

ABSTRACT
The formation of secondary bile acids by gut microbes is a current topic of considerable biomedical interest. However, a detailed understanding of the biology of anaerobic bacteria in the genus Clostridium that are capable of generating secondary bile acids is lacking. We therefore sought to determine the transcriptional responses of two prominent secondary bile acid producing bacteria, Clostridium hylemonae and Clostridium hiranonis to bile salts (in vitro) and the cecal environment of gnotobiotic mice. The genomes of C. hylemonae DSM 15053 and C. hiranonis DSM 13275 were closed, and found to encode 3,647 genes (3,584 protein-coding) and 2,363 predicted genes (of which 2,239 are protein-coding), respectively, and 1,035 orthologs were shared between C. hylemonae and C. hiranonis. RNA-Seq analysis was performed in growth medium alone, and in the presence of cholic acid (CA) and deoxycholic acid (DCA). Growth with CA resulted in differential expression (>0.58 log2FC; FDR < 0.05) of 197 genes in C. hiranonis and 118 genes in C. hylemonae. The bile acid-inducible operons (bai) from each organism were highly upregulated in the presence of CA but not DCA. We then colonized germ-free mice with human gut bacterial isolates capable of metabolizing taurine-conjugated bile acids. This consortium included bile salt hydrolase-expressing Bacteroides uniformis ATCC 8492, Bacteroides vulgatus ATCC 8482, Parabacteroides distasonis DSM 20701, as well as taurine-respiring Bilophila wadsworthia DSM 11045, and deoxycholic/lithocholic acid generating Clostridium hylemonae DSM 15053 and Clostridium hiranonis DSM 13275. Butyrate and iso-bile acid-forming Blautia producta ATCC 27340 was also included. The Bacteroidetes made up 84.71% of 16S rDNA cecal reads, B. wadsworthia, constituted 14.7%, and the clostridia made up <.75% of 16S rDNA cecal reads. Bile acid metabolomics of the cecum, serum, and liver indicate that the synthetic community were capable of functional bile salt deconjugation, oxidation/isomerization, and 7α-dehydroxylation of bile acids. Cecal metatranscriptome analysis revealed expression of genes involved in metabolism of taurine-conjugated bile acids. The in vivo transcriptomes of C. hylemonae and C. hiranonis suggest fermentation of simple sugars and utilization of amino acids glycine and proline as electron acceptors. Genes predicted to be involved in trimethylamine (TMA) formation were also expressed.
KEYWORDS: Gnotobiotic, RNA-Seq, clostridium, bile acid
Introduction
The complex interaction between the liver and the gut microbiota is mediated in part by bile acids. Bile acid metabolism by gut bacteria is of considerable biomedical interest with implications for liver1,2 and colon3,4 cancers, gallstone disease,5,6 cirrhosis of the liver,7,8 and Clostridioides difficile(C. diff) infection.9-11The liver is the only organ in the body that expresses all the enzymes required to generate both the dihydroxyl bile acid chenodeoxycholic acid (CDCA) and the trihydroxyl bile acid cholic acid (CA) from cholesterol through the neutral pathway of bile acid synthesis.12 In rodents, CDCA is 6α-hydroxylated resulting in formation of α-muricholic acid (α-MCA) and epimerization to β-muricholic acid (β-MCA). In mice, the majority of bile acids are taurine-conjugated. In humans, the ratio of glycine:taurine depends largely on diet, with vegetarian diets favoring glycine conjugation, and high animal protein/seafood diets favoring taurine,13,14 whereas in rodents, bile acids are conjugated primarily to taurine15 irrespective of diet.
The ~2 g bile acid pool of humans circulates 8–12 times a day with meal-induced contraction of gallbladder contents into the duodenum where conjugated bile salts aid in lipid digestion and absorption throughout the small bowel. In the terminal ileum, high affinity intestinal bile acid transporter (IBAT) and organic solute and steroid transporter (OSTalpha/beta) transport bile salts from the lumen into ileocytes and across the basolateral membrane where they are transported back to the liver via the portal vein. This recycling process, known as enterohepatic circulation (EHC), is ~95% efficient. The 400–800 mg of bile acids that escape this process daily are biotransformed by gut bacteria, primarily in the colon, into what are termed “secondary bile acids.”16,17
Deconjugation of bile salts is a widespread generalist function of the gut microbiota spanning the major bacterial phyla, and even the domain Archaea.18 Bile salt deconjugation is catalyzed by bile salt hydrolase (BSH), enzymes that hydrolyze the amide bond between the bile acid and taurine or glycine, generating “free bile acids” with a C-24 carboxylic acid group. Diverse gut bacteria also encode various hydroxysteroid dehydrogenases (HSDH) that catalyze the reversible oxidation-reduction of bile acid hydroxyl groups at C-3, C-7, and C-12.19,20 These enzymes are pyridine nucleotide-dependent and both regio-specific (e.g. 3α-HSDH vs. 7α-HSDH) and stereo-specific (e.g. 7α-HSDH vs. 7β-HSDH).21 Gut bacteria are capable of epimerizing host bile acids through the concerted action of enzymes sharing regio-specificity but differing in stereo-specificity, forming a stable oxo-bile acid intermediate.21,22
The gut microbome is the only “virtual” organ in the body that express enzymes capable of generating hydrophobic “secondary bile acids.”4 A small population of anaerobic Firmicutes in Clostridium clusters IV, XI, and XIVa harbor a multigene bile acid-inducible (bai) operon encoding enzymes involved in bile acid 7α-dehydroxylation.21 Human gut bacterial isolates, including Clostridium hylemonae and C. hiranonis that encode the bai operon convert host cholic acid (CA; 3α-,7α-,12α-trihydroxy-5β-cholan-24-oic acid) to deoxycholic acid (DCA; 3α-,12α-dihydroxy-5β-cholan-24-oic acid), as well as chenodeoxycholic acid (CDCA; 3α-,7α-dihydroxy-5β-cholan-24-oic acid) and ursodeoxycholic (UDCA; 3α-,7β-dihydroxy-5β-cholan-24-oic acid) to lithocholic acid (LCA; 7α-monohydroxy-5β-cholan-24-oic acid) through a series of enzymatic steps.23 DCA and LCA comprise the majority of fecal bile acids (>90%) in healthy individuals.24 The increased hydrophobicity of these compounds allows passive diffusion through colonocytes where DCA and LCA can accumulate in the bile of humans from 25% to upwards of 75% of the bile acid composition.21 By contrast, rodent livers are capable of 7α-hydroxylation of secondary bile acids to maintain a hydrophilic biliary pool.25 However, the germ-free mouse has become an important model in understanding host–microbe and microbe–microbe interactions.
Prior work by Narushima et al (2006) established minimal bacterial communities in germ-free BALB/c mice capable of converting TCA to DCA.26 Bacteria that composed these consortia included human fecal isolates, such as bile salt deconjugating strains of Bacteroides uniformis, Bacteroides vulgatus, and Parabacteroides distasonis, the taurine-respiring δ-Proteobacteria member Bilophila wadsworthia, and bile acid 7α-dehydroxylating Firmicutes C. hylemonae and C. hiranonis. Characterization of the Narushima model was limited to cecal bile acid analysis and culture-based viability counts of cecal community members. Revisiting this model with “omics” technologies has the potential to shed significant insight into the in vivo‘lifestyle’ of the defined community. Currently, very little is known about the biology of bile acid 7α-dehydroxylating Firmicutes beyond the enzymology and structural biology of bai pathway enzymes. The in vitro transcriptome of C. scindens ATCC 35704 in the presence and absence of bile acids was recently reported27; however, the in vitro transcriptional profile of other bile acid 7α-dehydroxylating bacteria such as C. hylemonae and C. hiranonis are important to generalize bile acid responses by clostridia capable of generating hydrophobic secondary bile acids. In addition, the in vivo transcriptome of 7α-dehydroxylating bacteria has yet to be reported.
A major focus of this study was to complete, analyze, and compare the genome of DCA-forming human gut isolates, C. hylemonae and C. hiranonis, and to perform transcriptome analysis in vitro in the presence and absence of CA or DCA. We repeated colonization with human bacterial isolates (type strains) reported in the Narushima model.26 We also included Blautia producta, which was recently shown to express 7β-HSDH28 and 3α- and 3β-HSDH29 and was included due to its ability to oxidize and epimerize bile acids as well as generation of butyrate important for epithelial and immune development. We refer to this bacterial consortium as the B4PC2 community (Figure 1). We also present data on 16S rDNA profile, cecal metatranscriptome, and bile acid metabolome throughout the EHC of C57BL/6 gnotobiotic mice harboring the B4PC2 defined cecal community. Comparative genomics and in vitro and in vivo RNA-Seq profiles provide novel insight into the in vivo lifestyles of two prominent bile acid 7α-dehydroxylating Firmicutes.
Figure 1.
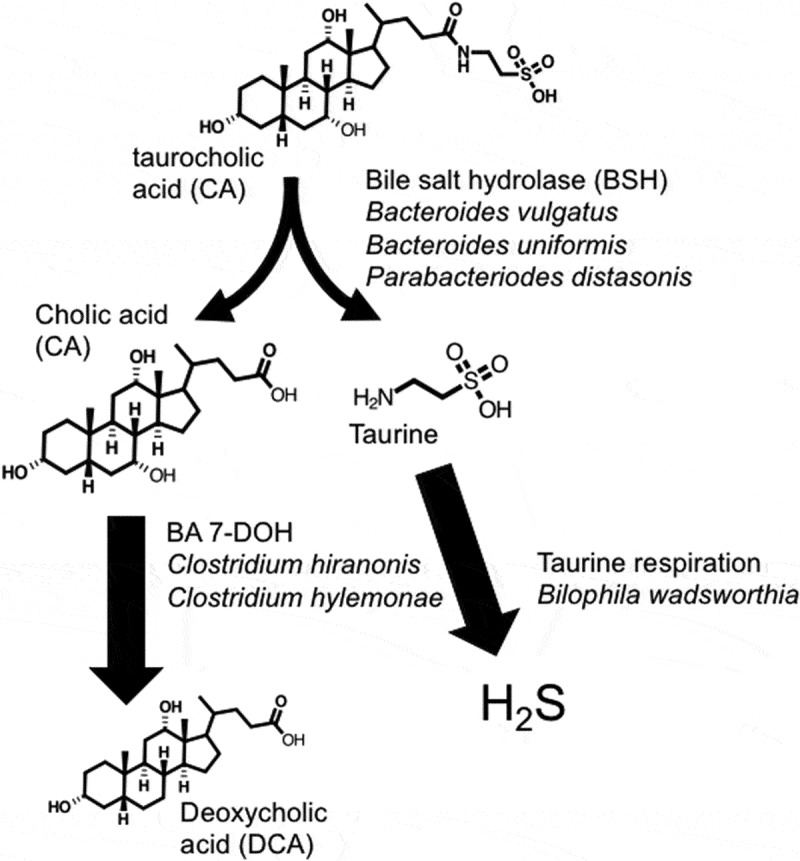
Conversion of taurocholic acid to deoxycholic acid and hydrogen sulfide by B4PC2 gnotobiotic consortium.
Materials and methods
Bacterial strains and chemical reagents
Bacteroides uniformis ATCC 8492, Bacteroides vulgatus ATCC 8482, Clostridium hylemonae DSM 15053, Clostridium hiranonis DSM 13275, Parabacteroides distasonis DSM 20701, and Blautia producta ATCC 27340 (B4PC2 consortium) were routinely cultured in brain heart infusion broth (BHI) supplemented with minimal salt solution, resazurin, vitamin K, hemin, yeast, and L-cysteine under anaerobic conditions. Bilophila wadsworthia DSM 11045 was cultured in the same medium, but also supplemented with taurine (50 mM). All seven strains were combined into glycerol stocks at 5 × 108 CFU of each strain per ml and frozen at −80◦ C until used for mouse study. Authentic reference bile acids (see Table S1) were purchased from the Sigma-Aldrich Corporation (St. Louis, MO) as follows: CA, CDCA, UDCA, DCA, LCA, GCA, GCDCA, GUDCA, GDCA, GLCA, TCA, TCDCA, TUDCA, TDCA, TLCA, HCA, GHCA, THCA, HDCA, GHDCA, and THDCA. The other bile acids were purchased from Steraloids (Newport, RI) as follows: The substances [2,2,4,4-d4]-CA (d4-CA, internal standard (IS) for unconjugated bile acids), [2,2,4,4-d4]-GCA (d4-GCA, IS for glycine-conjugated bile acids), and [2,2,4,4-d4]-TCA (d4-TCA, and IS for taurine-conjugated and double-conjugated bile acids) were obtained from C/D/N Isotopes, Inc. (Pointe-Claire, QC, Canada). T-α-MCA, α-MCA, T-β-MCA, β-MCA, T-ω-MCA, ω-MCA, and the 3-sulfates for CA, CDCA, UDCA, DCA, LCA, GCA, GCDCA, GUDCA, GDCA, GLCA, TCA, TCDCA, TUDCA, TDCA, and TLCA were gifts from Professor Takashi Iida (Nihon University, Tokyo, Japan). Ethanol, methanol, acetonitrile, and water were of high-performance liquid chromatography (HPLC) grade; ammonium acetate was analytical grade; and all were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan). An InertSep C18-B (100 mg/ml) solid phase extraction cartridge was obtained from GL Sciences (Tokyo, Japan).
