

ABSTRACT
Enteric bacterial pathogens cause significant morbidity and mortality globally. Studies in tissue culture and animal models shaped our initial understanding of these host–pathogen interactions. However, intrinsic shortcomings in these models limit their application, especially in translational applications like drug screening and vaccine development. Human intestinal enteroid and organoid models overcome some limitations of existing models and advance the study of enteric pathogens. In this review, we detail the use of human enteroids and organoids to investigate the pathogenesis of invasive bacteria Shigella, Listeria, and Salmonella, and noninvasive bacteria pathogenic Escherichia coli, Clostridium difficile, and Vibrio cholerae. We highlight how these studies confirm previously identified mechanisms and, importantly, reveal novel ones. We also discuss the challenges for model advancement, including platform engineering to integrate environmental conditions, innate immune cells and the resident microbiome, and the potential for pre-clinical testing of recently developed antimicrobial drugs and vaccines.
KEYWORDS: Human intestinal enteroids, organoids, bacterial pathogens, enteric bacteria
Introduction
Bacterial enteric pathogens are a significant contributor to global diarrheal disease burdens.1 Infectious diarrhea is among the leading causes of death for children in low – and middle-income countries (LMIC).2,3 In the United States, elderly and immunocompromised individuals are especially susceptible to enteric infections. Enteropathogens are commonly spread through contaminated food and water or poor hygiene. The alarming increase in antibiotic resistance among enteric bacteria has increased the urgent need for therapeutic and preventative interventions.4,5
To study the molecular mechanisms employed by pathogenic enteric bacteria, researchers have primarily used immortalized human cell lines such as T84, HT-29, and Caco-2, human tissue explants, or small animal models including mice, rats, rabbits, and pigs. These models have greatly advanced our understanding of molecular pathogenesis by identifying host receptors for bacterial/toxin engagement, host cell surface remodeling, bacterial/toxin internalization pathways, and immune responses. However, it has also been widely recognized that these models can be inconsistent with the physiology of the human intestine.6 Transformed cell lines do not manifest the diversity of epithelial cell types equivalent to native intestine, and they may change genotypically with increasing passage in cell culture; as such, they do not necessarily render a complete or stable physiological response to infection.6 Non-transformed human intestinal explants overcome the limitations associated with cancer-derived cell lines but are short-lived and must be continuously sourced. Small animal models may lack faithful replication of human receptors, infection susceptibility, antimicrobial peptides, and innate immune responses.7,8
The development of human intestinal organoid mini-gut models9,10 presents a highly human-relevant novel platform with the potential to revolutionize the study of enteric bacterial pathogenesis as well as the evaluation of new therapeutic and preventative interventions. The central advantage of organoid models is their multicellular composition of non-transformed human cells including absorptive enterocytes, mucus-producing Goblet cells, antimicrobial peptide-producing Paneth cells, hormone-secreting enteroendocrine cells, chemosensory tuft cells, antigen-sampling microfold cells, and multipotent proliferative stem cells. Collectively, organoids retain distinct features of human intestinal epithelium, including donor genetics, segmental specification, cell polarization, nutrient and ion transport, barrier function, mucus secretion, and microbicidal peptide production.11–13
The term organoid is broadly applied to ex vivo cultures, and requires additional distinction as to whether it refers to an intestinal tissue-derived organoid9 (enteroid, consisting only of epithelial cells) or a human induced pluripotent stem cell (iPSC)-derived intestinal organoid14 (HIO, containing both epithelial and mesenchymal lineages) (Figure 1). Due to the maintenance of segmental specificity, enteroids derived from the large intestine are referred to as colonoids. Enteroids can be studied as three-dimensional (3D) cultures embedded in extracellular matrix (ECM) or as two-dimensional (2D) monolayers on permeable tissue culture supports. Due to tissue-like complexity, HIOs are used exclusively as 3D cultures. Enteroids can be maintained as a crypt-like population or be directed to differentiate into surface/villus-like epithelium, whereas HIOs contain both crypt and surface/villus regions simultaneously. Each model has additional unique characteristics that have been comprehensively reviewed15,16 and continues to evolve through manipulation of culture conditions.
Figure 1.
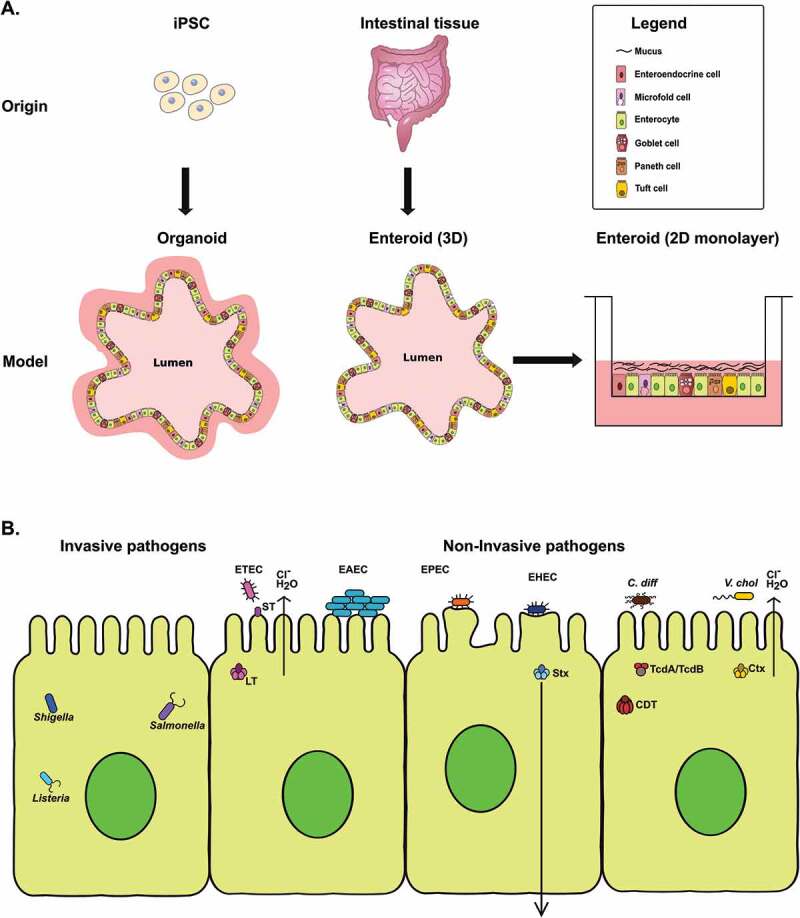
(a) Illustration of the origin and lineage composition of organoids and enteroids. Organoids are derived from induced pluripotent stem cells (iPSCs) and contain cells of epithelial and mesenchymal lineages. Enteroids are derived from intestinal tissue and contain cells of epithelial lineage only. Both enteroids and organoids contain multiple epithelial cell types. (b) Schematic diagram showing an outline of the molecular pathogenesis mechanisms of invasive and noninvasive enteric bacteria in organoids and enteroids discussed in this review. Invasive bacteria such as Shigella, Salmonella and Listeria infect and gain entry into the epithelial cells whereas noninvasive bacteria such as pathogenic E. coli, C. difficile and V. cholerae exert their effect on epithelial cells via toxins or effectors.
In the approximately ten-year period since the introduction of mini-gut culture methods, application to the study of enteropathogens has flourished. Some of the earliest host-pathogen human organoid studies demonstrated viral entry and replication by rotavirus,17–19 and bacterial and parasitic20 infection models soon followed. In this review, we will highlight findings that include the invasive bacteria Shigella, Listeria, and Salmonella, which enter host cells and subvert the surrounding environment to facilitate intracellular replication, as well as noninvasive bacteria, including pathogenic E. coli (enterotoxigenic E. coli, enterohemorrhagic E. coli, enteroaggregative E. coli, and enteropathogenic E.coli), Clostridium difficile, and Vibrio cholerae, which associate with the cell surface, inject molecular effectors, and/or secrete toxins (Table 1). Several bacterial enteropathogen studies have utilized enteroids established from mice, but these studies will not be discussed. We summarize the technical advances, innovative methodologies, and novel discoveries of host–pathogen interaction modeling in human enteroids and HIOs to demonstrate progress in understanding human-specific disease mechanisms and the potential for preclinical testing of new therapeutics and vaccines to ultimately decrease the global burden of diarrheal diseases.
