil migration into the intestine. This influx of neutrophils, and their killing associated antimicrobial proteins, was observed at the 12 hour and 30 hour time points. Thus, we propose that neutrophils drive intestinal inflammation and the initiation of diarrhea in C. jejuni-infected individuals.
Determining the role of epithelial cells in initiating immune responses
C. jejuni induces IL-8 in cultured epithelial cells in a CiaD dependent manner
The neutrophils detected in response to C. jejuni inoculation may have been recruited by chemoattractants produced from resident intestinal cells. To determine a possible source of the initial chemoattractants and to understand if this process happens in human cells, we tested the ability of C. jejuni to induce IL-8 from intestinal epithelial cells and macrophage-like cells. To take into account the role of cellular invasion in this process, we first tested if the C. jejuni were capable of invading human T84 (immortalized colon cells) cells using a gentamicin-protection assay. We found that the invasion of C. jejuni into T84 cells was dependent on the presence of CiaD, as the invasion of the ∆ciaD mutant was significantly less than the wild-type strain (Figure 9a, p < .05). The amount of invasion by the ∆ciaD mutant was equivalent to ∆flgL mutant, which lacks motility, a functional flagellar secretion apparatus, and does not invade at all (negative control). Complementation of the ∆ciaD mutant restored invasiveness in T84 cells. Furthermore, the amount of IL-8 secreted from the T84 cells was evaluated after a 24 hour incubation period (Figure 9b). The trends in the IL-8 production were similar to the invasion trends, where the C. jejuni wild-type strain resulted in more IL-8 in the supernatant than the ∆ciaD and ∆flgL mutants (p < .05). This indicates that C. jejuni invasion of the T84 cells participates in IL-8 production, which can promote the recruitment of neutrophils.32
Figure 9.
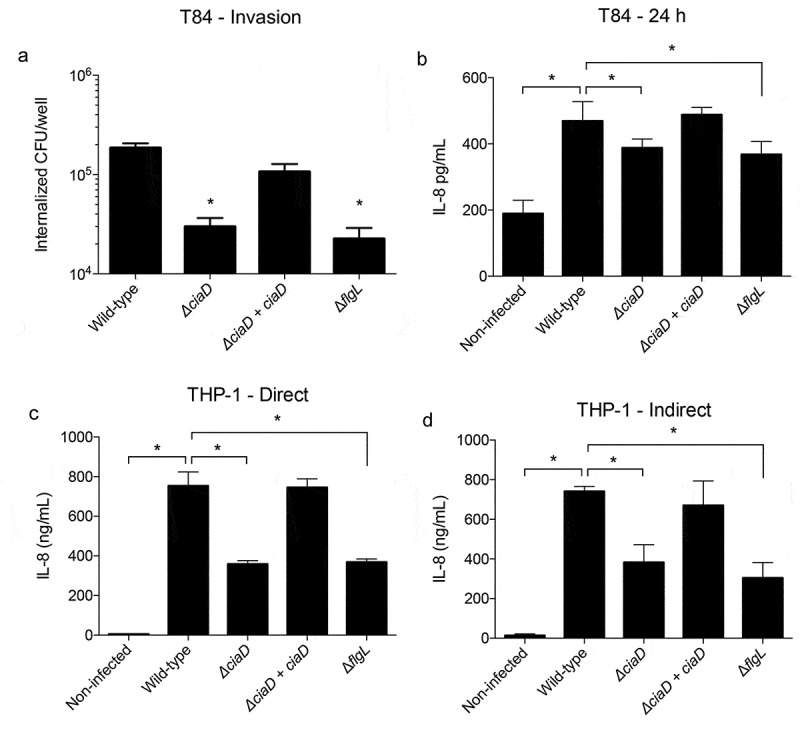
Trends in immune cytokine production are consistent between the pig model and cultured human cells. Panels: a: The invasion potential of C. jejuni into cultured human colonic (T84) cells was tested using a gentamicin protection assay. T84 cells were infected with a C. jejuni wild-type isolate, a ∆ciaD mutant, a complemented ∆ciaD mutant, and a ∆flgL mutant (negative control). The number of bacteria surviving gentamicin treatment (which kills extracellular bacteria) was counted and plotted. b: The amount of IL-8 induced by C. jejuni from T84 cells was measured after 24 hours of incubation. c: Human macrophage-like (THP-1) cells were infected with C. jejuni (direct infection) and the amount of IL-8 produced was measured after 24 hours. d: Cell-free conditioned medium from infected T84 cells (panel B) was used to treat THP-1 cells, and the amount of IL-8 induced was measured. It was found that the cell-free medium induces similar amounts of IL-8, as direct infection with C. jejuni. Bars indicate mean ± standard deviation, * indicates p < .05 by one-way ANOVA followed by Sidak’s multiple comparisons test.
Secreted factors from epithelial cells can induce IL-8 from THP-1 macrophage-like cells
To test if there were differences in the responses of epithelial cells and immune cells to C. jejuni, we infected differentiated THP-1 cells, which are macrophage-like cells that are commonly used to model human monocytes. THP-1 cells were directly infected with the C. jejuni wild-type strain, ∆ciaD mutant, and ∆flgL mutant for 24 hours, and secreted IL-8 was measured (Figure 9c). The trends observed were similar to that of T84 cells, where the wild-type strain resulted in a greater level of IL-8 than both the ∆ciaD and ∆flgL mutants, and the amount of IL-8 induced by the ∆ciaD and ∆flgL mutants was roughly equivalent. Complementation of the ∆ciaD mutant restored secretion of IL-8 compared to infection with the C. jejuni wild-type strain. As would be expected from immune cells, the quantity of IL-8 secreted from THP-1 cells is significantly higher than T84 cells, with secretion in the ng/mL range rather than the pg/mL range. These findings indicate that both epithelial cells and resident immune cells may act together to recruit neutrophils to the intestine.
To determine the extent by which epithelial cells and macrophages work together to promote inflammatory cytokine secretion, cell-free conditioned medium from T84 cells incubated with (or without) C. jejuni for 24 hours was added to THP-1 cells. The bacteria-free supernatant (no culturable C. jejuni) was able to stimulate the same responses in the THP-1 cells as direct infection with C. jejuni, suggesting that the C. jejuni-infected human T84 epithelial cells are capable of secreting factors that initiate IL-8 secretion from THP-1 cells (Figure 9d). This finding supports the proposal that C. jejuni does not need to directly interact with tissue-resident immune cells to promote IL-8 secretion, and that this process is correlated with the ability of C. jejuni to invade the epithelial cells. In total, this data is consistent with the data collected from the pig intestinal loops, where the C. jejuni ∆ciaD mutant causes less immune cytokine production and raises the possibility of an epithelial cell-driven mechanism of immune activation.
Discussion
An animal model for a human disease is fundamental to understanding the disease process and dissecting the pathogen-host interaction when it is not practical to do so in humans. In this study, we have used a pig ligated intestinal loop model to gain insight into the development of C. jejuni-mediated disease and to understand the events that initiate inflammation during C. jejuni infection. This model was chosen because the symptoms and pathologies that develop in pigs mimic human campylobacteriosis. We have found that C. jejuni replicate within the pig intestine, trigger the production of immune cytokines, and cause an influx of neutrophils. This process begins with a motile C. jejuni. The flagellum is essential for motility, however, unlike most other bacteria, the C. jejuni flagellum does not activate the TLR5 receptor.15,33 In combination with chemosensing proteins, there is directional motility of C. jejuni toward mucus and epithelial cells.34-37 C. jejuni reaches the basolateral surface of the epithelial cells by migrating between the cells (paracellular transmigration) utilizing the HtrA protease, which is secreted into the surrounding space and deposited in outer membrane vesicles. The HtrA protease cleaves the cellular tight junction proteins occludin and E-cadherin.38-40 C. jejuni then uses the CadF and FlpA adhesins41,42 to bind fibronectin on the basolateral surface of the cells.43,44 C. jejuni invade the IECs in a manner that is dependent on secreted effector proteins, including the Cia proteins and FedA.17,45-48 In particular, CiaD facilitates cellular invasion by activating the MAPK pathway, leading to cortactin phosphorylation and host cell membrane ruffling.49 The process of cellular invasion by C. jejuni has been coined the ‘binding and effector mechanism’.50 During the process of cellular invasion, C. jejuni stimulates the release of IL-8 (primarily from the basolateral surface) of cells.51-53 The increased concentration of IL-8 results in neutrophil influx, and ultimately leads to tissue damage (Figure 10). Although it is not clear where the neutrophils become activated, it is evident that there are activated neutrophils in the intestinal lumen based on the increase of CD11b (ITGAM) protein.54
Figure 10.
