

ABSTRACT
Background and Aims
Alcoholic hepatitis is the most severe form of alcohol-related liver disease. While the gut microbiome is known to play a role in disease development and progression, less is known about specific compositional changes of the gut bacterial microbiome associated with disease severity. Therefore, the aim of our study was to correlate gut microbiota features with disease severity in alcoholic hepatitis patients.
Methods
We used 16S rRNA gene sequencing on fecal samples from 74 alcoholic hepatitis patients, which were enrolled at 9 centers in Europe, the United States, and Mexico in a multi-center observational study. The relative abundance of gut bacterial taxa on genus level, as well as the microbiome diversity, was correlated to various clinical, laboratory, and histologic parameters.
Results
We observed a negative correlation between the model for end-stage liver disease score and Shannon diversity, independent of potentially confounding factors (Padjust = 0.046). Alcoholic hepatitis patients with more severe disease had significantly decreased relative abundances of Akkermansia while the relative abundance of Veillonella was increased. We observed a reduction in the Bacteroides abundance (Padjust = 0.048) and Shannon diversity (Padjust = 0.018) in antibiotic-treated patients and patients receiving steroids had an increase in Veillonella abundance (Padjust = 0.005), which was both independent of potentially confounding factors.
Conclusion
We observed distinct changes in the gut bacterial microbiome of alcoholic hepatitis patients with more severe disease. The gut bacterial microbiome might be an attractive target to prevent and treat this deadly disease.
KEYWORDS: Microbiome, 16S sequencing, alcohol-related liver disease, metagenomics
Introduction
Alcohol-associated liver disease includes a wide spectrum of hepatic clinical syndromes and pathologic findings associated with heavy alcohol consumption1. Approximately 1 in 20 deaths worldwide is attributed to alcohol abuse, and alcohol-associated liver disease resulted in over 22,000 deaths in the US alone in 2017.2 Alcohol-associated liver disease includes steatosis, fibrosis, cirrhosis, and alcoholic hepatitis. These clinical entities, despite discrete definition, have substantial overlap and are closely interrelated. Simple steatosis involves fatty infiltration of the liver and is typically asymptomatic with normal or mild elevation in liver transaminases. A subset of these patients will go on to develop liver fibrosis, and ultimately, alcoholic cirrhosis. Alcoholic hepatitis is its own separate entity, which is related to but does not lie within the linear spectrum of steatosis, fibrosis, and cirrhosis in any predictable way. Thirty to 40% of chronic heavy drinkers will develop alcoholic hepatitis, but there is no clear identifiable trigger.3 Within alcoholic hepatitis, there is a wide range of disease severity, ranging from chronic and clinically silent to a fulminant syndrome of inflammation and cholestasis. Prognosis in alcoholic hepatitis varies widely, but is generally poor, with 30-day mortality reaching up to 50%.4
Chronic alcohol use, even in the absence of significant liver disease, is known to alter the intestinal microbiome. The effects of alcohol on the intestinal, hepatobiliary, and immune system physiology, are all postulated to contribute to the observed intestinal dysbiosis. In addition to the hepatotoxic effects of alcohol, the microbiome itself is likely an integral factor for the development and progression of alcohol-associated liver disease.5 While there are several studies that have analyzed the compositional changes in the microbiome associated with alcohol-associated liver disease, few have utilized 16S rRNA gene sequencing data from the fecal samples of human subjects. These studies vary widely in methodology, and often grouped patients into a single cirrhosis category, regardless of etiology. Among the studies which purely looked at patients with alcohol-associated liver disease, few used 16S rRNA gene sequencing data of fecal samples from patients with alcoholic hepatitis.6,8
Moreover, little is known about how particular microbiome changes are associated with alcoholic hepatitis disease severity or specific clinical parameters. In 2016, Llopis et al. compared the intestinal microbiome of humans with moderate versus severe alcoholic hepatitis prior to transplanting the microbiome into germ-free and conventional mice. This single-center study enrolled a total of 38 patients (22 with alcoholic hepatitis) and classified severity by a histologic scoring system, specifically requiring a neutrophilic infiltrate for severe disease.7 Similarly, Smirnova et al. described the microbiome of alcoholic hepatitis by disease severity, but utilized model for end-stage liver disease (MELD) score rather than liver histology to classify severe disease. Patients in this study were enrolled at three centers in the US, 10 and 24 patients were classified as moderate and severe alcoholic hepatitis groups, respectively.9
The aim of our study was to characterize the gut microbiome of alcoholic hepatitis patients enrolled in an observational study at multiple centers worldwide.
Methods
Patients
The patient cohort has been described before.8,10,11 In brief, alcoholic hepatitis patients were enrolled within the InTeam Consortium (ClinicalTrials.gov identifier number: NCT02075918). Seventy-four patients with alcoholic hepatitis hospitalized in nine centers across the United States, Mexico, and Europe, which were enrolled between June 2014 and April 2017, were included in this study. The inclusion criteria for alcoholic hepatitis were as follows: in the last three months prior to enrollment, patients must (1) meet criteria for excessive alcohol use (>50 g/day for men and >40 g/day for women), (2) meet laboratory criteria of aspartate aminotransferase (AST) > alanine aminotransferase (ALT) and total bilirubin >3 g/dl, and (3) have liver biopsy and/or clinical picture consistent with alcoholic hepatitis (all liver biopsies were performed only if part of routine clinical care). Patients carrying diagnoses with other hepatobiliary pathology including autoimmune liver disease (ANA >1/320), chronic viral hepatitis, hepatocellular carcinoma, and complete portal vein thrombosis were excluded. Similarly, patients with other significant comorbidities such as diabetes, inflammatory bowel disease, any terminal condition, or clinically significant cardiac, pulmonary, or renal conditions were excluded. Other exclusion criteria included pregnancy and lack of written informed consent. The protocol was approved by the Ethics Committee of each participating center. Written informed consent was obtained from each subject.
Bacterial DNA extraction and 16S rRNA sequencing
16S rRNA sequencing of human stool samples was previously described.8 Raw 16S sequence reads are available for download in the NCBI Sequence Read Archive (SRA) associated with Bioproject PRJNA525701.8 A standardized protocol for fecal sample collection across all centers was used. Further processing of fecal samples was performed at one time concurrently, at one single center.
