

ABSTRACT
The pathogenesis of Crohn's disease (CD) is multifactorial and involves genetic susceptibility, environmental triggers and intestinal microbiota. Adherent-invasive Escherichia coli (AIEC) are flagellated bacteria more prevalent in CD patients than in healthy subjects and promote chronic intestinal inflammation. We aim at deciphering the role of flagella and flagellin modulation by intestinal conditions. AIEC flagellum expression is required for optimal adhesion to and invasion of intestinal epithelial cells. Interestingly, differential flagellin regulation was observed between commensal E. coli (HS) and AIEC (LF82) strains: flagellum expression by AIEC bacteria, in contrast to that of commensal E. coli, is enhanced under intestinal conditions (the presence of bile acids and mucins). Flagella are involved in the ability of the AIEC LF82 strain to cross a mucus layer in vitro and in vivo, conferring a selective advantage in penetrating the mucus layer and reaching the epithelial surface. In a CEABAC10 mouse model, a non-motile mutant (LF82-ΔfliC) exhibits reduced colonization that is restored by a dextran sodium sulfate treatment that alters mucus layer integrity. Moreover, a mutant that continuously secretes flagellin (LF82-ΔflgM) triggers a stronger inflammatory response than the wild-type strain, and the mutant's ability to colonize the CEABAC10 mouse model is decreased. Overexpression of flagellin in bacteria in contact with epithelial cells can be detrimental to their virulence by inducing acute inflammation that enhances AIEC clearance. AIEC pathobionts must finely modulate flagellum expression during the infection process, taking advantage of their specific virulence gene regulation to improve their adaptability and flexibility within the gut environment.
Introduction
Crohn's disease (CD) is an inflammatory bowel disease caused by an abnormal inflammatory response to intestinal microbiota in genetically susceptible hosts and is stimulated by environmental triggers.1-3 Although no pathogen has been consistently associated with inflammatory bowel disease, a subset of CD patients show increased prevalence of a unique enteropathogenic pathobiont that belongs to B2-phylotype Escherichia coli and is termed AIEC, for adherent-invasive E. coli.4,5
In a gut physiology context, luminal bacteria cannot access the surface of the intestinal epithelium because it is protected by several mechanisms. The gastrointestinal mucus system is the first line of defence against bacteria. The organization of the mucus layer varies along the digestive tract: the colon handles its large bacterial load with a two-layered mucus system; the inner layer normally remains impenetrable to bacteria, while in the small intestine, the mucus system is organized in such a way as to facilitate nutritional uptake. Thus, the mucus acts as a matrix loaded with antimicrobial peptides that are secreted by the Paneth cells, such as defensins, lysozyme C, phospholipases6 and secreted immunoglobulin A7, and forms an antimicrobial gradient towards the lumen. In this way, small intestine mucus hinders the ability of viable bacteria to reach epithelial cells, protecting crypts and stem cells. Defects in this protective system have been previously reported in patients with genetic susceptibilities to CD, e.g., polymorphisms affecting autophagy,8,9 the unfolded protein response,10 and bacterium sensing and clearance.11-13 Paneth cell defects and deficient secretion of antimicrobial molecules such as meprin β also play a role in CD pathogenesis.1,3,14,15
However, reports of the presence of intramucosal or mucosa-associated AIEC in CD patients in independent studies performed in Europe, North America and Australia suggest that these bacteria have developed adaptive mechanisms that allow them to colonize the mucosa.16,17 For that, AIEC bacteria must be able (i) to implant in the intestinal lumen within the resident microbiota, (ii) to cross the mucus layer and resist antimicrobial peptides to access the epithelium, (iii) to adhere to the epithelium and cross it via M cells, enterocytes or other mechanisms, and (iv) to finally interact with the resident macrophages in the lamina propria, within which they can survive and multiply. Virulence factors involved in early stages of mucosal invasion have been described in AIEC strains. A high level of resistance to the antimicrobial peptides (α- and β-defensins) has been described to occur in some clinical isolates of AIEC by the acquisition of arlA and arlC genes,18 and the protease Vat-AIEC, which is secreted by AIEC, promotes degradation of human secretory mucins, which leads to a loosened mucus layer that might affect the secretion of antimicrobial peptides.19 In addition, genes encoding propanediol utilization proteins, specifically the pdu operon, have been shown to significantly correlate with the AIEC pathotype and are rarely present in other E. coli strains.20 Altogether, expression of these virulence factors enhances AIEC fitness in the gut and gives a selective advantage to commensal strains in the lumen.
After implanting in the intestinal lumen and crossing the mucus layer, the bacterial adhesion process is mediated by flagella, both directly via mobility and indirectly by maintaining the expression of type 1 pili. The expression of flagella depends on the expression of 3 classes of genes belonging to flagellar regulon (class 1 or early, class 2 or middle, class 3 or late), which presents more than 50 genes. The expression of the structural protein FliC is controlled by FliA (sigma 28 subunit of RNA polymerase). In addition, FliA affects phase variation of fim operon encoding type 1 pili, which results in increased type 1 pili synthesis via a regulatory pathway involving the cyclic dimeric GMP (c-di-GMP).21 FimH adhesin located at the tip of type 1 pili binds to the mannose residues of glycoproteins, such as the carcinoembryonic antigen-related cell adhesion molecule 6 (CEACAM6). Type 1 pili expression was forced in the isogenic mutant of AIEC reference strain LF82, LF82-ΔfliC, in which fliC, encoding flagellin, is deleted. Results from experiments with this mutant indicated that flagella play a role in the invasion process of intestinal epithelial cells (IEC) but operate independently of type 1 pili.22 Moreover, flagellum expression is involved in the inflammatory process triggered by AIEC because AIEC strain LF82 is able to aggravate dextran sodium sulfate (DSS)-induced colitis in mice but this phenotype is lost in a mutant in which the flagellin gene is deleted.23 Indeed, flagellin is considered to be a microbe-associated molecular pattern (MAMP) that is recognized by the receptors of the innate immune system: the transmembrane Toll-like receptor 5 (TLR5)24-26 and the cytoplasmic receptor NLR family CARD domain-containing protein 4 (NLRC4).27 While flagella are present at the bacterial surfaces of both pathogenic and nonpathogenic E. coli strains, differential regulation of flagellin expression has been suggested to occur, notably under the control of the two-component EnvZ/OmpR regulatory system.28
In the present study, we focused on AIEC flagellum involvement in the early stages of AIEC pathogenic mechanisms and, more particularly, on deciphering the modulation of flagellin expression by environmental gut conditions in AIEC compared to commensal E. coli in order to better understand AIEC mucosa colonization and in vivo persistence. We showed that flagellum expression by AIEC bacteria, in contrast to commensal E. coli, is enhanced under intestinal conditions. This enhancement confers a selective advantage to AIEC bacteria to penetrate the mucus layer and reach the epithelial surface.
Results
Involvement of flagella in the AIEC phenotype
Flagella have been implicated in the adhesion and invasion capacities of the reference AIEC strain LF82.22 To extend these observations to a greater number of AIEC strains and to confirm the involvement of the flagella in the AIEC phenotype, we constructed mutants in which the fliC gene is deleted in 5 additional AIEC strains isolated from CD patients (Table S1). Deletion of fliC gene did not impair bacterial growth ability (Fig S1). The adhesion and invasion abilities of these AIEC isogenic mutants that lack fliC were determined using Caco-2 IECs. Since these mutants were non-motile, a centrifugation step was performed to bring bacterial and epithelial cells into close contact and to thereby enable bacteria to initiate infection. The adhesion levels of isogenic mutants were significantly lower, reaching only 15.7 to 50.2% of those of the corresponding WT strains (Fig. 1A), and the invasion levels of mutants were 15.5 to 54.9% of those of corresponding WT strains (which were taken as 100%, Fig. 1B). Of note, the invasion defect is related to the decrease of adhesion ability. These phenotypes were restored after complementation with fliC gene. In addition, since macrophage survival is one of the main characteristic of AIEC strains, the ability of AIEC-ΔfliC mutants to multiply within THP-1 human macrophages was analysed at 1 and 24 h post-infection. Deletion of fliC gene in AIEC strains impaired bacterial ability to replicate within macrophages (Fig. S2). Together, these results confirm that flagellin expression is required for the full AIEC phenotype, which includes the abilities to adhere to and invade IECs and to survive and replicate within macrophages.
Figure 1.
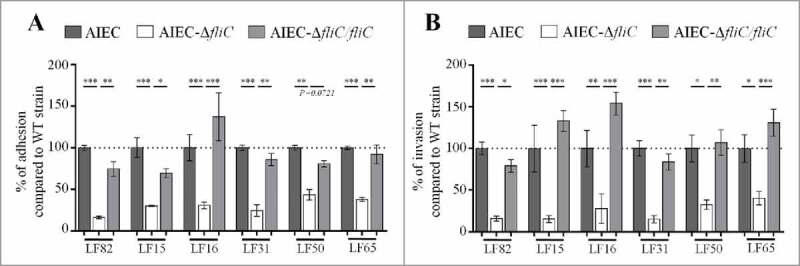
Deletion of fliC gene reduces AIEC ability to adhere to and invade Caco-2/TC7 cells. A: Cell-associated bacteria were quantified after a 3-h infection period. The results are expressed as the percentage of wild-type associated bacteria. B: Invasion ability was determined after gentamicin treatment for an additional hour. The results are expressed as the percentage of wild-type invasive bacteria. Each value is the mean ± standard error of mean (SEM) of at least three separate experiments. Statistical analysis were performed using one-way ANOVA with Tukey's multiple comparison test. P< 0.05 (*), P < 0.01 (**) and P < 0.001 (***).