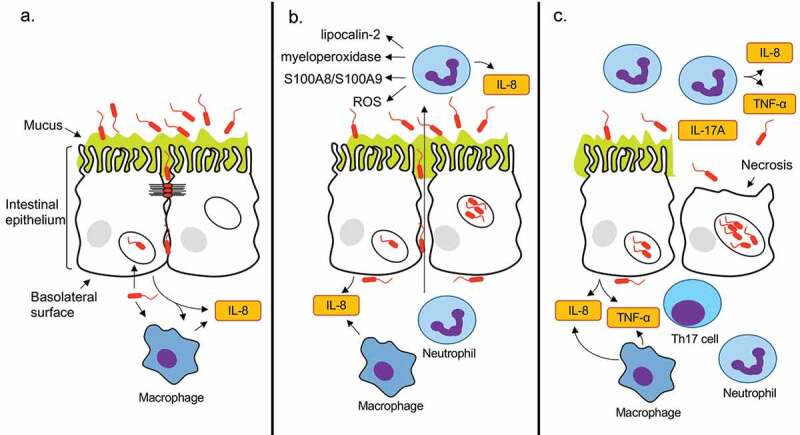
C. jejuni-mediated intestinal inflammation is driven by epithelial cell invasion. Panels: a: After penetrating the intestinal mucus, C. jejuni translocate to the basolateral surface of the epithelial cells by transiently disputing the tight junctions with the secreted protease HtrA. C. jejuni then invades the epithelial cells and triggers interleukin 8 (IL-8) secretion at the basolateral surface of the epithelial cells. Resident monocytes (macrophages) are also stimulated both directly and indirectly to secrete IL-8. b: Due to the elevated levels of IL-8, there is activated neutrophil influx to the lumen of the intestine. Antimicrobial neutrophil proteins, including lipocalin-2, myeloperoxidase, S100A8, S100A9, and an increase in reactive oxygen species (ROS) kill C. jejuni that are not internalized within epithelial cells. As the intestinal inflammation proceeds, intestinal barrier disruption is initiated, permitting more C. jejuni access to the basolateral surface where invasion occurs. c: As the infection continues, the amount of luminal TNF-α increases and C. jejuni multiply within intestinal epithelial cells resulting in necrosis and increased intestinal barrier disruption, as evidenced by the appearance of IL-17A. Ultimately, these processes are potentiated by C. jejuni cell invasion and lead to the development clinical symptoms in a host. Cytokines are indicated in yellow boxes.
A goal of this study was to determine whether the initiation of intestinal inflammation was dependent on the ability of C. jejuni to invade the IECs. In this regard, we found that the amount of both IL-8 and TNF-α cytokines in the intestinal lumen increased sooner after inoculation with the virulent C. jejuni wild-type strain compared to the ∆ciaD mutant. This result is consistent with the finding that a ciaD deletion mutant caused less IL-8 production from THP-1 and T84 cells. Furthermore, CiaD itself may be participating in the IL-8 induction, as prior studies have shown that ectopic expression of CiaD-EGFP in human epithelial cells causes IL-8 production.17 We also utilized proteomic approaches to determine the host response to C. jejuni infection and found that neutrophil markers dominated the changes in the intestinal lumen. Macrophage-specific markers CSF1R, CD68, and CD16355 did not increase in the intestine due to C. jejuni inoculation. Using an in vitro model, intestinal epithelial cells were tested for their ability to indirectly signal to immune cells to prompt cytokine production. Relevant to the findings with the animal model, the ability of T84 epithelial cells to induce IL-8 secretion from THP-1 macrophage-like cells was dependent on the presence of CiaD and the ability of C. jejuni to invade cells. Using this newly developed pig model of C. jejuni disease, we have gained significant insight into the intestinal environment during the early process of infection and the process of initiating intestinal inflammation during C. jejuni infection.
Given that human disease symptoms develop 48–72 hours post-ingestion, we targeted earlier time points to determine the specific events that begin the inflammatory process. We determined that inflammatory cytokine induction (IL-8 and TNF-α) was most pronounced during inoculation with the invasive C. jejuni wild-type strain. These cytokines increased in concentration much earlier than the onset of disease symptoms, as soon as 12 hours after infection. Because epithelial cells primarily secrete IL-8 and TNF-α at the basolateral surface, the luminal IL-8 and TNF-α is either the result of a disruption of the intestinal barrier or produced from immune cells recruited to the lumen.51,56 In humans, IL-8 and TNF-α are produced from C. jejuni-specific CD4 + T-cells and are thought to further potentiate infection due to the promotion of inflammation.57 At 30 hours post-infection, there were mild histopathological lesions and an increase in IL-17A, which is a cytokine associated with a ‘leaky gut’ and appears to be associated with the development of inflammatory bowel syndrome and intestinal fibrosis.58 Specifically, IL-17A is produced by differentiated Th17 cells, which further points to the beginning of an adaptive immune response being mounted against the C. jejuni. Compared to the intestinal pathology, which is quite mild at 30 hours after infection, the intestinal cytokines were significantly altered as early as 12 hours after infection. This difference in timing suggests that C. jejuni causes intestinal inflammation very early during colonization, and further indicates that the disease outcomes of invasive and attenuated C. jejuni diverge well before the necrosis caused by C. jejuni intracellular replication.
Proteomic analysis of the intestinal contents revealed key neutrophil markers, and the specific host antimicrobial factors that are deployed by the host to combat C. jejuni infection. We used Gene ontology (GO) term enrichment analysis to determine if there were relevant protein functions enriched in the proteomic data set. The top categories of proteins that increased in abundance were ‘positive regulation of establishment of protein localization to telomere’ and ‘azurophil granule.’ Despite the category names, most of these proteins are involved in neutrophil degranulation. Furthermore, analysis of the data set with the Reactome pathway database identified ‘Neutrophil degranulation’ as the most highly enriched pathway. While neutrophils are a key mediator of the innate immune response, they are also a cause of intestinal damage during inflammatory responses.59 This inflammation-driven intestinal damage is hypothesized to potentiate further intestinal infection by disrupting the barrier between the intestinal epithelial cells.60 The proteome data ultimately points to neutrophil infiltration into the intestine. This trend has been observed in human infections where elevated levels of the neutrophil proteins S100A8, S100A9, and S100A12 are detectible in the stool.61 The S100A8 and S100A9 proteins are antimicrobial factors and potent neutrophil chemoattractants;30,62 these proteins also promote neutrophil activation, and S100A9 potentiates IL-8 production from neutrophils in response to other stimuli.63 In addition to these three proteins, our study has uncovered additional host factors that appear in the intestine during C. jejuni infection, including antimicrobial neutrophil related proteins.
The immune responses to C. jejuni appear to coincide with the ability of C. jejuni to invade epithelial cells. When the pig intestinal loops were infected with a C. jejuni isolate lacking CiaD, a protein that is important for cellular invasion and IL-8 induction,17 the amount of TNF-α in the lumen of the intestinal loops was lower compared to loops infected with the C. jejuni wild-type strain. To investigate the mechanistic basis for the observation, we infected human colonic epithelial cells (T84) and macrophage-like cells (THP-1) with the C. jejuni wild-type strain and the ∆ciaD mutant. Direct infection of both cell types (T84 and THP-1) with the C. jejuni wild-type strain resulted in high levels of IL-8 induction and lower IL-8 induction by the ∆ciaD mutant. Furthermore, cell-free conditioned medium from infected T84 cells was able to induce an IL-8 response in THP-1 cells that was nearly identical to direct infection of the THP-1 cells. This suggests that both direct and indirect actions by C. jejuni are able to modulate immune cell responses. This finding supports a model whereby the intestinal epithelial cells act as the front-line defense against C. jejuni and have the ability to initiate neutrophil influx. We propose that the initial influx of neutrophils potentiates further immune cell recruitment and intestinal inflammation. In other words, the early events during C. jejuni colonization determine the course of disease symptoms. Because the C. jejuni wild-type strain is capable of invading epithelial cells, it may use this strategy to avoid being killed by neutrophils recruited to the intestine by inflammatory cytokines. We propose that the C. jejuni ∆ciaD mutant, which is attenuated in invasion, lacks the ability to evade intestinal immune cells and is killed by these cells. This proposal is further supported by the finding that the decrease in luminal C. jejuni from 12 to 30 hours is greater in the ∆ciaD mutant than the wild-type strain.