Table 1.
Invasive and noninvasive enteric bacteria that have been studied using human organoids and enteroids and the respective research focus. *Enteroid – no mesenchyme; from duodenum, jejunum, or ileum. **Colonoid – no mesenchyme; from colon or cecum. ***HIO: human induced pluripotent stem cell derived-intestinal organoid (epithelium and mesenchyme).
| Bacteria | Origin | Intestinal section | Type | Research focus | Refs |
|---|---|---|---|---|---|
| Invasive pathogens | |||||
| Shigella flexneri | Human Biopsy | Duodenum, Ileum, Colon | 2D Enteroid*, 2D Colonoid** | Invasion, intracellular replication, host gene expression | 21 |
| Human Biopsy | Duodenum, Jejunum, Ileum, Colon | 2D Enteroid, 2D Colonoid | Invasion, M cells, intracellular replication, mucus changes, cytokine secretion | 22 | |
| Human Biopsy | Cecum | 2D Colonoid | Testing bacteriophage therapy | 23 | |
| Human Biopsy | Colon | 2D Colonoid | Adherence factor expression | 24 | |
| Listeria monocytogenes | Human Biopsy | Ileum | 3D Enteroid | Reversing enteroid polarity to facilitate apical infection | 25 |
| Salmonella enterica serovar Typhimurium | iPSC (dermal fibroblasts) | n/a | 3D HIO*** | Invasion, infection-associated transcriptional changes | 26 |
| iPSC | n/a | 3D HIO | Organoids derived from healthy iPSC and infant IBD patient; role of IL-22 in controlling infection | 27 | |
| Human Biopsy | Ileum | 3D Enteroid | Reversing enteroid polarity to facilitate apical infection | 25 | |
| Salmonella enterica serovar Typhi | Human Biopsy | Ileum | 2D Enteroid | Cytoskeletal changes during infection | 28 |
| Noninvasive pathogens | |||||
| Enterotoxigenic E. coli | Human biopsy | Duodenum, jejunum | 3D Enteroid | Toxin-mediated inhibition of ion transporter NHE3 | 29 |
| Human biopsy | Jejunum | 2D Enteroid | Toxin-induced polarized cyclic nucleotide secretion | 30 | |
| Human biopsy | Jejunum | 2D Enteroid | ST-induced pathology in Intestine Chip model | 31 | |
| Human biopsy | Jejunum, ileum | 2D Enteroid | Minor adhesin-mediated adherence & cyclic nucleotide production | 32 | |
| Human biopsy | Duodenum, jejunum, colon | 2D Enteroid, 2D Colonoid | Effect of macrophage co-culture on infection and epithelial cell differentiation | 33 | |
| Enterohemorrhagic E. coli | Human biopsy | Colon | 2D Colonoid | Role of mucus and SPATE EspP in infection | 34 |
| Human biopsy | Colon | 2D Colonoid | SPATE EspP-induced ion transport | 35 | |
| iPSC | n/a | 3D HIO | Pathogen vs. commensal effect on epithelium; co-culture with neutrophils | 36 | |
| iPSC | n/a | 3D HIO | Shiga toxin effects and mesenchymal-epithelial cross talk | 37 | |
| Enteroaggregative E. coli | Human biopsy | Duodenum, ileum, colon | 2D Enteroid, 2D Colonoid | Adherence patterns by donor and intestinal segment type | 38 |
| Human biopsy | Colon | 2D Colonoid | SPATE SepA-induced ion transport | 35 | |
| Enteropathogenic E. coli | Human biopsy | Duodenum, jejunum, colon | 2D Enteroid, 2D Colonoid | Effect of macrophage co-culture on infection and epithelial cell differentiation | 33 |
| Clostridium difficile | iPSC | n/a | 3D HIO | Viable bacterial infection, toxin-induced pathology | 39 |
| Human biopsy | Colon | 3D Colonoid | Toxin receptor expression | 40 | |
| Human biopsy | Jejunum | 2D Enteroid | Toxin-induced pathology, toxin receptor expression | 41 | |
| Human biopsy | Colon | 3D Colonoid | Toxin binding to intestinal receptors | 42 | |
| iPSC | n/a | 3D HIO | Role of mucus in infection | 43 | |
| iPSC | n/a | 3D HIO | Dysregulation of ion transporter NHE3 and microbiota | 44 | |
| iPSC | n/a | 3D HIO | Human alpha-defensin-1 and toxin neutralization | 45 | |
| iPSC | n/a | 3D HIO | Bacitracin as defense against toxins | 46 | |
| Vibrio cholerae | Human biopsy | Duodenum, jejunum | 3D Enteroid | Toxin-mediated inhibition of ion transporter NHE3 | 29 |
| Human biopsy | Ileum, colon | 2D Enteroid, 2D Colonoid | Toxin-induced cyclic nucleotide production | 47 | |
| Human biopsy | Rectum | 3D Enteroid | Toxin inhibitors to reduce pathology | 48 | |
| Human biopsy | Rectum | 3D Enteroid | Toxin inhibitors to reduce pathology | 49 | |
| Human biopsy | Jejunum | 2D Enteroid | Toxin inhibitors targeting a non-canonical binding site | 50 | |
| Extraintestinal pathogenic E. coli (ExPEC) | Human biopsy | Jejunum | 2D Enteroid | Pilus-mediated adherence & invasion | 51 |
| Bacteria causing necrotizing enterocolitis (NEC) | Human biopsy, fetal | Duodenum | 2D & 3D Enteroid | Immune response, maturation, & barrier function in fetal vs. adult intestine | 52 |
| Human biopsy, fetal | Ileum | 3D Enteroid | Anti-inflammatory metabolite as probiotic | 53 | |
| Human biopsy, fetal | Ileum | 3D Enteroid | Human milk oligosaccharides as probiotic | 54 | |
| Human biopsy, fetal | Not specified | 3D Enteroid | Cell stress and apoptosis of intestinal stem cells | 55 | |
| Human biopsy, fetal | Ileum | 3D Enteroid | Impaired signaling and its role in dysfunction of intestinal regeneration | 56 | |
Invasive enteric bacteria
Invasive pathogens penetrate the host epithelium directly through a variety of different mechanisms. These bacteria evade elimination by the host cell and use effector molecules for downstream pathogenesis. Some bacteria then spread from cell to cell while others gain access through the basolateral side of the epithelium. Invasion ultimately dysregulates intestinal cell processes and destroys the epithelium, leading to diarrheal disease.
Shigella
Shigella is an invasive, Gram-negative bacterial pathogen that causes acute diarrheal disease in humans. It is associated with significant morbidity and mortality, particularly in children below 5 years of age and immunocompromised individuals.2,57 Current standard of care treatment includes antibiotic administration; however, the emergence of multiple antibiotic-resistant strains of Shigella is narrowing therapeutic options. While several vaccine candidates are in different stages of development, a licensed vaccine is not currently available.
The molecular pathogenesis mechanisms of Shigella have been reviewed extensively.58–61 Shigella reaches the gastrointestinal tract via ingestion of contaminated food or water or by person-to-person contact. The bacterium is able to subvert the mucus layer and reach the epithelial surface.62 However, Shigella does not infect epithelial cells efficiently via the apical surface; it exploits the microfold cells, or M cells, to transcytose the intestinal epithelium.63–65 Virulent Shigella injects over 30 effector proteins into the host cell to aid its entry.66 The resident macrophages at the base of the M cells phagocytose incoming bacteria, but Shigella escapes the phagocytic vacuole to gain access to the cytoplasm. Shigella then induces pyroptosis to escape the macrophage and access the basolateral surface of the epithelium.67–69 Intracellular replication70,71 and cell-to-cell spread of Shigella72,73 trigger the secretion of pro-inflammatory cytokines, including IL-8, by the epithelium,74,75 which recruit neutrophils to the site of the infection. Neutrophils initially exacerbate the infection by causing severe inflammation but eventually help eliminate the bacteria.76–80
Two independent studies using human intestinal colonoids to study S. flexneri pathogenesis were published in tandem in 2019.21,22 Both studies used colonoids derived from healthy human colon and established that human colonoids are a viable model to study Shigella pathogenesis. Using 2D colonoid monolayers plated on permeable membrane scaffolds, it was reaffirmed that basolateral infection by S. flexneri is significantly more efficient than apical infection.21,22 Using differentiated colonoids containing mucus-producing goblet cells, Shigella infection was shown to cause increased production of the major intestinal mucin glycoprotein MUC2.22
M cells are thought to be one of the ports of entry for Shigella across the epithelium.64,81 Complex cell culture models have been developed to induce an M cell-like phenotype in tissue culture.82 Using TNFα and RANKL, ileal enteroids were differentiated to include M cells83,84 which facilitated increased apical infection by S. flexneri.22 However, the transcytosis of bacterial cargo via the M cell was not visualized, potentially due to the long infection time periods used in the study.