Complete genome sequencing
Genomic DNA was isolated by a modification of a previously reported method.27 We utilized a combination of Oxford Nanopore which provides a small number of long reads with Illumina MiSeq which provides short but numerous and accurate reads to complete the genomes of C. hylemonae and C. hiranonis. Briefly, pipetting of aqueous solution was replaced with decanting to avoid shearing DNA. Phenol/chloroform solutions were removed from 50 ml Falcon conical tubes by 20 cc syringe puncture. Aqueous layer was then ethanol precipitated. The shotgun genomic libraries were prepared with the Hyper Library construction kit from Kapa Biosystems. Phix DNA is used as a spike-in control for MiSeq runs. The library was quantitated by qPCR and sequenced on one MiSeq flowcell for 251 cycles from each end of the fragments using a MiSeq 500-cycle Micro sequencing kit version2. Adaptors were trimmed from the 3ʹ-end of the reads. Adaptor sequence in read1: AGATCGGAAGAGCACACGTCTGAACTCCAGTCACNNNNNNNNATCTCGTATGCCGTCTTCTGCTTG (NNNNNNNN = 8nt index). AT Adaptor sequence in read2: AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGCCGTAT. Sequence of adaptors used to make the libraries (used portion in bold font for adaptor trimming). Fastq files were generated and demultiplexed with the bcl2fastq v2.17.1.14 Conversion Software (Illumina).
For Oxford Nanopore sequencing, 1 μg of genomic DNA were sheared in a gTube (Covaris, Woburn, MA) for 1min at 6,000 rpm in an Eppendorf MiniSpin plus microcentrifuge (Eppendorf, Hauppauge, NY). The sheared DNA was converted into a Nanopore library with the Nanopore Sequencing kit SQK-LSK108 and the Native 1D Barcoding kit EXP-NBD103 (Oxford Nanopore). The libraries were pooled in equimolar concentration and the pool was sequenced on a SpotON Flowcell MK I R9.5 flowcell for 24 h, using a MinION MK 1B sequencer. Initial base-calling and demultiplexing were performed in real time with the Metrichor Agent V2.45.3 using the 1D BaseCalling plus Barcoding for FLO-MIN_106 450bp workflow. Poretools v-0.5.1 software was used to extract sequences from Oxford Nanopore MinION output file folders and converts them to fastq format. FastQC v-0.11.2 software was also used to further access quality scores and other attributes of the data set. An in-house Perl script was used to trim 60 bases from the both ends of raw nanopore reads. Then, the reads that are longer than 1001 bps were selected for assembling purposes. For MiSeq data sets, Trimmomatic-0.36 was used to remove quality scores less than 30, and the reads with 50 bps and more were kept. 22,498 trimmed nanopore reads and 2,940,368 MiSeq reads from C. hiranonis, and 44,846 trimmed nanopore reads and 2,064,248 Miseq reads from C. hylemonae were used to construct assemblies through Spades-3.10.1 software separately. The two Spades runs produced two assemblies successfully. Circlator-1.4.1(https://github.com/sanger-pathogens/circlator) was used to circularize the two genome assemblies. For C. hiranonis assembly, Node1 contig was chosen to run Circlator. For C. hylemonae assembly, the ends of the two scaffolds overlapped, therefore Node1 and Node2 contigs were merged prior to running Circlator. The nanopore error corrected reads that were used in Circlator software were from Canu assembly runs for the two samples. Nucleotide level comparisons between the newly constructed assemblies and their reference genomes from NCBI were done with the dnadiff program from MUMmer v-3.23. Mummerplot from MUMmer was used to create a graphical representation of the alignments for the newly constructed assemblies and publicly available reference genomes. Annotation comparisons between the newly constructed assembles and their reference genomes were made with Prokka v-1.12.
Gnotobiotic mice
All experiments were completed with the guidelines of the Institutional Animal Care and Use Committees of the Mayo Clinic (Rochester, MN) (Protocol# A00001902-16). Mice were housed in the Mayo Clinic Gnotobiotic Facility within shoebox polysulfone cages on P.J. Murphy Sani-Chips. Autoclaved LabDiet 5K67 was provided ad libitum through wire bar feeders. Water was provided ad libitum. The cage components (cages, bedding, wire bars, cage card holder, and water bottles) were autoclaved using longer cycles with multiple indicators to accommodate gnotobiotic mice.
Six-week old C57Bi/6N mice (N = 6) were purchased from Taconic Farms (Germantown, NY) and orally gavaged with 200 μl of glycerol stock containing ~108 CFU of each member of the B4PC2 consortium. On day 27, mice were euthanized by CO2 asphyxiation followed by cervical dislocation. Serum, liver, a portion of cecal contents and tissues were stored at −80°C for subsequent bile acid analysis. A separate portion of cecal contents was snap frozen and stored at −80°C for microbial community analysis.
Microbiome community profiling
Genomic DNA was extracted from cecum samples using a Maxwell® 16 Tissue DNA Purification Kit, with extractions conducted on a Maxwell16 automated extraction system (Promega, Madison, WI). Genomic DNA was PCR amplified with primers 515F-modified and 926R30 targeting the V4-V5 variable region of the microbial small subunit ribosomal RNA gene using a two-stage “targeted amplicon sequencing (TAS)” protocol.31,32 The primers contained 5ʹ common sequence tags (known as common sequence 1 and 2, CS1 and CS2) as described previously.33 The CS1_515F and CS2_926R primer sequences were 5ʹ-ACACTGACGACATGGTTCTACAGTGYCAGCMGCCGCGGTAA-3ʹ and 5ʹ-TACGGTAGCAGAGACTTGGTCTCCGYCAATTYMTTTRAGTTT-3ʹ, respectively, with the underlined regions indicating the common sequence tags. First stage PCR amplifications were performed in 10 µl reactions in 96-well plates, using the MyTaq HS 2X mastermix (Bioline, Taunton, MA). PCR conditions were 95°C for 5 min, followed by 28 cycles of 95°C for 30 sec, 50°C for 60 sec and 72°C for 90 sec.
Subsequently, a second PCR amplification was performed in 10 µl reactions in 96-well plates. A mastermix for the entire plate was made using the MyTaq HS 2X mastermix. Each well received a separate primer pair with a unique 10-base barcode, obtained from the Access Array Barcode Library for Illumina (Fluidigm, South San Francisco, CA; Item# 100–4876), as well as 1 µl of first-stage PCR product. These AccessArray primers contained the CS1 and CS2 linkers at the 3ʹ ends of the oligonucleotides. Cycling conditions were as follows: 95°C for 5 min, followed by 8 cycles of 95°C for 30 sec, 60°C for 30 sec, and 72°C for 30 sec. A final, 7 min elongation step was performed at 72°C. Samples were pooled in equal volume using an EpMotion5075 liquid handling robot (Eppendorf, Hamburg, Germany). The pooled library was purified using an AMPure XP cleanup protocol (0.6X, vol/vol; Agencourt, Beckmann-Coulter) to remove fragments smaller than 300 bp. The pooled libraries, with a 20% phiX spike-in, were loaded onto an Illumina MiniSeq mid-output flow cell (2 × 150 paired-end reads). Based on the distribution of reads per barcode, the amplicons (before purification) were repooled to generate a more balanced distribution of reads. The re-pooled library was purified using AMPure XP cleanup, as described above. The re-pooled libraries, with a 15% phiX spike-in, were loaded onto a MiSeq v3 flow cell, and sequenced using an Illumina MiSeq sequencer. Fluidigm sequencing primers, targeting the CS1 and CS2 linker regions, were used to initiate sequencing. Library preparation, pooling, and MiniSeq sequencing were performed at the DNA Services (DNAS) facility, Research Resources Center (RRC), University of Illinois at Chicago (UIC).
Bile acid-induction and RNA isolation from cultures of clostridium hylemonae and clostridium hiranonis
Clostridium hylemonae was grown to mid-log phase in DM (control) or in DM supplemented with 100 µM CA or DCA as previously described.27 Clostridium hiranonis was cultivated in PYG medium or PYG with 100 µM CA or DCA. Cells were quenched with RNAprotect (Qiagen) and stored at −80°C until further processing. Total RNA was recovered from cells following disruption by bead beating in the presence of acid phenol as previously reported.34 In brief, stored cells were dissolved in lysis buffer (200 mM NaCl, 20 mM EDTA, and diethylpyrocarbonate-treated water) and transferred to 2-ml screw-cap bead-beating tubes (Sarstedt, Germany). Cells were washed with the same buffer and resuspended in 500 µl of lysis buffer. To each tube, 200 μl of zirconium beads, 210 μl of 20% SDS (Ambion), and 1 ml of 5:1 acid phenol were added. Cells were disrupted on a Mini-BeadBeater (Biospec Products, Bartlesville, OK) at maximum speed twice for 1 min, with tubes kept on ice in between treatments. The aqueous and phenol phases were separated by centrifugation at 5,000 rpm for 1 min, and the aqueous phase was transferred to a new tube and washed once with 1 ml of 5:1 acid phenol. Nucleic acids in the aqueous phase were precipitated at −80°C for 60 min by addition of 1/10 volume of 5 M ammonium acetate (Ambion), 1 μl of Glycoblue (Ambion), 1 µl of RNase inhibitor (NEB) and 1 volume of ice-cold isopropanol, followed by centrifugation at 13,600 g for 30 min. Precipitated nucleic acids were treated with RNase-free TURBO DNase (Ambion) according to the manufacturer’s instructions to remove contaminated genomic DNA. Total RNA was purified using the MEGAclear kit (Ambion) according to the manufacturer’s instructions. RNA purity and integrity were checked spectrophotometrically by the A260:A280 ratio and by separating 16S and 23S rRNA on a 1% agarose gel, respectively.
In vitro RNA-Seq analysis
Stranded RNASeq libraries were constructed and sequenced at the DNA Services laboratory of the Roy J. Carver Biotechnology Center at Illinois using the TruSeq Stranded RNA Sample Preparation Kit (Illumina San Diego, CA). Briefly, total RNA was quantitated by Qubit (Life Technologies) and checked for integrity on a 2% eGel (Life Technologies). Starting with 1 μg of RNA, ribosomal RNA was removed from the total RNA using the Ribo-Zero™ Magnetic Bacteria kit (Illumina, CA). First-strand synthesis was generated with a random hexamer and SuperScript II (Life Technologies). Double stranded DNA was blunt-ended, 3ʹ-end A-tailed and ligated to unique dual-indexed adaptors. The adaptor-ligated double-stranded cDNA was amplified by PCR for 8 cycles with the Kapa HiFi polymerase (Kapa Biosystems, Wilmington, MA). The final libraries were quantitated on Qubit and the average size determined on the AATI Fragment Analyzer (Advanced Analytics, Ames, IA). Libraries were pooled evenly, and the pool was cleaned one additional time using a 50:50 ratio with AxyPrep Mag PCR Cleanup beads (Axygen, Inc. Union City, CA) to ensure removal of primer and adaptor dimers, then evaluated on AATI Fragment Analyzer. The final pool was diluted to a 5 nM concentration and further quantitated by qPCR on a BioRad CFX Connect Real-Time System (Bio-Rad Laboratories, Inc. Hercules, CA).
The final pool containing 12 libraries was loaded onto 1 lane of an 8-lane flowcell each for cluster formation on the cBOT and then sequenced on an Illumina HiSeq4000 with version 1 SBS sequencing reagents from one end of the molecules for a total read length of 100 nt. The run generates bcl files which are converted into adaptor-trimmed demultiplexed fastq files using bcl2fastq v2.20 Conversion Software (Illumina).
Cecal RNA-Seq analysis
Total RNA was extracted from mouse cecum using the EZ1 RNA tissue kit (Qiagen, Germantown, MD) conducted on an EZ1 automated extraction instrument. Prior to extraction, tissue was homogenized with a FastPrep-24 5G bead-beating device (MP Biomedicals, Solon, OH) at 6 m/s for 40 sec. Total RNA extracts were subjected to an additional DNase treatment using the RNeasy Mini Kit (Qiagen) based on an RNeasy Total RNA Cleanup Protocol (W.M. Keck Foundation Biotechnology Resource Laboratory at Yale University). Microbial RNA-seq libraries were prepared using the ScriptSeq v2 RNA-Seq Library Prep kit (Illumina, San Diego, CA). 250 ng of total RNA was prepared for library preparation by ribosomal RNA depletion using both mouse and bacterial Ribo-Zero rRNA Removal Kit (Illumina) according to the manufacturer’s instructions. The resulting double depleted RNA was used as input for ScriptSeq library preparation according to the manufacturer’s instructions. Resulting libraries were pooled and sequenced on an Illumina NextSeq500 instrument using paired-end 2 × 150 base reads. Extraction, library preparation and sequencing were performed at the DNAS facility, University of Illinois at Chicago.