Analysis of the C. jejuni within the intestine indicated unique in vivo responses to the pig ligated loop. More specifically, the early C. jejuni transcriptome indicated that C. jejuni was not responding to bile, but rather host cells and other intestinal components. It is possible that some of the in vivo responses, which included reduced expression of iron acquisition genes (chuA, chuB, chuD, ceuB, cfbpA, cfbpB, cfrA, and cj1658) and oxidative stress response genes (katA), were due to the intestinal mucus. It has been observed that incubating C. jejuni with mammalian mucus results in similar changes in gene expression when compared to avian mucus.64 In contrast, it has been found that in the chicken cecum, there is increased expression of chuA, chuB, chuD, and katA compared to growth in MH broth.65 This further indicates that C. jejuni initiates different transcriptional responses during mammalian disease compared to colonizing a commensal avian host.
In the present study, we have defined an important animal model to study the host and bacterial factors that initiate intestinal inflammation during C. jejuni infection. The key advantage of this model is the ability to study immune responses that are likely to mimic the human infection process. Using this system, we have found that C. jejuni induces an immune response as early as 12 hours after infection that is characterized by neutrophil influx, highlighting a key time during the infection process. Furthermore, the early immune responses in the intestine are dependent on a virulent C. jejuni. This data supports a model where virulent C. jejuni cause rapid cytokine production from resident intestinal cells (Figure 10), while an attenuated C. jejuni ∆ciaD mutant results in a delayed cytokine response. Using the immune signaling platform established by the proteomic evaluation of the intestinal loops, it will be possible to specifically target C. jejuni virulence determinants to determine the mechanisms of disease progression in the intestine.
Materials and methods
Bacterial strains
The C. jejuni F38011 wild-type strain was grown on Mueller-Hinton agar plates supplemented with 5% citrated bovine blood (MHB) or in MH broth in a microaerobic atmosphere (5% O2, 10% CO2, 10% H2, 75% N2) at 37°C. The C. jejuni ∆ciaD mutant and complemented isolates were generated as described previously.17 When needed, MHB agar plates were supplemented with 128 µg/mL of tetracycline or 8 µg/mL of chloramphenicol. The isolates were routinely passaged every 24 to 48 hours on MHB agar. Colony-forming unit (CFU) determination from fecal and intestinal samples (described below) was done by serially diluting the samples and plating on Campy-Cefex agar,66 a selective medium, and incubating for 48 to 72 hours.
Pig acquisition and transport
This study was performed in accordance with the National Research Council’s Guide for the Care and Use of Laboratory Animals (8th edition) and approved by the Washington State University Institutional Animal Care and Use Committee under protocol ASAF 6236. Pigs (Sus scrofa domesticus or Minnesota mini pigs) of both sexes were obtained from the National Swine Research and Resource Center located at the University of Missouri-Columbia. All twelve pigs, three per experimental time point, were culture-negative for C. jejuni and Campylobacter coli prior to shipment, as judged by streaking fecal samples onto Campy-Cefex agar plates and incubating the plates at 37°C in a microaerobic atmosphere for 72 hours. At the time of arrival, the pigs were 20–23 weeks of age and weighed between 47 and 82 lbs. Pigs were given feed [Mazuri Mini Pig Active Adult (Land O’Lakes, Inc., Arden Hills, Minnesota)] and water ad libitum. The pigs were given oral ampicillin (20 mg/kg) 5 and 7 days prior to surgery to enhance Campylobacter colonization.67 At the time of the experiments, the pigs were between 27 and 39 weeks of age and between 70 and 90 lbs.
We chose to utilize Minnesota minipigs from the University of Missouri National Swine Resource and Research Center (NSRRC) after unsuccessful attempts to use locally sourced Yorkshire cross pigs. Regardless of the time of year (May to November), we were unable to obtain Yorkshire pigs locally that were free from C. coli. Although the amount of culturable C. coli in the stool was less than 1 × 104 CFU/g and was reduced to undetectable levels by treatment with ampicillin, there were still detectable levels of C. coli in the distal region of the intestine by culture. In studies up to 24 hours in duration, we did not observe any C. jejuni induced disease symptoms in the locally sourced pigs that were C. coli positive. To avoid the confounding factor of resident Campylobacter, we obtained pigs that were free of resident C. coli from the NSRRC.
Anesthesia
All animals were fasted from solid food for 12 hours before the surgery. For sedation, the pigs received an intramuscular injection of 0.05 mg/kg of dexmedetomidine (Dexdomitor® 0.5 mg/mL, Orion Corporation, Finland) and 10 mg/kg of ketamine (Ketaset® 100 mg/ml, Zoetis Inc., Spain). The animals were transported to the surgical area when non-responsive to handling. If more sedation was needed, an additional 5 mg/kg ketamine was given intramuscularly. The pigs were placed on the surgical table at sternal recumbency. A face mask was applied to the animal’s snout for additional muscle relaxation with 5% isoflurane (isoflurane USP, Akorn Inc, USA) in 100% oxygen. Promptly, the animals were orotracheally intubated with a cuffed endotracheal tube. Once the endotracheal tube was secured and pressure checked, it was connected to a mechanical ventilator (Tafonius junior, Hallowell EMC, USA) to maintain normocapnia. Anesthesia was maintained with isoflurane (1–2%) in 50% oxygen and 50% medical air delivered via a circle rebreathing system. During the entire duration of the anesthesia, the animals were monitored continuously using multiparametric monitoring equipment (DPM™6, Mindray DS, USA). The cardiovascular and pulmonary variables monitored were: heart rate via electrocardiography, respiratory rate, blood oxygen saturation, pulse rate, ETCO2, noninvasive arterial blood pressure (i.e., systolic, diastolic, and mean arterial blood pressures), and body temperature. A heating pad was used to maintain the pig’s body temperature above 98 °F (HotDog™ Veterinary Warming Controller, Augustine Biomedical and Design, USA). To maintain normotension, vasopressors were given at a constant rate of infusion when mean arterial blood pressure was less than 60 mmHg. The vasopressors available were dopamine 5–15 mcg/kg/min (dopamine HCl USP, 40 mg/mL, Hospira, USA), and norepinephrine 0.1–1 mcg/kg/min (norepinephrine bitartrate injection USP, 1 mg/mL, Claris, USA). Fluid therapy was provided with lactated Ringer’s solution (USP, Hospira, USA) at 10 mL/kg/hour for the entire duration of anesthesia. Blood glucose was monitored every 6 hours and maintained at levels above least 100 mg/dL by intravenous infusion of 2.5% dextrose (dextrose USP 0.5 g/mL, Hospira, USA). For analgesia, a lidocaine constant rate of infusion of 25–50 mcg/kg/minute was started after the administration of a 1 mg/kg lidocaine loading bolus intravenously (Lidocaine HCl injection USP, 2%, Hospira, USA). After the indicated duration (from 3 to 30 hours), the animal’s small intestine was surgically removed. Euthanasia was performed with an intravenous injection of pentobarbital sodium and phenytoin sodium at 1 mL for every 4.5 kg of body weight (Euthasol, Virbac AH, Inc., USA).