Statistical analysis
Results are expressed as median and range unless stated otherwise. The bacterial sequence reads were normalized to get the proportional, relative abundance for further statistical analysis. Two groups were compared using the Mann-Whitney-Wilcoxon rank-sum test for continuous and Fisher’s exact test for categorical variables. Spearman’s correlations were conducted to correlate the relative bacterial abundance at genus level with clinical parameters. Partial Spearman was used to adjust correlations between two numeric variables for cofactors. Multivariate analysis by linear models (MaAsLin), a multivariate statistical framework, was used to detect independent associations between clinical metadata and microbial community abundance.12 Only significant associations (P < .05) were plotted in the respective graphs. Bray–Curtis dissimilarity matrices were determined for the principal coordinate analysis (PCoA) and P values were determined by permutational multivariate analysis of variance (PERMANOVA) while adjusting for potentially confounding factors. Multivariate logistic regression analysis was further used to adjust associations for potentially confounding factors. All statistical tests were two-sided. For all analyses, P values of 0.05 or less were considered to be statistically significant. Statistical analysis was performed using R statistical software, R version 3.5.1, 2018 the R Foundation for Statistical Computing.
Results
The median age of the study cohort was 49 and 67% were male. One-third received steroid treatment, 49% were treated with antibiotics (including prophylactic antibiotics) and the median MELD score was 24 (Table 1). Patients with severe alcoholic hepatitis, stratified based on a MELD score higher than 21, had significantly higher stages of fibrosis, but no significant differences were observed in terms of age, sex, ethnicity, and geographic origin (Supplementary Table 1).
Table 1.
Characteristics of patients with alcoholic hepatitis (n = 74).
| Demographics | ||
|---|---|---|
| Sex (% male), n (%), n = 73 | 49 (67.1%) | |
| Age (y), n = 73 | 49.2 (31.3–74.8) | |
| BMI (kg/m2), n = 68 | 27.1 (16.3–48.3) | |
| Mean Alcohol Intake (g/day), n = 63 | 104 (113.5*) | |
| Ethnicity, n = 73 | ||
| Hispanic, n (%) | 13 (17.8) | |
| Non-Hispanic, n (%) | 60 (82.2) | |
| Geographic Location, n = 74 | ||
| USA, n (%) | 43 (58.1) | |
| Mexico, n (%) | 6 (8.1) | |
| Europe, n (%) | 25 (33.8) | |
| Infections and treatment | ||
| Infections, n (%), n = 57 | 13 (22.8) | |
| Steroids, n (%), n = 72 | 22 (30.6) | |
| Pentoxifylline, n (%), n = 70 | 8 (11.4) | |
| Antibiotics, n (%), n = 72 | 35 (48.6) | |
| Proton Pump Inhibitor, n (%), n = 37 | 5 (6.8) | |
| Laboratory parameters | ||
| Creatinine (mg/dL), n = 73 | 0.8 (0.3–8.1) | |
| Bilirubin (mg/dL), n = 73 | 14.1 (2.5–38.6) | |
| AST (IU/L), n = 73 | 136.0 (38.0–456) | |
| ALT (IU/L), n = 73 | 48.0 (15.0–216.0) | |
| Albumin (g/dL), n = 69 | 2.4 (1.3–4.1) | |
| INR, n = 72 | 1.8 (1.0–4.4) | |
| GGT (IU/L), n = 34 | 242.5 (33.0–3632.0) | |
| Platelet count (109/L), n = 70 | 126.0 (21.0–447.0) | |
| Liver histology | ||
| Stage of Fibrosis, n (%), n = 41 | 0/1/2/3/4 | 2 (4.9)/0 (0.0)/6 (14.6)/8 (19.5)/25 (61.0) |
| Lobular fibrosis, n (%), n = 40 | 0/1/2/3 | 4 (10.0)/6 (15.0)/2 (5.0)/28 (70.0) |
| Pericellular fibrosis, n (%), n = 40 | 0/1 | 9 (22.5)/31 (77.5) |
| Grade of steatosis, n (%), n = 41 | 0/1/2/3 | 0 (0.0)/16 (39.0)/12 (29.3)/13 (31.7) |
| Mallory bodies, n (%), n = 40 | 0/1 | 6 (15.0)/34 (85.0) |
| Bilirubinostasis, n (%), n = 40 | 0/1/2/3 | 14 (35.0)/18 (45.0)/1 (2.5)/7 (17.5) |
| Ballooning, n (%), n = 40 | 0/1 | 27 (37.5)/13 (32.5) |
| Giant mitochondria, n (%), n = 37 | 0/1 | 32 (86.5)/5 (13.5) |
| PMN infiltration, n (%), n = 41 | 0/1/2 | 9 (22.0)/18 (43.9)/14 (34.1) |
| Inflammatory grade, n (%), n = 41 | 0/1/2 | 11 (26.8)/27 (65.9)/3 (7.3) |
| Clinical scores and outcome | ||
| MELD, median (range), n = 72 | 23.8 (11.7–43.0) | |
| MELD > 21, n (%) | 54 (75) | |
| Child-Pugh stage, n (%), n = 71 | A/B/C | 1 (1.4)/22 (31.0)/48 (67.6) |
Antibiotics include prophylactic antibiotics. Values are presented as median (range or *interquartile range) for continuous variables or number (percentage) for categorical variables. Percentages are calculated based on the actual number of patients in each group where the respective data was available. The number of subjects for which the respective data was available is indicated in the first column. Fibrosis stage, 0 no fibrosis, 1 portal fibrosis, 2 expansive periportal fibrosis, 3 bridging fibrosis, 4 cirrhosis. Lobular fibrosis, 0 no fibrosis, 1 zone 3 (centrilobular) fibrosis, 2 zone 2 + 3 (midzonal) fibrosis, 3 panlobular fibrosis. Pericellular fibrosis, 0 absent, 1 present. Steatosis, 1 mild <33%, 2 moderate <33–66%, 3 marked >66%. Mallory bodies, 0 absent, 1 present. Bilirubinostasis, 0 no, 1 hepato-canalicular, 2 cholangiolar, 3 both. Ballooning, 0 occasional hepatocellular, 1 marked hepatocellular, 2 none present. Megamitochondria, 0 absent, 1 present. PMN infiltration, 0 no, 1 mild, 2 severe. Inflammation, 0 no, 1 mild, 2 severe.
BMI, body mass index; AST, aspartate aminotransferase; ALT, alanine aminotransferase; INR, international-normalized ratio; GGT, gamma-glutamyl transferase; MELD, model for end-stage liver disease; PMN, polymorphonuclear infiltration.