Bioinformatics
The QIIME v1.8 pipeline35 was utilized to generate operational taxonomic units (OTU) from 16S rDNA reads in a de novo manner using UCLUST method.36 Taxonomic annotations were assigned to each OUT using UCLUST and greengenes_13_8.97 reference database.37 Quality control of raw RNA-seq reads was performed using the FastQC 0.11.5. Reads that had Q scores of <32 were used for further analysis. Raw reads from in vitro RNA-Seq and cecal RNA-Seq were aligned with ribosomal RNA sequences prepared from respective bacterial genomes using bowtie2 (v2.3.3.1). Unaligned files were saved and realigned with the respective bacterial genomes using the same tool. Output sam files were converted to the bam format using Samtools (v0.1.6) and were name-sorted prior to input into HTSeq (v0.9.1). HTSeq counting was performed in union mode against a Gene Feature Format (GFF) annotations of each bacterium. Reads were counted against coding DNA sequences (CDS) of the organism. Differential gene expression analysis was performed using the edgeR38 in R package. Low-expression reads were filtered from analysis, and a minimum Bonfferoni corrected P-value (false discovery rate (FDR) value) of <0.05 was accepted as indicating differentially expressed (DE) genes. Genes were binned according to known functionality, and totals were generated for up-regulated and down-regulated genes. Category analysis was performed using the eggNOG eggnog-mapper (web interface with: Bacteria as taxonomic scope and DIAMOND as search program).39,40
Sample preparation for bile acid metabolomics
Sample preparation and the LC-MS/MS for bile acid analysis were essentially based on the previously developed method24 with modifications as follows: Cecum contents were lyophilized before extraction. To the dried matter (10 mg), 90% ethanol (2 ml) was added and bile acids were extracted by ultra-sonication at room temperature for 1 h. The supernatant was separated by centrifugation at 2,500 rpm for 5 min and collected in a glass test tube. This procedure was repeated twice and, the second and third extracts were combined to the first extract. Ethanol was evaporated under an N2 stream. Liver (300–400 mg) was homogenized with cold water (500 µl). To each homogenate, 20 mg/ml Proteinase K solution (50 µl) was added and the liver was completely digested at 55℃ for 16 h. Bile acids were extracted using 90% ethanol by the same manner as described for cecum. Serum (50 µl) was added to acetonitrile (5 ml) and evaporated to dryness under and N2 stream.
Thus, prepared crude bile acid extracts were resuspended in 90% ethanol (1ml) by ultra-sonication and, deuterium-labeled internal standards, d4-CA, d4-GCA, and d4-TCA were added at each concentration at 100 nmol/ml. For the cecum and liver extracts, an aliquot (100 µl) was diluted with water (900 µl) and then applied on a solid-phase extraction cartridge (InertSep C18-B 100 mg sorbent weight, preconditioned with 1 ml of methanol and 3 ml of water). The column was washed with water (1 ml) and then desired bile acids were eluted with 90% ethanol (1 ml). For the serum extract, the whole suspension (1 ml) was diluted with water (9 ml) and, applied on a solid-phase extraction column. The column was eluted in the same manner. After evaporation of the solvent, the residue was dissolved in 20% acetonitrile (1 ml), and aliquot (5 µl) of the solution was analyzed by the LC/ESI-MS/MS.
LC/ESI-MS/MS analysis
The LC/ESI-MS/MS system comprised a LCMS-8050 tandem mass spectrometer, equipped with an ESI probe and Nexera X2 ultra high-pressure liquid chromatography system (Shimadzu, Japan). A separation column, InertSustain C18 (150 mm × 2.1 mm ID, 3 μm particle size; GL Sciences, Tokyo, Japan), was employed at 40°C. A mixture of 10 mM ammonium acetate and acetonitrile was used as the eluent, and the separation carried out by linear gradient elution at a flow rate of 0.2 ml/min. The mobile phase composition was gradually changed as follows: ammonium acetate-acetonitrile (86:14, v/v) for 0.5 min, ammonium acetate-acetonitrile (78:22, v/v) for 0.5–5 min, ammonium acetate-acetonitrile (72:28, v/v) for 5–28 min, ammonium acetate-acetonitrile (46:54, v/v) for 28–55 min, ammonium acetate-acetonitrile (2:98, v/v) for 55–66 min, and ammonium acetate-acetonitrile (2:98, v/v) for 4 min. The total run time was 70 min.
To operate the LC/ESI-MS/MS, the MS parameters were as follows: Spray voltage; 3000V, heating block temperature; 400°C, nebulizing gas flow; 3 L/min, drying gas flow; 10 L/min, heating gas flow; 10 L/min, interface temperature 300°C, collision gas (argon) pressure; 1.3 mm Torr, collision energy; 11–85 eV, all in the negative ion mode.
Accession numbers
Genomic and RNA-Seq datasets for C. hylemonae BioProject PRJNA523213 and PRJNA523217 for C. hiranonis. Cecal RNA-Seq datasets were deposited as Bioproject PRJNA523415.
Results
Comparative genomics of bile acid 7α-dehydroxylating bacteria
Draft genomes of Clostridium hylemonae DSM 15053 and Clostridium hiranonis DSM 13275 were reported as part of the Human Microbiome Project (Bioprojects PRJNA30369 and PRJNA28657). The genomes of Clostridium hylemonae DSM 15053 and Clostridium hiranonis DSM 13275 were closed through a combination of Oxford Nanopore and Illumina MiSeq sequencing. C. hiranonis MiSeq 250 single-end bp reads totaled 4,303,190 and when combined with 22,498 trimmed Nanopore reads, resulted in a single scaffold. C. hylemonae MiSeq analysis resulted in 3,573,168 single-end bp reads and when combined with 48,846 trimmed Nanopore reads, also resulted in a single scaffold (Table S2).
Comparative genomics determined that C. hylemonae contained 1,105 paralogs while C. hiranonis contained 449 paralogs, or 3.30 and 3.03 paralogs per paralog-containing group, respectively (Figure 2(a)). C. hylemonae presented 2,479 single-copy genes, while C. hiranonis had 1,790. There were 1,035 orthologous groups shared between C. hylemonae and C. hiranonis, involving 1,199 and 1,108 genes, respectively (Figure 2(b)). C. hiranonis encodes 2,363 predicted genes (of which 2,239 are protein-coding) and C. hylemonae encodes 3,647 genes (3,584 protein-coding).
Figure 2.
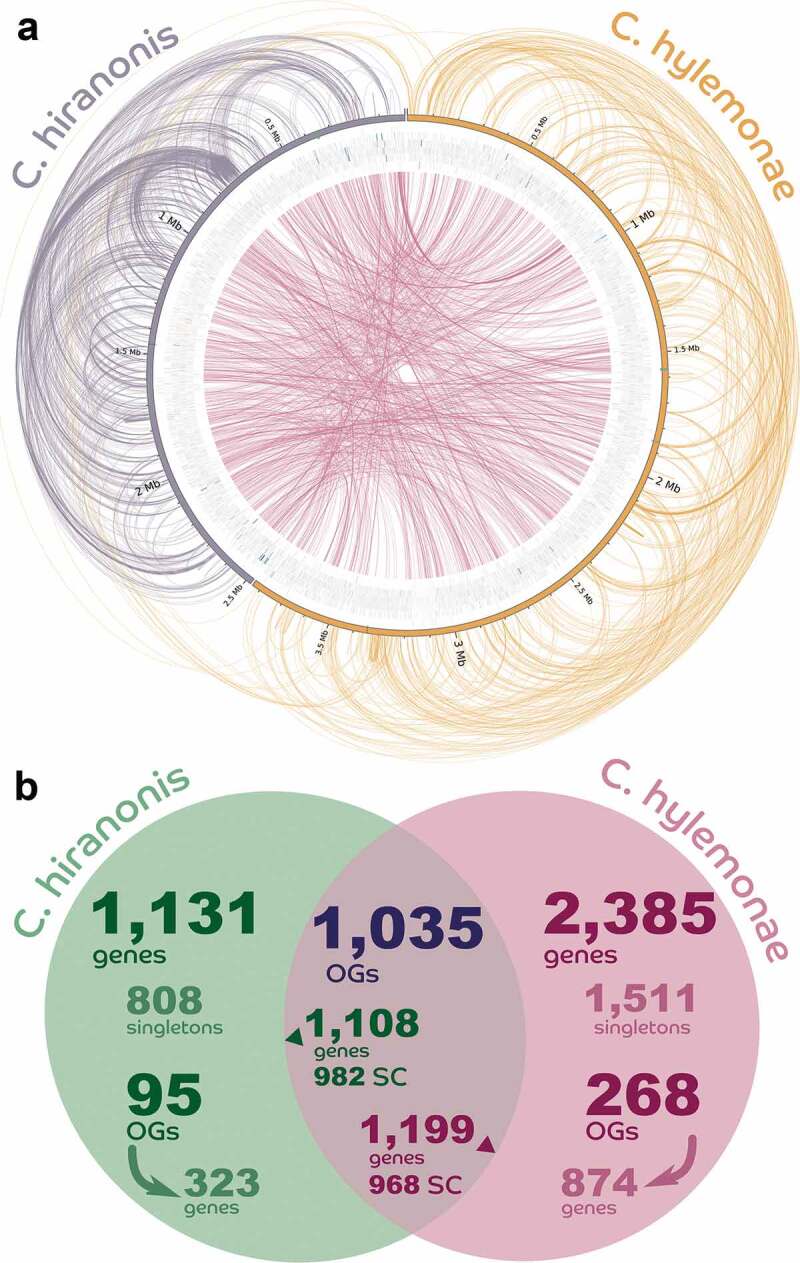
Comparative genomics between Clostridium hylemonae and Clostridium hiranonis. (a) Helios plot of orthologs and paralogs inferred by the comparison between the proteomes of the two species (colored lines linking the genes) and all predicted genes (with protein-coding genes in gray, CRISPR repeat regions in black, tRNA genes in red, and rRNA genes in blue); colored highlights over the bar representing each sequence are the locations of the NADP-dependent 7α-hydroxysteroid dehydrogenase and the bai operon and other related genes in green. (b) Venn Diagram showing both core genes shared between the two clostridia, as well as unique genes. Protein coding genes (PCGs), orthologous groups (OGs).
We performed orthologous group (OG) analysis on the total deduced proteome of C. hylemonae and C. hiranonis (Figure 3(a); Supplementary Dataset 1) and also the deduced proteome exclusive to each bacterium (Figure 3(b); Supplementary Dataset 2). Significant differences in OG categories between total protein datasets include G (Carbohydrate metabolism and transport), J (Translation), L (replication and repair), N (cell motility), none (proteins unique to each organism), and T (signal transduction). Differences in category N relate to C. hylemonae being motile39 and harboring numerous genes encoding flagellar proteins (Figure 3(a)). C. hiranonis differs from C. hylemonae in category L, encoding many additional genes predicted to encode transposases (53 genes). Intriguingly, C. hylemonae had significant increases in categories C (energy production and conversion), E (amino acid metabolism), and G (carbohydrate metabolism and transport) suggesting increased metabolic capacity and recognition of growth substrates.
Figure 3.

eggNOG category analysis of protein coding genes between Clostridium hylemonae and Clostridium hiranonis. (a) orthologous group (OG) analysis on the total deduced proteome of C. hiranonis (red), and C. hylemonae (blue). (b) Orthologous group category of deduced proteome exclusive to C. hiranonis (green) and C. hylemonae (orange). Orthologous group categories displayed on the X-axis are as follows: (a) RNA processing and modification, (b) chromatin structure and dynamics, (c) energy conversion, (d) cell cycle control and mitosis, (e) amino acid metabolism and transport, (f) nucleotide metabolism and transport, (g) carbohydrate metabolism and transport, (h) coenzyme metabolism, (i) lipid metabolism, (j) translation, (k) transcription, (l) replication and repair, (m) cell wall/membrane/envelope biogenesis, (n) cell motility, (NH) no hits, unknown function (o) posttranslational modification, protein turnover, chaperone functions, (p) inorganic ion transport and metabolism, (q) secondary structure, (s) function unknown, (t) signal transduction, (u) intracellular trafficking and secretion, and (v) defense mechanisms. The asterisks mark the categories for which there is significant difference, according to the Fisher exact test, between the counts of proteins: one asterisk means significant at alpha < 0.00238 (which is 0.05/21, with the Bonferroni correction due to the 21 tests being performed), two asterisks mean significant at alpha < 2.38 × 10−6 and three asterisks mean significant at alpha < 2.38 × 10−9.