Surgery
The pigs were positioned in dorsal recumbency, and following a standard surgical scrub, a 7 cm ventral midline celiotomy was performed to expose the intestine. Prior to the loop construction, the lumen of the ileum and distal jejunum was gently washed with a phosphate-buffered saline (PBS) injection to remove the intestinal contents. Ligated intestinal loops were constructed starting 10 cm proximal of the ileocecal junction using monofilament suture. Ligation was done by a circumferential ligature through the mesentery without damaging grossly visible mesenteric vascular arcades, thus maintaining full blood supply for both the C. jejuni-inoculated loops and control (inter-loop) segments. C. jejuni was grown to mid-log phase, and the cells pelleted by centrifugation. The cells were resuspended in PBS to an OD540 of 0.35. Loops were inoculated with approximately 1 × 1010 bacterial cells suspended in 10 mL of PBS using a 21-gauge needle. The inter-loop segments were mock-inoculated with sterile PBS as a negative control. Care was taken to avoid over-distension of loops. A 16 G, 5.25-inch catheter was surgically placed in the urinary bladder to drain the bladder during the incubation period and then attached to a fluid extension to monitor the urinary output of the animal. The abdominal incision was closed, and the pigs were maintained under anesthesia for the remainder of the experiment. After incubation, the abdominal incision was opened, and the ligated section of the intestine was removed. The luminal contents and intestinal tissues were recovered and processed for further analysis. The animals were euthanized, as outlined above.
Sample collection
Luminal contents, mucosal scrapings, and intestinal tissues were collected and processed for analysis. All loops were subjected to gross and histopathologic evaluation, plating of serial dilutions (to measure bacterial numbers), measurement of cytokine secretion, proteomic analysis, and analysis of the microbiota associated with the luminal contents and tissues. Loops were separated by cutting between the double ligation. Luminal contents were collected by injecting each loop with 5 mL of PBS and collecting the resulting fluid. The fluids were centrifuged at 14,000 x g for one minute to collect the supernatants and pellets. Tissue scrapings were collected from a 1 inch2 tissue section using a tissue-culture scraper. Mucosa was suspended in 5 mL of PBS and collected by centrifugation. Processed luminal contents and tissue scrapings were flash-frozen in liquid nitrogen immediately after collection. Tissue samples for histology were immediately transferred to 10% neutral buffered formalin for fixation before histochemical processing.
IPEC-J2 cell culture and RNA-Seq sample collection
The IPEC-J2 cells were kindly provided by Dr. Anthony Blikslager at North Carolina State University.68 The IPEC-J2 cells were routinely cultured in a 1:1 mixture of Dulbecco’s modified Eagle medium and Ham’s F12 Nutrient Mixture (DMEM/F12) supplemented with 10% fetal bovine serum (FBS; GE Healthcare Life Sciences, HyClone Characterized Fetal Bovine Serum US Origin cat. # SH30071), 1% insulin-transferrin-selenium supplement (Thermo Fisher Scientific, Waltham MA), and 2.5 µg/mL epidermal growth factor (Thermo Fisher Scientific). The samples for RNA-Seq analysis were generated as follows. C. jejuni were first inoculated at an OD540 of 0.05 in MH broth and incubated with shaking for 18 hours. A liquid culture of C. jejuni was then adjusted to an OD540 of 0.3 in DMEM/F12 supplemented with 1% FBS and incubated with the IPEC-J2 cells for 2.5 or 4 hours at 37°C with 5% CO2. The sample used to infect the cells was collected as the ‘input’ sample. The supernatant was collected, and the bacteria were recovered by centrifugation and collected in 1/10 volume of ice-cold stop solution (5% phenol, 95% ethanol), flash-frozen in liquid nitrogen, and stored at −80°C until RNA extraction. For IPEC-J2 samples, RNA was processed as described previously 22. RNA from pig loops was extracted with the SV Total RNA Isolation System (Promega) with 3 mg/mL lysozyme, treated with Turbo DNAse AM1907 (Thermo Fisher), and rRNA was depleted using 1/8th reaction volumes of Ribo-Zero Gold rRNA Removal Kit-Epidemiology (Illumina, San Diego, CA) following manufacturer’s instructions with ethanol purification.
RNA-Sequencing
Illumina MiSeq libraries were prepared using the KAPA stranded RNAseq kit (Kapa Biosystems, Wilmington, MA), following the manufacturer’s instructions except for the following changes: 1–187 ng RNA was sheared for 6 min at 85°C. Standard desalted TruSeq HT primers (Integrated DNA Technologies, Coralville, IA) were used at 25–50 nM final concentration based on starting RNA amount. The PCR step was performed for six to thirteen cycles. Libraries were quantified using the KAPA Library Quantification Kit (Kapa), except with 10 µL volume and a 90 second annealing/extension PCR. Libraries were pooled and normalized to 4 nM. The pooled libraries were re‐quantified by ddPCR on a QX200 system (Bio‐Rad, Hercules, CA) using the Illumina TruSeq ddPCR Library Quantification Kit and following manufacturer’s protocols. The libraries were sequenced in two 2 × 76 bp paired-end v3 runs on a MiSeq instrument at 13.5 pM, following manufacturer’s protocols. Fastq files were generated for each sample by the MiSeq Instrument Software. Reads were mapped to the reference genome of C. jejuni subsp. jejuni F38011 (CP006851) using Bowtie2 (version 2.2.5) and reads were assigned to genes using featureCounts (version 1.5.0). Differential expression analysis was performed using DESeq2 (version 1.10.1).
To construct the hierarchical clustering of expression data from the pig, deoxycholate (DOC), and IPEC-J2 cells, the data were analyzed where each individual sample was compared to its respective control (input) sample grown in MH broth. The data were filtered such that genes were only kept if they had a Benjamini-Hochberg adjusted p-value (q-value) of ≤ 0.05, and had a log2(fold change) of greater than 1.5 (or less than −1.5) in at least four of the six compared samples (two time points per condition). These filters were established to select the genes that are significantly and meaningfully changing in our samples, and to find genes that were changing at both time points in each of the samples.
Determination of cytokine levels
TNF-α, IL-8, and IL-17A concentrations were assessed in C. jejuni-infected and non-infected samples collected from pig intestinal ligated loops. Cytokine levels were analyzed by using commercially available enzyme-linked immunosorbent assay (ELISA) kits (R & D systems cat #s PTA00 and P8000, and Thermo Fisher Scientific cat # ESIL17A). All ELISAs were performed according to the manufacturer’s protocol.
Microbiome analysis
Genomic DNA was isolated from mucosal and luminal samples using the MagAttract PowerMicrobiome 96-well DNA/RNA kit (Qiagen, Germantown, MD) following the manufacturer’s instructions. The V4 region of the bacterial 16S rRNA gene was PCR amplified and sequenced on the MiSeq platform (Illumina) as previously described.69
The 16S rRNA gene sequences were processed in R version 3.5.1 using the Dada2 package version 1.8.0.70 Forward reads were truncated at 220 bp and reverse reads at 210 bp. A maximum expected error of 2 was assigned for both paired reads with no ambiguous bases allowed. Reads were merged, chimeric sequences removed, and taxonomy assigned at 100% similarity using the naive Bayesian RDP classifier71 and the SILVA SSU database release 13272 with a 50% bootstrap confidence threshold. Although control loops were confirmed to be Campylobacter negative after selective culture (see above), some Campylobacter 16S rRNA gene reads were detected in some control samples. Because of cross-contamination between adjacent samples in 96-well DNA extraction formats has been previously observed,73 as well as sequencing barcode bleed-through in multiplexed sequencing runs,74 only control samples with 0.5% or fewer Campylobacter reads were retained for analysis. Ordination plots were made with PRIMER software (v7).75
Proteomics sample preparation
Both intestinal pellets and supernatants were analyzed by LC-MS/MS for protein content. Proteins in the supernatant samples were purified following the Metabolite, Protein and Lipid Extraction (MPLEx) method as described previously.76 In brief, samples were mixed with cold (−20°C) chloroform and methanol at a volume ratio of 10:5:3. The mixture was chilled on ice for 5 minutes, vortexed for 1 minute, and centrifuged for 10 minutes at 4°C at 16,260 × g. The interphase that contained the proteins was collected and washed twice with cold (−20°C) methanol. Recovered protein pellets were then denatured in 100 μL of 8 M urea in 50 mM NH4HCO3 (pH 8.0). Dithiothreitol (DTT, Thermo Fisher Scientific) was added to a final concentration of 5 mM prior to incubation at 60°C for 30 minutes with shaking at 850 rpm. After DTT reduction, iodoacetamide (IAA, Thermo Fisher Scientific) was added to a final concentration of 40 mM to alkylate free thiol groups, followed by incubation at 37°C for 1 hour with shaking at 850 rpm in the dark. Samples were then diluted ten-fold with 100 mM NH4HCO3 (pH 8.0) and adjusted to a CaCl2 concentration of 1 mM. Trypsin was added at a 1:50 enzyme-to-protein ratio to digest proteins at 37°C for 3 hours. The digestion was stopped by adding trifluoroacetic acid to a concentration of 0.1%, followed by centrifugation at 10,000 g for 2 minutes to remove precipitates. Peptides were cleaned using DSC-18 solid phase extraction tubes (Supelco Inc., Bellefonte, PA) and dried in a SpeedVac vacuum concentrator. The final peptide concentration was quantified by a PierceTM BCA protein Assay Kit (Thermo Fisher Scientific) and adjusted to 0.1 μg/μL with Milli-Q water for proteomic analysis.