AIEC flagellum expression is required to reach the epithelium surface in mono-colonized mice
To assess the importance of flagella in vivo, germ-free C57Bl/6 mice were challenged with AIEC LF82 or LF82-ΔfliC. Mono-colonized mice were sacrificed two weeks post-infection, and their colonic tissues were observed by confocal microscopy after mucin immunostaining and using FISH to detect bacteria (Fig. 2A). The distance between bacteria and the epithelial cell monolayer was measured. Wild-type AIEC LF82 was found at a mean distance of 5.5 µm from the epithelium surface, while the LF82-ΔfliC mutant was observed significantly farther away, at a mean distance of 15.5 µm; these distances indicate that in contrast with the WT LF82 strain, the LF82-ΔfliC mutant is impaired in its ability to reach the epithelial cell surface (Fig. 2B). The commensal E. coli strain HS, which is highly motile in soft agar plates (Fig. S3), is not observed in contact with epithelial cell surface (mean distance 24.9 µm). This result could be explained by the down-regulation of flagellum expression in commensal E. coli in vivo. We therefore quantified flagellum expression in E. coli associated with ileal (Fig 2C and D) and colonic (Fig S4) tissues in mono-colonized mice. The fliC mRNA levels indicated that in ileal tissue expression of the flagellin gene in the commensal strain was weaker than that in the wild-type AIEC LF82 strain, explaining the former strain's inability to cross the mucus layer (Fig. 2C). Moreover, type 1 pili expression is downregulated in the commensal E. coli strain HS, whereas AIEC LF82 expressed high levels of type 1 pili in a ileal environment, as determined by fimA mRNA quantitation (Fig. 2D). Thus, flagella are necessary to access the surface of the ileal epithelium, but regulation of flagellum expression differs between a commensal strain and the AIEC LF82 strain in the intestinal microenvironment. Taken together, these features confer a selective advantage to AIEC pathobionts in the colonization of ileal mucosa, natural tropism for AIEC bacteria.
Figure 2.
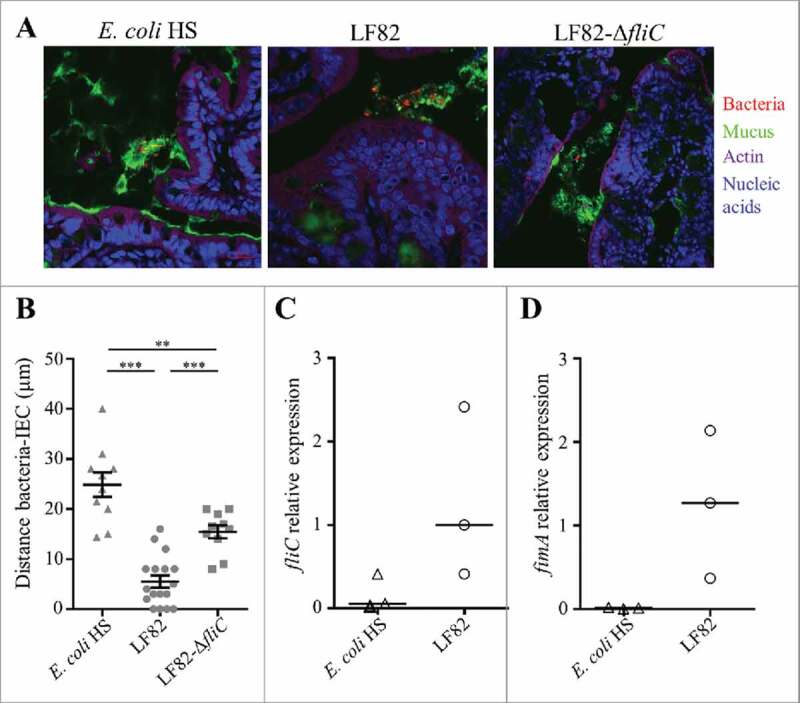
AIEC LF82 strain can reach the intestinal epithelium of mono-colonized mice by flagellum expression, in contrast with the commensal E. coli strain HS. A: Confocal microscope examinations of intestinal tissues of mono-colonized mice for 2 weeks with wild-type LF82, LF82-ΔfliC or the commensal HS strain. Histological sections were immunostained for mucus (green), bacteria (red), actin (purple) and nucleic acids (blue). B: Distance between bacteria and epithelial cell monolayer was determined by using Zen 2011 version 7.1 software on immunostained sections. The data are presented as the mean ± SEM. Statistical analysis was performed using one-way ANOVA with Tukey's multiple comparison test. P < 0.01 (**) and P < 0.001 (***). C and D: Quantification of fliC and fimA expression by RT-qPCR in ileal tissue. The 16S rRNA gene was used as an internal standard to normalize the data. Bar indicates median.
Flagellum expression in AIEC is favoured by intestinal conditions
In germ-free mice, commensal E. coli strain HS, which is highly motile in soft agar plates, expressed flagella at a low level in the gut environment and did not reach the epithelium surface (unlike LF82 bacteria). To investigate the regulation of flagellum expression in these strains, we measured fliC promoter activity in AIEC and commensal genetic backgrounds by using a β-galactosidase assay. The fliC promoter was cloned upstream of a lacZ reporter gene in a pRS550 plasmid, and the final construct was transformed into the LF82 strain and the commensal E. coli strain HS. We measured β-galactosidase activity under various conditions that mimic the gut environment. The presence of bile acids is a host signal that enteric bacteria encounter as they travel through the gastrointestinal tract, and enteric pathogens, including E. coli, respond to bile acids by increasing their expression of virulence factors.19,29-31 In a medium supplemented with 1% bile acids (compared to the medium without supplementation), the fliC promoter is activated in AIEC LF82 strain by 2.6-fold (3 h) and 3.4-fold (6 h). In contrast, no significant activation was observed in the commensal E. coli strain HS (Fig. 3A).
Figure 3.
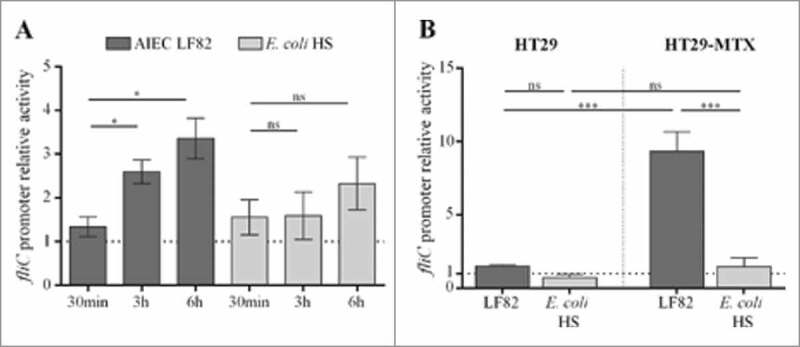
Contact with mucus-hyperproducing HT-29-MTX cells activates fliC promoter in AIEC but not in commensal E. coli strain. The LF82 fliC gene promoter was cloned upstream of lacZ in the pRS550 plasmid, and the pRS550-pfliC construct was transformed into the LF82 and the commensal E. coli HS strains. Background β-galactosidase activity that was generated by a promoterless pRS550 construct was subtracted at each time point. A: Activation of fliC promoter by 1% cholic and deoxycholic acids (50/50) after 30 min, 3 h and 6 h in AIEC LF82 and E. coli HS strains harbouring the pRS550-pfliC plasmid. The results are expressed as the ratio of β-galactosidase activity in the presence of bile salts to those in the basal medium (20% 5x M9 broth, 20% LB and 60% Tris buffer). B: Promoter activity of LF82 or HS E. coli harbouring the pRS550-pfliC plasmid and associated with HT29 epithelial cells or mucus-hyperproducing HT29-MTX cells. The results are expressed as the ratio of β-galactosidase activity of associated bacteria to those of bacteria in the cell culture medium. The data are presented as the mean ± SEM of at least five independent experiments Statistical analysis were performed using one-way ANOVA with Tukey's multiple comparison test. P< 0.05 (*) and P< 0.005 (***). ns, not significant.
We next investigated the impact of mucins on fliC promoter activity. The mucin-hyperproducing HT29-MTX cells and the closely related HT29 cells, which do not produce significant levels of mucins, were infected with AIEC LF82 and commensal HS strains that both harboured the reporter plasmid. The β-galactosidase activity of cell-associated bacteria was measured at three hours post-infection and compared to the β-galactosidase activity of bacteria in the cell culture medium alone. As shown in Fig. 3B, contact with mucus-nonproducing cells has no significant impact on fliC promoter activity in AIEC LF82 and commensal HS strains. In contrast, the fliC promoter in AIEC LF82 bacteria associated with mucin-producing cells is significantly activated (9.3-fold), whereas no activation is observed in the commensal strain (Fig. 3B). We performed adhesion experiments on HT29 and HT29-MTX cells for the AIEC LF82 and E. coli HS strains. The level of AIEC LF82 adhesion was similar regardless of the cells, indicating that this strain is able in vitro to cross the mucus layer to adhere (Fig S5). In contrast, the level of E. coli HS strain adhesion is significantly decreased (1.6-fold) in the presence of mucus (HT29-MTX cells compared to HT29 cells). These results indicate that AIEC and commensal E. coli strains use different systems to regulate the expression of flagella, suggesting that AIEC strains express their flagella when they contact mucus to become able to cross it.