The bile acid-inducible gene cluster (baiBCDEAFGH) consists of a bile acid CoA-ligase (baiB; KGNDJEFE_00401), NAD+-dependent 7α-3-dehydro-4-cholenoic acid oxidoreductase (baiCD; KGNDJEFE_00402), bile acid 7α-dehydratase (baiE, KGNDJEFE_00403), NAD+-dependent 3α-hydroxysteroid dehydrogenase (baiA; KGNDJEFE_00404), bile acid CoA-transferase (baiF; KGNDJEFE_00405), H+-dependent bile acid transporter (baiG; KGNDJEFE_00406), and NAD+-dependent 7β-3-dehydro-4-cholenoic acid oxidoreductase (baiH; KGNDJEFE_00407). A conserved bai promoter is located upstream of the baiB gene and a putative transcription factor with a helix-turn-helix domain is encoded by the first ORF upstream of the promoter, but on the opposite strand.41 Downstream of the putative transcriptional regulator is a 20.8 kDa hypothetical protein, followed by a gene encoding a 21.6-kDa SnoaL homolog predicted to have bile acid 7β-dehydratase activity (baiI).21,42 Unlike C. scindens strains, C. hiranonis DSM 13275 only has one baiA copy. The bai operon in C. hylemonae DSM is also distinct from C. scindens and C. hiranonis in its organization (baiBCDEFGHI; LAJLEIBI_01433-LAJLEIBI_01439), with baiA (LAJLEIBI_01704) downstream of the baiJKL operon.41,43 A baiJ homolog (KGNDJEFE_00624) encoding a predicted flavin-dependent oxidoreductase in C. hiranonis was identified. The genome of C. hiranonis encodes a predicted bile salt hydrolase (KGNDJEFE_01093). Bile salt hydrolase catalyzes the hydrolysis of C24 amide bond linking taurine or glycine to bile acids. In one study, C. hiranonis was reported to express BSH activity in vitro, but not in vivo.26
In vitro RNA-Seq analysis of bile acid induction of C. hylemonae
Next, we wanted to determine the effect of induction with either 100 μM CA or 100 μM DCA on the transcriptional profiles (Illumina HiSeq 4000) of C. hiranonis and C. hylemonae. Growth of C. hylemonae in defined medium27 was not affected by bile acids at 100 μM; however, we did observe a small, but significant decline in growth of C. hiranonis in the presence of CA and DCA (Figure S1). We obtained an average of 12,496,992 mapped reads per sample for C. hylemonae and 10,984,255 mapped reads for C. hiranonis (Figure S2).
Growth of C. hylemonae in defined medium in the presence of 100 μM CA resulted in 334 genes significantly up-regulated >1.5 fold (P <.05) (Figure 4(a); Extended Dataset 3). Cultivation of C. hylemonae in DM + 100 μM DCA significantly modulated the expression of 332 genes >1.5 fold (p value <0.05) (Figure 4(a); Extended Dataset 4). There was overlap (216 genes) between CA and DCA datasets owing perhaps to general responses to bile acids, or due to conversion of CA to DCA at the time of mRNA collection (Figure 4(b)). Expression of genes involved in bile acid 7α-dehydroxylation of CA were significantly upregulated, including the baiB (835.9-fold; P = a1.44E-241; FDR = 5.15E-238), baiCD (609.1-fold; P = 9.57E-225; FDR = 1.14E-221), baiE (1,416.2-fold; P = 5.81E-51; FDR = 2.98E-48), baiF (431-fold; P = 1.35E-206; FDR = 9.68E-204), and baiG (263.3-fold; P = 1.38E-213; FDR = 1.24E-210). At the 3ʹ-end of the bai operon are two genes, baiH (176.8-fold; P = 1.04E-229; FDR = 1.87E-226), and baiI (5.1-fold; P = 2.32E-47; FDR = 1.04E-44) involved in 7β-dehydroxylation of ursodeoxycholic acid to lithocholic acid (Figure 5(a)).
Figure 4.
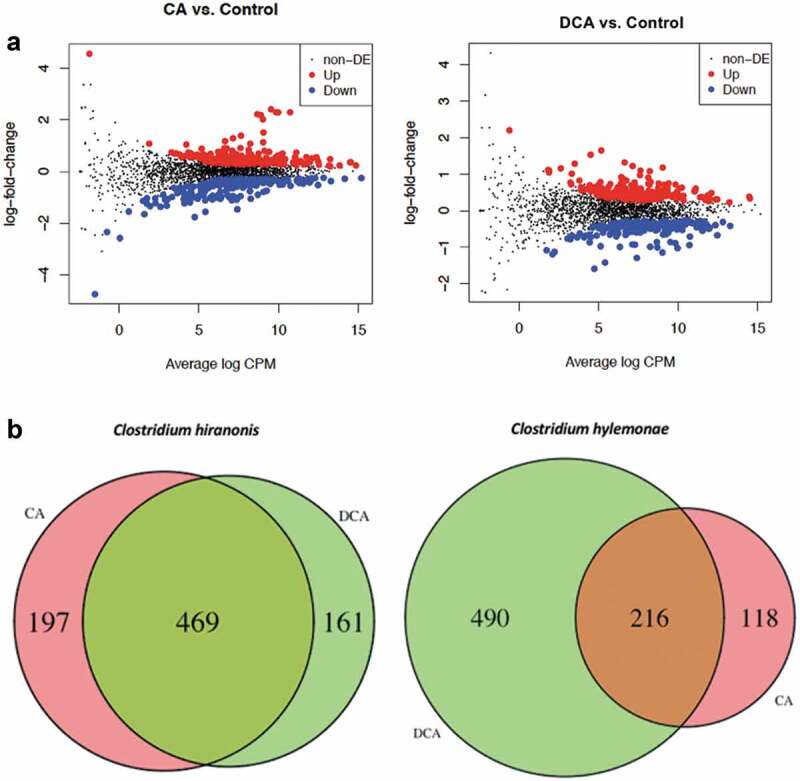
Log2 fold change vs. log counts per million (CPM) scatterplot and Venn Diagram of differentially expressed genes after cholic acid (CA) or deoxycholic acid (DCA) induction of C. hylemonae and C. hiranonis. (a) Left: scatterplot of CA vs. control. Right: Scatterplot of DCA vs. control for C. hiranonis. Upregulated genes are presented by a red dot, down-regulated genes by a blue dot, and a black dot represents genes not differentially expressed (see Extended Dataset 2, 3). (b) Venn Diagram of CA vs. DCA differentially expressed genes in C. hiranonis and C. hylemonae. Up- and down-regulated genes were defined at a threshold of P <.05 and a log2FC >(-)0.58.
Figure 5.
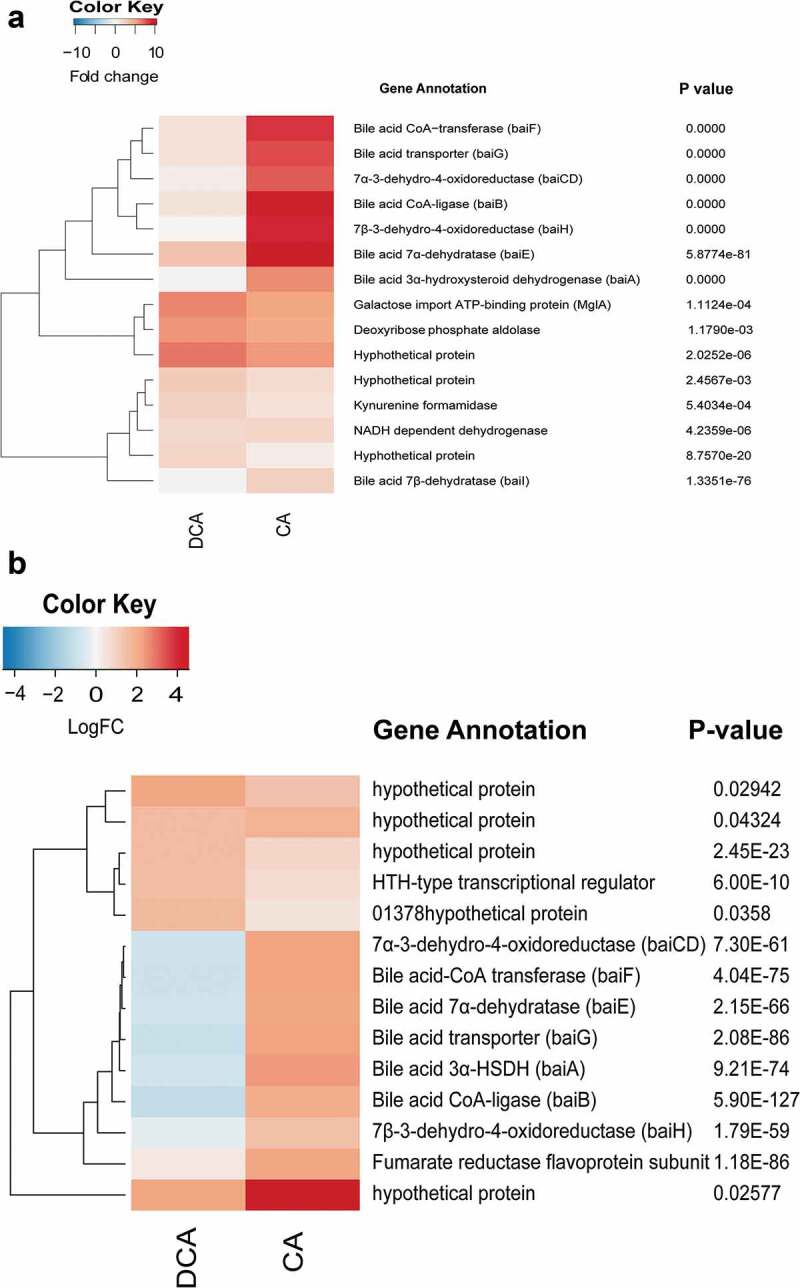
Heat maps of genes up-regulated by CA and DCA in Clostridium hylemonae and Clostridium hiranonis. (a) Heat map of differential expression for C. hylemonae in the presence of 100 μM CA or 100 μM DCA. B. Heat map of differential expression for C. hiranonis in the presence of 100 μM CA or 100 μM DCA. Up- and downregulated genes were defined at a threshold of P <.05 and a log2FC >(-)0.58. P values displayed to the right of each gene.
NAD(H) is an essential cofactor in enzyme reactions catalyzed by BaiA,44,45 BaiCD/BaiH,42 and presumably, by the three reductive steps in the pathway (3-dehydro-4,6-DCA~CoA → 3-dehydro-4-DCA~CoA → 3-dehydro-DCA~CoA → DCA~CoA), therefore upregulation of pyridine nucleotide synthesis in the presence of bile acids may be expected. Two putative kynurenine formamidases (LAJLEIBI_00016 and LAJLEIBI_02677) involved in the conversion of tryptophan to NAD(H) were up-regulated 1.37 log2FC (P =.02) and 1.32 log2FC (P =.04), respectively, by CA. A kynureninase (LAJLEIBI_01991) was also up-regulated by CA 0.96 log2FC (P = 8.6E-05; FDR = 6.4E-03), as was 2,5-dihydroxypyridine-5,6-dioxygenase (LAJLEIBI_03152; 0.80 log2FC; P =.02 (Figure 5(a)). In C. hylemonae, DCA addition resulted in far lower expression of bai genes. The baiE gene was expressed 3.2 log2FC by DCA vs. 10.46 log2FC by CA (P = 5.88E-81; FDR = 3.0E-78). Deoxycholic acid induced a number of stress-response genes (Table S3). C. perfringens was previously shown to express genes involved in early-to-late sporulation and to form endospores in the presence of DCA.46
In vitro RNA-Seq analysis of bile acid induction of C. hiranonis
The nutritional requirements for C. hiranonis DSM 13275 have yet to be determined, and since growth was not observed in DM, the strain was cultivated in peptone-yeast-glucose broth (PYG medium). The baiBCDAEFGH gene cluster was upregulated 5.3 fold to 2.85 fold in the presence of CA (P = 4.09E-28 to 9.72E-50; FDR = 1.76E-29 to 1.09E-46) (Figure 5(b); Extended Datasets 6 and 7). These results may seem surprising given that in the CA 7α-dehydroxylation activity assay suggests a significantly higher rate of conversion of CA to DCA by C. hiranonis than C. hylemonae, which has been reported previously.47 However, C. hiranonis basal bai gene expression in PYG (but not PYG + DCA) is on par with expression levels of C. hylemonae grown in the presence of CA (Table S4; Extended Dataset 3 and 4). Alternatively, basal expression of bai genes in C. hiranonis are intrinsically higher than that of C. hylemonae. Also expressed is a predicted urocanate reductase (KGNDJEFE_00624) upregulated 1.04 log2FC (P = 9.96E-09; FDR = 5.44E-07) by CA but not DCA, consistent with recently reported bile acid-induced transcriptome analysis in C. scindens ATCC 35704 (Figure 5(b)).27 The urocanate reductase is a baiJ homolog. The baiJ encodes a predicted flavoprotein and is located upstream of baiK which was shown previously to encode a bile acid CoA transferase.41,43
There were 58 genes significantly down-regulated in C. hiranonis in the presence of DCA including 26 hypothetical proteins (Extended Dataset 8). The bai genes were among the most highly downregulated genes in the presence of DCA (Figure 5(b)). This may indicate a feedback inhibition during the accumulation of the toxic end-product DCA.