Pellet samples were resuspended in 200 μL of 50 mM NH4HCO3 (pH 8.0) and subjected to bead beating with 0.1 mm Zirconia/Silica beads (BioSpec Products, Bartlesville, OK) using five cycles consisting of a 1 minute vortex with a 30 second break. After bead beating, samples were moved to new tubes and mixed with powdered urea to a final concentration of 8 M. The denatured samples were mixed with DTT to a final concentration of 10 mM and incubated at 37°C for 1 hour with shaking at 750 rpm. The samples were then mixed with IAA to a final concentration of 40 mM and incubated at 37°C for 1 hour in the dark. Samples were diluted ten-fold with 50 mM NH4HCO3 (pH 8.0) containing 1.1 mM CaCl2. Trypsin was added at a 1:50 enzyme-to-protein ratio, and the mixture was incubated at 37°C for 3 hours. Digested peptides were cleaned using DSC-18 solid phase extraction tubes, dried in a SpeedVac vacuum concentrator, and finally adjusted to 0.1 μg/μL for proteomic analysis.
Proteomics analysis
The digested proteins were analyzed with liquid chromatography-tandem mass spectrometry (LC-MS/MS). The liquid chromatography (LC) consisted of a Waters nanoAcquity UPLC system equipped with a custom analytical column packed with 3 µm Jupiter C18 media (Phenomenex, Torrance, CA) into 70 cm × 75 µm (ID) fused silica (Polymicro Technologies, Phoenix, AZ). 5 µL of each digested sample was loaded onto the LC system, and peptides were separated at 300 nL/minute at room temperature. Mobile phases comprised mobile phase A (0.1% formic acid in water) and mobile phase B (0.1% formic acid in acetonitrile). The supernatant peptides were separated with the following gradient: 1–8% B for 2 minutes, 8–12% B for 18 minutes, 12–30% B for 55 minutes, 30–45% B for 22 minutes, 45–95% B for 3 minutes, maintained at 95% for 10 minutes, and equilibrated at 1% B. The pellet peptides were analyzed with the following gradient: 1–8% B for 4 minutes, 8–12% B for 32 minutes, 12–30% B for 96 minutes, 30–45% B for 40 minutes, 45–95% B for 5 minutes, maintained at 95% for 10 minutes, and equilibrated at 1% B.
The mass spectrometry (MS) analysis was conducted in a Q Exactive Plus mass spectrometer (Thermo Fisher Scientific) outfitted with a custom nano-electrospray ionization (ESI) interface.77 Spray voltage and capillary temperature were controlled at 2 kV and 325°C, respectively. Full MS scans were acquired from 300–2000 m/z at a resolution of 35,000. Top 12 data-dependent tandem MS scans were performed at a resolution of 17,500, isolation window of 2.0 m/z, normalized collision energy of 30, and dynamic exclusion of 30 seconds.
Proteomics data analysis
The supernatant and pellet RAW datafiles were searched separately with MaxQuant (v1.6.0.16)78 against a database comprising the Uniprot Sus scrofa proteome and C. jejuni F38011 proteome. Carbamidomethylation was set as a fixed modification, and methionine oxidation and N-term acetyl were set as variable modifications. A matching window within 1.5 minutes was applied to the samples. False discovery rate (FDR) at the peptide level and protein level was set less than 0.01. Trypsin was set as the digestion enzyme. Default settings were used for the rest of the MaxQuant search parameters. Proteins were quantified using label-free quantification.
The MaxQuant output containing proteins and corresponding label-free quantification was processed with R package RomicsProcessor v0.1.0, which is available on both Github (https://github.com/PNNL-Comp-Mass-Spec/RomicsProcessor) and Zenodo (https://zenodo.org/record/3386527). Briefly, the protein features were filtered to retain proteins with more than 80% of detection among biological replicates under infected conditions or control conditions. The remaining missing values were imputed with the underlying assumption that values were distributed typical of low abundances levels.79 In the statistical analysis, Student’s paired t-test was applied to determine if the mean abundance of a protein was different in the infected loops and in the control loops. The infected loops were paired with their adjacent control loops. The null hypothesis was that the mean protein abundances were the same was rejected if the p-value was less than 0.05. The proteins with significantly different expression levels (p < .05) had enrichment analyses performed for Gene Ontology (GO) terms, KEGG pathways, and Reactome pathways with an online tool g:Profiler.80 The log2 fold change of protein abundance due to infection was calculated by normalizing the mean protein abundance in the infected loops to the mean protein abundance in the control loops.
T84 and THP-1 cell culture
T84 cells (ATCC CCL-248) were routinely cultured in Eagles MEM (Gibco, Waltham, MA) supplemented with 10% FBS (Cat# 97068–091, VWR, Radnor, PA) at 37°C in a 5% CO2 incubator. THP-1 cells were maintained as described previously.81 Three days prior to the experiments, the THP-1 cells were differentiated using 100 nM phorbol 12-myristate 13-acetate (PMA; Sigma Aldrich) treatment for 24 hours.
Determining adherent and internalized C. jejuni
The number of adherent and internalized C. jejuni was determined as described previously.82 Briefly, the number of adherent bacteria was determined by infecting T84 cells in a 24-well dish with C. jejuni at an OD540 of 0.03 in MEM supplemented with 1% FBS and incubating for three hours at 37°C in a 5% CO2 incubator. The cells were rinsed with PBS three times, lysed with 0.1% Triton X-100, and lysates were serially diluted and plated on MHB agar. To determine the number of internalized bacteria, T84 cells were infected with C. jejuni for three hours, rinsed three times with PBS, and then treated with 250 µg/mL gentamicin for three hours. The cells were then rinsed, lysed with 0.1% Triton X-100, and lysates were serially diluted and plated to determine the number of bacteria that survived the gentamicin treatment.
Detection of human IL-8 cytokines from cultured cells
Cultured cells, either T84 or THP-1, were treated with C. jejuni at an OD540 of 0.03 or conditioned medium for 24 hours. The supernatants were collected and centrifuged at 13,000 x g for 5 minutes to remove any cells or cell debris. The amount of IL-8 in the supernatant was determined by a commercial ELISA kit following the manufacturer’s instructions (Cat# 555244, BD Biosciences, San Jose, CA).
Data availability
RNA-Seq data is available at the NCBI Gene Expression Omnibus database with the identifier GSE147629. Proteomics data is available at MassIVE (Mass spectrometry Interactive Virtual Environment) with accession number MSV000079501. The 16S rRNA gene sequence data is available under the NCBI BioProject PRJNA615040.
Supplementary Material
Acknowledgments
We thank the following individuals for their assistance in the pig surgeries: Dr. Lucas Nolazco, Dr. Kyle Heaton, Dr. Joseph Davis, Amy Shields, Teri Olson, Jane Hommel, Gary Turner, Dr. Andrew Jones, Abby Scarlett, Dr. Camila Souza, and Dr. Tania Perez-Jimenez. The following individuals assisted in facilitating experiments, monitoring the animals, or sample collection: Colby Corneau, Kyrah Turner, Prabhat Talukdar, Courtney Klappenbach, Marcelo Patara, and Lisa Orfe. We also thank Kristin M. Engbrecht for proteomics sample preparation, and Karl K. Weitz and Vanessa L. Paurus for mass spectrometer operation.