Koestler et al. observed donor-to-donor differences in infection and intracellular replication of S. flexneri.21 In contrast to the predominantly colon-restricted pathology observed in natural cases of shigellosis in humans, S. flexneri was able to infect human enteroids derived from all four segments of the intestine equally.21,22 Differences were observed in the intracellular replication of S. flexneri with the lowest doubling rate in colonoids compared to enteroids derived from small intestinal segments.22 It was hypothesized that the dominant colonic pathology observed in vivo may be attributed to other factors such as peristalsis, nutrient availability, mucus layer, and microbiome that are not yet sufficiently modeled in the 2D enteroid monolayer system.
Human colonoids derived from healthy adults were used to study metabolism and immune response to infection. Koestler et al. used qRT-PCR to show upregulation of IL-8, TNFα, IFNβ and TNFAIP3 in colonoids apically infected with S. flexneri.21 Shigella infection is known to induce an amino acid starvation response and promote mTOR-mediated xenophagy in host cells.85,86 However, transformed cell lines with altered amino acid transport mechanisms are not suitable models to study this effect. Using qRT-PCR, Koestler et al. showed the upregulation of amino acid starvation response gene SLC7A5 upon Shigella infection and emphasized the use of colonoids with unaltered physiology to study changes in host cell metabolism upon infection.21 Researchers are examining the potential use of human enteroids as a preclinical model to test therapeutics against human-restricted pathogens such as Shigella. Llanos-Chea et al. used human cecum-derived colonoids to assess the efficacy of a bacteriophage against Shigella serotypes infection.23 The administration of bacteriophage significantly reduced the ability of S. flexneri to infect cells. Chanin et al. showed that exposure of Shigella to glucose and bile salts upregulated the expression of multiple adherence structures. The authors used colonoids to show that these adherence structures on the bacteria were important for their initial attachment to the epithelial surface.24
Salmonella
Salmonella enterica subsp. enterica includes over 1400 serotypes with a wide range of pathogenic features and clinical outcomes. The most important clinical divisions are typhoidal and non-typhoidal. The four serovars Typhi, Paratyphi A, Paratyphi B and Paratyphi C constitute the group of typhoidal salmonellae that cause a serious systemic disease called typhoid or enteric fever in humans. Typhoidal Salmonellae are a major global health concern as they infect more than 27 million people worldwide and lead to 200,000 deaths annually.87 All remaining serotypes are grouped together as non-typhoidal salmonellae (NTS) which generally cause self-limited gastroenteritis, accounting for ~94 million cases and 155,000 deaths globally.88 Among the NTS, Salmonella enterica serovar Typhimurium is the most studied serotype.
Researchers studying Salmonella have primarily used tissue culture and small animal models to study host–pathogen interactions.89 S. Typhimurium has a broad host range including humans, livestock, small rodents, and birds; however, S. Typhimurium infection presents differently in each host. For instance, S. Typhimurium causes only localized gastroenteritis in humans but manifest as a systemic infection in mice. Typhoidal Salmonellae are particularly difficult to study as they are exclusively human-restricted pathogens. Humanized mice and chimpanzees have been used to study S. Typhi, but there is widespread recognition that a more amenable model to study Salmonella pathogenesis is vital for research.89
In order to develop an alternative model for studying Salmonella pathogenesis, Forbester et al. used HIOs to study the interaction of S. Typhimurium with the intestinal epithelium.26 In this study, HIOs were infected apically by microinjection with either S. Typhimurium SL1344 (wildtype strain) or a SL1344∆invA strain. Using a modified gentamicin protection assay, the authors showed that the invA mutant strain was ~30-fold less invasive than the wildtype counterpart, thus confirming previous studies that invA is critical for the bacterium’s ability to infect epithelial cells.90 The authors used a transcriptomic approach to acquire a global snapshot of the effect of S. Typhimurium infection in HIOs. RNA-seq of HIOs infected with S. Typhimurium for 3 hours showed differential regulation of several genes involved in cytokine signaling, extracellular matrix reorganization and innate immune responses.26 Gene expression studies showed that proinflammatory cytokines such as IL-23, IL-1β, IL-8, CXCL2 and TNFα were upregulated upon infection with S. Typhimurium. Cytokine measurements confirmed increases in IL-6, IL-8 and TNFα in the supernatant of infected HIOs. Using transmission electron microscopy (TEM), the authors showed that S. Typhimurium resided in vacuoles within the epithelial cells, confirming a hallmark of Salmonella infection.
In a follow-up study, Forbester et al. investigated the role of IL-22 in priming the antimicrobial defense of the human intestine to counteract S. Typhimurium infection.27 Organoids were generated using stem cells from a patient with infantile inflammatory bowel disease, harboring a loss-of-function mutation in the IL-10Rβ gene that encodes IL-10R2. As IL-10R2 is a component of the IL-22 receptor, patients with this mutation are non-responsive to a number of cytokines including IL-22. The authors showed that HIOs generated from healthy donor iPSCs expressed IL-22 receptors on their basolateral surface and responded to exogenous IL-22 treatment, whereas HIOs derived from the patient with a loss-of-function mutation in IL-10Rβ were non-responsive to IL-22.27 Healthy HIOs responded to IL-22 by upregulating the expression of genes involved in antimicrobial response such as REG3A and REG1B, mucin genes MUC1 and MUC4, IFN-regulated genes IFITM1, IFITM2 and IFITM3, and genes involved in maturation of epithelial oxidase complex DUOX2, DUOXA1 and DUOXA2. When healthy HIOs were infected with S. Typhimurium concomitant with IL-22 treatment, genes involved in host defense mechanisms such as DUOX2, LCN2 and CXCL2 were further upregulated. Such IL-22-dependent antimicrobial responses were not observed in the HIOs derived from the IL10Rβ-mutated iPSC donor. IL-22 induced a protective phenotype in HIOs as IL-22 treated HIOs were more resistant to initial invasion by S. Typhimurium compared to the IL-22 non-responsive HIOs. The long-term survival of S. Typhimurium was also reduced in IL-22 pretreated HIOs due to calgranulin A-mediated increase in phagosome-lysosome fusion, a process that is strategically evaded by S. Typhimurium.91 This study shows the advantage of using HIOs to illustrate the effects of a cytokine receptor mutation on infection outcomes.
3D enteroids and HIOs require microinjection to introduce pathogenic material to the apical surface of epithelial cells, a tedious process that requires precise technique. Based on previous work which showed that the epithelial polarity of 3D MDCK spheroids is driven by their exposure to extracellular matrix components (ECM),92 Co et al. generated ‘apical out’ enteroids by growing 3D enteroid spheres in suspension instead of embedding them in ECM.25 These ‘apical out’ enteroids polarized with villin localizing on the outer edge of the spheres marking the apical surface and β-catenin along cell junctions and the inner edge marking the basolateral surface. Other than the lack of a lumen and fewer goblet cells upon differentiation, the ‘apical out’ enteroids were comparable to ECM-embedded 3D enteroids. This technical advancement of ‘apical-out’ enteroids allows for easy access to the apical surface of enteroids and was used to show the preferential apical infection by S. Typhimurium and basolateral infection by L. monocytogenes. Some hallmarks of S. Typhimurium infection, such as actin ruffling and infected cell extrusion, were also modeled in this system. The authors speculated that sites of cell extrusion could serve as a vulnerable site for pathogens to infect. Human enteroids serve as a valuable model to answer these early pathogenesis mechanisms.