Cecal gut microbial community analysis
We then colonized germ-free C57BL/6 mice with the B4PC2 synthetic gut community, which includes C. hylemonae and C. hiranonis. After 27 days of colonization, DNA and RNA was isolated from cecal contents. We obtained on average 49,965.8 ± 3,439.9 assembled MiSeq reads from cecal DNA from six mice harboring B4PC2. The Bacteroidetes made up 84.71% of 16S rDNA cecal reads comprised of B. vulgatus (39.06%), B. uniformis (34.65%), and P. distasonis (11%). The taurine-respiring member of the δ-proteobacteria, B. wadsworthia, constituted 14.7% of the gut microbial community. The bile acid 7α-dehydroxylating Firmicutes, C. hylemonae, and C. hiranonis, made up only 0.41% and 0.14% of the microbial community, respectively, abundances that reflect levels <1% observed in human fecal samples and in prior gnotobiotic studies.5,26,48 Blautia producta made up 0.007% relative abundance. The microbial community composition was consistent across all six C57BL6 mice (Figure 6).
Figure 6.
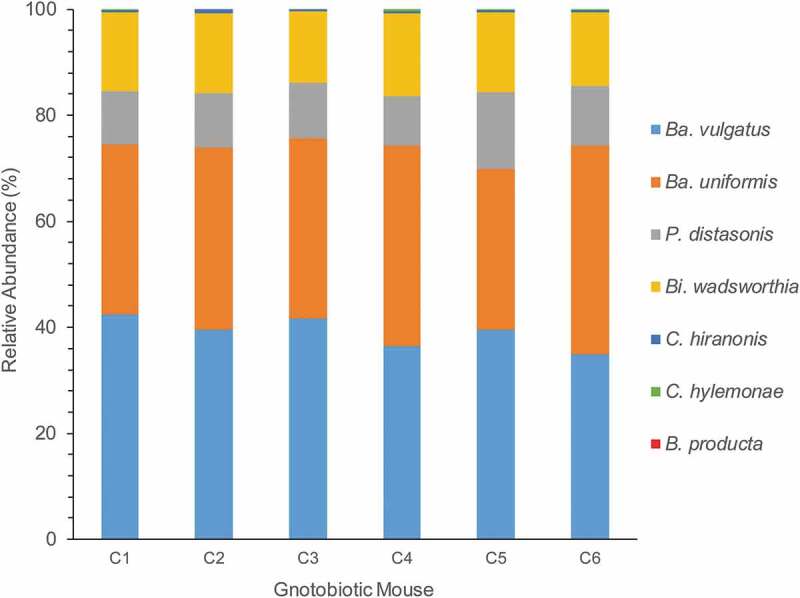
Relative abundance of B4PC2 community in cecum of gnotobiotic mice. The X-axis displays each mouse (C1–C5), and the y-axis displays Illumina MiSeq counts of the 16S rDNA gene from each organism in the community.
Bile acid metabolomics analysis of gnotobiotic mice
To determine the extent of functional bile acid metabolism by the B4PC2 community, we analyzed the bile acid profile in the cecum, serum, and liver. The bile acid panel was composed of 100 bile acids including taurine and glycine conjugates, sulfated bile acids, unconjugated cholate and chenodeoxycholate derivatives, as well as bile acid precursors and C-23 and C-27 bile acids (Table S1).
We detected and quantified 14 bile acid metabolites in the cecum of mice colonized with the B4PC2 community. Total cecal bile acids averaged 1.29 ± 0.29 μmol/g (Figure 7(a)). Unconjugated bile acids comprised 84.0 ± 35.0% of cecal bile acids, conjugated bile acids were 7.15 ± 2.24% of the total cecal bile acid pool, indicating functional bile salt deconjugation by cecal bacteria. Sulfated bile acids (3-sulfocholic acid, 3-sulfoursodeoxycholyltaurine) were also detected in the cecal bile acid pool (5.4 ± 2.99%). Of the unconjugated bile acids, 54.5 ± 25.7% were primary bile acids while 29.46 ± 9.26% were secondary bile acids (DCA or LCA and their metabolites). Of the primary bile acids, the vast majority (0.74 μmol/g) are muricholic acids (alpha or beta), which are not converted to secondary murideoxycholic (MDCA) acids by human bile acid 7α-dehydroxylating isolates such as C. hylemonae and C. hiranonis.26,49 MDCA was detected at low levels in the cecum of two mice (0.01 µmol/g); however, the mouse liver is capable of converting LCA formed from bacterial 7α-dehydroxylation of CDCA or 7β-dehydroxylation of UDCA to MDCA.12 3-sulfocholic acid made up 42% of the primary cholic acid derivatives, which is presumably not metabolized by C. hylemonae or C. hiranonis due to lack of bacterial sulfatase in the B4PC2 community capable of recognizing this substrate. DCA was a major bile acid detected in cecum, composing 27.1 ± 7.9% of total bile acids and 78.2% of CA derivatives. These results indicate that C. hylemonae and C. hiranonis were highly efficient at converting primary bile acids to secondary bile acids in vivo. A prior report on combinations of bile salt deconjugating and 7α-dehydroxylating communities colonizing gnotobiotic BALB/c mice reported DCA at 20–48% of cecal bile acids.26 These results confirm functional bile acid metabolism by the B4PC2 consortium.
Figure 7.

Bile acid metabolomics in cecum, serum, and liver of gnotobiotic mice colonized with B4PC2 human gut bacterial consortium. (a) Bile acid profile (umol g−1) of mouse ceca (S.E.M, N = 6). (b) Bile acid profile (umol L−1) of mouse sera. (c) Bile acid profile of mouse livers (nmol g−1). Left panel of each profile displays ratio of conjugated/unconjugated bile acids (top) and secondary bile acids/primary bile acids (bottom).
Serum total bile acids were detected at a concentration of 7.38 ± 3.33 μmol/L. There was near equal concentration of serum conjugated bile acids (51.0 ± 25.3%) and serum unconjugated bile acids (49.9 ± 30.9%) reflecting absorption of host conjugated and microbial bile acid metabolites. Of total unconjugated bile acids (3.93 μmol/L), 30.9% are products of bile acid 7α-dehydroxylation (2.30 μmol/L), showing that in this model, microbial bile acid products are being absorbed from the gut, and returned to the liver. Of the bile acid 7α-dehydroxylation products in serum, LCA and its derivatives predominate, comprising 79.9% of secondary bile acids and 23% of total serum bile acids (Figure 7(b)). Lithocholic acid derivatives included isolithocholic acid (3β-monohydroxy-5β-cholan-24-oic acid) and allo-isolithocholic acid (3β-monohydroxy-5α-cholan-24-oic acid). The formation of 3β-isomers of LCA can be explained by expression of 3α-HSDH and 3β-HSDH by B. producta.29 Bile acid 7α-dehydroxylating bacteria are capable of forming allo-secondary bile acids from host primary bile acids.41,50 Deoxycholic acid (0.52 ± 0.51 μmol/L) and taurodeoxycholic acid (0.38 ± 0.27 μmol/L) were also detected in serum.
In the liver, total bile acids were measured at 187.79 ± 55.35 nmol/g, of which 56.4% were taurine conjugates (Figure 7(c)). TCA and T-β-MCA were in near equal proportion and constituted ~95% of bile acid conjugates. β-MCA (68.86 ± 17.59 nmol/g) was the major unconjugated bile acid measured in liver, constituting 84% of unconjugated bile acids. Notable minor bile acid metabolites include glycine (1.27 ± 0.31 nmol/g) and taurine (1.01 ± 0.28 nmol/g) conjugates 3-sulfoUDCA, MDCA (0.52 ± 0.27 nmol/g), as well as TDCA (1.66 ± 0.8 nmol/g). Taken together, these results demonstrate that the B4PC2 defined microbial consortium was established and functionally active in altering host bile acids. Moreover, it is important to note the disproportionate role that low abundance members, such as the bile acid 7α-dehydroxylating bacteria and Blautia, contribute to metabolism.
Expression of microbial bile acid-metabolizing genes in the gnotobiotic mouse cecum
The data presented in the preceding sections established both the successful colonization of TCA metabolizing bacteria, but also the capability of the B4PC2 community to functionally metabolize bile acids. Next, we wanted to extend previous work on the B4PC2 community by measuring microbial gene expression in the gnotobiotic mouse cecum, in part to observe RNA levels of functional bile acid metabolizing genes, but also to gain insight into the in vivo gene expression profile of bile acid-metabolizing bacteria. After filtering rRNA reads, we obtained an average of 24 million RNA reads per sample (N = 6) that were mapped to the seven genomes.
Cecal RNA-Seq predicted functional bile acid metabolism (Table 1, Figure 8). Bile salt hydrolase expression by Bacteroidetes species was relatively low, with the exception of BACUNI_RS1345 (811 ± 453 TPM). Genes encoding BSH are generally constitutively expressed.21 Despite the low level of bsh gene expression in Bacteroides spp., bile acid metabolomics confirm BSH activity. By contrast, genes involved in taurine respiration by B. wadsworthia were highly expressed (Table 1). Coupled with bile acid metabolomics data, this indicates that taurine is being deconjugated from bile acids and metabolized by Bilophila.51-53 Genes encoding bai enzymes were expressed in the mouse cecum (Table 1). The level of expression observed for bai genes from C. hylemonae in vivo are comparable to those observed during treatment with 100 μM CA in defined medium (Supplementary Dataset 3). A putative cholylglycine hydrolase (KGNDJEFE_01093; 18.3 ± 13.4 TPM) was not expressed, nor was a homolog of NADP-dependent 7α-hydroxysteroid dehydrogenase (KGNDJEFE_01338; 2.68 ± 1.4 TPM) characterized previously in C. scindens.54 Expression across the bai operon of C. hiranonis is not uniform with two genes within the expression range (TPM) observed in PYG medium + 100 μM CA (baiE, baiA), but with others at lower expression levels comparable to in vitro control data. The low expression level (relative to in vitro data) is likely reflects the low level of primary bile acids, such as CA and CDCA, measured in the cecum at the time of cecal content preservation, which are necessary for induction of bai genes.27 This may indicate the necessity to observe transcriptional responses to diurnal digestive processes since mice forage during the night, and cecal content was collected during the day.
Table 1.
Expression of bile acid metabolizing genes in the gnotobiotic mouse cecum.
| Organism | Gene | Locus | Transcripts Per kilobase Million (TPM) |
|---|---|---|---|
| Taurine Deconjugation | |||
| Ba. vulgatus | bile salt hydrolase 1 | BVU_RS05230 | 87 ± 58 |
| Ba. vulgatus | bile salt hydrolase 2 | BVU_RS13575 | 35.5 ± 23 |
| Ba. vulgatus | bile salt hydrolase 3 | BVU_RS20010 | 86.7 ± 29.4 |
| Ba. uniformis | bile salt hydrolase 1 | BACUNI_RS03925 | 149 ± 50 |
| Ba. uniformis | bile salt hydrolase 2 | BACUNI_RS13450 | 811 ± 453 |
| P. distasonis | bile salt hydrolase 1 | BDI_RS01760 | 57 ± 55 |
| P. distasonis | bile salt hydrolase 2 | BDI_RS07735 | 31 ± 35 |
| P. distasonis | bile salt hydrolase 3 | BDI_RS03790 | 89 ± 218 |
| Taurine Metabolism | |||
| Bi. wadsworthia | alanine dehydrogenase | T370_RS0108865 | 24,569 ± 6,057 |
| Bi. wadsworthia | taurine-pyruvate aminotransferase | T370_RS0108870 | 7,380 ± 2,225 |
| Bi. wadsworthia | dissimilatory-type sulfite reductase subunit alpha | T370_RS0109480 | 12,217 ± 1,762 |
| Bi. wadsworthia | dissimilatory-type sulfite reductase subunit beta | T370_RS0109485 | 2,131 ± 516 |
| Bi. wadsworthia | sulfite reductase | T370_RS0115475 | 1,542 ± 818 |
| Bi. wadsworthia | Isethionate sulfite-lyase(IslA) | T370_RS0117375 | 9,975.3 ± 6,140.5 |
| Deoxycholic acid Formation | |||
| C. hylemonae | bile acid transporter (baiG) | LAJEIBI_01437 | 184 ± 174 |
| C. hylemonae | bile acid CoA ligase (baiB) | LAJEIBI_01433 | 327 ± 220 |
| C. hylemonae | 3alpha-hydroxysteroid dehydrogenase (baiA) | LAJEIBI_01704 | 673 ± 484 |
| C. hylemonae | bile acid 7alpha-3-oxo-4-oxidase (baiCD) | LAJEIBI_01434 | 228 ± 143 |
| C. hylemonae | bile acid 7alpha-dehydratase (baiE) | LAJEIBI_01435 | 44.5 ± 28 |
| C. hylemonae | bile acid CoA transferase (baiF) | LAJEIBI_01436 | 103 ± 59 |
| C. hylemonae | bile acid 7beta-3-oxo-4-oxidase (baiH) | LAJEIBI_01438 | 206 ± 141 |
| C. hylemonae | bile acid 7beta-dehydratase (baiI) | LAJEIBI_01439 | 104 ± 60 |
| C. hylemonae | 3-oxo-4,6-reductase (baiN) | LAJEIBI_01670 | 138 ± 28 |
| C. hylemonae | 12alpha-hydroxysteroid dehydrogenase | LAJEIBI_01869 | 1,368 ± 620 |
| C. hiranonis | bile acid transporter (baiG) | KGNDJEFE_00406 | 192 ± 238 |
| C. hiranonis | bile acid CoA ligase (baiB) | KGNDJEFE_00401 | 69 ± 127 |
| C. hiranonis | 3alpha-hydroxysteroid dehydrogenase (baiA) | KGNDJEFE_00404 | 2,436 ± 4,031 |
| C. hiranonis | 3-oxo-4-oxidase (baiCD) | KGNDJEFE_00402 | 207 ± 324 |
| C. hiranonis | bile acid 7alpha-dehydratase (baiE) | KGNDJEFE_00403 | 802 ± 1429 |
| C. hiranonis | 3-oxo-4,6-reductase (baiN) | KGNDJEFE_00567 | 62 ± 75 |
| C. hiranonis | 12alpha-hydroxysteroid dehydrogenase | KGNDJEFE_00859 | 42 ± 35 |
Figure 8.