Funding Statement
This work was supported with funds awarded to Dr. Konkel from the School of Molecular Biosciences, WSU Graduate School, and National Institutes of Health (NIH, Award Number [R01AI125356 and R21AI124144]. This research was also supported, in part, by a grant from the National Institutes of Health GM094623 to Dr. Adkins. Work was partially performed in the Environmental Molecular Sciences Laboratory (EMSL), a DOE-BER national scientific user facility at Pacific Northwest National Laboratory (PNNL). PNNL is a multi-program national laboratory operated by Battelle Memorial Institute for the DOE under contract DE-AC05-76RLO 1830. This work was also supported by the United States Department of Agriculture, Agricultural Research Service CRIS project 2030-42000-051-00D and 5030-31320-004-00D. Dr. Gourley was supported by funds from the School of Molecular Biosciences and National Institutes of Health through the Infectious Diseases and Microbial Immunology Post-doctoral Training Program at Washington State University Award Number [5T32AI007025]. Dr. Bose was supported by National Institutes of Health grants [R01AI083387 and R21AI124144]. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.Madara JL, Stafford J.. Interferon-gamma directly affects barrier function of cultured intestinal epithelial monolayers. J Clin Invest. 1989;83(2):724–727. doi: 10.1172/JCI113938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilson CL, Ouellette AJ, Satchell DP, Ayabe T, Lopez-Boado YS, Stratman JL, Hultgren SJ, Matrisian LM, Parks WC. Regulation of intestinal alpha-defensin activation by the metalloproteinase matrilysin in innate host defense. Science. 1999;286(5437):113–117. doi: 10.1126/science.286.5437.113. [DOI] [PubMed] [Google Scholar]
- 3.Bhinder G, Stahl M, Sham HP, Crowley SM, Morampudi V, Dalwadi U, Ma C, Jacobson K, Vallance BA. Intestinal epithelium-specific MyD88 signaling impacts host susceptibility to infectious colitis by promoting protective goblet cell and antimicrobial responses. Infect Immun. 2014;82(9):3753–3763. doi: 10.1128/IAI.02045-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gewirtz AT, Navas TA, Lyons S, Godowski PJ, Madara JL. Cutting edge: bacterial flagellin activates basolaterally expressed TLR5 to induce epithelial proinflammatory gene expression. J Immunol. 2001;167(4):1882–1885. doi: 10.4049/jimmunol.167.4.1882. [DOI] [PubMed] [Google Scholar]
- 5.Guerrant RL, Steiner TS, Lima AA, Bobak DA. How intestinal bacteria cause disease. J Infect Dis. 1999;179(Suppl 2):S331–S337. doi: 10.1086/513845. [DOI] [PubMed] [Google Scholar]
- 6.Blaser MJ, Berkowitz ID, LaForce FM, Cravens J, Reller LB, Wang WL. Campylobacter enteritis: clinical and epidemiologic features. Ann Intern Med. 1979;91(2):179–185. doi: 10.7326/0003-4819-91-2-179. [DOI] [PubMed] [Google Scholar]
- 7.Ellstrom P, Feodoroff B, Hanninen ML, Rautelin H. Lipooligosaccharide locus class of Campylobacter jejuni: sialylation is not needed for invasive infection. Clin Microbiol Infect. 2014;20(6):524–529. doi: 10.1111/1469-0691.12382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Godschalk PC, Kuijf ML, Li J, St Michael F, Ang CW, Jacobs BC, Karwaski MF, Brochu D, Moterassed A, Endtz HP, et al. Structural characterization of Campylobacter jejuni lipooligosaccharide outer cores associated with Guillain-Barre and Miller Fisher syndromes. Infect Immun. 2007;75(3):1245–1254. doi: 10.1128/IAI.00872-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jacobs BC, O’Hanlon GM, Breedland EG, Veitch J, van Doorn PA, Willison HJ. Human IgM paraproteins demonstrate shared reactivity between Campylobacter jejuni lipopolysaccharides and human peripheral nerve disialylated gangliosides. J Neuroimmunol. 1997;80(1–2):23–30. doi: 10.1016/S0165-5728(97)00130-6. [DOI] [PubMed] [Google Scholar]
- 10.Dhillon AS, Shivaprasad HL, Schaberg D, Wier F, Weber S, Bandli D. Campylobacter jejuni infection in broiler chickens. Avian Dis. 2006;50(1):55–58. doi: 10.1637/7411-071405R.1. [DOI] [PubMed] [Google Scholar]
- 11.Allos BM. Campylobacter jejuni infections: update on emerging issues and trends. Clin Infect Dis. 2001;32(8):1201–1206. doi: 10.1086/319760. [DOI] [PubMed] [Google Scholar]
- 12.Black RE, Levine MM, Clements ML, Hughes TP, Blaser MJ. Experimental Campylobacter jejuni infection in humans. J Infect Dis. 1988;157(3):472–479. doi: 10.1093/infdis/157.3.472. [DOI] [PubMed] [Google Scholar]
- 13.Chowdhury MN. Campylobacter jejuni enteritis; a review. Trop Geogr Med. 1984;36:215–222. [PubMed] [Google Scholar]
- 14.van Spreeuwel JP, Duursma GC, Meijer CJ, Bax R, Rosekrans PC, Lindeman J. Campylobacter colitis: histological immunohistochemical and ultrastructural findings. Gut. 1985;26(9):945–951. doi: 10.1136/gut.26.9.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stephenson HN, Mills DC, Jones H, Milioris E, Copland A, Dorrell N, Wren BW, Crocker PR, Escors D, Bajaj-Elliott M. Pseudaminic acid on Campylobacter jejuni flagella modulates dendritic cell IL-10 expression via Siglec-10 receptor: a novel flagellin-host interaction. J Infect Dis. 2014;210(9):1487–1498. doi: 10.1093/infdis/jiu287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maue AC, Mohawk KL, Giles DK, Poly F, Ewing CP, Jiao Y, Lee G, Ma Z, Monteiro MA, Hill CL, et al. The polysaccharide capsule of Campylobacter jejuni modulates the host immune response. Infect Immun. 2013;81(3):665–672. doi: 10.1128/IAI.01008-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Samuelson DR, Eucker TP, Bell JA, Dybas L, Mansfield LS, Konkel ME. The Campylobacter jejuni CiaD effector protein activates MAP kinase signaling pathways and is required for the development of disease. Cell Commun Signal. 2013;11(1):79. doi: 10.1186/1478-811X-11-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Taylor DJ, Olubunmi PA. A re-examination of the role of Campylobacter fetus subspecies coli in enteric disease of the pig. Vet Rec. 1981;109(6):112–115. doi: 10.1136/vr.109.6.112. [DOI] [PubMed] [Google Scholar]
- 19.Price AB, Jewkes J, Sanderson PJ. Acute diarrhoea: campylobacter colitis and the role of rectal biopsy. J Clin Pathol. 1979;32(10):990–997. doi: 10.1136/jcp.32.10.990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Babakhani FK, Bradley GA, Joens LA. Newborn piglet model for campylobacteriosis. Infect Immun. 1993;61(8):3466–3475. doi: 10.1128/IAI.61.8.3466-3475.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Santaolalla R, Fukata M, Abreu MT. Innate immunity in the small intestine. Curr Opin Gastroenterol. 2011;27(2):125–131. doi: 10.1097/MOG.0b013e3283438dea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Negretti NM, Gourley CR, Clair G, Adkins JN, Konkel ME. The food-borne pathogen Campylobacter jejuni responds to the bile salt deoxycholate with countermeasures to reactive oxygen species. Sci Rep. 2017;7(1):15455. doi: 10.1038/s41598-017-15379-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weiner IM, Lack L. Absorption of bile salts from the small intestine in vivo. Am J Physiol. 1962;202(1):155–157. doi: 10.1152/ajplegacy.1962.202.1.155. [DOI] [PubMed] [Google Scholar]
- 24.Palyada K, Sun YQ, Flint A, Butcher J, Naikare H, Stintzi A. Characterization of the oxidative stress stimulon and PerR regulon of Campylobacter jejuni. BMC Genomics. 2009;10(1):481. doi: 10.1186/1471-2164-10-481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stroncek DF, Skubitz KM, McCullough JJ. Biochemical characterization of the neutrophil-specific antigen NB1. Blood. 1990;75(3):744–755. doi: 10.1182/blood.V75.3.744.744. [DOI] [PubMed] [Google Scholar]