Nickerson et al. primarily used an ex vivo human intestinal biopsy model to understand the early events of S. Typhi pathogenesis and confirmed key results using enteroids derived from human ileum.28 This study confirmed that upon S. Typhi infection, there was a change in cytoskeleton and microtubule dynamics of the host epithelium. The authors used inhibitors of actin polymerization and microtubule reorganization to show that invasion by S. Typhi decreased when cytoskeletal remodeling was inhibited.
Noninvasive enteric bacteria
In contrast to invasive pathogens, noninvasive pathogens do not directly penetrate the host epithelium but instead attach to host intestinal epithelium to inject toxins and/or effector molecules. Intoxication of epithelial cells disrupts homeostatic signaling for a variety of cellular processes, resulting in diarrhea.
Enterotoxigenic E. coli (ETEC):
Enterotoxigenic Escherichia coli (ETEC) is a leading cause of diarrheal disease in young children, particularly in less industrialized regions,2 and is a major causative agent of traveler’s diarrhea. ETEC adheres to the intestinal epithelium and secretes heat-stable enterotoxin (ST) and/or heat-labile enterotoxin (LT), eliciting cGMP or cAMP-mediated signaling, respectively, to dysregulate ion and water transport across the epithelium.93 ST is structurally homologous to the paracrine hormones guanylin and uroguanylin and activates the intestinal guanylate cyclase C (GC-C) receptor.94 Upon binding of ST, the cytoplasmic catalytic domain of GC-C converts GMP to cGMP, which leads to the phosphorylation and activation of the cystic fibrosis transmembrane conductance regulator (CFTR), an apical channel permeable to chloride and bicarbonate.94 NHE3, the brush border Na+/H+ exchanger important for intestinal absorption of sodium and water, is inhibited by ST exposure.29 Upon binding of LT to the receptor GM1, LT is endocytosed and activates adenylyl cyclase, leading to cAMP-induced activation of the CFTR and inhibition of NHE3.95
Foulke-Abel et al. determined that exposure to ETEC enterotoxins induced jejunal enteroid monolayers to secrete cyclic nucleotides in a polarized manner.30 LT exposure induced significant apical secretion of cAMP, while addition of ST significantly increased basolateral secretion of cGMP. Interestingly, intracellular cGMP accumulation was markedly restricted by phosphodiesterase PDE5-mediated degradation in enteroids and colonoids, whereas Caco-2 and T84 polarized monolayers incubated with ST yielded high amounts of both intracellular and extracellular cGMP. Toxin delivery by ETEC is known to be enhanced by several virulence factors,96 and this was reinforced in the observed dependence on expression of adhesins CFA/I and EtpA and the mucinase EatA by ETEC H10407 to elicit ST-induced cGMP production in enteroids.
ST was also used to probe the contribution of fluid flow and mechanical stretch to functional responses using microfluidic Intestine Chips, an innovative tool in the advancement of the enteroid model.31 The chip establishes an enteroid monolayer within a flexible polymer chamber containing channels that enable luminal surface fluid flow and application of culture membrane deformation to mimic peristalsis-like forces. Under continuous flow conditions, exposure of jejunal enteroid monolayers to ST increased the secreted but not intracellular cGMP concentrations and increased expression of cyclic nucleotide transporter MRP4, compared to monolayers under static conditions. Repetitive stretch did not have an additive effect on cGMP levels. When considering the aforementioned studies,30,31 the choice of experimental model, such as enteroids vs. immortalize cell lines or application of static vs. shear fluid forces, can give different results, and it is not yet clear which model is to be considered most reflective of native intestinal physiology or pathophysiology of ETEC infection.
As an example of using enteroids to interrogate mechanisms related to disease epidemiology, Kumar and Kuhlmann et al. demonstrated that the ETEC adhesin EtpA targets polysaccharides with the blood group A surface antigen.32 The study was driven by a meta-analysis of volunteer challenge studies that suggested ETEC caused more severe disease in individuals bearing the A type antigen. Blood group A donor jejunal and ileal enteroid monolayers were colonized more extensively by ETEC H10407 than monolayers derived from blood group B or O donors. H10407 lacking EtpA expression induced significantly decreased cyclic nucleotide levels in type A enteroids relative to wild type H10407, suggesting that enhanced colonization and toxin delivery supported by EtpA correlates with disease severity.
ETEC infection of epithelium alongside innate immune cell effectors was studied using a co-culture of enteroid or colonoid monolayers and human monocyte-derived macrophages.33 The enteroid monolayer and basolaterally engrafted macrophages affected each other as evidenced by the morphological changes in both cell types. Increased basal production of innate cytokines, including IL-8, IFN-γ, and IL-6, increased cell height, and transepithelial resistance (TER) across the monolayer was also observed.33 Following ETEC infection, cytokine production did not significantly increase; however, macrophages extended dendritic-like projections through the permeable culture membrane and paracellular space of the monolayer to contact adherent ETEC on the apical surface. Fewer ETEC bacteria were found adhered to the enteroids co-cultured with macrophages, as early as 30 min post infection, compared to enteroid-only cultures. The authors hypothesized that this decrease may be due to phagocytosis of ETEC by the macrophages. This co-culture system is the first primary human macrophage-enteroid model established to study host–pathogen interactions. Future studies aim to incorporate other immune cell types, including neutrophils, dendritic cells, and T cells, to study mechanisms of infection clearance by the immune system.
Enterohemorrhagic E. coli (EHEC):
Enterohemorrhagic Escherichia coli (EHEC) causes foodborne illness worldwide, with the serotype O157:H7 associated with most E. coli food poisoning outbreaks in the U.S.97 EHEC preferentially colonizes the colon, injects virulence factors through a type 3 secretion system (T3SS) to form attaching/effacing (A/E) lesions, and remodels the actin cytoskeleton into pedestal-like structures.98 Shiga toxin is released into the lumen, where it binds the epithelial receptor Gb3/CD77, induces cell death through inhibition of protein synthesis, and is disseminated to other tissues.99 Infection is characterized by abdominal cramping and bloody diarrhea; however, severe disease can affect multiple organ systems, including the circulatory and renal systems, resulting in hemolytic uremic syndrome (HUS).100 HUS is characterized by clot formation, destruction of red blood cells, platelet depletion, and acute kidney failure.
The enteroid model has confirmed and extended understanding of many classical features of EHEC pathogenesis. In et al. used colonoid monolayers to demonstrate that EHEC preferentially adhere to differentiated monolayers that produce mucus and subsequently degrade the mucus for attachment.34,101 Microscopy revealed that EHEC form the characteristic A/E lesions and actin pedestals via membrane remodeling. EHEC-induced damage of intestinal cells was not limited to cells in direct contact with EHEC A/E lesions. Another hallmark of EHEC infection is the loss of microvilli; however, the molecular mechanism is poorly understood. In et al. showed that the serine protease autotransporter of Enterobacteriaceae (SPATE) EspP, produced by EHEC, specifically targeted protocadherin 24 (PCDH24), a major building block of intermicrovillar bridges, resulting in brush border effacement and remodeling. EHEC infection also induced the redistribution of tight junction protein occludin in these colonoid monolayers. While EspP has been shown to drive initial contact and membrane remodeling during EHEC infection, Tse et al. described a new role for EspP as an enterotoxin that alters electrolyte transport, using colonoid monolayers.35 EspP stimulated short circuit current in a dose-dependent and serine protease-independent mechanism in monolayers. Many bacterial enterotoxin mechanisms involve the activation of the CFTR channel.102 However, EspP-stimulated current was shown to be independent of CFTR in colonic monolayers.35 These studies suggest that EspP may be an important therapeutic target.
Karve et al. used 3D human organoids (HIOs) to study the pathogenic effects of Shiga toxin-producing E. coli O157:H7 in direct contrast to E. coli commensals to understand intestinal tolerance to commensals.36 Luminal infection with E. coli O157:H7 via microinjection resulted in increased bacterial replication and damage to the HIOs, characterized by extensive actin rearrangement and mucus depletion. After the induction of the bacterial SOS stress response, E. coli O157:H7 infection specifically induced reactive oxygen species (ROS) followed by secretion of Shiga toxin into the lumen of the HIOs. Commensal bacteria ECOR13 also replicated comparably but remained contained within the lumen and did not induce ROS production or cause cellular damage, supporting the role of the mucus layer as an innate defense barrier.