Flagella promote crossing of the mucus layer
The colonization of gut mucosa by CD-associated AIEC requires the penetration of bacteria through the protective mucus barrier, which is impenetrable in a physiological context. The AIEC LF82 strain produces a secreted mucinase, Vat-AIEC that favours adhesion to IECs covered by mucus. However, the implications of AIEC flagella in the crossing of mucus layers are not known.
To assess the consequences of the deletion of the fliC gene in the ability of the AIEC LF82 strain to adhere to IECs covered by mucus, we infected mucin-hyperproducing HT29-MTX cells and the closely related HT29 cells, which do not produce significant levels of mucins. As the adhesion ability of LF82-ΔfliC was defective compared to that of the WT strain, we measured the ability of each strain to adhere to mucin-hyperproducing HT29-MTX cells and mucin-nonproducing HT29 cells. The LF82 strain has the same capacity to adhere to IECs in the presence or absence of mucins (Fig. 4A). In contrast, the ability of the non-motile mutant to adhere to HT29-MTX cells decreases a significant 40% when they produce mucus, suggesting that motility promotes bacterial access to epithelial cells.
Figure 4.
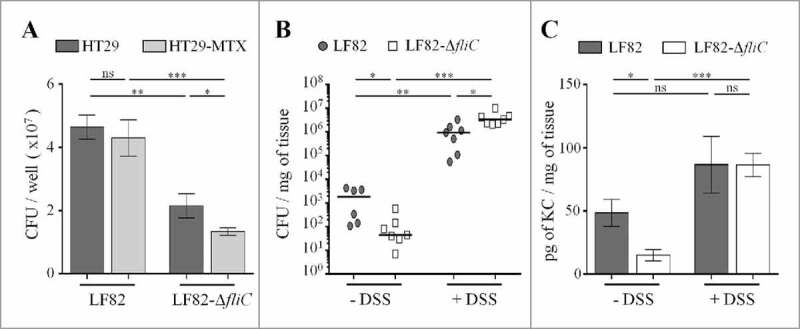
Flagella enhance LF82 access to intestinal epithelial cells in vitro and in vivo, A: Quantification of bacteria associated with intestinal epithelial HT29 cells or with mucus-hyperproducing intestinal epithelial HT29-MTX cells after 3 h of infection. Each value is the mean ± SEM of at least five independent experiments. Statistical analysis were performed using one-way ANOVA with Tukey's multiple comparison test. P< 0.05 (*). B and C: After antibiotic treatment, CEABAC10 transgenic mice were treated or not treated with 0.5% DSS. Mice were orally challenged for 3 consecutive days with 109 LF82 or LF82-ΔfliC CFUs, and (B) the number of mucosa-associated bacteria and (C) the amount of colonic pro-inflammatory keratinocyte-derived chemokine (KC) secretion were determined 4 days after the last infection. Statistical analysis were performed using Kruskal-Wallis with Dunn's multiple comparison test for B panel or one-way ANOVA with Tukey's multiple comparison test for C panel. P< 0.05 (*), P < 0.01 (**) and P < 0.001 (***). ns, not significant.
We hypothesized that AIEC flagella may favour close contact to IEC surfaces and gut colonization in SPF mice. SPF CEABAC10 transgenic mice were treated (or not) with a low dose of DSS (0.5%) to alter the mucus integrity and test this hypothesis.32-34 Mice were orally challenged three times with WT LF82 or LF82-ΔfliC, and the number of mucosa-associated bacteria was determined 4 days after the final infection. In the absence of DSS treatment, the ability of LF82-ΔfliC to colonize colonic tissue in CEABAC10 mice was significantly less than that of the WT LF82 strain (45.1 versus 1846 colony-forming units (CFUs) per milligram of tissue, respectively) (Fig. 4B). This result aligned with the significantly lower secretion of the pro-inflammatory cytokine KC by colonic mucosa after LF82-ΔfliC infection (compared to that of the WT LF82 strain) in the absence of DSS treatment (12.5 versus 42.2 pg/mg of tissue, respectively) (Fig. 4C). Moreover, when mice were treated with DSS, mucosal colonization was strongly enhanced (more than 3-log) in both strains, probably due to the decreases of the mucus layer, but also possibly by additional effects of DSS on the intestinal mucosa. Importantly, such a low dose of DSS was sufficient to abolish the colonization defect that was previously observed in LF82-ΔfliC (compared to LF82 WT) and to restore IL-8 secretion to the LF82 WT level (Fig. 4C). This finding further highlights that AIEC flagella play a role in bacterial penetration of the mucus layer and are hence dispensable in conditions in which mucus layer integrity is altered (Fig. 4B). Under these conditions, the non-flagellated mutant is even more persistent in the mucosa than the WT strain (3.33 × 106 vs 9.33× 105 CFU/mg of tissue, respectively), perhaps because of less efficient clearance by the immune system.
Modulation of flagellum expression is essential for AIEC persistence
According to our results, flagellum expression in the intestinal lumen could favour AIEC virulence by facilitating access to the epithelium surface; nevertheless, exaggerated expression at the mucosal level could be harmful to bacteria as it would induce acute inflammation that would lead to their elimination. This relation led us to hypothesize that AIEC bacteria may modulate the expression of flagella, depending on the stage of the infectious process. To study this hypothesis, we constructed a mutant invalidated for the flgM gene, which encodes the anti-sigma28 factor that controls the transcription of late genes in the flagellar operon; this mutation led to constitutive expression of flagellin. The virulence of this mutant, which was unable to control flagellum expression, was investigated in vitro and in vivo. The deletion of the flgM gene in AIEC strain LF82 did not interfere with bacterial growth since growth curves for the WT strain LF82 and the LF82-ΔflgM mutant in LB medium were similar (Fig. S6). However, the LF82-ΔflgM mutant showed a decrease in motility in soft agar and a decrease in the ability to agglutinate Saccharomyces cerevisiae yeast, indicating that through type 1 pili/flagellum co-regulation, the functionality of the type 1 pili is altered in this mutant (Fig. S3). This conclusion is confirmed by the presence of decreased adhesion and invasion capacities in IECs Caco-2/TC7 (Figs. 5A and B). Moreover, this mutant secretes massive amounts of soluble flagellin in the supernatant due to the absence of regulation of late flagellar gene transcription by FlgM (Fig. 5C and Fig S7). This high flagellin secretion probably explains the higher secretion of the chemokine interleukin-8 (IL-8) by IECs that are in contact with the LF82-ΔflgM mutant compared to those in contact with the WT strain (after a 3 h infection period, 20.2 and 1.3 pg/mL, respectively) (Fig. 5D). Expression of flgM induced by arabinose in transcomplemented LF82-ΔflgM mutant leads to restoration of the WT phenotype (Fig 5).
Figure 5.
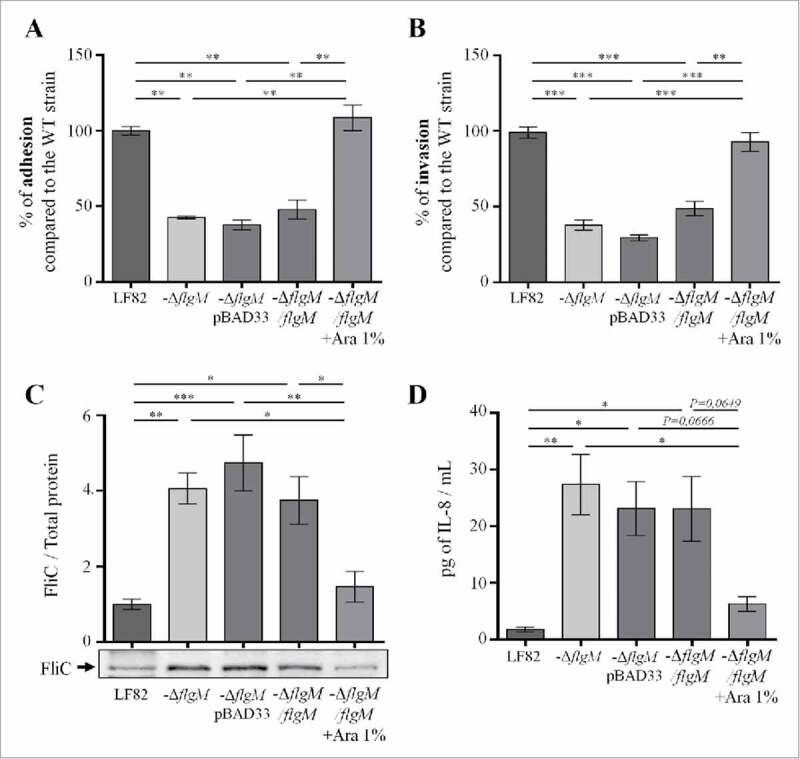
Deletion of flgM gene reduces LF82 adhesion to and invasion of intestinal epithelial Caco-2/TC7 cells but increases their interleukin-8 response, A: The numbers of cell-associated bacteria were quantified after a 3-h infection period. The results are expressed as the percentage of adherent wild-type LF82 bacteria. B: Invasion ability was determined after gentamicin treatment for an additional hour. The results are expressed as the percentage of invasive wild-type LF82 bacteria taken as 100%. C: Representative western blot analysis of flagellin release in the culture supernatant at 24h. Quantification of FliC band intensity standardize to total protein is shown in the top panel (representative from 4 independent experiments). D: Pro-inflammatory chemokine interleukin-8 secretion by Caco-2/TC7 cells after a 3-h infection. The data are presented as the mean ± SEM of at least four independent experiments Statistical analysis were performed using one-way ANOVA with Tukey's multiple comparison test. P < 0.05 (*),P < 0.01 (**) and P < 0.005 (***).