To get a first overview about potential associations between the gut bacterial microbiome and clinical, laboratory and histologic parameters in alcoholic hepatitis patients, we performed a heatmap analysis, showing color-coded Spearman correlations between the most abundant gut bacterial taxa at genus level, as well as the overall microbiome diversity with clinical, laboratory, and histological parameters. The most notable associations were related to parameters associated with more disease severity (MELD score, bilirubinemia, albumin levels), treatment modalities (antibiotics and steroids), and inflammatory hepatic changes on liver histology. A higher mean self-reported intake of alcohol was associated with higher abundances of Enterococcus and Lactobacillus but not with differences in the bacterial diversity (Figure 1).
Figure 1.
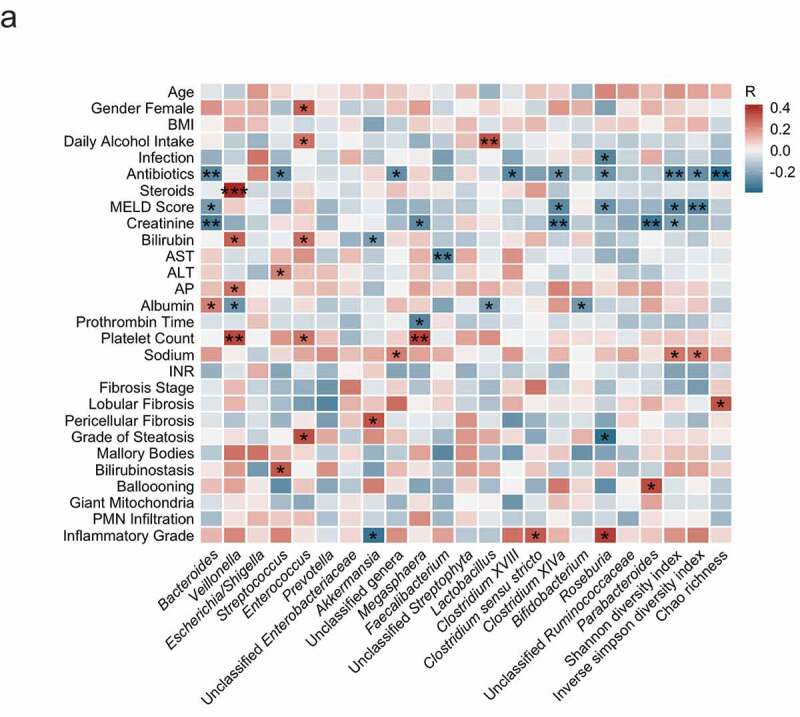
Intestinal bacteria correlate with clinical parameters in alcoholic hepatitis.
Heat map representing color-coded Spearman’s correlations of clinical parameters. Red color indicates positive- and blue color negative correlation. All variables are coded from low to high- i.e. red color in antibiotics means antibiotic use is positively correlated with the respective bacterial taxon. *P < .05, P > .01, ** P < .01, P > .001, *** P < .001. PMN, polymorphonuclear infiltration; INR, international-normalized ratio; ALT, alanine aminotransferase; AST, aspartate aminotransferase; AP, alkaline phosphatase; BMI, body mass index. MELD, Model for End-stage Liver Disease.
Severity of alcoholic hepatitis associated with alterations in the composition and diversity of the gut bacterial microbiome
To investigate if liver disease severity in alcoholic hepatitis patients is associated with alterations in the gut bacterial microbiome, we grouped patients according to a MELD score higher than 21, which is a commonly used cutoff to distinguish severe from non-severe alcoholic hepatitis. Since these groups were very uneven in regard to patient numbers (n = 54 in high MELD vs. n = 18 in low MELD), we additionally compared two groups based on the median bilirubin level of 14.1 mg/dl.
We compared the overall gut microbiome composition between groups by performing a principal coordinate analysis including 323 gut bacterial taxa at genus level. After adjustment for treatment with antibiotics, steroids and pentoxifylline, ethnicity as well as the region where the patient was enrolled, the high bilirubin patient group (higher than 14.1 mg/dl, n = 36) clustered significantly different (Padjust = 0.010) when compared with the lower bilirubin group (less or equal 14.1 mg/dl, n = 37) (Figure 2a, upper left panel). Prominent univariate differences were an increase in Veillonella and Enterococcus, as well as a reduction in Akkermansia in the group with more severe disease (Figure 2a, upper right panel). A higher Veillonella abundance was significantly associated with bilirubin levels as continuous variable, independent of antibiotics, steroids, pentoxifylline, ethnicity, and center origin (Padjust = 0.026, Figure 2a, lower left panel). MaAsLin analysis revealed significant independent associations between several bacterial taxa and high bilirubin levels, whereas a significantly reduced Akkermansia abundance in the high bilirubin group was one major finding (Figure 2a, lower right panel). The Shannon index did not significantly differ among patients with high versus low bilirubin (Padjust = 0.289).
Figure 2.
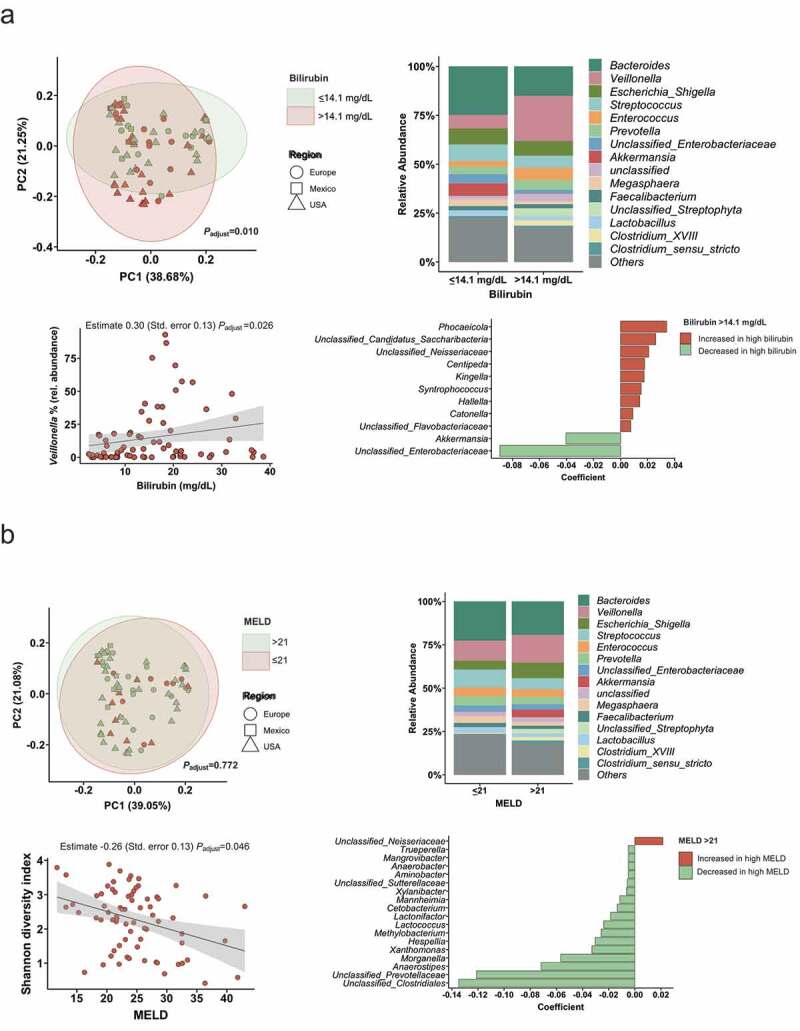
Association of disease severity with the intestinal microbiota.