Elevated neutrophil counts are associated with the development of HUS and mortality due to EHEC infection.103 Karve et al. designed a co-culture system with the HIOs and human polymorphonuclear cells (PMNs) to compare outcomes of E. coli O157:H7 or commensal infection.36 Introducing E. coli O157:H7 to the media resulted in barrier function loss, PMN recruitment to the basolateral side of HIOs, and low numbers of neutrophils that transcytosed into the HIO lumen, whereas introduction of a commensal did not cause barrier function loss or recruitment of PMNs. Global transcriptional analysis using RNA-seq demonstrated that HIO infection with E. coli O157:H7 or the commensal strain upregulated over 70 genes, including those involved in growth factor receptor signaling and iron responses. However, infection with E. coli O157:H7 exclusively upregulated inflammatory responses, including IL-8 and IL-18, both key to neutrophil recruitment. This co-culture HIO model validates the recruitment of neutrophils in EHEC infection and provides a system to investigate this host–pathogen interaction at a more mechanistic level.
The early effects of Shiga toxin on human intestine are not well described. Using HIOs exposed via luminal microinjection or basolateral addition to cell culture media, Pradhan et al. demonstrated that purified Shiga toxin, Stx2a, disrupts epithelial barrier function after 48 hours, resulting in cell death via both necrosis and apoptosis.37 Transcytosis of Stx2a across the epithelium occurred while the epithelial barrier was still intact within 24 hours. Pradhan et al. also explored the effect of Stx2a on mesenchymal–epithelial transition in the HIOs, an established response in wound healing. Following Stx2a exposure, vimentin-positive mesenchymal cells in the HIOs expressed E-cadherin, an adhesion molecule normally restricted to epithelial cells, suggesting transition to an epithelial phenotype. Interestingly, this transition was also observed in response to other bacterial toxins, including C. difficile toxins TcdA and TcdB, but not in response to LPS, suggesting that this event is stimulated by general cellular damage, not by specific toxins. This is the first human cell-based study that demonstrates the role of mesenchymal cells in maintaining the integrity of the epithelial barrier upon Shiga toxin exposure.
Enteroaggregative E. coli (EAEC)
Enteroaggregative Escherichia coli (EAEC) is an enteric pathogen that can cause both acute and persistent diarrhea, growth faltering, and death, particularly in children in developing countries.104 EAEC, named after the aggregative “stacked-brick” adherence phenotype, primarily adheres to the intestine using aggregative adherence fimbriae (AAFs), although atypical strains also exist.105 Infection with EAEC may manifest as a diverse array of symptoms and duration, as strains can express a varied combination of enterotoxins and virulence factors.
Rajan et al. used enteroid and colonoid monolayers derived from three donors to understand donor-dependent effects on EAEC adherence phenotypes.38 The authors observed that EAEC adherence to monolayers was independent of AafA fimbriae, whereas the aggregative phenotype was dependent on AafA, suggesting distinct mechanisms for the two behaviors. EAEC adhered to enteroid monolayers derived from duodenum and ileum in an aggregative pattern with donor-dependent differences in the phenotype. In contrast, EAEC adhered to colonoid monolayers in a mesh-like pattern and exhibited minimal adherence to jejunal monolayers. This study distinguished the aggregative phenotype of EAEC as having multiple morphologies and emphasized the contribution of the host to the disease.
EAEC SPATEs such as SepA and Pic are established effectors of colonization.106–108 Using colonoid monolayers, Tse et al. found that SepA also stimulated CFTR-dependent chloride secretion.35 This is in contrast to the EHEC SPATE, EspP, which stimulated CFTR-independent chloride secretion, or the EAEC SPATE, Pic, that did not affect ion transport.35 This study provided the first evidence of the contribution of SepA to EAEC-induced diarrhea.
Enteropathogenic Escherichia coli (EPEC)
Enteropathogenic Escherichia coli (EPEC) causes diarrheal disease in humans, especially in young children. Similar to EHEC, initial attachment by EPEC is mediated by induction of the attaching and effacing (A/E) lesions that are mediated by the T3SS. Both pathotypes also affect intestinal ion transporters using various effector proteins.109 Previous experiments in rat jejunum suggested that EPEC extracellular serine protease C (EspC) acts as a highly potent enterotoxin and induces chloride secretion.110 Noel et al. studied interactions of EPEC with the host using a human enteroid monolayer-macrophage co-culture model.33 Following EPEC adherence to the enteroid monolayer, human monocyte-derived macrophages displayed phagocytic phenotypes including actin projections to interact with bacteria. EPEC infection resulted in significantly more adherent macrophages and projections across the monolayer. This is the first study of host–pathogen interaction with EPEC in a highly human-relevant model.
Other Escherichia coli
Extraintestinal pathogenic Escherichia coli (ExPEC) include E. coli strains that have translocated from the intestine to distal locations, including the renal system, nervous system, and vascular system to induce urinary infections, neonatal meningitis, and septicemia, respectively.111 Many studies have investigated virulence factors involved in these distal infections; however, few studies have investigated the initial adherence and subsequent translocation of ExPEC from the intestine. Jejunal enteroid and Caco-2 monolayers were used to investigate the role of the adhesin FimH, a mannose-binding type 1 pilus tip protein in ExPEC.51 Poole et al. determined that wildtype and FimH-deficient ExPEC strains adhered to the monolayers at similar levels. However, subsequent invasion of the monolayers was partially dependent on FimH. These findings contrasted experiments in Caco-2 monolayers in which both adherence and invasion were FimH dependent. This study suggested that targeting FimH could attenuate ExPEC infection.
Untreated necrotizing enterocolitis (NEC) contributes to massive intestinal inflammation and necrosis with a high mortality rate in preterm infants.112 Previous studies suggested that early bacterial colonization of immature and underdeveloped fetal intestine contributes to inflammation. Senger et al. modeled NEC in infants using fetal enteroids from early (11 weeks) and late (22.5 weeks) gestational stages and compared the results with adult duodenal enteroids.52 Following addition of LPS or commensal E. coli HS to properly developed early fetal, late fetal, or adult enteroids, RNA-seq revealed that gene expression in early fetal enteroids lacked key inflammatory cytokines, including TNFα, IL-8, and IFN-γ, as well as NFkB expression. Late fetal enteroid cytokine levels were similar to adult enteroids. These data confirmed the immature immune response in infants and the subsequent negative impact upon bacterial colonization. A subsequent publication demonstrated that the molecule indole-3-lactic acid (ILA), a metabolite secreted by Bifidobacterium longum subspecies infantis, a bacterial species commonly found in breastmilk, had anti-inflammatory effects on IL-1β-treated fetal enteroids from early and late gestational stages.53 Treatment with ILA may serve as a probiotic for premature infants at risk for NEC. In accordance with the discovery of probiotics to combat NEC in premature infants, Wu et al. demonstrated that exposure to human milk oligosaccharides, also isolated from human breastmilk, increased intestinal crypt budding and the production of the mucin MUC2 in 3D human fetal ileal enteroids.54
NEC is characterized by the loss of intestinal stem cells by apoptosis. Previous studies demonstrated that the presence of NEC in premature human fetal enteroids was associated with increased ER stress and increased apoptosis within the intestinal crypts, compared to healthy premature and full-term fetal enteroids.55 The renewal of these stem cells, controlled primarily by the Wnt/β-catenin pathway, is required for gut regeneration and maintenance in response to this acute injury. Using 3D human fetal ileal enteroids isolated from infants with active NEC, Li et al. confirmed the previous observation of the loss of intestinal stem cells and the decreased proliferation, as compared to healthy premature fetal enteroids. Additionally, these fetal enteroids with NEC had impaired endogenous Wnt signaling at both the RNA and protein levels.56 The loss of stem cells and lack of regeneration in fetal enteroids with NEC was directly affected by Wnt activity as administration of Wnt rescued these phenotypes.56 As there is limited access to human fetal tissues and appropriate NEC models, enteroids provide a tool to investigate aspects that allow NEC to manifest in premature infants and to develop therapeutics.