- 26.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2018;46(D1):D649–D655. doi: 10.1093/nar/gkx1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hessian PA, Edgeworth J, Hogg N. MRP-8 `and MRP-14, two abundant Ca(2+)-binding proteins of neutrophils and monocytes. J Leukoc Biol. 1993;53(2):197–204. doi: 10.1002/jlb.53.2.197. [DOI] [PubMed] [Google Scholar]
- 28.Bachman MA, Miller VL, Weiser JN. Mucosal lipocalin 2 has pro-inflammatory and iron-sequestering effects in response to bacterial enterobactin. PLoS Pathog. 2009;5(10):e1000622. doi: 10.1371/journal.ppat.1000622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Klebanoff SJ, Kettle AJ, Rosen H, Winterbourn CC, Nauseef WM. Myeloperoxidase: a front-line defender against phagocytosed microorganisms. J Leukoc Biol. 2013;93(2):185–198. doi: 10.1189/jlb.0712349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryckman C, Vandal K, Rouleau P, Talbot M, Tessier PA. Proinflammatory activities of S100: proteins S100A8, S100A9, and S100A8/A9 induce neutrophil chemotaxis and adhesion. J Immunol. 2003;170(6):3233–3242. doi: 10.4049/jimmunol.170.6.3233. [DOI] [PubMed] [Google Scholar]
- 31.Krause-Gruszczynska M, Rohde M, Hartig R, Genth H, Schmidt G, Keo T, Konig W, Miller WG, Konkel ME, Backert S. Role of the small Rho GTPases Rac1 and Cdc42 in host cell invasion of Campylobacter jejuni. Cell Microbiol. 2007;9(10):2431–2444. doi: 10.1111/j.1462-5822.2007.00971.x. [DOI] [PubMed] [Google Scholar]
- 32.Hammond ME, Lapointe GR, Feucht PH, Hilt S, Gallegos CA, Gordon CA, Giedlin MA, Mullenbach G, Tekamp-Olson PIL-8. induces neutrophil chemotaxis predominantly via type I IL-8 receptors. J Immunol. 1995;155:1428–1433. [PubMed] [Google Scholar]
- 33.de Zoete MR, Keestra AM, Wagenaar JA, van Putten JP. Reconstitution of a functional Toll-like receptor 5 binding site in Campylobacter jejuni flagellin. J Biol Chem. 2010;285(16):12149–12158. doi: 10.1074/jbc.M109.070227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chang C, Miller JF. Campylobacter jejuni colonization of mice with limited enteric flora. Infect Immun. 2006;74(9):5261–5271. doi: 10.1128/IAI.01094-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yao R, Burr DH, Guerry P. CheY-mediated modulation of Campylobacter jejuni virulence. Mol Microbiol. 1997;23(5):1021–1031. doi: 10.1046/j.1365-2958.1997.2861650.x. [DOI] [PubMed] [Google Scholar]
- 36.Hugdahl MB, Beery JT, Doyle MP. Chemotactic behavior of Campylobacter jejuni. Infect Immun. 1988;56(6):1560–1566. doi: 10.1128/IAI.56.6.1560-1566.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Day CJ, King RM, Shewell LK, Tram G, Najnin T, Hartley-Tassell LE, Wilson JC, Fleetwood AD, Zhulin IB, Korolik V. A direct-sensing galactose chemoreceptor recently evolved in invasive strains of Campylobacter jejuni. Nat Commun. 2016;7(1):13206. doi: 10.1038/ncomms13206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boehm M, Hoy B, Rohde M, Tegtmeyer N, Baek KT, Oyarzabal OA, Brondsted L, Wessler S, Backert S. Rapid paracellular transmigration of Campylobacter jejuni across polarized epithelial cells without affecting TER: role of proteolytic-active HtrA cleaving E-cadherin but not fibronectin. Gut Pathog. 2012;4(1):3. doi: 10.1186/1757-4749-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elmi A, Nasher F, Jagatia H, Gundogdu O, Bajaj-Elliott M, Wren B, Dorrell N. Campylobacter jejuni outer membrane vesicle-associated proteolytic activity promotes bacterial invasion by mediating cleavage of intestinal epithelial cell E-cadherin and occludin. Cell Microbiol. 2016;18(4):561–572. doi: 10.1111/cmi.12534. [DOI] [PubMed] [Google Scholar]
- 40.Harrer A, Bucker R, Boehm M, Zarzecka U, Tegtmeyer N, Sticht H, Schulzke JD, Backert S. Campylobacter jejuni enters gut epithelial cells and impairs intestinal barrier function through cleavage of occludin by serine protease HtrA. Gut Pathog. 2019;11(1):4. doi: 10.1186/s13099-019-0283-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Konkel ME, Christensen JE, Keech AM, Monteville MR, Klena JD, Garvis SG. Identification of a fibronectin-binding domain within the Campylobacter jejuni CadF protein. Mol Microbiol. 2005;57(4):1022–1035. doi: 10.1111/j.1365-2958.2005.04744.x. [DOI] [PubMed] [Google Scholar]
- 42.Konkel ME, Larson CL, Flanagan RC. Campylobacter jejuni FlpA binds fibronectin and is required for maximal host cell adherence. J Bacteriol. 2010;192(1):68–76. doi: 10.1128/JB.00969-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pogacar MS, Klancnik A, Mozina SS, Cencic A. Attachment, invasion, and translocation of Campylobacter jejuni in pig small-intestinal epithelial cells. Foodborne Pathog Dis. 2010;7(5):589–595. doi: 10.1089/fpd.2009.0301. [DOI] [PubMed] [Google Scholar]
- 44.Konkel ME, Mead DJ, Hayes SF, Cieplak W Jr.. Translocation of Campylobacter jejuni across human polarized epithelial cell monolayer cultures. J Infect Dis. 1992;166(2):308–315. doi: 10.1093/infdis/166.2.308. [DOI] [PubMed] [Google Scholar]
- 45.Barrero-Tobon AM, Hendrixson DR. Identification and analysis of flagellar coexpressed determinants (Feds) of Campylobacter jejuni involved in colonization. Mol Microbiol. 2012;84(2):352–369. doi: 10.1111/j.1365-2958.2012.08027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Neal-McKinney JM, Konkel ME. The Campylobacter jejuni CiaC virulence protein is secreted from the flagellum and delivered to the cytosol of host cells. Front Cell Infect Microbiol. 2012;2:31. doi: 10.3389/fcimb.2012.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Monteville MR, Konkel ME. Fibronectin-facilitated invasion of T84 eukaryotic cells by Campylobacter jejuni occurs preferentially at the basolateral cell surface. Infect Immun. 2002;70(12):6665–6671. doi: 10.1128/IAI.70.12.6665-6671.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Christensen JE, Pacheco SA, Konkel ME. Identification of a Campylobacter jejuni-secreted protein required for maximal invasion of host cells. Mol Microbiol. 2009;73(4):650–662. doi: 10.1111/j.1365-2958.2009.06797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Samuelson DR, Konkel ME. Serine phosphorylation of cortactin is required for maximal host cell invasion by Campylobacter jejuni. Cell Commun Signal. 2013;11(1):82. doi: 10.1186/1478-811X-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Talukdar PK, Negretti NM, Turner KL, Konkel ME. Molecular dissection of the Campylobacter jejuni CadF and FlpA virulence proteins in binding to host cell fibronectin. Microorganisms. 2020;8(3):389. doi: 10.3390/microorganisms8030389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eckmann L, Kagnoff MF, Fierer J. Epithelial cells secrete the chemokine interleukin-8 in response to bacterial entry. Infect Immun. 1993;61(11):4569–4574. doi: 10.1128/IAI.61.11.4569-4574.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hickey TE, Baqar S, Bourgeois AL, Ewing CP, Guerry P. Campylobacter jejuni-stimulated secretion of interleukin-8 by INT407 cells. Infect Immun. 1999;67(1):88–93. doi: 10.1128/IAI.67.1.88-93.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eucker TP, Samuelson DR, Hunzicker-Dunn M, Konkel ME. The focal complex of epithelial cells provides a signalling platform for interleukin-8 induction in response to bacterial pathogens. Cell Microbiol. 2014;16(9):1441–1455. doi: 10.1111/cmi.12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kinhult J, Egesten A, Benson M, Uddman R, Cardell LO. Increased expression of surface activation markers on neutrophils following migration into the nasal lumen. Clin Exp Allergy. 2003;33(8):1141–1146. doi: 10.1046/j.1365-2222.2003.01682.x. [DOI] [PubMed] [Google Scholar]