(A) The upper left panel shows the principal coordinate analysis, that was used to show ß-diversity between the groups based on the abundance of 323 bacterial taxa at the genus level. The axes represent the two most discriminating axes using the Bray–Curtis distance metric. The P value was determined by permutational multivariate analysis of variance (PERMANOVA). The upper right graph demonstrates the mean relative abundance of sequence reads in each genus for each group (alcoholic hepatitis patients were grouped according to the median bilirubin level of 14.1 mg/dl in the cohort). The lower left panel shows the Spearman correlation between bilirubin levels and the relative abundance of Veillonella. The lower right panel shows MaAsLin (Multivariate analysis by linear models), including all 323 bacterial taxa on genus level. Bilirubin >14.1 mg/dl: n = 37; <14.1 or equal: n = 36. (B) The upper left panel shows the principal coordinate analysis, that was used to show ß-diversity between the groups based on the abundance of 323 bacterial taxa at the genus level. Alcoholic hepatitis patients were grouped according to a MELD score higher or lower/equal than 21. The axes represent the two most discriminating axes using the Bray–Curtis distance metric. The P value was determined by PERMANOVA. The upper right graph demonstrates the mean relative abundance of sequence reads in each genus for each group. The lower left panel shows the negative Spearman correlation between the Shannon index and the MELD score. The lower right panel shows MaAsLin results. All analyses except for the upper right panels were adjusted for treatment with antibiotics, steroids, pentoxifylline, ethnicity, and the center origin. MELD >21 mg/dl: n = 54; <21 or equal: n = 18.
Only 18 patients had a MELD score lower or equal than 21 compared with 54 patients with a MELD higher than 21. The principal coordinate analysis did not cluster significantly different when comparing the overall microbiome composition between alcoholic hepatitis patients with high versus low MELD (Figure 2b, upper left panel). When we looked into associations between the MELD as continuous with diversity, we found that a higher MELD score was significantly associated with a lower bacterial diversity, as measured by the Shannon index, which was independent of treatment with antibiotics, steroids and pentoxifylline, ethnicity, and the center origin (estimate −0.26, standard error 0.13, Padjust = 0.046, Figure 2b, lower left panel). In the MaAsLin analysis, a lower relative abundance of unclassified Clostridales, unclassified Prevotellaceae, and Anaerostipes independently characterized the high MELD patients (Figure 2b, lower right panel).
Overall, these data indicate that a more pronounced disease severity is associated with compositional changes and a reduced bacterial diversity in alcoholic hepatitis patients.
Gut bacterial alterations associated with antibiotic and steroid treatment
Next, we associated treatment with antibiotics and steroids with alterations in the diversity and composition of the gut bacterial microbiome in alcoholic hepatitis patients. The overall gut microbiome composition significantly (Padjust<0.01) differed among treated groups when performing principal coordinate analyses including the relative abundance of all detected taxa at genus level, adjusted for other treatment (steroids/antibiotics/pentoxifylline respectively), the center origin, levels of bilirubin, albumin, creatinine, the international-normalized ratio (INR), and platelet counts as markers for disease severity (Figures 3 and 4, upper left panel, respectively). When we compared the mean relative abundance of the 15 most abundant taxa at genus level between antibiotic-treated and antibiotic-naïve patients, the most remarkable change was a reduced abundance of Bacteroides and an increased abundance of Escherichia/Shigella in patients treated with antibiotics (mean relative Bacteroides abundance: 26% in the antibiotic-naïve group compared with 12% in the antibiotic-treated patient group, Padjust = 0.048, Figure 3, upper right and middle right panel). In the MaAsLin analysis, antibiotic use was independently associated with lower levels of Blautia, Clostridium spp., Roseburia, Dorea, and several other taxa (Figure 3, lower left panel). Further, we observed a reduced bacterial diversity, as measured by the Shannon index (Figure 3, lower right panel, Padjust = 0.015).
Figure 3.
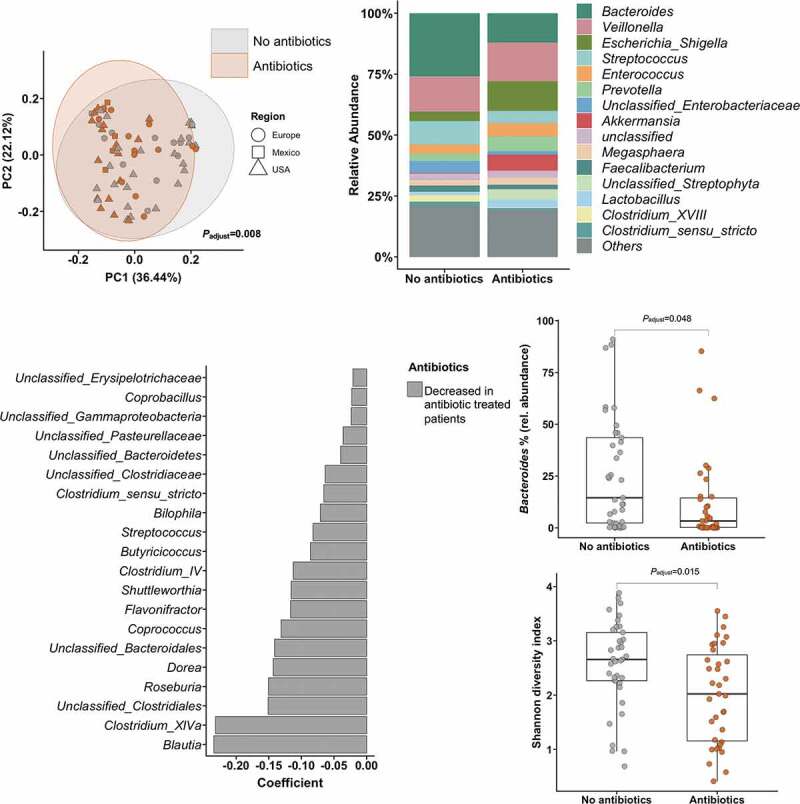
Association of antibiotic treatment with intestinal bacteria.