Identification of functional bile acid metabolizing genes in mouse cecal metatranscriptome of B4PC2 community.
The gene encoding NADP(H)-dependent bile acid 12α-HSDH was highly expressed by C. hylemonae (1,368.95 ± 620.9 TPM), but was not expressed in C. hiranonis (42 ± 14.4 TPM) (Table 1). Expression of the baiA gene (LAJLEIBI_01704) in C. hylemonae, responsible for oxidizing C3 in the BA7 pathway was measured to be 673.5 ± 484 TPM. By contrast, the baiJKLN operon (LAJLEIBI_01701-01703) was not expressed in vivo. The baiN gene (LAJLEIBI_01670), recently reported to encode a flavin-dependent 3-oxo-4,6-reductase5556 and constitutively expressed in C. scindens ATCC 3570427 showed expression levels of 138.9 ± 28.75 TPM. These results indicate expression of bile acid metabolizing genes by human gut microbes in the mouse cecum.
The in vivo “lifestyle” of bile acid 7α-dehydroxylating bacteria
Carbohydrate metabolism by bile acid 7α-dehydroxylating Firmicutes appears to be largely restricted to simple monosaccharides and disaccharides.27,39 A gene cluster (LAJLEIBI_01095-LAJLEIBI_01102) predicted to be involved in D-galactose import and metabolism was highly expressed. A gene predicted to encode D-galactose-binding periplasmic protein (LAJLEIBI_01095; 62,483.9 ± 9,167.6 TPM) and with galactose import ATP-binding protein mglA (LAJLEIBI_01096; 4,776.5 ± 675.5 TPM), and galactose transport system permease mglC (LAJLEIBI_01097; 1,559.0 ± 240.96 TPM) were highly upregulated. Interestingly, a gene encoding a putative neopullalanase (tvaI) (LAJLEIBI_00506; 243 ± 76.6 TPM), a 1,4-α-glucan branching enzyme (glgB) (LAJLEIBI_00659; 129 ± 51.8 TPM), and an acetylxylan esterase (LAJLEIBI_00751; 392.1 ± 143.8 TPM) were also expressed in the cecum, suggesting the potential for some limited metabolism of complex carbohydrates.
Many clostridia are capable of coupling amino acid fermentations resulting in regeneration of NAD+ and formation of ATP through substrate-level phosphorylation in a process known as Stickland fermentation.56,57 A gene cluster in C. hylemonae encoding putative oligopeptide transport ATP-binding subunits oppC (LAJLEIBI_03275; 1,357.2 ± 244.2 TPM), oppD (LAJLEIBI_03274; 609.0 ± 157.8 TPM) and oppF (LAJLEIBI_03273; 622.6 ± 120.8 TPM) were highly expressed as well as flanking nickel transport permease nikB (LAJLEIBI_03276; 680.4 ± 213.1 TPM) and oligopeptide-binding protein appA (LAJLEIBI_03277; 1,280.3 ± 164.4 TPM). A predicted peptidase close to this gene cluster was also expressed (LAJLEIBI_03265; 1,390.2 ± 98.5 TPM). Several genes in C. hylemonae are predicted to encode enzymes involved in oxidizing threonine to L-2-amino-3-oxobutyrate catalyzed by threonine-3-dehydrogenase followed by cleavage to glycine and acetyl-CoA by glycine C-acetyltransferase. C. hylemonae expresses serine/threonine transporter (LAJLEIBI_00191; 654.085 ± 271.96 TPM) L-threonine-3-dehydrogenase (LAJEIBI_00192; 1446.68 ± 568.2 TPM), glycine C-acetyltransferase (LAJEIBI_00193; 534.09 ± 271.53 TPM) leading to acetate and ammonia. A second possible route of threonine fermentation is through cleavage to glycine and acetaldehyde by threonine aldolase (LAJLEIBI_00890; 109.329 ± 72.577 TPM).
Several enzymes involved in Stickland fermentation require selenocysteine residues.58 Genes encoding putative enzymes and tRNA involved in selenocysteine formation were identified and highly expressed in C. hylemonae in vivo (Figure S3). D-proline reductase is also composed of selenoproteins,59 and is also highly expressed in C. hylemonae (Figure S4). The first step, the conversion of naturally occurring L-proline to the D-imino form, is catalyzed by proline racemase (LAJLEIBI_00783; 38,853.7 ± 16,983.7 TPM), which was one of the most highly expressed genes measured from C. hylemonae in the mouse cecum. Genes predicted to encode subunits of D-proline reductase are located adjacent to proline racemase in a gene cluster (Figure S4). A separate gene cluster expressing putative D-proline reductase PrdA_1 and PrdA_2 was also identified (LAJLEIBI_00538; 286.76 ± 38 TPM, LAJLEIBI_00539; 216.45 ± 46.06 TPM). An electron transport complex subunit RnfC is adjacent to these genes (LAJLEIBI_00785; 2,993.84 ± 1,105 TPM).
Glycine reductase complex B subunits α and β (grdE_2) (LAJLEIBI_01338; 1,021.33 ± 461.2 TPM), glycine reductase complex component B subunit gamma (grdB_3) (LAJLEIBI_01339; 524.86 ± 208.8 TPM), glycine reductase A (grdA) (LAJLEIBI_01342; 921.53 ± 534.96 TPM), thioredoxin reductase (trxB_2) (LAJEIBI_01340; 1,921.8 ± 709.2 TPM), thioredoxin (trxA_1) (LAJLEIBI_01341; 2,682.8 ± 1,162.1 TPM), glycine reductase A1 (grdA1) (LAJLEIBI_10343; 1,382.65 ± 573.271 TPM), glycine reductase C subunit β (LAJLEIBI_01344; 648.67 ± 307.16 TPM), and glycine reductase C subunit α (grdD) (LAJLEIBI_01345; 129.5 ± 82.3 TPM) were highly expressed as a gene-cluster (Figure S5). A second cluster encoding glycine reductase was also detected encoding putative glycine reductase complex component B subunits α and β (grdE_1) (LAJLEIBI_00090; 133.0 ± 59.57 TPM), glycine reductase complex component B subunits gamma (grdB_1) (LAJLEIBI_00091; 369 ± 180.4 TPM), glycine reductase complex component B subunits gamma (grdB_2) (LAJLEIBI_00092; 79.9 ± 188.9). A glycine/betaine transporter (BetL) was also identified (LAJLEIBI_00333; 67.75 ± 26.24 TPM). This suggests that C. hylemonae is utilizing glycine as an electron acceptor in the mouse cecum.
A gene cluster encoding putative thioredoxin and glycine reductase complex enzymes was highly expressed in C. hiranonis in vivo. Thioredoxin (KGNDJEFE_01781; 2,414.3 ± 341.7 TPM) and thioredoxin reductase (KGNDJEFE_01782) were co-expressed as a gene cluster. Genes encoding D-proline reductase were also highly expressed by C. hiranonis. Proline racemase (KGNDJEFE_00584) was expressed at 8,958.9 ± 1,475.1 TPM as part of a gene cluster with four copies of D-proline reductase proprotein A (prdA) (KGNDJEFE_00583, 533.2 ± 127.0 TPM; KGNDJEFE_00582, 179.1 ± 81.2 TPM; KGNDJEFE_00578, 1,569.9 ± 201.4 TPM; KGNDJEFE_00577, 2,478.9 ± 304.3 TPM). A proline iminopeptidase was also highly expressed (KGNDJEFE_00878, 778.2 ± 83.8 TPM). These results indicate the likelihood that these clostridia utilize the amino acids glycine and proline as electron acceptors during fermentation.
Predicted stress pathways were expressed by C. hiranonis. Rurerythrin (rbr) was highly expressed (KGNDJEFE_02033; 46,555.5 ± 13,778.4 TPM), as was flanking universal stress protein (KGNDJEFE_02034; 5,534.3 ± 1,865.3 TPM). Reverse rubrerythrin (rbr3A) was expressed in vivo (KGNDJEFE_01702; 1,803.8 ± 289.9 TPM) as was high molecular weight rubredoxin (KGNDJEFE_01844; 1,583.76 ± 209.0 TPM). A putative peroxiredoxin (bcp; KGNDJEFE_01200; 432.6 ± 102.9 TPM) involved in detoxification of H2O2 and organoperoxides was also expressed. Osmoprotectant import ATP-binding protein (osmV) was also expressed (KGNDJEFE_01813; 1,394.3 ± 279.3 TPM).
Trimethylamine (TMA) is a microbial metabolite generated by diverse gut microbial taxa from dietary quaternary amines such as choline and carnitine.60 TMA is absorbed by the intestinal epithelium and oxidized to trimethylamine N-oxide (TMAO). TMAO is associated with atherosclerosis. The genes responsible include a specific glycyl radical enzyme, the choline TMA-lyase (CutC) and its activator (CutD).61 We detected the genes encoding CutD (KGNDJEFE_00505; 1,059.0 ± 189 TPM) and CutC (KGNDJEFE_00506; 3,595.9 ± 838.3 TPM), which were expressed in the mouse cecum, indicating that C. hiranonis may contribute significantly to TMA production by the gut microbiota. C. hylemonae encodes four additional putative cutC gene copies, although these genes were expressed at a much lower level.
Discussion
The metabolism of bile acids by intestinal bacteria is of considerable interest due to the effect of bile acids on gut microbial community structure62 and effects on host physiology and pathophysiology63. The formation of certain secondary bile acids, such as DCA, in the context of host health appears to be a double-edged sword; the balance between benefit and harm are likely concentration and context dependent. Indeed, low fecal concentrations of secondary bile acids are associated with inflammatory bowel disease,63 cirrhosis of the liver,8,64,65 and antibiotic-induced C. difficile infection.9,10 Excess concentrations of secondary bile acids such as DCA and LCA in the GI tract are associated with subsets of cholesterol gallstone patients,6 liver cancer,1,2 and colorectal cancer.4 Thus, understanding the biology of relatively low abundant gut bacteria capable of converting primary host bile acids into secondary bile acids will be important in developing strategies to alter gut microbial-bile acid-host axis.
The study of the formation of DCA from CA has a relatively long history, and has taken two routes. Both routes required the isolation of bile acid 7α-dehydroxylating bacteria from feces. The first is understanding the association and conversion of defined communities capable of bile acid deconjugation and bile acid 7α-dehydroxylation. In the late 1950’s and early 1960’s, Samuelsson proposed a two-step model of bile acid 7α-dehydroxylation based on radioisotope studies in rabbits and rats.66 In the late 1960s and early 1970s, isolates of bile acid 7α-dehydroxylating bacteria, now lost, were collected in Sweden and associated in germ-free mice and rats along with other pure cultures of facultative and strict anaerobes to determine bile acid conversion.67,68 A prior study examined in vitro and in vivo bile acid metabolism by B. distasonis strain K-5 and C. hiranonis sp. strain TO-931.69 C. hiranonis was reported to deconjugate TCA and convert CA to DCA in monoculture, as well as coculture with B. distasonis. By contrast, B. distasonis deconjugated <30% of TCA in culture and produced no DCA. Mono-association of gnotobiotic mice with C. hiranonis did not yield observable deconjugation or bile acid 7α-dehydroxylation.69 Mono- and bi-association with B. distasonis similarly resulted in low levels of unconjugated cecal bile acids and undetectable amounts of DCA.69 Subsequently, Narushima et al (2006) reported DCA formation in gnotobiotic mice colonized with human fecal isolates including B. vulgatus, B. uniformis, P. distasonis, C. hylemonae, C. hiranonis, and B. wadsworthia.26 Our results confirm the ability of this consortium, in our case utilizing C57BL/6 mice and the type strains of each species, rather than the fecal isolates obtained in that prior study, to colonize GF mice and metabolize bile acids that mice share with humans (TCA, TCDCA). The reduced complexity of this model enabled transcript profiling of C. hylemonae and C. hiranonis in vivo. Expression of the bai operon has been observed, and our results indicate the possible utilization of galactose as electron donor, and the utilization of L-proline and L-glycine as electron acceptors in vivo. Bile acid metabolomics indicates a large proportion of muricholic acids that are deconjugated, but not 7-dehydroxylated, consistent with other in vitro and in vivo studies with human bile acid 7-dehydroxylating bacteria.26,70 The reduced complexity of this consortium may be important for determining the biological function of low abundance gut microbes in animal models of human disease.