The behaviour of the LF82-ΔflgM mutant was analysed in vivo in CEABAC10 transgenic mice that were orally challenged with LF82 or LF82-ΔflgM at day 0 and day 1. Following infection, the mice that were colonized by the WT strain showed a strong weight loss compared to the mice infected with the mutant strain (Fig. 6A). At days 4 and 6, quantification of bacteria in stool samples revealed a two-log reduction of the LF82-ΔflgM mutant compared with WT LF82 (Fig. 6B). This result could be explained by the decreased mobility of the mutant, which would reduce its ability to access the epithelium, as well as its decreased adhesion and invasion capacities as observed in vitro; however, the result could also be caused by better clearance by the host. At day 6, the mice were again orally challenged with LF82 or LF82-ΔflgM. The isogenic mutant, unable to control its flagellin secretion, induced a higher mortality rate than the WT strain (Fig. 6C). Moreover, the surviving mice of the LF82-ΔflgM group exhibited a high level of serum antibodies that bind to the FliC protein and their colonic mucosa strongly produced the KC chemokine, the equivalent of human IL-8 secreted following flagellin/TLR5 interaction and leading to immune cells recruitment (Figs. 6D and E). Mutant AIEC bacteria constitutively producing the flagellin induce acute inflammation, whereas the WT strain persists without inducing an exacerbated inflammatory response. Altogether, these observations suggest that AIEC bacteria are able to finely regulate flagellum production in a FlgM-dependent manner to avoid over-activation of the immune system, allowing their persistence in the gut.
Figure 6.
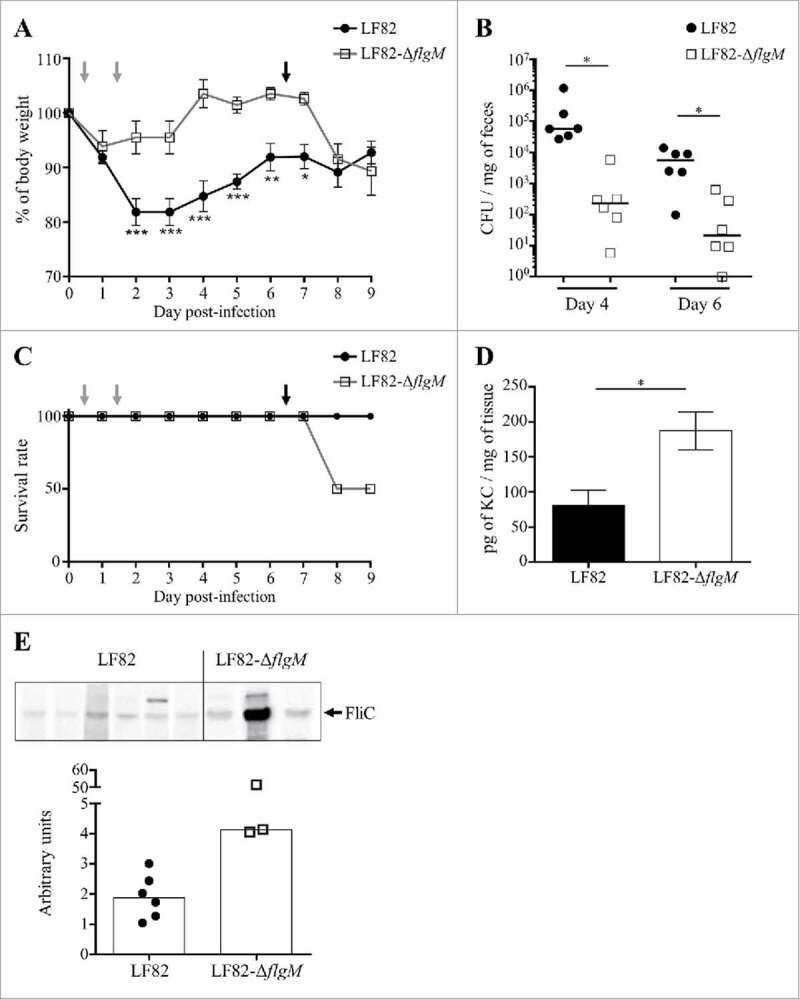
Modulation of flagellin synthesis by FlgM allows LF82 persistence and limits host inflammatory response. A and B: After antibiotic treatment, CEABAC10 transgenic mice were treated with 0.25% DSS. Their body weights were measured every day (A). Statistical analysis was performed using two-way ANOVA with Bonferroni's multiple comparison test. P< 0.05 (*),P< 0.01 (**) and P< 0.005 (***). Mice were orally challenged for two consecutive days with 109 LF82 or LF82-ΔflgM CFUs (grey arrows on graphs A and C). The colonization was assessed by quantifying bacteria in stool samples, which were counted on LB agar plates at days 4 and 6 (B). Bar indicates median. Statistical analysis was performed using Kruskal-Wallis with Dunn's multiple comparison test. P< 0.05. Mice were orally challenged again at day 6 (black arrow). C: Survival rate of mice during the experiment. D: After sacrifice, the pro-inflammatory keratinocyte-derived chemokine (KC) secretion by colonic tissue was measured. The data are presented as the mean ± SEM. Statistical analysis was performed using unpaired Student t test. P< 0.05 (*). E: Measure of serum antibodies raised against FliC in infected mice. Purified LF82 flagellin was electrophoresed in each lane of a 12% SDS-polyacrylamide gel and blotted to a nitrocellulose membrane. Diluted serum was used as the primary antibody. Quantification of band intensity was realized using the Image Lab software.
Discussion
The search for specific pathogens involved in CD has identified several candidates in the intestinal mucosa of patients; AIEC is the candidate that is supported by much evidence, as it has both a pro-inflammatory phenotype and invasiveness properties that could contribute to the pathogenesis of CD.35-38 In the digestive tract, a number of mechanisms protect the intestine from pathogens and against the resident intestinal microbiota. The outermost of these mechanisms is secreted mucus, which entraps bacteria and prevents their access to epithelial cells and their translocation into the intestinal mucosa. Host defence peptides secreted by colonocytes and Paneth cells also play a key role in innate host defences in the gut. Thus, colonization of the intestinal mucosa by AIEC requires the penetration of bacteria through the mucus layer, which is impenetrable in the physiological context. Comparative genomic analysis of AIEC strains from different collections (Europe, North America and Australia) show that this pathobiont does not harbour an exclusive molecular signature,17,37,39 suggesting that AIEC strains have evolved from commensal E. coli by different mechanisms to favour their implantation in genetically susceptible CD patients. The presence of a mucus-degrading protease, called Vat-AIEC, in the AIEC LF82 strain has recently been described to confer a competitive advantage in mucosal colonization.19,40,41 Since the abundances of some antimicrobial peptide classes (β-defensins 2 and 3) increase in inflamed CD lesions,42,43 mucosa-associated AIEC bacteria require mechanisms to resist antimicrobial peptide defences in the inflamed gut. A genomic island, named PI-6 and harbouring arlA and arlC genes, in CD-associated strain NRG857c confers resistance to a vast range of human and synthetic antimicrobial peptides. This mechanism is one that could permit chronic host colonization by AIEC.18 The flagellum is also a key element in the virulence of AIEC strains, which is similar for other pathogens, such as the uropathogenic E. coli that need mobility during their ascension in the urinary tract.44 In this study, we extended observations that were previously made of AIEC reference strain LF82 to AIEC strains that were isolated from CD patients and confirmed that flagellum expression is required for optimal adhesion to and invasion of IECs. In addition, the decrease in survival observed in macrophages is likely not a direct consequence of lack of motility, but may be related to a decreased expression of flhDC (master regulon) and fliA, that will subsequently impact the expression of other virulence factors.21 Moreover, a recent study reported that the flagellar master operon flhDC is a pleiotropic regulator involved in motility and virulence of Enterobacteria.22,45 We also demonstrated that AIEC flagella are expressed in the presence of bile acids or in contact with mucus and that they are involved in the ability of the AIEC LF82 strain to cross the protective mucus barrier in vivo, which allows these bacteria to reach the epithelium surface.