The upper left panel shows the principal component analysis, that was used to show ß-diversity between the groups based on the abundance of 323 bacterial taxa at the genus level. The axes represent the two most discriminating axes using the jaccard distance metric. The P value was determined by the permutational multivariate analysis of variance (PERMANOVA). The upper right graph demonstrates the mean relative abundance of sequence reads in each genus for each group (alcoholic hepatitis patients were grouped according to the treatment with antibiotics, including prophylactic antibiotics (n = 35 (49%)). The middle right panels show the decreased abundance of Bacteroides and the decreased Shannon diversity index in patients receiving antibiotics. Wilcoxon-Mann-Whitney test. The lower left panel shows MaAsLin (Multivariate analysis by linear models) results, including all 323 bacterial taxa on genus level. All analyses except for the upper right panel were adjusted for treatment with steroids and pentoxifylline, the center origin, levels of bilirubin, albumin, creatinine, the international-normalized ratio (INR), and platelet counts as markers for disease severity.
Figure 4.
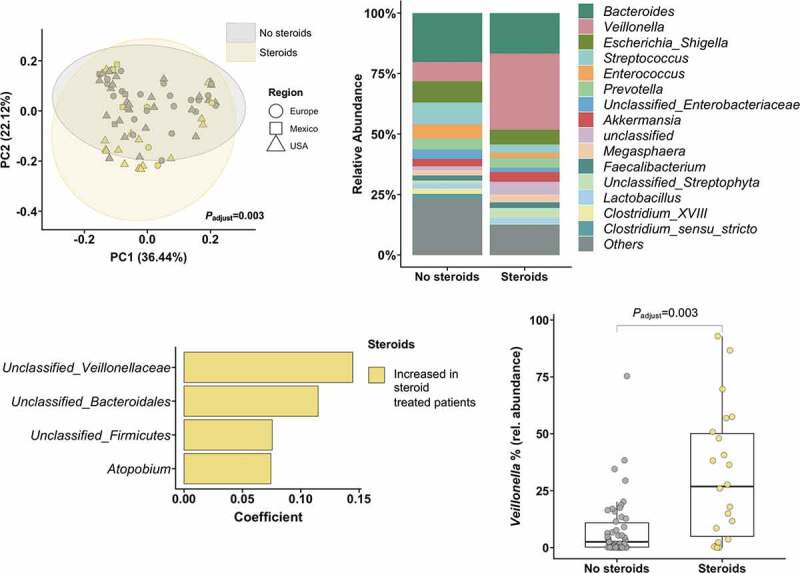
Association of steroid treatment with intestinal bacteria.
The upper left panel shows the principal component analysis, that was used to show ß-diversity between the groups based on the abundance of 323 bacterial taxa at the genus level. The axes represent the two most discriminating axes using the jaccard distance metric. The P value was determined by permutational multivariate analysis of variance (PERMANOVA). The upper right graph demonstrates the mean relative abundance of sequence reads in each genus for each group (alcoholic hepatitis patients were grouped according to the treatment with steroids at admission (n = 22 (31%)). The lower right panel shows the increased abundance of Veillonella in patients receiving steroids. Wilcoxon-Mann-Whitney test. The lower left panel shows MaAsLin (Multivariate analysis by linear models) results, including all 323 bacterial taxa on genus level. All analyses except for the upper right panel were adjusted for treatment with antibiotics, the center origin, levels of bilirubin, albumin, creatinine, the international-normalized ratio (INR), and platelet counts as markers for disease severity.
In steroid-treated patients, the most remarkable difference compared with patients that did not receive steroids was related to an increased Veillonella abundance (mean relative Veillonella abundance: 8% in the steroid-naïve group compared with 27% in the steroid-treated patient group, Padjust = 0.003, Figure 4, upper and lower right panel), independent of disease severity (levels of bilirubin, albumin, creatinine, INR, and platelet counts), concomitant antibiotic use, ethnicity, or the center origin. The Shannon index did not significantly differ among steroid-treated patients and steroid-naïve patients (Padjust = 0.820).
These data indicate that treatment with antibiotics and steroids might lead to alterations in the gut microbiome of alcoholic hepatitis patients.
Microbiome associations with histologic changes on liver biopsy
We next investigated if liver histology parameters are associated with specific alterations in the composition of the gut bacterial microbiome. Fecal samples from alcoholic hepatitis patients with the highest grade of steatosis contained more Enterococcus compared with patients with lower steatosis grades (Figure 5a, upper panel). In the MaAsLin analysis, Bifidobacterium, Lactococcus, Oribacterium, Desulfovibrio, Enterococcus, and Veillonella were the most increased taxa in patients with grade 3 steatosis (Figure 5a, lower panel). Akkermansia was lower abundant in patients with a higher degree of polymorphonuclear infiltration (Figure 5b, upper panel) and a higher degree of histological inflammation (Figure 5c, upper panel). We further observed a higher Veillonella abundance in patients with more severe inflammation on liver biopsy (Figure 5c).
Figure 5.