The second strategy, complementary to the first, is to work out the molecular biology and biochemistry of the bile acid 7α-dehydroxylation pathway. The original model suggested by Samuelsson66 predicted diaxial trans-elimination of water (removal of 7α-hydroxy group and 6β-hydrogen) followed by reduction of the C6 = C7 bond. Subsequent research detected multiple bile acid inducible (bai) proteins after addition of CA during growth71,72 and at least seven stable bile acid intermediates have been observed during the biotransformation of CA to DCA.50 Cloning and characterization of recombinant proteins or purification of native Bai enzymes over the course of several decades defined the bai genes and accessory bile acid metabolizing enzymes21,23 that now allows detection of bai genes in bacteria genomes and measurement of their expression in vivo. This highlights the importance of furthering biochemical investigations into bacterial metabolism of host endogenous, dietary, and pharmaceutical compounds, which allows interpretation of metagenomic and metatranscriptomic data.
The human fecal isolate “NB-12”, isolated in the Narushima study shared 99.6% 16S rDNA identity with B. wadsworthia26 and was reported to have high BSH activity in vitro, and when omitted from the gnotobiotic consortium, the percentage of deconjugated bile acids in the cecum significantly declined. There are no annotated bile salt hydrolase genes in NCBI genome assemblies of B. wadsworthia strains (ATCC 49260, 3_1_6) and BLAST searches using amino acid sequences of BSH genes from Bacteroides or Clostridium spp. do not yield promising targets. Further work is needed to determine the role of B. wadsworthia strains in bile salt deconjugation. Given the relatively low expression observed among known BSH genes (Table 1), it may be that Bilophila BSH activity is responsible for a significant proportion of bile salt deconjugation in this model. Bilophila wadsworthia has been shown previously to expand during the feeding of milk fat, which is high in taurine, or during taurocholic acid feeding.52 Bilophila wadsworthia is a δ-Proteobacterium closely related to Desulfovibrio spp..51 Unlike sulfate-reducing bacteria that utilize inorganic sulfate, B. wadsworthia respires organic sulfonates, including taurine.51 Metabolomic analysis of cecum, serum, and liver clearly demonstrate efficient bile salt deconjugation, indicating abundant release of taurine into the cecum. In our study, RNA-Seq analysis revealed high expression of B. wadsworthia genes involved in taurine respiration. The regulation of these genes is still poorly understood, as is the requirement of sulfonates for persistence in vivo.
Our study represents, to our knowledge, the first in vivo transcriptome analysis of bile acid 7α-dehydroxylating bacteria. The genomes of C. scindens ATCC 35704,27 C. hylemonae DSM 15053 and C. hiranonis DSM 13275 have now been completed. We recently reported development of a defined medium and bile acid transcriptome analysis of the high activity bile acid 7α-dehydroxylating bacterium, C. scindens ATCC 35704.27 Here, we determined bile acid transcriptomes of C. hylemonae and C. hiranonis. It is interesting to note, as shown in the Venn diagram, that the number of single-copy genes (SCG) in the shared orthologous genes is about the same in the two bacteria, even though one of them has 60% more genes than the other. That would suggest that these SCGs are part of the genetic core for these distantly related clostridia. Org-specific single-copy genes (the singletons), on the other hand, showed much more variation. These singletons may indicate niche specificity, important for their cohabitation.
The physiological role of the bai pathway and the formation of toxic secondary bile acids in the colonization and persistence of the bacteria that encode these genes still remains to be determined. Several selection pressures may explain the evolution of this complex pathway. A proximate cause is that bile acid intermediates serve as electron acceptors, allowing regeneration of NAD+7373 Distal causes for the utility of the bai pathway include generation of a more toxic end-product (e.g. DCA + LCA) that reduces the growth of competitors in the environment.9,62 Bile acids are nutrient signaling hormones, and it is thus also possible that the bai pathway evolved as an effective mode of interkingdom signaling.74,75 Our results confirm that a small population of gut Clostridium spp. is capable of quantitatively converting host primary bile acids to secondary bile acids such as DCA and LCA. The B4PC2 gnotobiotic model is thus a valuable resource for the future study of bile acid metabolism by gut bacteria.
Supplementary Material
Acknowledgments
We gratefully acknowledge the financial support provided to J.M.R. for new faculty startup through the Department of Animal Sciences at the University of Illinois at Urbana-Champaign (grant Hatch ILLU-538-916) as well as Illinois Campus Research Board RB18068. This work was also supported by grants (JMR, HRG) 1RO1 CA204808-01, NIH R01CA179243, College of ACES 2017 FIRE grant (JMR, HRG) and the Young Investigators Grant for Probiotic Research (JMR; Danone, Yakult). L.L. is supported by a Graduate Research Fellowship through the National Science Foundation. P.G.W is supported by the UIC Cancer Education and Career Development Training Program Administered by the Institute for Health Research and Policy at the University of Illinois at Chicago with funding by the National Cancer Institute (Grant No. T32CA057699).
Funding Statement
This work was supported by the College of Agricultural, Consumer and Environmental Sciences, University of Illinois at Urbana-Champaign [RB18068]; National Cancer Institute [NIH R01CA179243]; National Cancer Institute [1RO1 CA204808-01]; National Cancer Institute [1RO1 CA204808-01]; National Institute of Food and Agriculture [ILLU-538-916]; Young Investigators Grant for Probiotics Research.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, Agdashian D, Terabe M, Berzofsky JA, Fako V, et al. Gut microbiome-mediated bile acid metabolism regulates liver cancer via NKT cells. Science. 2018;360(6391):eaan5931. doi: 10.1126/science.aan5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et al. Obesity-induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature. 2013;499(7456):97–101. doi: 10.1038/nature12347. [DOI] [PubMed] [Google Scholar]
- 3.Pai R, Tarnawski AS, Tran T.. Deoxycholic acid activates β-catenin signaling pathway and increases colon cell cancer growth and invasiveness. Mol Biol Cell. 2004;15:2156–2163. doi: 10.1091/mbc.e03-12-0894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ridlon JM, Wolf PG, Gaskins HR. Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes. 2016;22:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wells JE, Berr F, Thomas LA, Dowling RH, Hylemon PB. Isolation and characterization of cholic acid 7α-dehydroxylating fecal bacteria from cholesterol gallstone patients. J Hepatol. 2000;32:4–10. [DOI] [PubMed] [Google Scholar]
- 6.Berr F, Kullak-Ublick GA, Paumgartner G, Münzing W, Hylemon PB. α-dehydroxylating bacteria enhance deoxycholic acid input and cholesterol saturation of bile in patients with gallstones. Gastroenterology. 1996;111:1611–1620. [DOI] [PubMed] [Google Scholar]
- 7.Kakiyama G, Hylemon PB, Zhou H, Pandak WM, Heuman DM, Kang DJ, Takei H, Nittono H, Ridlon JM, Fuchs M, et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2014;306(11):G929–37. doi: 10.1152/ajpgi.00315.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, Takei H, Muto A, Nittono H, Ridlon JM, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol. 2013;58(5):949–955. doi: 10.1016/j.jhep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015;517(7533):205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Theriot CM, Bowman AA, Young VB. Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere. 2016;1(1):e00045–15. doi: 10.1128/mSphere.00045-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sorg JA, Sonenshein AL. Bile salts and glycine as co-germinants for Clostridium difficile spores. J Bacteriol. 2008;190:2505–2512. doi: 10.1128/JB.01765-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiang JY. Bile acids: regulation of synthesis. J Lipid Res. 2009;50:1955–1966. doi: 10.1194/jlr.R900010-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hardison WG. Hepatic taurine concentration and dietary taurine as regulators of bile acid conjugation with taurine. Gastroenterology. 1978;75:71–75. [PubMed] [Google Scholar]
- 14.Sjövall J. Dietary glycine and taurine on bile acid conjugation in man: bile acids and steroids 75. Proc Soc Exp Biol Med. 1959;100:676–678. doi: 10.3181/00379727-100-24741. [DOI] [PubMed] [Google Scholar]
- 15.Swann JR, Want EJ, Geier FM, Spagou K, Wilson ID, Sidaway JE, Nicholson JK, Holmes E. Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci U S A. 2011;108(Suppl 1):4523–4530. doi: 10.1073/pnas.1006734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vlahcevic ZR, Heuman DM, Hylemon PB. Physiology and pathophysiology of enterohepatic circulation of bile acids. In: Zakim D, Boyer J, editors. Hepatology: a textbook of liver disease. 3rd edition. Vol. 1. Philadelphia (PA): Saunders; 1996. p. 376–417. [Google Scholar]
- 17.Hofmann AF. The continuing importance of bile acids in liver and intestinal disease. Arch Intern Med. 1999;159:2647–2658. doi: 10.1001/archinte.159.22.2647. [DOI] [PubMed] [Google Scholar]
- 18.Jones BV, Begley M, Hill C, Gahan CG, Marchesi JR. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc Natl Acad Sci USA. 2008;105(36):13580–13585. doi: 10.1073/pnas.0804437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mythen SM, Devendran S, Méndez-García C, Cann I, Ridlon JM. Targeted Synthesis and characterization of a gene cluster encoding NAD(P)H-dependent 3α-, 3β-, and 12α-hydroxysteroid dehydrogenases from Eggerthella CAG:298, a Gut Metagenomic Sequence. Appl Environ Microbiol. 2018;84(7):e02475–17. doi: 10.1128/AEM.02475-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Song C, Wang B, Tan J, Zhu L, Lou D. Discovery of tauroursodeoxycholic acid biotransformation enzymes from the gut microbiome of black bears using metagenomics. Sci Rep. 2017;7:45495. doi: 10.1038/srep45495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ridlon JM, Kang DJ, Hylemon PB. Bile salt biotransformations by human intestinal bacteria. J Lipid Res. 2006;47(2):241–259. doi: 10.1194/jlr.R500013-JLR200. [DOI] [PubMed] [Google Scholar]
- 22.Devlin AS, Fischbach MA. A biosynthetic pathway for a prominent class of microbiota-derived bile acids. Nat Chem Biol. 2015;11(9):685–690. doi: 10.1038/nchembio.1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ridlon JM, Harris SC, Bhowmilk S, Kang DJ, Hylemon PB. Consequences of bile salt metabolism by intestinal bacteria. Gut Microbes. 2016;7(1):22–39. doi: 10.1080/19490976.2015.1127483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kakiyama G, Muto A, Takei H, Nittono H, Murai T, Kurosawa T, Hofmann AF, Pandak WM, Bajaj JS. A simple and accurate HPLC method for fecal bile acid profile in healthy and cirrhotic subjects: validation by GC-MS and LC-MS. J Lipid Res. 2014;55(5):978–990. doi: 10.1194/jlr.D047506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chiang JYL. Bile acid metabolism and signaling in liver disease and therapy. Liver Res. 2017;1(1):3–9. doi: 10.1016/j.livres.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Narushima S, Itoha K, Miyamoto Y, Park SH, Nagata K, Kuruma K, Uchida K. Deoxycholic acid formation in gnotobiotic mice associated with human intestinal bacteria. Lipids. 2006;41:835–843. [DOI] [PubMed] [Google Scholar]