Clostridium difficile
Clostridium difficile is an anaerobic, spore-forming enteric bacteria that is responsible for a high burden of antibiotic-induced diarrhea and colitis in humans.113 Much of C. difficile pathogenesis has been examined using the primary toxins TcdA and TcdB, which modify cellular Rho GTPases and stimulate several negative downstream cellular effects. Hypervirulent strains also secrete C. difficile transferase toxin (CDT) that leads to actin – and microtubule-mediated deformations in the cell surface, increasing adherence and colonization.114
Leslie et al. performed the first experiments to demonstrate that viable obligate anaerobic C. difficile can persist in the lumen of HIOs for up to 12 hours and cause loss of barrier function in a toxin-dependent manner.39 Both TcdA and TcdB induced increased epithelial permeability over time, disrupting the cytoskeleton and cellular junctions. TcdA exerted additional effects on redistribution of E-cadherin, zonula occludens 1 (ZO-1), and actin, as compared to TcdB-injected HIOs. These findings confirmed previously identified toxin mechanisms.
Several cell surface proteins are proposed receptors for TcdB, including chondroitin-sulfate proteoglycan 4 (CSPG4), poliovirus receptor-like 3 (PVRL3), and frizzled-1/2/7 (FZD1/2/7).115 Schottelndreier et al. detected mRNA for each of these receptors in 3D colonoids from four human donors and in transformed intestinal cell lines, including Caco-2 and HT-29 cells, each with varying levels of expression.40 Engevik et al. examined cell rounding in jejunal enteroid monolayers exposed to the toxins, demonstrating TcdA to be 10-fold more effective than TcdB at inducing cytoskeletal rearrangement.41 Compared to immortalized cell lines, enteroids were found to express higher levels of the toxin receptors yet demonstrated less sensitivity to the toxins; this was attributed to the protective extracellular mucus layer in enteroids that is absent in other models. Mileto et al. demonstrated that TcdB induced pathogenic effects, particularly stem cell death, through both FZD7-dependent and – independent mechanisms in 3D colonoids, supporting evidence that receptors in addition to FZD7 are essential for toxin-induced pathology.42
The interaction of C. difficile with intestinal mucus and subsequent colonization in humans has not been thoroughly investigated. Engevik et al. reported that patients with C. difficile infection have disrupted intestinal mucus, and biopsies from infected patients exhibited almost complete loss of the loose outer MUC2 layer as assessed by confocal microscopy, exposing the firmly attached inner MUC1 layer.43 Loss of the MUC2 layer was recapitulated in HIOs injected with C. difficile alone or infected patient stool and revealed that C. difficile interacted with remaining MUC1. Engevik et al. also investigated components within the mucus layer during C. difficile infection. Stool from infected patients had an altered microbiota with increased levels of the phyla Bacteroidetes and Proteobacteria as well as differential expression of oligosaccharides, including GlcNAc, GalNAC, and galactose, compared to healthy donor stool. As C. difficile colonizes the mucus layer, it is hypothesized that the bacteria bind to these specific mucus oligosaccharides and proliferate; however, the levels of these oligosaccharides in mucus during C. difficile infection of HIOs were not significantly different than those in the uninfected HIOs. The complex mucus layers in the HIO model facilitated increased understanding for the role of mucus in C. difficile infection.
Other effects associated with C. difficile infection include toxin-induced dysregulation of ion channel function. Early in vitro studies found that C. difficile infection resulted in the internalization of the NHE3,116 and loss of NHE3 in animal models is known to yield chronic diarrhea, disrupted microbiota, and dysregulated luminal electrolytes. Engevik et al. demonstrated that biopsies from patients with C. difficile infection exhibited altered intestinal structure, decreased brush border NHE3, elevated fecal sodium, and differential microbiome composition.44 HIOs injected with viable C. difficile or infected stool supernatant had significantly decreased NHE3 mRNA and protein expression.
Many therapeutic strategies against C. difficile infection target TcdA and TcdB, and enteroids have provided a platform to assess preclinical efficacy. As hypoalbuminemic patients experience increased morbidity from C. difficile infection, di Masi et al. hypothesized that human serum albumin (HSA) could have a neutralizing effect on clostridial toxins.117 TcdA and TcdB induced epithelial damage, particularly in the crypt-like regions of HIOs; however, HIOs incubated with toxins and HSA had reduced pathology. While the mechanism is not completely understood, the authors speculate that HSA might bind to the toxins in the bloodstream and induce conformational changes, leading to toxin autoproteolysis. In another study, Fischer et al. investigated potential protection conferred by human alpha-defensin-1 against C. difficile toxins TcdA, TcdB, and CDT, finding reduced toxicity, less cytoskeletal disruption, and minimal E-cadherin mislocalization in HIOs exposed to a toxin cocktail.45 In vitro studies showed that human alpha-defensin-1 induced TcdA aggregation, but further work is required to characterize potential inhibition of downstream toxin-induced signaling.
In addition to antibiotic properties, bacitracin was previously demonstrated to protect cultured human cells from intoxication by anthrax lethal toxin and CDT.118 Zhu et al. demonstrated that bacitracin protected Caco-2 and HIOs from TcdB activity, reducing downstream glucosylation of the target protein Rac1 and preventing depolymerization of F-actin in HIOs.46 These experiments suggest that bacitracin, an approved drug for clinical use, has potential application to a combination therapy against C. difficile infection.
Vibrio cholerae
Vibrio cholerae is responsible for a high burden of diarrheal morbidity and mortality globally.119 The primary virulence factor cholera toxin is an AB5 structured protein consisting of an enzymatically active A subunit that triggers ADP-ribosylation and immunogenic B subunits that mediate host cell binding.119 Surface receptor GM1 is a canonical binding site for the B subunit, yielding toxin internalization and cyclic nucleotide-dependent fluid hypersecretion via ion channels and transporters, such as CFTR and NHE3.102 Cholera toxin exposure inhibited NHE3 activity in 3D duodenal and jejunal enteroids, confirming a functional response in the human enteroid model.29 Cholera toxin exposure also resulted in the production of the cyclic nucleotide cAMP in 2D ileal enteroids and colonoids.47 Interestingly, enteroids and colonoids isolated from blood group O individuals exhibited consistently higher levels of cAMP upon cholera toxin exposure, as compared to blood group A individuals, suggesting a mechanism behind the association of severe cholera disease in blood group O individuals.47
In addition to the two approved vaccines for cholera, toxin inhibitors have been engineered as a therapeutic strategy. Many inhibitors are multivalent and target GM1 to prevent toxin binding.120 Previously, the potency of these inhibitors was evaluated by classical tests and model systems in the field, including GM1-ELISA or rabbit ileal loop assays.119 Zomer-van Ommen et al. adopted the 3D enteroid swelling assay as an alternative method to test inhibitor potency.48 Compared to maximal toxin-induced swelling in 3D rectal enteroids, GM1 inhibitors limited swelling by varying degrees. Strikingly, the tetravalent and pentavalent GM1 inhibitors were most potent against toxin-induced swelling. Haksar et al. also adopted this swelling assay using 3D rectal enteroids.49 The ligand meta-nitrophenyl α-galactoside (MNPG), previously shown to be a promising ligand for cholera toxin, and synthesized ligand derivatives were each conjugated to three multivalent polymers, each chosen for their potency and low cost. Some of these compounds exhibited increased inhibition of enteroid swelling upon dose-dependent cholera toxin exposure, emphasizing the potential for effective and economically practical prophylactics against cholera.49 An alternative strategy is to target a noncanonical site on cholera toxin with affinity for fucosylated moieties such as histo-blood group antigens. Cervin et al. designed polymers to target the fucosylated non-canonical binding site, finding they blocked the binding of cholera toxin to 2D jejunal enteroid monolayers; however, polymers designed to target both the canonical and non-canonical binding sites of cholera toxin were observed to block binding, intoxication, and ion secretion.50 Using human enteroids as a tool for inhibitor potency assays surpasses animal models in the ability to model human-specific cell surface glycosylation patterns.
Enteroid and organoid models have validated many previously identified mechanisms for noninvasive bacteria and provided insight on unrecognized pathogenic mechanisms.