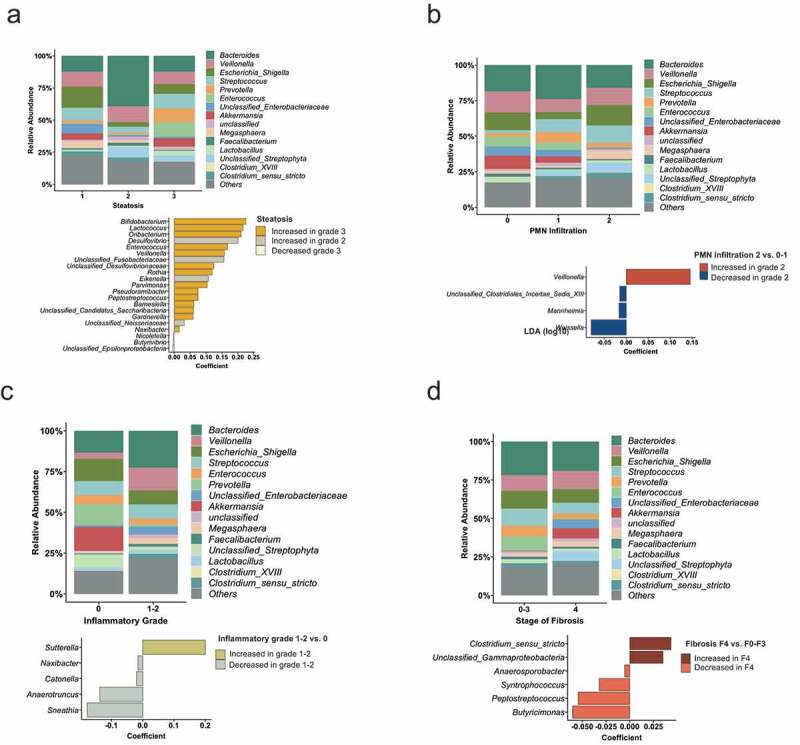
Liver histology features associated with the intestinal microbiota.
(A) Mean relative abundance of sequence reads in each genus for each group (alcoholic hepatitis patients were grouped according to steatosis on liver histology (1, n = 16; 2, n = 12, 3, n = 13)). The lower panel shows MaAsLin (Multivariate analysis by linear models) results, including all 323 bacterial taxa on genus level. (B) Alcoholic hepatitis patients were grouped according to PMN (polymorphonuclear) infiltration on liver histology (1, n = 9; 2, n = 18, 3, n = 14). Mean relative abundance of sequence reads and MaAsLin results, including all 323 bacterial taxa on genus level. (C) Mean relative abundance of sequence reads in each genus for each group (alcoholic hepatitis patients were grouped according to the inflammatory grade on liver histology (0, n = 11; 1–2, n = 30)) and MaAsLin results. (D) Mean relative abundance of sequence reads in each genus for each group (alcoholic hepatitis patients were grouped according to the liver fibrosis stage on liver histology (0–3, n = 16; 4, n = 25)). The lower panel shows MaAsLin results including all 323 bacterial taxa.
Although some lower abundant gut bacterial taxa were associated with less fibrosis, we did not observe major alterations when comparing alcoholic hepatitis patients with cirrhosis (n = 25) versus non-cirrhotic patients (n = 16) (Figure 5d).
Discussion
This study describes alterations in the fecal microbiota of alcoholic hepatitis patients associated with the severity of the disease, antibiotic and steroid use, and histologic changes on liver biopsy. With increasing data suggesting that the microbiome plays an important role in the pathogenesis of liver disease, there is a need to better characterize particular alterations related to findings commonly encountered in clinical practice.
Prognosis in alcoholic hepatitis is highly variable, but mortality is especially high in severe disease. In the present study, MELD score and degree of hyperbilirubinemia were used as surrogates for disease severity. In patients with more severe liver disease, there was an increased relative abundance of Veillonella. Veillonella carries an unknown degree of pathogenicity and has been found to be more prevalent in patients with liver disease.13,14 Kakiyama et al. found increased Veillonellaceae at the family level in patients with cirrhosis, and an even more significant increase when the etiology of cirrhosis is alcohol-induced.14,15
A diverse variety of microorganisms in the gut are essential for a healthy symbiotic relationship between the microbiome and human host.5 Reduced intestinal bacterial diversity has been linked with various negative outcomes, such as obesity and inflammatory bowel disease.16,17 While it has been shown that patients with liver cirrhosis tend to have reduced bacterial diversity, there are little data regarding the severity of alcohol-associated liver disease and the heterogeneity of the microbiome.13 Ciocan et al. found that patients with severe alcoholic hepatitis had slightly reduced diversity compared to alcoholic controls without liver disease, though not reaching statistical significance. They defined severe disease using a histologic scoring system, although they did not compare with alcoholic hepatitis patients with less severe disease.6 In the present study, we found a significant negative correlation between MELD scores and bacterial diversity, independent of antibiotic or steroid treatment. Overall, these findings suggest that severe disease is associated with less bacterial diversity in the microbiome of alcoholic hepatitis patients.
Few studies have described microbiome associations with disease severity in alcoholic hepatitis. A recent study by Smirnova et al. did not find significant differences in the gut microbiota composition or diversity among alcoholic hepatitis patients with moderate versus severe alcoholic hepatitis (defined by MELD of 21 or greater); however, patient numbers were low with only 10 and 24 patients, respectively,9 which limits the ability to reach statistical significance. There were some significant differences in individual bacterial taxa between moderate and severe alcoholic hepatitis patients, including an increased abundance of Actinomycetaceae and Fusobacteriaceae. The authors formulated a microbiome predictive model utilizing MELD score in all alcohol-consuming groups (regardless of the presence of alcoholic hepatitis) and identified the top 20 genera contributing to the model’s ability to accurately predict a MELD score greater than 20. Veillonellaceae Veillonella, Lachnospiracea incertae sedis, Bacteroidaceae Bacteroides, and Streptococcaceae Streptococcus were the most abundant bacterial taxa in this group.9 In our study, we did not see these specific expansions in the high MELD group.
Llopis et al. used a histologic scoring system (alcoholic hepatitis score) to determine alcoholic hepatitis severity and also identified an expansion of Streptococci, as well as Bifidobacteria, and Enterobacteria in patients with severe disease. Streptococci and Enterobacteria were both positively correlated with the alcoholic hepatitis score, and Enterobacteria was also positively correlated with serum bilirubin level.7 Lastly, Puri et al. examined the circulating microbiome in patients with alcoholic hepatitis with severity determined by MELD greater than 20. They noted an expansion of Brevibacterium and Staphylococcus in the circulating microbiome of alcoholic hepatitis patients with severe disease compared to moderate disease. Regression analysis including all alcohol-consuming patients found a negative correlation between MELD score and Janthinobacterium and Enhydrobacter, two genera of the Proteobacteria phylum.18 We did not see these associations in our analyses, though it is important to note that the study by Puri et al. uses 16S rRNA gene sequencing from whole blood samples.