- 27.Devendran S, Shrestha R, Alves JMP, Wolf PG, Ly L, Hernandez AG, Mendez C, Inboden A, Wiley J, Paul O, et al. Clostridium scindens ATCC 35704: integration of nutritional requirements, the complete genome sequence, and global transcriptional responses to bile acids. Appl Environ Microbiol. 2019. February 8;019:pii: AEM.00052–19. doi: 10.1128/AEM.00052-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee JY, Arai H, Nakamura Y, Fukiya S, Wada M, Yokota A. Contribution of the 7β-hydroxysteroid dehydrogenase from Ruminococcus gnavus N53 to ursodeoxycholic acid formation in the human colon. J Lipid Res. 2013;54(11):3062–3069. doi: 10.1194/jlr.M039834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edenharder R, Pfützner A, Hammann R. Characterization of NAD-dependent 3α- and 3β-hydroxysteroid dehydrogenase and of NADP-dependent 7β-hydroxysteroid dehydrogenase from Peptostreptococcus productus. Biochim Biophys Acta. 1989;1004:230–238. doi: 10.1016/0005-2760(89)90272-5. [DOI] [PubMed] [Google Scholar]
- 30.Walters W, Hyde ER, Berg-Lyons D, Ackermann G, Humphrey G, Parada A, Gilbert JA, Jansson JK, Caporaso JG, Fuhrman JA, et al. Improved bacterial 16S rRNA gene (V4 and V4-5) and fungal internal transcribed spacer marker gene primers for microbial community surveys. mSystems. 2015;1:pii: e00009–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Green SJ, Venkatramanan R, Naqib A. Deconstructing the polymerase chain reaction: understanding and correcting bias associated with primer degeneracies and primer-template mismatches. PLoS One. 2015;10(5):e0128122. doi: 10.1371/journal.pone.0128122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bybee SM, Bracken-Grissom H, Haynes BD, Hermansen RA, Byers RL, Clement MJ, Udall JA, Wilcox ER, Crandall KA. Targeted amplicon sequencing (TAS): A scalable next-gen approach to multilocus, multitaxa phylogenetics. Genome Biol Evol. 2011;3:1312–1323. doi: 10.1093/gbe/evr106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Moonsamy PV, Williams T, Bonella P, Holcomb CL, Höglund BN, Hillman G, Goodridge D, Turenchalk GS, Blake LA, Daigle DA, et al. High throughput HLA genotyping using 454 sequencing and the fluidigm access array™ system for simplified amplicon library preparation. Tissue Antigens. 2013;81(3):141–149. doi: 10.1111/tan.12071. [DOI] [PubMed] [Google Scholar]
- 34.Ridlon JM, Ikegawa S, Alves JM, Zhou B, Kobayashi A, Iida T, Mitamura K, Tanabe G, Serrano M, De Guzman A, et al. Clostridium scindens: A human gut microbe with a high potential to convert glucocorticoids into androgens. J Lipid Res. 2013;54(9):2437–2449. doi: 10.1194/jlr.M038869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al. Qiime allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7(5):335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26(19):2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- 37.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. Isme J. 2012;6(3):610–618. doi: 10.1038/ismej.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huerta-Cepas J, Szklarczyk D, Heller D, Hernández-Plaza A, Forslund SK, Cook H, Mende DR, Letunic I, Rattei T, Jensen LJ, et al. eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019;47(D1):D309–D314. doi: 10.1093/nar/gky1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12(1):59–60. doi: 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- 41.Ridlon JM, Kang DJ, Hylemon PB. Isolation and characterization of a bile acid inducible 7alpha-dehydroxylating operon in Clostridium hylemonae TN271. Anaerobe. 2010;16(2):137–146. doi: 10.1016/j.anaerobe.2009.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kang DJ, Ridlon JM, Moore DR. 2nd, Barnes S, Hylemon PB. Clostridium scindens baiCD and baiH genes encode stereo-specific 7alpha/7beta-hydroxy-3-oxo-delta4-cholenoic acid oxidoreductases. Biochim Biophys Acta. 2008;1781(1–2):16–25. doi: 10.1016/j.bbalip.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ridlon JM, Hylemon PB. Identification and characterization of two bile acid coenzyme A transferases from Clostridium scindens, a bile acid 7α-dehydroxylating intestinal bacterium. J Lipid Res. 2012;53(1):66–76. doi: 10.1194/jlr.M020313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mallonee DH, Lijewski MA, Hylemon PB. Expression in Escherichia coli and characterization of a bile acid-inducible 3alpha-hydroxysteroid dehydrogenase from Eubacterium sp. strain VPI 12708. Curr Microbiol. 1995;30:259–263. [DOI] [PubMed] [Google Scholar]
- 45.Bhowmik S, Jones DH, Chiu HP, Park IH, Chiu HJ, Axelrod HL, Farr CL, Tien HJ, Agarwalla S, Lesley SA. Structural and functional characterization of BaiA, an enzyme involved in secondary bile acid synthesis in human gut microbe. Proteins. 2014;82(2):216–229. doi: 10.1002/prot.24353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yasugi M, Okuzaki D, Kuwana R, Takamatsu H, Fujita M, Sarker MR, Miyake M. Transcriptional profile during deoxycholate-induced sporulation in a Clostridium perfringens isolate causing foodborne illness. Appl Environ Microbiol. 2016;82(10):2929–2942. doi: 10.1128/AEM.00252-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Doerner KC, Takamine F, LaVoie CP, Mallonee DH, Hylemon PB. Assessment of fecal bacteria with bile acid 7 alpha-dehydroxylating activity for the presence of bai-like genes. Appl Environ Microbiol. 1997;63:1185–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, Relman DA. Diversity of the human intestinal microbial flora. Science. 2005;308(5728):1635–1638. doi: 10.1126/science.1110591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Narushima S, Ito K, Kuruma K, Uchida K. Composition of cecal bile acids in ex-germfree mice inoculated with human intestinal bacteria. Lipids. 2000;35:639–644. [DOI] [PubMed] [Google Scholar]
- 50.Hylemon PB, Melone PD, Franklund CV, Lund E, Björkhem I. Mechanism of intestinal 7alpha-dehydroxylation of cholic acid: evidence that allo-deoxycholic acid is an inducible side-product. J Lipid Res. 1991;32:89–96. [PubMed] [Google Scholar]
- 51.Laue H, Denger K, Cook AM. Taurine reduction in anaerobic respiration of Bilophila wadsworthia RZATAU. Appl Environ Microbiol. 1997;63:2016–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Devkota S, Wang Y, Musch MW, Leone V, Fehlner-Peach H, Nadimpalli A, Antonopoulos DA, Jabri B, Chang EB. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10-/- mice. Nature. 2012;487(7405):104–108. doi: 10.1038/nature11225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Peck SC, Denger K, Burrichter A, Irwin SM, Balskus EP, Schleheck D. A glycyl radical enzyme enables hydrogen sulfide production by the human intestinal bacterium Bilophila Wadsworthia. Proc Natl Acad Sci U S A. 2019;Epub [ahead of print]. doi: 10.1073/pnas.1815661116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baron SF, Franklund CV, Hylemon PB. Cloning, sequencing, and expression of the gene coding for bile acid 7alpha-hydroxysteroid dehydrogenase from Eubacterium sp. strain VPI 12708. J Bacteriol. 1991;173(15):4558–4569. doi: 10.1128/jb.173.15.4558-4569.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harris SC, Devendran S, Alves JMP, Mythen SM, Hylemon PBRidlon JM.. Identification of a gene encoding a flavoprotein involved in bile acid metabolism by the human gut bacterium clostridium scindens atcc 35704. Biochim Biophys Acta Mol Cell Biol Lipids. 2018;1863(3):276–283. doi: 10.1016/j.bbalip.2017.12.001. [DOI] [PubMed] [Google Scholar]
- 56.Mead GC. The amino acid-fermenting clostridia. J Gen Microbiol. 1971;67:47–56. doi: 10.1099/00221287-67-1-47. [DOI] [PubMed] [Google Scholar]
- 57.Fonknechten N, Chaussonnerie S, Tricot S, Lajus A, Andreesen JR, Perchat N, Pelletier E, Gouyvenoux M, Barbe V, Salanoubat M, et al. Clostridium sticklandii, a specialist in amino acid degradation: revisiting its metabolism through its genome sequence. BMC Genomics. 2010;11:555. doi: 10.1186/1471-2164-11-S4-S6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stadtman TC. Selenocysteine. Annu Rev Biochem. 1996;65:83–100. doi: 10.1146/annurev.bi.65.070196.000503. [DOI] [PubMed] [Google Scholar]
- 59.Kabisch UC, Gräntzdörffer A, Schierhorn A, Rücknagel KP, Andreesen JR, Pich A . Identification of D-Proline Reductase from Clostridium sticklandii as a Selenoenzyme and Indications for a Catalytically Active Pyruvoyl Group Derived from a Cysteine Residue by Cleavage of a Proprotein. J Biol Chem. 1999;274(13):8445–8454. [DOI] [PubMed] [Google Scholar]
- 60.Rath S, Heidrich B, Pieper DH, Vital M. Uncovering the trimethylamine-producing bacteria of the human gut microbiota. Microbiome. 2017;5(1):54. doi: 10.1186/s40168-017-0271-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Craciun S, Marks JA, Balskus EP. Characterization of choline trimethylamine-lyase expands the chemistry of glycyl radical enzymes. ACS Chem Biol. 2014;9(7):1408–1413. doi: 10.1021/cb500113p. [DOI] [PubMed] [Google Scholar]
- 62.Islam KB, Fukiya S, Hagio M, Fujii N, Ishizuka S, Ooka T, Ogura Y, Hayashi T, Yokota A. Bile acid is a host factor that regulates the composition of the cecal microbiota in rats. Gastroenterology. 2011;141(5):1773–1781. doi: 10.1053/j.gastro.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 63.Joyce SA, Gahan CGM. Disease-associated changes in bile acid profiles and links to altered gut microbiota. Dig Dis. 2017;35:169–177. doi: 10.1159/000450907. [DOI] [PubMed] [Google Scholar]
- 64.Bajaj JS, Heuman DM, Sanyal AJ, Hylemon PB, Sterling RK, Stravitz RT, Fuchs M, Ridlon JM, Daita K, Monteith P, Noble NA, White MB, Fisher A, Sikaroodi M, Rangwala H, Gillevet PM. Modulation of the metabiome by rifaximin in patients with cirrhosis and minimal hepatic encephalopathy. PLoS One. 2013;8(4):e60042. doi: 10.1371/journal.pone.0060042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ridlon JM, Kang DJ, Hylemon PB, Bajaj JS. Gut microbiota, cirrhosis, and alcohol regulate bile acid metabolism in the gut. Dig Dis. 2015;33(3):338–345. doi: 10.1159/000371678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bergstrom S, Lindstedt S, Samuelsson B. Bile acids and steroids. LXXXII. On the mechanism of deoxycholic acid formation in the rabbit. J Biol Chem. 1959;234:2022–2025. [PubMed] [Google Scholar]
- 67.Gustaffson BE, Midtvedt T, Norman A. Metabolism of cholic acid in germfree animals after the establishment in the intestinal tract of deconjugating and 7α-dehydroxylating bacteria. Acta Path Microbiol Scandinav. 1968;72:433–443. doi: 10.1111/j.1699-0463.1968.tb00457.x. [DOI] [PubMed] [Google Scholar]
- 68.Dickinson AB, Gustafsson BE, Norman A. Determination of bile acid conversion potencies of intestinal bacteria by screening in vitro and subsequent establishment in germfree rats. Acta Path Microbiol Scandinav Sec B. 1971;79:691–698. [DOI] [PubMed] [Google Scholar]
- 69.Narushima S, Itoh K, Takamine F, Uchida K. Absence of cecal secondary bile acids in gnotobiotic mice associated with two human intestinal bacteria with the ability to dehydroxylate bile acids in vitro. Microbiol Immunol. 1999;43:893–897. [DOI] [PubMed] [Google Scholar]
- 70.Marion S, Studer N, Desharnais L, Menin L, Escrig S, Meibom A, Hapfelmeier S, Bernier-Latmani R. In vitro and in vivo characterization of Clostridium scindens bile acid transformations. Gut Microbes. 2018;1–23. doi: 10.1080/19490976.2018.1549420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.White BA, Lipsky RL, Fricke RJ, Hylemon PB. Bile acid induction specificity of 7alpha-dehydroxylase activity in an intestinal Eubacterium species. Steroids. 1980;35:103–109. [DOI] [PubMed] [Google Scholar]
- 72.Hylemon PB, Cacciapuoti AF, White BA, Whitehead TR, Fricke RJ. 7alpha-Dehydroxylation of cholic acid by cell extracts of Eubacterium species V.P.I. 12708. Am J Clin Nutr. 1980;33(11 Suppl):2507–2510. doi: 10.1093/ajcn/33.11.2507. [DOI] [PubMed] [Google Scholar]
- 73.White BA, Paone DA, Cacciapuoti AF, Fricke RJ, Mosbach EH, Hylemon PB. Regulation of bile acid 7-dehydroxylase activity by NAD+ and NADH in cell extracts of Eubacterium species V.P.I. 12708. J Lipid Res. 1983;24:20–27. [PubMed] [Google Scholar]
- 74.Nagahashi M, Takabe K, Liu R, Peng K, Wang X, Wang Y, Hait NC, Wang X, Allegood JC, Yamada A, et al. Conjugated bile acid-activated S1P receptor 2 is a key regulator of sphingosine kinase 2 and hepatic gene expression. Hepatology. 2015;61(4):1216–1226. doi: 10.1002/hep.27592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhou H, Hylemon PB. Bile acids are nutrient signaling hormones. Steroids. 2014;86:62–68. doi: 10.1016/j.steroids.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.