Conclusions and future perspectives
The bacterial pathogenesis studies discussed in this review, in addition to efforts using viral pathogens and parasites, demonstrate the functionality of enteroid and organoid models for infectious disease research. Many infection responses that were previously identified in cell lines or animals have been confirmed in enteroids/organoids, but some direct comparisons have revealed new model-specific phenotypes. This has been illustrated in the enhanced receptor expression yet diminished response to C. difficile toxins in enteroids compared to T84 and HT-29 cells,41 and the level and polarized secretion pattern of cGMP upon ETEC infection in enteroids compared to T84 and Caco-2 cells.30 The utility of enteroids/organoids is further justified in the novel mechanistic findings such as those related to intestinal segment tropism (Shigella, EAEC), histo-blood group antigens (ETEC), cell surface glycosylations (cholera toxin), a complex mucus layer (Shigella, EAEC, EHEC, C. difficile), and the ability to compare fetal and adult human tissue states (NEC). Each of these features is not directly mimicked in animal or immortalized cell line experiments.
While enteroids impart sophistication to epithelial tissue culture models, additional features of the GI tract such as oxygen gradients, physical forces, a diverse microbiome, vascularization, the enteric nervous system, and immune components are not yet fully represented in the model. The challenges in constructing more complex tissue culture models lay in the development of culture platforms to provide individual material support to multiple cell populations yet allow cells the freedom to interact seamlessly. Platforms should also provide access for on-demand sampling of secreted materials. The microfluidic Intestine-Chip seeded with enteroids from the small intestine121-124 or colon125 offers integrated manipulation of mechanical stretch, anaerobic compartmentalization, and endothelial interfacing. Silk scaffolding provides a highly permeable support network for enteroid monolayer communications with lamina propria.126 Other cell support materials such as those based on synthetic127 or extracellular matrix-based polymer networks128 remain to be implemented for enteroid co-cultures.
The engraftment of non-epithelial cell types will expand the mechanistic understanding of infection and enhance their utility for therapeutic or vaccine screening. Immune cell populations are crucial to modeling innate and adaptive responses to pathogenic antigens and provide antimicrobials to combat infection. The enteric nervous system is an understudied contributor to infectious diarrheal diseases, releasing neurotransmitters that impact epithelial ion transport and may affect other cell populations. The causal or protective role of the gut microbiome in infectious disease severity remains unclear, and interactions between competing microbes in addition to the effects of microbial metabolites on host barrier and immune function require further evaluation.
As co-culture models evolve, they present the opportunity for screening new antimicrobial compounds and vaccine candidates for toxicity and efficacy before transitioning to human clinical trials. Also, as enteroid models maintain the genetic background and segmental specificity from the original human donor, this system aligns with the advancement of personalized medicine. Patient-derived enteroids can be screened following specific therapy for early indications of treatment success.
Previous and current studies modeling bacterial pathogenesis in the human enteroid and organoid models have proven to be invaluable in the understanding the pathogenesis of both invasive and noninvasive bacteria. For some bacteria, this model has been used to study important research questions in the field; however, there are other bacteria for which this model is in early phase exploration or remains untested. These studies have provided a strong foundation to confirm pathogenic mechanisms as well as reveal novel findings and should be considered as the most relevant option to understand interactions of bacteria with the human intestine. The use of the enteroid and organoid models is expected to continue transforming the study of bacterial enteropathogens and accelerate development of critical therapeutic and preventative interventions.
Funding Statement
National Institute of Allergy and Infectious Diseases [U19 AI142725]; National Institute of Allergy and Infectious Diseases [P01 AI125181]; National Institute of Allergy and Infectious Diseases [U19 AI109776]; National Institute of Diabetes and Digestive and Kidney Diseases [K01 DK113043]; National Institute of Diabetes and Digestive and Kidney Diseases [P30 DK089502].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- 1.Troeger C, Blacker BF, Khalil IA, Rao PC, Cao S, Zimsen SRM, Albertson SB, Stanaway JD, Deshpande A, Abebe Z, et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: a systematic analysis for the global burden of disease study 2016. Lancet Infect Dis. 2018;18(11):1211–20. doi: 10.1016/S1473-3099(18)30362-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet. 2013;382(9888):209–222. doi: 10.1016/S0140-6736(13)60844-2. [DOI] [PubMed] [Google Scholar]
- 3.Liu J, Platts-Mills JA, Juma J, Kabir F, Nkeze J, Okoi C, Operario DJ, Uddin J, Ahmed S, Alonso PL, et al. Use of quantitative molecular diagnostic methods to identify causes of diarrhoea in children: a reanalysis of the GEMS case-control study. Lancet. 2016;388(10051):1291–1301. doi: 10.1016/S0140-6736(16)31529-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang W, Luo Y, Li J, Lin L, Ma Y, Hu C, Jin S, Ran L, Cui S.. Wide dissemination of multidrug-resistant Shigella isolates in China. J Antimicrob Chemother. 2011;66(11):2527–2535. doi: 10.1093/jac/dkr341. [DOI] [PubMed] [Google Scholar]
- 5.Hazen TH, Michalski J, Nagaraj S, Okeke IN, Rasko DA. Characterization of a large antibiotic resistance plasmid found in enteropathogenic Escherichia coli strain B171 and its relatedness to plasmids of diverse E. coli and Shigella strains. Antimicrob Agents Chemother. 2017;61. doi: 10.1128/AAC.00995-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Le Ferrec E, Chesne C, Artusson P, Brayden D, Fabre G, Gires P, Guillou F, Rousset M, Rubas W, Scarino ML. In vitro models of the intestinal barrier. The report and recommendations of ECVAM workshop 46. European centre for the validation of alternative methods. Altern Lab Anim. 2001;29:649–668. doi: 10.1177/026119290102900604. [DOI] [PubMed] [Google Scholar]
- 7.Hackam DG, Redelmeier DA. Translation of research evidence from animals to humans. JAMA. 2006;296:1731–1732. doi: 10.1001/jama.296.14.1731. [DOI] [PubMed] [Google Scholar]
- 8.Martic-Kehl MI, Schibli R, Schubiger PA. Can animal data predict human outcome? Problems and pitfalls of translational animal research. Eur J Nucl Med Mol Imaging. 2012;39:1492–1496. doi: 10.1007/s00259-012-2175-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sato T, Stange DE, Ferrante M, Vries RG, Van Es JH, Van den Brink S, van Houdt WJ, Pronk A, van Gorp J, Siersema PD, et al. Long-term expansion of Epithelial organoids from human Colon, Adenoma, Adenocarcinoma, and Barrett’s Epithelium. Gastroenterology. 2011;141(5):1762–1772. doi: 10.1053/j.gastro.2011.07.050. [DOI] [PubMed] [Google Scholar]
- 10.Spence JR, Mayhew CN, Rankin SA, Kuhar MF, Vallance JE, Tolle K, Hoskins EE, Kalinichenko VV, Wells SI, Zorn AM, et al. Directed differentiation of human pluripotent stem cells into intestinal tissue in vitro. Nature. 2011;470(7332):105–109. doi: 10.1038/nature09691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sato T, Clevers H. Growing self-organizing mini-guts from a single intestinal stem cell: mechanism and applications. Science. 2013;340(6137):1190–1194. doi: 10.1126/science.1234852. [DOI] [PubMed] [Google Scholar]