Antibiotic use has also been shown to reduce gut bacterial diversity in healthy human subjects.19 Also, in our study, antibiotic treatment was associated with a lower bacterial diversity in alcoholic hepatitis patients. The greatest reduction in specific bacterial taxa was seen in genus Bacteroides, which is largely considered to be a beneficial organism within the intestinal environment.20 While some studies have shown an increased abundance in family Bacteroidaceae amongst cirrhotics,14,21 the association appears to invert and show a reduction of Bacteroidaceae when the analysis includes or is limited to alcoholic cirrhosis.13,15,22 The antibiotic-treated group in our study also had a significant increase in anaerobes Bilophila and Flavonifractor, both of which are rare clinical pathogens, but have been associated with systemic inflammation and/or colorectal malignancy.23,26 However, whether antibiotic use is causatively linked to these observations, can not be answered with our study design.
We found a marked increase in the relative abundance of Veillonella in alcoholic hepatitis patients that had been treated with steroids. In clinical practice, steroid use is generally reserved for severe alcoholic hepatitis, often defined by Maddrey’s Discriminant Function (DF) Score exceeding 32 and if no contraindications are present. Since we had already identified Veillonella to be more prevalent in patients with severe hyperbilirubinemia (which is a component of the Maddrey’s DF score), we deduced that at least some of this association was likely attributable to disease severity. However, the association between a higher Veillonella abundance and steroid use remained significant after performing a multivariate logistic regression analysis controlling for parameters associated with disease severity (including bilirubin level). While further mechanistic studies are needed to elucidate the potential pathogenic function of Veillonella in alcoholic hepatitis, this study corroborates that Veillonella is also positively correlated with disease severity and may be independently expanded by the use of steroids.
The fecal microbiota in alcoholic patients were also evaluated for associations with various liver histologic changes. Severe steatosis is associated with an increase in genera Enterococcus, which are largely regarded as pathologic organisms. When we evaluated inflammatory histologic changes, such as PMN infiltration and inflammatory grade, Akkermansia abundance was inversely correlated with increased inflammation and also reduced in patients with higher bilirubin levels. Akkermansia is a well-described beneficial inhabitant of the human microbiome and is important for fatty acid metabolism. Its depletion has been implicated in obesity and fatty liver disease, and supplementation of Akkermansia has even been shown to decrease serum triglyceride and alanine aminotransferase levels in obese mice.27 Recently, Addolorato et al. published their findings of significant Akkermansia reduction in patients with alcohol use disorder, irrespective of the degree of liver disease.28 In line, Grander et al. found a depletion of Akkermansia in fecal samples from alcoholic hepatitis patients and Akkermansia supplementation protected mice against liver injury in experimental alcoholic liver disease.29 Our study suggests that the reduction in Akkermansia abundance is related to the degree of liver inflammation as seen on liver biopsy in patients with alcoholic hepatitis.
The main strength of our study is its international multicenter design, which aims to control for various geographical and ethnic factors that may affect microbiome composition. To our knowledge, our study sequenced fecal samples from the largest number of alcoholic hepatitis patients, that were also enrolled in multiple centers including Europe, the United States, and Mexico. However, our study is descriptive and limited by its inability to determine causation. Further, the use of medications other than antibiotics, steroids, and pentoxifylline was incompletely reported; therefore, we can not rule out that other concomitant medication use might have influenced our results. Further mechanistic studies are required to better understand the role of the microbiome in the pathogenesis and progression of alcoholic hepatitis.
In summary, we observed compositional alterations, such an increased Veillonella abundance, and a lower bacterial diversity among alcoholic hepatitis patients with more severe disease. We further observed a reduction in Bacteroides in antibiotic-treated patients and an enrichment of Veillonella in patients treated with steroids. The gut bacterial microbiome might be an attractive target to prevent and treat this deadly disease.
Supplementary Material
Funding Statement
S.L. was supported by a DFG fellowship (LA 4286/1-1). This study was supported in part by a Biocodex Microbiota Foundation Grant, NIH grants R01 AA020703, R01 AA24726, U01 AA026939, by Award Number BX004594 from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development (to B.S.), and services provided by P30 DK120515 and P50 AA011999.
Author contributions
S.L. was responsible for data analysis, interpretation of data and editing of the manuscript, B.F. was responsible for the interpretation of data and writing of the manuscript, B.G. provided assistance with data analysis and editing the manuscript, X.Z. provided assistance with statistical analysis, Y.D. was responsible for fecal DNA extraction and edited the manuscript, D.E.F. was responsible for 16 S rRNA sequencing, B.S. was responsible for the concept, design, and supervision of the study and editing of the manuscript.
Disclosure of potential conflicts of interest
B.S. has been consulting for Ferring Research Institute, Intercept Pharmaceuticals, HOST Therabiomics and Patara Pharmaceuticals. B.S.’s institution UC San Diego has received grant support from BiomX, NGM Biopharmaceuticals, CymaBay Therapeutics, Synlogic Operating Company and Axial Biotherapeutics.
Supplementary material
Supplemental data for this article can be accessed here.