Concerning the regulation of flagellum synthesis, the regulation of their expression by the EnvZ/OmpR two-component system is known to differ in adherent-invasive and commensal E. coli.28 In addition, high osmolarity induces a significant increase in the ability of AIEC LF82 bacteria to interact with Intestine-407 cells, which correlates with increased OmpC expression. Here, we showed that contact with mucus-producing cells activates flagellum synthesis in the LF82 AIEC strain, but not in commensal E. coli strain HS. This lack of synthesis may contribute to the inability of commensal E. coli strains to cross the mucus, protecting the epithelium. E. coli MG1655 adapts rapidly to the intestine of germ-free mice by losing mobility, which is due to single point mutations in its EnvZ/OmpR two-component signal transduction system.46 In addition, when mice are fed the commensal E. coli strain MG1655, selection in the intestine leads to a loss of bacterial motility by the deletion of segments of the flhDC gene, the major regulator of the flagellar operon; this selection occurs in 45 to 50% of bacteria after 3 days and 80 to 90% after 15 days.47 By saving the energy that is necessary for the synthesis and function of the flagella, bacteria can grow more rapidly in a nutritionally competitive environment (e.g., the intestinal lumen). Interestingly, the authors suggest that the 10 to 20% of the bacteria that remain motile may occupy a particular niche in contact with the epithelium and require flagella to implant and remain despite the renewal of mucus. AIEC strains have been isolated in close contact to or within the mucosa of patients with CD, which is consistent with the finding in this study that AIEC keep their motility and regulate flagellum expression more finely than the commensal E. coli. This particular mode of flagellar regulation could result from patho-adaptation of AIEC strains. Indeed, in contrast to classical infectious agents, AIEC strains can be considered pathobionts because they promote inflammatory diseases through their adaptive genomic evolution within a susceptible host.36,48-50 In particular, patho-adaptive mutations in the FimH adhesin gene in AIEC increase the affinity of type-1 pili for the CEACAM6 receptor, favouring interactions between bacteria and the intestinal mucosa.51
The bacterial flagellum plays dual roles during infection since flagellin is a MAMP that induces an innate immune response against bacteria. Whereas flagella represent a selective advantage in crossing the mucus layer for AIEC, elevated flagellin production in bacteria in contact with intestinal mucosa would activate innate immunity, leading to an acute inflammatory response and subsequent elimination of this pathobiont. As chronic inflammatory agents, AIEC bacteria must persist and probably negatively regulate the expression of their flagellum when in contact with the mucosa. In this study, we observed that a mutant whose flagellin gene is deleted, LF82-ΔfliC, exhibits better persistence in a DSS-treated mouse model in which the implantation of bacteria is facilitated by the alteration of intestinal barrier (reduction of the mucus layer, but also additional effects on the intestinal epithelium mimicking IBD susceptibility). In this condition, the expression of fliC is no longer necessary to confer full virulence to AIEC strains. This indicates that flagella expression in the virulence of AIEC bacteria is crucial in the early stages of colonization. Then, the expression of flagella could be regulated negatively for optimal persistence following colonization of the intestinal mucosa by AIEC strains. This hypothesis was confirmed by the constitutive production of flagellin by the LF82-ΔflgM mutant. Indeed, the expression of the structural protein FliC is controlled by FliA (sigma 28 subunit of RNA polymerase) and an efficient way to force flagellin expression is to invalidate the corresponding anti-sigma factor, FlgM.52 The flgM negative mutant induces a stronger inflammatory response than the WT strain. This behaviour could explain the higher mortality of infected mice and contribute to the decreased amounts of bacteria in faeces through more efficient clearance. The expression of flagella in Salmonella enterica serovar Typhimurium (S. typhimurium) is regulated depending on the compartment: high expression occurs during invasion through Peyer's Patches and is followed by inhibition within macrophages and in mesenteric lymph nodes and spleen so that dendritic cell-mediated activation of FliC-specific CD4+ T cells is restricted.53,54 Thus, the ability to switch off flagellum expression is associated with invasive human infections.55
Flagella represent a potential therapeutic target because of their accessibility and essential role in access to the epithelium. Several strategies could be considered, especially an action targeting the global regulators of transcription in bacteria. The two-component quorum-sensing B and C regulator (QseBC) system in E. coli detects and responds to microbiota-generated signals and host-derived catecholamines, and the absence of qseC suppresses LF82 flagellum expression. Perturbation of QseC by the biochemical inhibitor LED20956,57 induces a loss of mobility and virulence in AIEC strain LF82 in vitro and attenuates experimental colitis in mice. Because of the different regulation of the flagellar operon by OmpR in the LF82 strain and the commensal E. coli K-12 MG1655 strain,28 this regulator could also constitute a specific target to inhibit flagella and, therefore, colonization by AIEC strains. Another strategy is to directly target flagella with either antibodies to prevent their movement or natural compounds that have various mechanisms of action. For example, phloretin, a natural component of apple and pear, efficiently decreases S. typhimurium colonization in the caecum of phloretin-supplemented mice by repressing flagellar genes.58 Curcumin, a dietary polyphenol, reduces the motility of S. typhimurium by shortening the flagellar filament and decreasing its density.59 Thus, these compounds could efficiently reduce AIEC colonization. To conclude, AIEC bacteria have developed specific virulence gene regulation to improve their adaptability and flexibility within the gut environment (compared to commensal E. coli), and a better understanding of the pathogenicity mechanisms of AIEC strains aid in developing specific inhibitors that interfere with bacteria/cell interactions.
Materials and methods
Bacterial strains, plasmids and media
The bacterial strains, plasmids and primers that were used in this study are listed in Table S1. Bacteria were grown at 37°C without shaking in Lysogenic Broth (LB) medium supplemented with the appropriate antibiotic (ampicillin 100 µg/mL, erythromycin 20 µg/mL, chloramphenicol 25 µg/mL or kanamycin 50 µg/mL).
Construction of isogenic mutants and transcomplementation
Isogenic mutants with fliC or flgM gene deletions were generated by using a PCR product and the Red recombinase system described by Datsenko and Wanner60 and modified by Chaveroche.61 The basic strategy was to replace a chromosomal sequence with a selectable kanamycin resistance gene that was generated by PCR.
The fliC or flgM gene was amplified by PCR from AIEC genomic DNA using Cp-fliC or Cp-flgM primers (Table S2). The amplified DNA was purified and cloned into pHSG575 expression vector or into arabinose-inducible expression vector pBAD33 (Table S1). The resulting constructs were used to transform the AIEC-ΔfliC isogenic mutants or the LF82-ΔflgM isogenic mutant.
Cell lines and cultures
Human intestinal epithelial Caco-2/TC7 (kindly supplied by Dr. Rousset, INSERM U505, Paris, France), HT29 (HTB-38, obtained from the ATCC) and HT29-MTX cells (with mucus hyperproduction, a kind gift from Thecla Lesuffleur, INSERM UMR S 938, Paris, France)62 were maintained in an atmosphere containing 5% CO2 at 37°C in Dulbecco's-modified Eagle's medium (DMEM) with 4.5 g/L glucose and supplemented with foetal calf serum (Lonza, 10% (v/v) for HT29 lines and 20% (v/v) for Caco-2/TC7 line), 2 mM L-glutamine (Life Technologies), 100 U of penicillin, 100 mg of streptomycin and 0.25 mg of amphotericin B per litre. HT29-MTX and HT-29, as controls, were seeded for 21 days in order to allow mucin production. For adhesion assays, cells were seeded at a density of 2 × 105 cells per square centimetre in culture plates (Falcon).
The human monocytic THP-1 cell line (TIB-202, obtained from the ATCC) was maintained in an atmosphere containing 5% CO2 at 37°C in RPMI-1640 medium supplemented with 10% (v/v) foetal calf serum (Lonza) and 2 mM L-glutamine (Life Technologies). THP-1 monocytes were seeded in a 24-well tissue culture plate at a density of 2 × 105 cells/well and differentiated into macrophages by treatment with 20 ng/mL phorbol myristate acetate (PMA) for 18 h.
In vitro cell adhesion assay
Adhesion and invasion assays were performed as previously described.37 For adhesion assays, bacteria were grown overnight at 37°C without shaking in LB, centrifuged at 3000 g for 10 min and then resuspended in cell culture medium. After 2 washes with phosphate-buffered saline (PBS), cells were infected for 3 h at a multiplicity of infection (MOI) of 10 bacteria per seeded cell. A centrifugation step (900 g for 10 min) was performed at the beginning of the infection period in experiments with Caco-2 and THP-1 cells but not with HT-29 and HT-29 MTX cells so as not to force the passage of bacteria through the mucus. At the end of the infection, cells were washed three times in PBS and then lysed with 1% Triton X-100 (Sigma) in deionized water. This concentration of Triton X-100 has no effect on bacterial viability for at least 30 min. Finally, the samples were diluted and plated onto LB agar plates to determine the number of CFUs corresponding to the total number of cell-associated bacteria (adherent and intracellular bacteria). The effectiveness of the bacterial invasion was quantified using the gentamicin protection assay: fresh cell culture medium containing 100 µg/ml of gentamicin (Sigma) was added for 1 h after the 3-h infection period to kill extracellular bacteria. Cells were then lysed with 1% Triton X-100, and the bacteria were quantified as described above.
Macrophage survival assay
The numbers of intracellular bacteria in macrophages were determined by using the gentamicin protection assay 63. Briefly, THP-1 macrophages were infected at an MOI of 100 and centrifuged for 10 min at 900 g. Infected cells were incubated for 10 min at 37°C and then washed and incubated with culture medium containing gentamicin at 100 μg/mL for 40 min (1 h post-infection) or 24 h (24 h post-infection). Survival was expressed as the mean fold increase of intracellular bacteria recovered after 24 h of incubation compared to the number of bacteria recovered at 1 h post-infection, which was defined as 1.