- 55.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011;11:723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Haller D, Bode C, Hammes WP, Pfeifer AM, Schiffrin EJ, Blum S. Non-pathogenic bacteria elicit a differential cytokine response by intestinal epithelial cell/leucocyte co-cultures. Gut. 2000;47(1):79–87. doi: 10.1136/gut.47.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fimlaid KA, Lindow JC, Tribble DR, Bunn JY, Maue AC, Kirkpatrick BD. Peripheral CD4+ T cell cytokine responses following human challenge and re-challenge with Campylobacter jejuni. PLoS One. 2014;9(11):e112513. doi: 10.1371/journal.pone.0112513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ray S, De Salvo C, Pizarro TT. Central role of IL-17/Th17 immune responses and the gut microbiota in the pathogenesis of intestinal fibrosis. Curr Opin Gastroenterol. 2014;30(6):531–538. doi: 10.1097/MOG.0000000000000119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mumy KL, McCormick BA. The role of neutrophils in the event of intestinal inflammation. Curr Opin Pharmacol. 2009;9(6):697–701. doi: 10.1016/j.coph.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Konig J, Wells J, Cani PD, Garcia-Rodenas CL, MacDonald T, Mercenier A, Whyte J, Troost F, Brummer RJ. Human intestinal barrier function in health and disease. Clin Transl Gastroenterol. 2016;7(10):e196. doi: 10.1038/ctg.2016.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shank JM, Kelley BR, Jackson JW, Tweedie JL, Franklin D, Damo SM, Gaddy JA, Murphy CN, Johnson JG. The host antimicrobial protein calgranulin C participates in the control of Campylobacter jejuni growth via zinc sequestration. Infect Immun. 2018;86(6). doi: 10.1128/IAI.00234-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Raquil MA, Anceriz N, Rouleau P, Tessier PA. Blockade of antimicrobial proteins S100A8 and S100A9 inhibits phagocyte migration to the alveoli in streptococcal pneumonia. J Immunol. 2008;180(5):3366–3374. doi: 10.4049/jimmunol.180.5.3366. [DOI] [PubMed] [Google Scholar]
- 63.Simard JC, Noel C, Tessier PA, Girard D. Human S100A9 potentiates IL-8 production in response to GM-CSF or fMLP via activation of a different set of transcription factors in neutrophils. FEBS Lett. 2014;588(13):2141–2146. doi: 10.1016/j.febslet.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 64.Looft T, Cai G, Choudhury B, Lai LX, Lippolis JD, Reinhardt TA, Sylte MJ, Casey TA. Avian intestinal mucus modulates Campylobacter jejuni gene expression in a host-specific manner. Front Microbiol. 2018;9:3215. doi: 10.3389/fmicb.2018.03215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Taveirne ME, Theriot CM, Livny J, DiRita VJ. The complete Campylobacter jejuni transcriptome during colonization of a natural host determined by RNAseq. PLoS One. 2013;8(8):e73586. doi: 10.1371/journal.pone.0073586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Oyarzabal OA, Macklin KS, Barbaree JM, Miller RS. Evaluation of agar plates for direct enumeration of Campylobacter spp. from poultry carcass rinses. Appl Environ Microbiol. 2005;71(6):3351–3354. doi: 10.1128/AEM.71.6.3351-3354.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Loughlin JL, Samuelson DR, Braundmeier-Fleming AG, White BA, Haldorson GJ, Stone JB, Lessmann JJ, Eucker TP, Konkel ME. The intestinal microbiota influences Campylobacter jejuni colonization and extraintestinal dissemination in mice. Appl Environ Microbiol. 2015;81(14):4642–4650. doi: 10.1128/AEM.00281-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schierack P, Nordhoff M, Pollmann M, Weyrauch KD, Amasheh S, Lodemann U, Jores J, Tachu B, Kleta S, Blikslager A, et al. Characterization of a porcine intestinal epithelial cell line for in vitro studies of microbial pathogenesis in swine. Histochem Cell Biol. 2006;125(3):293–305. doi: 10.1007/s00418-005-0067-z. [DOI] [PubMed] [Google Scholar]
- 69.Johnson TA, Sylte MJ, Looft T. In-feed bacitracin methylene disalicylate modulates the turkey microbiota and metabolome in a dose-dependent manner. Sci Rep. 2019;9(1):8212. doi: 10.1038/s41598-019-44338-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJ, Holmes SP. DADA2: high-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581–583. doi: 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007;73(16):5261–5267. doi: 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glockner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41(Database issue):D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Minich JJ, Sanders JG, Amir A, Humphrey G, Gilbert JA, Knight R. Quantifying and understanding well-to-well contamination in microbiome research. mSystems. 2019;4(4). doi: 10.1128/mSystems.00186-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Costello M, Fleharty M, Abreu J, Farjoun Y, Ferriera S, Holmes L, Granger B, Green L, Howd T, Mason T, et al. Characterization and remediation of sample index swaps by non-redundant dual indexing on massively parallel sequencing platforms. BMC Genomics. 2018;19(1):332. doi: 10.1186/s12864-018-4703-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Clarke K, Gorley R. PRIMER v7: user Manual/Tutorial. Plymouth (UK): PRIMER-E Ltd; 2015.
- 76.Nakayasu ES, Nicora CD, Sims AC, Burnum-Johnson KE, Kim YM, Kyle JE, Matzke MM, Shukla AK, Chu RK, Schepmoes AA, et al. MPLEx: a robust and universal protocol for single-sample integrative proteomic, metabolomic, and lipidomic analyses. mSystems. 2016;1(3):e00043-16. doi: 10.1128/mSystems.00043-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kelly RT, Page JS, Luo Q, Moore RJ, Orton DJ, Tang K, Smith RD. Chemically etched open tubular and monolithic emitters for nanoelectrospray ionization mass spectrometry. Anal Chem. 2006;78(22):7796–7801. doi: 10.1021/ac061133r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Tyanova S, Temu T, Cox J. The MaxQuant computational platform for mass spectrometry-based shotgun proteomics. Nat Protoc. 2016;11(12):2301–2319. doi: 10.1038/nprot.2016.136. [DOI] [PubMed] [Google Scholar]
- 79.Tyanova S, Temu T, Sinitcyn P, Carlson A, Hein MY, Geiger T, Mann M, Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat Methods. 2016;13(9):731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 80.Raudvere U, Kolberg L, Kuzmin I, Arak T, Adler P, Peterson H, Vilo J. g:Profiler: a web server for functional enrichment analysis and conversions of gene lists (2019 update). Nucleic Acids Res. 2019;47(W1):W191–W198. doi: 10.1093/nar/gkz369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pokharel SM, Shil NK, Gc JB, Colburn ZT, Tsai SY, Segovia JA, Chang TH, Bandyopadhyay S, Natesan S, Jones JCR, et al. Integrin activation by the lipid molecule 25-hydroxycholesterol induces a proinflammatory response. Nat Commun. 2019;10(1):1482. doi: 10.1038/s41467-019-09453-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Negretti NM, Konkel ME. Methods to study Campylobacter jejuni adherence to and invasion of host epithelial cells. Methods Mol Biol. 2017;1512:117–127. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-Seq data is available at the NCBI Gene Expression Omnibus database with the identifier GSE147629. Proteomics data is available at MassIVE (Mass spectrometry Interactive Virtual Environment) with accession number MSV000079501. The 16S rRNA gene sequence data is available under the NCBI BioProject PRJNA615040.