References
- 1.Masarone M, Rosato V, Dallio M, Abenavoli L, Federico A, Loguercio C, Persico M.. Epidemiology and natural history of alcoholic liver disease. Rev Recent Clin Trials. 2016;11(3):167–12. doi: 10.2174/1574887111666160810101202. [DOI] [PubMed] [Google Scholar]
- 2.Kochanek KDMS, Xu JQ, Arias E. Deaths: final data for 2017. National vital statistics reports. Hyattsville (MD): National Center for Health Statistics; 2017. [Google Scholar]
- 3.Osna NA, Donohue TM Jr., Kharbanda KK. Alcoholic liver disease: pathogenesis and current management. Alcohol research: Current Reviews. 2017;38:147–161. [PMC free article] [PubMed] [Google Scholar]
- 4.Singal AK, Bataller R, Ahn J, Kamath PS, Shah VH. ACG clinical guideline: alcoholic liver disease. Am J Gastroenterol. 2018;113(2):175–194. doi: 10.1038/ajg.2017.469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hartmann P, Seebauer CT, Schnabl B. Alcoholic liver disease: the gut microbiome and liver cross talk. Alcohol Clin Exp Res. 2015;39:763–775. doi: 10.1111/acer.12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ciocan D, Rebours V, Voican CS, Wrzosek L, Puchois V, Cassard AM, Perlemuter G. Characterization of intestinal microbiota in alcoholic patients with and without alcoholic hepatitis or chronic alcoholic pancreatitis. Sci Rep. 2018;8:4822. doi: 10.1038/s41598-018-23146-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Llopis M, Cassard AM, Wrzosek L, Boschat L, Bruneau A, Ferrere G, Puchois V, Martin JC, Lepage P, Le Roy T. Intestinal microbiota contributes to individual susceptibility to alcoholic liver disease. Gut. 2016;65(5):830–839. doi: 10.1136/gutjnl-2015-310585. [DOI] [PubMed] [Google Scholar]
- 8.Duan Y, Llorente C, Lang S, Brandl K, Chu H, Jiang L, White RC, Clarke TH, Nguyen K, Torralba M, et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature. 2019;575(7783):505–511. doi: 10.1038/s41586-019-1742-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Smirnova E, Puri P, Muthiah MD, Daitya K, Brown R, Chalasani N, Liangpunsakul S, Shah VH, Gelow K, Siddiqui MS, et al. Fecal microbiome distinguishes alcohol consumption from alcoholic hepatitis but does not discriminate disease severity. Hepatology. 2020. doi: 10.1002/hep.31178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lang S, Duan Y, Liu J, Torralba MG, Kuelbs C, Ventura‐Cots M, Abraldes JG, Bosques‐Padilla F, Verna EC, Brown RS, et al. Intestinal fungal dysbiosis and systemic immune response to fungi in patients with alcoholic hepatitis. Hepatology. 2020;71(2):522–538. doi: 10.1002/hep.30832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao B, Lang S, Duan Y, Wang Y, Shawcross DL, Louvet A, Mathurin P, Ho SB, Stärkel P, Schnabl B, et al. Serum and fecal oxylipins in patients with alcohol-related liver disease. Dig Dis Sci. 2019;64(7):1878–1892. doi: 10.1007/s10620-019-05638-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 2012;13(9):R79. doi: 10.1186/gb-2012-13-9-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen Y, Yang F, Lu H, Wang B, Chen Y, Lei D, Wang Y, Zhu B, Li L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology. 2011;54(2):562–572. doi: 10.1002/hep.24423. [DOI] [PubMed] [Google Scholar]
- 14.Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, Takei H, Muto A, Nittono H, Ridlon JM. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol. 2013;58(5):949–955. doi: 10.1016/j.jhep.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kakiyama G, Hylemon PB, Zhou H, Pandak WM, Heuman DM, Kang DJ, Takei H, Nittono H, Ridlon JM, Fuchs M, et al. Colonic inflammation and secondary bile acids in alcoholic cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2014;306(11):G929–37. doi: 10.1152/ajpgi.00315.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, et al. A core gut microbiome in obese and lean twins. Nature. 2009;457(7228):480–484. doi: 10.1038/nature07540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKenna P, Hoffmann C, Minkah N, Aye PP, Lackner A, Liu Z, Lozupone CA, Hamady M, Knight R, Bushman FD, et al. The macaque gut microbiome in health, lentiviral infection, and chronic enterocolitis. PLoS Pathog. 2008;4(2):e20. doi: 10.1371/journal.ppat.0040020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Puri P, Liangpunsakul S, Christensen JE, Shah VH, Kamath PS, Gores GJ, Walker S, Comerford M, Katz B, Borst A, et al. The circulating microbiome signature and inferred functional metagenomics in alcoholic hepatitis. Hepatology. 2018;67(4):1284–1302. doi: 10.1002/hep.29623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dethlefsen L, Huse S, Sogin ML, Relman DA, Eisen JA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11):e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wexler HM. Bacteroides: the good, the bad, and the nitty-gritty. Clin Microbiol Rev. 2007;20(4):593–621. doi: 10.1128/CMR.00008-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bajaj JS, Heuman DM, Hylemon PB, Sanyal AJ, White MB, Monteith P, Noble NA, Unser AB, Daita K, Fisher AR, et al. Altered profile of human gut microbiome is associated with cirrhosis and its complications. J Hepatol. 2014;60(5):940–947. doi: 10.1016/j.jhep.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mutlu EA, Gillevet PM, Rangwala H, Sikaroodi M, Naqvi A, Engen PA, Kwasny M, Lau CK, Keshavarzian A. Colonic microbiome is altered in alcoholism. Am J Physiol Gastrointest Liver Physiol. 2012;302(9):G966–78. doi: 10.1152/ajpgi.00380.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Garrett WSOA. Bacteroides, prevotella, porphyromonas, and fusobacterium species (and other medically important anaerobic gram-negative bacilli). In: Mandell GLBJ, Dolin R, editors. Mandell, Douglas and Bennett’s principles and practice of infectious diseases. New York (NY): Churchill Livingston; 2015. p. 2773–2780. [Google Scholar]
- 24.Feng Z, Long W, Hao B, Ding D, Ma X, Zhao L, Pang X. A human stool-derived Bilophila wadsworthia strain caused systemic inflammation in specific-pathogen-free mice. Gut Pathog. 2017;9(1):59. doi: 10.1186/s13099-017-0208-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dahmus JD, Kotler DL, Kastenberg DM, Kistler CA. The gut microbiome and colorectal cancer: a review of bacterial pathogenesis. J Gastrointest Oncol. 2018;9(4):769–777. doi: 10.21037/jgo.2018.04.07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gupta A, Dhakan DB, Maji A, Saxena R, P KV, Mahajan S, Pulikkan J, Kurian J, Gomez AM, Scaria J, et al. Association of flavonifractor plautii, a flavonoid-degrading bacterium, with the gut microbiome of colorectal cancer patients in India. mSystems. 2019;4(6):e00438-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim S, Lee Y, Kim Y, Seo Y, Lee H, Ha J, et al. Akkermansia muciniphila prevents fatty liver disease, decreases serum triglycerides, and maintains gut homeostasis. Appl Environ Microbiol. 2020;86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Addolorato G, Ponziani FR, Dionisi T, Mosoni C, Vassallo GA, Sestito L, Petito V, Picca A, Marzetti E, Tarli C. Gut microbiota compositional and functional fingerprint in patients with alcohol use disorder and alcohol-associated liver disease. Liver Int. 2020;40(4):878–888. doi: 10.1111/liv.14383. [DOI] [PubMed] [Google Scholar]
- 29.Grander C, Adolph TE, Wieser V, Lowe P, Wrzosek L, Gyongyosi B, Ward DV, Grabherr F, Gerner RR, Pfister A, et al. Recovery of ethanol-induced Akkermansia muciniphila depletion ameliorates alcoholic liver disease. Gut. 2018;67(5):891–901. doi: 10.1136/gutjnl-2016-313432. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.