Promoter activity assay
The fliC promoter of AIEC LF82 and E. coli HS are not identical, however, the transcription of the fliC gene is dependent on the binding of the factor σ28 FliA (subunit of the RNA polymerase) at the regions -10 and -35, which are both strictly identical between the two strains. In addition, the distance between these two consensus sequences is 11 nucleotides for both promoters, indicating a similar binding of FliA on these promoters.64,65 Thus, the AIEC LF82 fliC promoter was amplified by PCR using BamHI-pfliC-F and EcoRI-pfliC-R primers (Table S2). The PCR product was ligated into a pRS550 plasmid,66 which was then designated pRS550-pfliC (Table S1). Promoter constructs were transformed into AIEC LF82 and E. coli HS bacteria. β-Galactosidase activity was determined by using the Miller method,67 which is based on o-nitrophenyl-β-D-galactopyranoside (ONPG) hydrolysis in strains harbouring pRS550-pfliC or the promoterless plasmid pRS550 (used to standardize the background of β-galactosidase activity which reflects the activity of cells, bacteria and possibly leaks plasmid). Overnight bacterial cultures were resuspended in a medium containing 20% 5x M9 broth, 20% LB and 60% Tris buffer19 at a pH of 7.5 or in the same medium containing 1% bile salts (∼50% cholic acid sodium salt and ∼50% deoxycholic acid sodium salt) (Sigma). ONPG hydrolysis and bacterial concentrations were determined after 30 min and 3 h by measuring absorbance at 420 and 620 nm, and the data are presented as the ratio between β-galactosidase activity in the presence of bile salts and in the basal medium.
To measure promoter activity of bacteria in contact with mucin-hyperproducing cells, infections with LF82 and E. coli HS cells harbouring pRS550-pfliC or the promoterless pRS550 were realized in HT29 and HT29-MTX cells as described in the in vitro cell adhesion assay protocol. After the 3-h infection period, the number of cell-associated bacteria was determined, and the β-galactosidase activity of these bacteria was measured. The absorbance at 620 nm was replaced by the number of cell-associated bacteria in the calculation of the β-galactosidase ratio between the numbers of cell-associated bacteria and bacteria in the cell culture medium after a 3-h incubation period.
CEABAC10 mice infections
Ten- to 12-week-old C57Bl/6 CEABAC10 male mice 68 were housed in specific pathogen-free (SPF) conditions at the animal care facility of the Université Clermont Auvergne (Clermont-Ferrand, France). To enhance E. coli strain colonization, mice were treated with 500 mg/l metronidazole (Sigma-Aldrich), 500 mg/l vancomycin (Sigma-Aldrich), 1 g/l neomycin (Sigma-Aldrich) and 1 g/l ampicillin (Euromedex) in drinking water for 5 days, as previously described 69. When needed, the animals received 0.25 to 0.5% (wt/vol) DSS (molecular mass, 36,000 to 50,000 Da; MP Biomedicals, LOT M7191) in their drinking water, starting 1 day before infection. Mice were orally challenged with 109 CFUs of the WT LF82, LF82-ΔfliC or LF82-ΔflgM strains on 2 or 3 consecutive days according to the experimental protocol. Mice were sacrificed at day 7 post-infection to compare the ability of strains to reach the epithelium surface with or without a DSS treatment and at day 10 post-infection to study the importance of negative regulation of flagellar genes by FlgM. The persistence of bacteria throughout the experiment was determined in faecal samples, and the number of intestinal mucosa-associated bacteria was assessed in colonic tissue after sacrifice. After mechanical homogenization and serial dilutions of faeces and colonic mucosa samples, they were plated on LB agar plates containing 100 µg/ml ampicillin and 20 µg/ml erythromycin (Euromedex) to isolate LF82 bacteria or on LB agar plates containing 50 µg/ml kanamycin (Euromedex) to isolate LF82 mutants and incubated at 37°C overnight. Before sacrifice, mice were anaesthetized with isoflurane, and blood was collected by cardiac puncture. To quantify keratinocyte-derived cytokine KC release, colonic tissue samples were placed in DMEM with antibiotics (gentamicin 50 µg/ml; penicillin 100 U; streptomycin 100 mg) and incubated for 24 h at 37 °C. The cytokine levels were quantified in the supernatant using ELISA kit from R&D systems according to the manufacturer's instructions.
FliC purification and FliC-directed antibody titration
Flagella were isolated as in Brett et al.70 with some modifications. Briefly, LF82 bacteria were grown overnight in 500 ml of LB and centrifuged, and the bacterial pellets were frozen at −20°C overnight. Thawed pellets were next resuspended in 20 ml of PBS, and flagella were sheared off with a homogenizer (low speed setting for 4 min). Cell debris was removed by centrifugation (6,000 g, 10 min, 4°C), and flagella were precipitated overnight from the supernatant with ammonium sulfate (final concentration 5%). The precipitate was centrifuged (12,000 g, 30 min, 4°C), and the supernatant was discarded. The pellet, containing flagella, was dissolved in 1 ml of PBS and centrifuged again (16,900× g, 10 min, 4°C). Flagellar filaments in the sediment were solubilized in 8 M urea; insoluble debris was removed by centrifugation (10,000 g, 1 min), and the solubilized flagellin was desalted with a Slide-A-Lyzer Mini Dialysis Device (3,5K MWCO, Thermo Fisher Scientific), using PBS as eluent. Flagellin was stored at −20°C. The purity and the molecular mass of flagellin were verified in 12% SDS-polyacrylamide gels that were stained with Coomassie blue R-250 (Thermo Fisher Scientific).
To detect FliC-directed antibodies, 2 µg of extracted LF82 flagellin was electrophoresed in each lane of a 12% SDS-polyacrylamide gel and blotted to a nitrocellulose membrane. Each lane was cut and incubated overnight with diluted serum (1:500) as primary antibodies. Secondary horse anti-rabbit IgG, HRP-linked antibody (Cell Signaling, 7076S) was used at a 1:15,000 dilution. Detection was realized using the ChemiDoc XRS system. Quantification of band intensity was realized with Image Lab software.
FliC detection in the supernatant
Overnight cultures were diluted 1:100 into LB and grown without shaking to compare flagellin secretion by the WT LF82 strain and the LF82-ΔflgM mutant. At the indicated time, 500 µL of the bacterial cultures at the same OD620nm were centrifuged at 3000 g for 10 min, and 20 µL of supernatants were electrophoresed in a 12% SDS-polyacrylamide gel and blotted to nitrocellulose membrane. Primary rabbit anti-FliC antibody (Abcam, ab93713) was used at a 1:10,000 dilution. Secondary goat anti-rabbit IgG HRP-linked antibody (Cell Signaling, 7074S) was used at a 1:15,000 dilution. Detection was realized using the ChemiDoc XRS system. Quantification of band intensity at 24 h was realized with Image Lab software and standardized with total protein on gel stained with silver nitrate (SilverQuest, Invitrogen).
Mono-colonization of germ-free mice
Germ-free C57Bl/6 mice were obtained from Taconic Inc. and maintained under germ-free conditions at Georgia State University using Park Bioservices isolators. AIEC, reference strain LF82, the LF82-ΔfliC mutant and commensal HS E. coli were grown overnight in 200 mL of LB at 37°C without agitation. Bacterial suspensions with an OD620nm of 2.0 were placed in the water bottles of germ-free C57Bl/6 mice, which had been placed in an isolated ventilated caging system (Isocage) that prevents exogenous bacterial contamination 71. Two weeks later, mice were euthanized, and their organs were collected for downstream analysis.
Immunostaining of mucins and localization of bacteria by FISH
Mucus immunostaining was paired with fluorescent in situ hybridization (FISH), as previously described.72,73 Briefly, proximal colon samples were placed in methanol-Carnoy's fixative solution (60% methanol, 30% chloroform, and 10% glacial acetic acid) for 1 week at 4°C. Tissues were then washed in methanol twice for 30 min, ethanol twice for 15 min, ethanol/xylene (1:1) for 15 min and xylene twice for 15 min, followed by their embedding in Paraffin in a vertical orientation. Five-µm sections were cut and dewaxed (60°C for 10 min, xylene at 60°C for 10 min, xylene for 10 min and 99.5% ethanol for 10 min). The hybridization step was performed at 50°C overnight with the EUB338 probe (5′-GCTGCCTCCCGTAGGAGT-3′, labelled at the 5′-end with Alexa 647) in hybridization buffer (20 mM Tris–HCl, pH 7.4, 0.9 M NaCl, 0.1% SDS, and 20% formamide). After washing (20 mM Tris–HCl, pH 7.4, and 0.9 M NaCl), a PAP pen (Sigma) was used to mark around the section, block solution (5% foetal bovine serum in PBS) was added, and the sample was incubated for 30 min at 4°C. Mucin-2 primary antibody (rabbit H-300, Santa Cruz Biotechnology) was diluted 1:1500 in block solution and applied overnight at 4°C in the dark. After the samples were washed three times for 10 min in PBS, block solution containing anti-rabbit Alexa 488 secondary antibody (diluted 1:1500), phalloidin-tetramethylrhodamine B isothiocyanate (Sigma, 1 µg/mL) and Hoechst 33258 (Sigma, 10 µg/mL) were applied to the section for 2 h at 4°C in the dark. After being washed three times for 10 min in PBS, slides were mounted using Prolong anti-fade mounting media (Life Technologies). Observations were performed with a Zeiss LSM 700 confocal microscope with Zen 2011 software, version 7.1. This software was used to determine the distance between bacteria and the epithelial cell monolayer.
Extraction and quantification of mRNA
RNAs were extracted from mono-colonized mice ileum and colon with a previously described phenol-chloroform method with a few modifications. Briefly, tissues were cut finely in liquid nitrogen. Next, 500 μl of Buffer A (200 mM NaCl, 200 mM Tris, and20 mM EDTA), 210 μl of 20% SDS (filter sterilized) and 500 μl of acid phenol:chloroform (5:1, pH 4.5, Ambion) were added to the samples. The mixture was then beaten with 0.1-mm glass beads from a Precellys lysing kit (VK01) three times for 45 s each (Precellys 24, Bertin technologies) and centrifuged at 4°C (3 min at 13000 rpm). The aqueous phase was recovered and mixed with 500 μl of acid phenol:chloroform. The mixture was centrifuged again at 4°C (3 min at 13,000 rpm), and the aqueous phase was recovered. A 1/10 volume of 3 M sodium acetate (pH 5.2) and 1 volume of −20°C chilled ethanol were added to the aqueous phase. The resulting solution was then mixed by gentle inversion and incubated for 20 min on ice. The mixture was then centrifuged at 4°C (20 min at 13,000 rpm). The resulting pellet was recovered and washed twice in 500 μl of cold 70% ethanol. The mixture was centrifuged at 4°C (5 min at 13,000 rpm), and the RNA pellet was recovered, air-dried and then resuspended in nuclease-free water. The RNA extracts were then purified using an RNeasy Mini kit (QIAGEN) according to the manufacturer's protocol. DNA in the RNA extracts was digested by using TURBO DNase (Ambion) according to the manufacturer's instructions, and mRNA expression levels were quantified by reverse transcribing this RNA using the PrimeScript™ RT reagent kit (Takara Bio). Quantitative RT-PCR was performed using iTaq Universal SYBR green Master Mix (Biorad) and the CFX96 Real Time system (Biorad) with the specific primers indicated in Table S2. Fold-inductions were calculated using the Ct method as follows: ΔΔCt = (Ct target gene- Ct housekeeping gene)treatment – (Ct target gene- Ct housekeeping gene)nontreatment, and the final data were derived from 2−ΔΔ CT.
Statistical analysis
Statistical analyses were performed using Graph Pad Prism 7 software. D'Agostino-Pearson omnibus test was used to verify the normal distribution of data. Comparisons of two groups of data were performed with an unpaired Student parametric t test. One-way ANOVA with Tukey's multiple comparison test for normally distributed data or Kruskal-Wallis with Dunn's multiple comparison were used for multiple group analysis. Two-way ANOVA with Bonferroni's multiple comparison test was used for the analysis of mice body weight. All in vitro experiments were repeated at least three times. P values less than or equal to 0.05 were considered statistically significant.
Acknowledgments
We thank Dr Abdelkrim Alloui for animal care (Animal facilities, Clermont-Ferrand, France), and Maëva Meynier and Melissa Chervy for experimental help. We also thank the Imagerie Confocale Clermont-Ferrand (ICCF) platform (Université d'Auvergne, Clermont-Ferrand, France) for electron microscopy.
Funding Statement
This study was supported by the Ministère de la Recherche et de la Technologie, Inserm (U1071), INRA (USC-2018) and by grants from the Association F. Aupetit (AFA), from Région Auvergne (Nouveau Chercheur), from ANR in the frame of JCJC Nutribiote (N. Barnich).
Competing interests
None.
Ethics statement
Animal protocols were approved by the committee for ethical issues, CEMEA Auvergne (Permit Number: CEMEAA, 2015032716314007), and all animals were used in accordance with the European Community Directive in the care and use of animals (86/609/CEE) Animal experiments performed at Georgia State University were under institutionally-approved protocols, Institutional Animal Care and Use Committee, IACUC #A14033.
References
- 1.Chassaing, B & Darfeuille-Michaud, A.. The commensal microbiota and enteropathogens in the pathogenesis of inflammatory bowel diseases. Gastroenterology 2011;140:1720–1728. [DOI] [PubMed] [Google Scholar]
- 2.Kaser, A, Zeissig, S & Blumberg, RS. Inflammatory Bowel Disease. Annu. Rev. Immunol. 2010;28:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xavier, RJ & Podolsky, DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007;448:427–434. [DOI] [PubMed] [Google Scholar]
- 4.Barnich, N & Darfeuille-Michaud, A. Role of bacteria in the etiopathogenesis of inflammatory bowel disease. World J. Gastroenterol. WJG 2007;13:5571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Darfeuille-Michaud, A, et al. Presence of adherent Escherichia coli strains in ileal mucosa of patients with Crohn's disease. Gastroenterology 1998;115:1405–1413. [DOI] [PubMed] [Google Scholar]
- 6.Bevins, CL & Salzman, NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat. Rev. Microbiol. 2011;9:356–368. [DOI] [PubMed] [Google Scholar]
- 7.Mantis, NJ & Forbes, SJ. Secretory IgA: Arresting Microbial Pathogens at Epithelial Borders. Immunol. Invest. 2010;39:383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cadwell, K, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature 2008;456:259–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu, B, et al. Irgm1-deficient mice exhibit Paneth cell abnormalities and increased susceptibility to acute intestinal inflammation. Am. J. Physiol. Gastrointest. Liver Physiol. 2013;305:G573–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaser, A, et al. XBP1 links ER stress to intestinal inflammation and confers genetic risk for human inflammatory bowel disease. Cell 2008;134:743–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hugot, JP, et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 2001;411:599–603. [DOI] [PubMed] [Google Scholar]
- 12.Ogura, Y, et al. A frameshift mutation in NOD2 associated with susceptibility to Crohn's disease. Nature 2001;411:603–606. [DOI] [PubMed] [Google Scholar]
- 13.Wehkamp, J, et al. NOD2 (CARD15) mutations in Crohn's disease are associated with diminished mucosal α-defensin expression. Gut 2004;53:1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Abraham, C & Cho, JH. Inflammatory bowel disease. N. Engl. J. Med. 2009;361:2066–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vazeille, E, et al. Role of meprins to protect ileal mucosa of Crohn's disease patients from colonization by adherent-invasive E. coli. PloS One 2011;6:e21199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinez-Medina, M & Garcia-Gil, LJ. Escherichia coli in chronic inflammatory bowel diseases: An update on adherent invasive Escherichia coli pathogenicity. World J. Gastrointest. Pathophysiol 2014;. 5:213–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Brien, CL, et al. Comparative genomics of Crohn's disease-associated adherent-invasive Escherichia coli. Gut 2016; (). doi: 10.1136/gutjnl-2015-311059. [DOI] [PubMed] [Google Scholar]
- 18.McPhee, JB, et al. Host defense peptide resistance contributes to colonization and maximal intestinal pathology by Crohn's disease-associated adherent-invasive Escherichia coli. Infect. Immun 2014; 82:3383–3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gibold, L, et al. The Vat-AIEC protease promotes crossing of the intestinal mucus layer by Crohn's disease-associated Escherichia coli. Cell. Microbiol 2015;n/a-n/a. doi: 10.1111/cmi.12539. [DOI] [PubMed] [Google Scholar]
- 20.Dogan, B, et al. Inflammation-associated adherent-invasive Escherichia coli are enriched in pathways for use of propanediol and iron and M-cell translocation. Inflamm. Bowel Dis. 2014;20:1919–1932. [DOI] [PubMed] [Google Scholar]
- 21.Claret, L, et al. The Flagellar Sigma Factor FliA Regulates Adhesion and Invasion of Crohn Disease-associated Escherichia coli via a Cyclic Dimeric GMP-dependent Pathway. J. Biol. Chem. 2007;282:33275–33283. [DOI] [PubMed] [Google Scholar]
- 22.Barnich, N, Boudeau, J, Claret, L & Darfeuille-Michaud, A. Regulatory and functional co-operation of flagella and type 1 pili in adhesive and invasive abilities of AIEC strain LF82 isolated from a patient with Crohn's disease. Mol. Microbiol. 2003;48:781–794. [DOI] [PubMed] [Google Scholar]
- 23.Carvalho, FA, et al. Crohn's disease adherent-invasive Escherichia coli colonize and induce strong gut inflammation in transgenic mice expressing human CEACAM. J. Exp. Med. 2009;206:2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gewirtz, AT, Navas, TA, Lyons, S, Godowski, PJ & Madara, JL. Cutting Edge Bacterial Flagellin Activates Basolaterally Expressed TLR5 to Induce Epithelial Proinflammatory Gene Expression. J. Immunol. 2001;167:1882–1885. [DOI] [PubMed] [Google Scholar]
- 25.Carvalho, FA, et al. Transient inability to manage proteobacteria promotes chronic gut inflammation in TLR5-deficient mice. Cell Host Microbe 2012;12:139–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chassaing, B, Koren, O, Carvalho, FA, Ley, RE & Gewirtz, AT. AIEC pathobiont instigates chronic colitis in susceptible hosts by altering microbiota composition. Gut 2013. doi: 10.1136/gutjnl-2013-304909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao, Y & Shao, F. The NAIP-NLRC4 inflammasome in innate immune detection of bacterial flagellin and type III secretion apparatus. Immunol. Rev. 2015;265:85–102. [DOI] [PubMed] [Google Scholar]
- 28.Rolhion, N, Carvalho, FA & Darfeuille-Michaud, A. OmpC and the sigma(E) regulatory pathway are involved in adhesion and invasion of the Crohn's disease-associated Escherichia coli strain LF82. Mol. Microbiol. 2007;63:1684–1700. [DOI] [PubMed] [Google Scholar]
- 29.Chassaing, B, Etienne-Mesmin, L, Bonnet, R & Darfeuille-Michaud, A. Bile salts induce long polar fimbriae expression favouring Crohn's disease-associated adherent-invasive Escherichia coli interaction with Peyer's patches. Environ. Microbiol. 2013;15:355–371. [DOI] [PubMed] [Google Scholar]
- 30.Pope, LM, Reed, KE & Payne, SM. Increased protein secretion and adherence to HeLa cells by Shigella spp. following growth in the presence of bile salts. Infect. Immun 1995;. 63:3642–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wibbenmeyer, JA, Provenzano, D, Landry, CF, Klose, KE & Delcour, AH. Vibrio cholerae OmpU and OmpT porins are differentially affected by bile. Infect. Immun. 2002;70:121–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johansson, M. E. V., et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014;63:281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petersson, J, et al. Importance and regulation of the colonic mucus barrier in a mouse model of colitis. Am. J. Physiol. – Gastrointest. Liver Physiol. 2011;300:G327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schreiber, O, et al. iNOS-Dependent Increase in Colonic Mucus Thickness in DSS-Colitic Rats. PLoS ONE 2013;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Baumgart, M, et al. Culture independent analysis of ileal mucosa reveals a selective increase in invasive Escherichia coli of novel phylogeny relative to depletion of Clostridiales in Crohn's disease involving the ileum. ISME J 2007;. 1:403–418. [DOI] [PubMed] [Google Scholar]
- 36.Conte, MP, et al. Adherent-invasive Escherichia coli (AIEC) in pediatric Crohn's disease patients: phenotypic and genetic pathogenic features. BMC Res. Notes 2014;7:748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Darfeuille-Michaud, A, et al. High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn's disease. Gastroenterology 2004;127:412–421. [DOI] [PubMed] [Google Scholar]
- 38.Martinez-Medina, M, et al. Molecular diversity of Escherichia coli in the human gut: new ecological evidence supporting the role of adherent-invasive E. coli (AIEC) in Crohn's disease. Inflamm. Bowel Dis. 2009;15:872–882. [DOI] [PubMed] [Google Scholar]
- 39.Sepehri, S, Kotlowski, R, Bernstein, CN & Krause, DO. Phylogenetic analysis of inflammatory bowel disease associated Escherichia coli and the fimH virulence determinant. Inflamm. Bowel Dis. 2009;15:1737–1745. [DOI] [PubMed] [Google Scholar]
- 40.Harrington, SM, et al. The Pic protease of enteroaggregative Escherichia coli promotes intestinal colonization and growth in the presence of mucin. Infect. Immun. 2009;77:2465–2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kobayashi, RK, Gaziri, LC & Vidotto, MC. Functional activities of the Tsh protein from avian pathogenic Escherichia coli (APEC) strains. J. Vet. Sci. 2010;11:315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Aldhous, MC, Noble, CL & Satsangi, J. Dysregulation of human beta-defensin-2 protein in inflammatory bowel disease. PloS One 2009;4:e6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meisch, JP, et al. Human β-defensin 3 peptide is increased and redistributed in Crohn's ileitis. Inflamm. Bowel Dis. 2013;19:942–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lane, MC, Alteri, CJ, Smith, SN & Mobley, H. L. T.. Expression of flagella is coincident with uropathogenic Escherichia coli ascension to the upper urinary tract. Proc. Natl. Acad. Sci. U. S. A. 2007;104:16669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jozwick, A. K. S., Graf, J & Welch, TJ. The flagellar master operon flhDC is a pleiotropic regulator involved in motility and virulence of the fish pathogen Yersinia ruckeri. J. Appl. Microbiol. 2017;122:578–588. [DOI] [PubMed] [Google Scholar]
- 46.Giraud, A, et al. Dissecting the genetic components of adaptation of Escherichia coli to the mouse gut. PLoS Genet 2008;. 4:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gauger, EJ, et al. Role of motility and the flhDC Operon in Escherichia coli MG1655 colonization of the mouse intestine. Infect. Immun. 2007;75:3315–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miquel, S, et al. Complete genome sequence of Crohn's disease-associated adherent-invasive E. coli strain LF82. PloS One 2010;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nash, JH, et al. Genome sequence of adherent-invasive Escherichia coli and comparative genomic analysis with other E. coli pathotypes. BMC Genomics 2010;11:667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schippa, S, et al. A potential role of Escherichia coli pathobionts in the pathogenesis of pediatric inflammatory bowel disease. Can. J. Microbiol 2012;. 58:426–432. [DOI] [PubMed] [Google Scholar]
- 51.Dreux, N, et al. Point mutations in FimH adhesin of Crohn's disease-associated adherent-invasive Escherichia coli enhance intestinal inflammatory response. PLoS Pathog 2013;. 9:e1003141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hughes, KT & Mathee, K. The anti-sigma factors. Annu. Rev. Microbiol. 1998;52:231–286. [DOI] [PubMed] [Google Scholar]
- 53.Alaniz, RC, Cummings, LA, Bergman, MA, Rassoulian-Barrett, SL & Cookson, BT. Salmonella typhimurium coordinately regulates FliC location and reduces dendritic cell activation and antigen presentation to CD4+ T cells. J. Immunol. Baltim. Md 1950 2006;177:3983–3993. [DOI] [PubMed] [Google Scholar]
- 54.Cummings, LA, Barrett, S. L. R., Wilkerson, WD, Fellnerova, I & Cookson, BT. FliC-specific CD4+ T cell responses are restricted by bacterial regulation of antigen expression. J. Immunol. Baltim. Md 1950 2005;174:7929–7938. [DOI] [PubMed] [Google Scholar]
- 55.Yim, L, et al. Repression of flagella is a common trait in field isolates of Salmonella enterica serovar Dublin and is associated with invasive human infections. Infect. Immun 2014;. 82:1465–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Curtis, MM, et al. QseC inhibitors as an antivirulence approach for Gram-negative pathogens. mBio 2014;5:e02165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rasko, DA, et al. Targeting QseC Signaling and Virulence for Antibiotic Development. Science 2008;321:1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shuai-Cheng, W, et al. Subinhibitory concentrations of phloretin repress the virulence of Salmonella typhimurium and protect against Salmonella typhimurium infection. Antonie Van Leeuwenhoek 2016;109:1503–1512. [DOI] [PubMed] [Google Scholar]
- 59.Marathe, SA, et al. Curcumin Reduces the Motility of Salmonella enterica serovar Typhimurium by Binding to the Flagella thereby Leading to Flagellar Fragility and Shedding. J. Bacteriol. 2016; JB:00092–16. doi: 10.1128/JB.00092-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Datsenko, KA & Wanner, BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 2000;97:6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chaveroche, MK, Ghigo, JM & d'Enfert, C. A rapid method for efficient gene replacement in the filamentous fungus Aspergillus nidulans. Nucleic Acids Res 2000; 28:E97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lesuffleur, T, et al. Differential expression of the human mucin genes MUC1 to MUC5 in relation to growth and differentiation of different mucus-secreting HT-29 cell subpopulations. J. Cell Sci. 1993;106(Pt 3):771–783. [DOI] [PubMed] [Google Scholar]
- 63.Bringer, M.-A., Barnich, N, Glasser, A.-L., Bardot, O & Darfeuille-Michaud, A. HtrA stress protein is involved in intramacrophagic replication of adherent and invasive Escherichia coli strain LF82 isolated from a patient with Crohn's disease. Infect. Immun. 2005;73:712–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aldridge, P, Gnerer, J, Karlinsey, JE & Hughes, KT. Transcriptional and Translational Control of the Salmonella fliC Gene. J. Bacteriol. 2006;188:4487–4496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ide, N, Ikebe, T & Kutsukake, K. Reevaluation of the promoter structure of the class 3 flagellar operons of Escherichia coli and Salmonella. Genes Genet. Syst. 1999;74:113–116. [DOI] [PubMed] [Google Scholar]
- 66.Simons, RW, Houman, F & Kleckner, N. Improved single and multicopy lac-based cloning vectors for protein and operon fusions. Gene 1987;53:85–96. [DOI] [PubMed] [Google Scholar]
- 67.Miller, J. Experiments in Molecular Genetics. .NY: Cold Spring Harbor Laboratory Press. Cold Spring Harb; 1972. [Google Scholar]
- 68.Chan, C. H. F. & Stanners, CP. Novel mouse model for carcinoembryonic antigen-based therapy. Mol. Ther. J. Am. Soc. Gene Ther. 2004;9:775–785. [DOI] [PubMed] [Google Scholar]
- 69.Dalmasso, G, et al. The PepT1-NOD2 signaling pathway aggravates induced colitis in mice. Gastroenterology 2011;141:1334–1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brett, PJ, Mah, DC & Woods, DE. Isolation and characterization of Pseudomonas pseudomallei flagellin proteins. Infect. Immun. 1994;62:1914–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hecht, G, et al. A simple cage-autonomous method for the maintenance of the barrier status of germ-free mice during experimentation. Lab. Anim. 2014;48:292–297. [DOI] [PubMed] [Google Scholar]
- 72.Johansson, M. E. V. & Hansson, GC. Preservation of mucus in histological sections, immunostaining of mucins in fixed tissue, and localization of bacteria with FISH. Methods Mol. Biol. Clifton NJ 2012;842:229–235. [DOI] [PubMed] [Google Scholar]
- 73.Chassaing, B, et al. Dietary emulsifiers impact the mouse gut microbiota promoting colitis and metabolic syndrome. Nature 2015;519:92. [DOI] [PMC free article] [PubMed] [Google Scholar]