- 12.In JG, Foulke-Abel J, Estes MK, Zachos NC, Kovbasnjuk O, Donowitz M. Human mini-guts: new insights into intestinal physiology and host-pathogen interactions. Nat Rev Gastroenterol Hepatol. 2016;13:633–642. doi: 10.1038/nrgastro.2016.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zachos NC, Kovbasnjuk O, Foulke-Abel J, In J, Blutt SE, de Jonge HR, Estes MK, Donowitz M. Human Enteroids/Colonoids and intestinal organoids functionally recapitulate normal Intestinal physiology and pathophysiology. J Biol Chem. 2016;291:3759–3766. doi: 10.1074/jbc.R114.635995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stelzner M, Helmrath M, Dunn JC, Henning SJ, Houchen CW, Kuo C, Lynch J, Li L, Magness ST, Martin MG, et al. A nomenclature for intestinal in vitro cultures. Am J Physiol Gastrointest Liver Physiol. 2012;302(12):G1359–63. doi: 10.1152/ajpgi.00493.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dedhia PH, Bertaux-Skeirik N, Zavros Y, Spence JR. Organoid models of human gastrointestinal development and disease. Gastroenterol. 2016;150:1098–1112. doi: 10.1053/j.gastro.2015.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kechele DO, Wells JM. Recent advances in deriving human endodermal tissues from pluripotent stem cells. Curr Opin Cell Biol. 2019;61:92–100. doi: 10.1016/j.ceb.2019.07.009. [DOI] [PubMed] [Google Scholar]
- 17.Saxena K, Blutt SE, Ettayebi K, Zeng XL, Broughman JR, Crawford SE, Karandikar UC, Sastri NP, Conner ME, Opekun AR, et al. Human Intestinal Enteroids: a new model to study human rotavirus infection, host restriction, and pathophysiology. J Virol. 2016;90:43–56. doi: 10.1128/JVI.01930-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zou WY, Blutt SE, Crawford SE, Ettayebi K, Zeng XL, Saxena K, Ramani S, Karandikar UC, Zachos NC, Estes MK. Human intestinal enteroids: new models to study gastrointestinal virus infections. Methods Mol Biol (Clifton, NJ). 2019;1576:229–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Finkbeiner SR, Zeng XL, Utama B, Atmar RL, Shroyer NF, Estes MK. Stem cell-derived human intestinal organoids as an infection model for rotaviruses. mBio. 2012;3:e00159–12. doi: 10.1128/mBio.00159-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Heo I, Dutta D, Schaefer DA, Iakobachvili N, Artegiani B, Sachs N, Boonekamp KE, Bowden G, Hendrickx APA, Willems RJL, et al. Modelling Cryptosporidium infection in human small intestinal and lung organoids. Nat Microbiol. 2018;3(7):814–823. doi: 10.1038/s41564-018-0177-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koestler BJ, Ward CM, Fisher CR, Rajan A, Maresso AW, Payne SM, Young VB. Human Intestinal Enteroids as a model system of Shigella Pathogenesis. Infect Immun. 2019;87(4). doi: 10.1128/IAI.00733-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ranganathan S, Doucet M, Grassel CL, Delaine-Elias B, Zachos NC, Barry EM, Young VB. Evaluating Shigella flexneri Pathogenesis in the human Enteroid model. Infect Immun. 2019;87(4). doi: 10.1128/IAI.00740-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Llanos-Chea A, Citorik RJ, Nickerson KP, Ingano L, Serena G, Senger S, Lu TK, Fasano A, Faherty CS. Bacteriophage therapy testing against Shigella flexneri in a novel human intestinal organoid-derived infection model. J Pediatr Gastroenterol Nutr. 2019;68:509–516. doi: 10.1097/MPG.0000000000002203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chanin RB, Nickerson KP, Llanos-Chea A, Sistrunk JR, Rasko DA, Kumar DKV, de la Parra J, Auclair JR, Ding J, Li K, et al. Shigella flexneri adherence factor expression in in vivo-like conditions. mSphere. 2019;4(6). doi:10.1128/mSphere.00751-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Co JY, Margalef-Catala M, Li X, Mah AT, Kuo CJ, Monack DM, Amieva MR. Controlling epithelial polarity: a human enteroid model for host-pathogen interactions. Cell Rep. 2019;26:2509–20 e4. doi: 10.1016/j.celrep.2019.01.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Forbester JL, Goulding D, Vallier L, Hannan N, Hale C, Pickard D, Mukhopadhyay S, Dougan G. Interaction of Salmonella enterica Serovar Typhimurium with Intestinal Organoids derived from Human Induced Pluripotent stem cells. Infect Immun. 2015;83(7):2926–2934. doi: 10.1128/IAI.00161-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Forbester JL, Lees EA, Goulding D, Forrest S, Yeung A, Speak A, Clare S, Coomber EL, Mukhopadhyay S, Kraiczy J, et al. Interleukin-22 promotes phagolysosomal fusion to induce protection against Salmonella enterica Typhimurium in human epithelial cells. Proc Natl Acad Sci U S A. 2018;115(40):10118–10123. doi: 10.1073/pnas.1811866115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nickerson KP, Senger S, Zhang Y, Lima R, Patel S, Ingano L, Flavahan WA, Kumar DKV, Fraser CM, Faherty CS, et al. Salmonella Typhi Colonization provokes extensive transcriptional changes aimed at evading host mucosal immune defense during early infection of human intestinal tissue. EBioMedicine. 2018;31:92–109. doi: 10.1016/j.ebiom.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foulke-Abel J, In J, Yin J, Zachos NC, Kovbasnjuk O, Estes MK, de Jonge H, Donowitz M. Human Enteroids as a model of upper small intestinal ion transport physiology and pathophysiology. Gastroenterology. 2016;150(3):638–49 e8. doi: 10.1053/j.gastro.2015.11.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Foulke-Abel J, Yu H, Sunuwar L, Lin R, Fleckenstein JM, Kaper JB, Donowitz M. Phosphodiesterase 5 (PDE5) restricts intracellular cGMP accumulation during enterotoxigenic Escherichia coli infection. Gut Microbes. 2020; 1–12. in press. doi: 10.1080/19490976.2020.1752125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sunuwar L, Yin J, Kasendra M, Karalis K, Kaper J, Fleckenstein J, Donowitz M. Mechanical stimuli affect E. coli heat stable enterotoxin (ST)-cyclic GMP signaling in a human enteroid intestine-chip model. Infect Immun. 2020;IAI:19–00866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumar P, Kuhlmann FM, Chakraborty S, Bourgeois AL, Foulke-Abel J, Tumala B, Vickers TJ, Sack DA, DeNearing B, Harro CD, et al. Enterotoxigenic Escherichia coli–blood group A interactions intensify diarrheal severity. J Clin Invest. 2018;128(8):3298–3311. doi: 10.1172/JCI97659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Noel G, Baetz NW, Staab JF, Donowitz M, Kovbasnjuk O, Pasetti MF, Zachos NC. A primary human macrophage-enteroid co-culture model to investigate mucosal gut physiology and host-pathogen interactions. Sci Rep. 2017;7(1):45270. doi: 10.1038/srep45270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.In J, Foulke-Abel J, Zachos NC, Hansen AM, Kaper JB, Bernstein HD, Halushka M, Blutt S, Estes MK, Donowitz M, et al. Enterohemorrhagic Escherichia coli reduce mucus and intermicrovillar bridges in human stem cell-derived colonoids. Cell Mol Gastroenterol Hepatol. 2016;2:48–62 e3. doi: 10.1016/j.jcmgh.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tse CM, In JG, Yin J, Donowitz M, Doucet M, Foulke-Abel J, Ruiz-Perez F, Nataro JP, Zachos NC, Kaper JB, et al. Enterohemorrhagic E. coli (EHEC)-secreted serine protease EspP stimulates electrogenic ion transport in human colonoid monolayers. Toxins. 2018;10(9). doi: 10.3390/toxins10090351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Karve SS, Pradhan S, Ward DV, Weiss AA. Intestinal organoids model human responses to infection by commensal and Shiga toxin producing Escherichia coli. PLoS One. 2017;12:e0178966. doi: 10.1371/journal.pone.0178966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pradhan S, Karve SS, Weiss AA, Hawkins J, Poling HM, Helmrath MA, Wells JM, McCauley HA. Tissue responses to Shiga toxin in human intestinal organoids. Cell Mol Gastroenterol Hepatol. 2020;10(1):171–190. doi: 10.1016/j.jcmgh.2020.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rajan A, Vela L, Zeng XL, Yu X, Shroyer N, Blutt SE, Poole NM, Carlin LG, Nataro JP, Estes MK, et al. Novel segment- and host-specific patterns of Enteroaggregative Escherichia coli adherence to human intestinal Enteroids. mBio. 2018;9(1). doi: 10.1128/mBio.02419-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leslie JL, Huang S, Opp JS, Nagy MS, Kobayashi M, Young VB, Spence JR. Persistence and toxin production by Clostridium difficile within human intestinal organoids result in disruption of epithelial paracellular barrier function. Infect Immun. 2015;83(1):138–145. doi: 10.1128/IAI.02561-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schottelndreier D, Seeger K, Grassl GA, Winny MR, Lindner R, Genth H. Expression and (lacking) internalization of the cell surface receptors of clostridioides difficile toxin B. Front Microbiol. 2018;9:1483. doi: 10.3389/fmicb.2018.01483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Engevik MA, Danhof HA, Chang-Graham AL, Spinler JK, Engevik KA, Herrmann B, Endres BT, Garey KW, Hyser JM, Britton RA, et al. Human intestinal enteroids as a model of Clostridioides difficile-induced enteritis. Am J Physiol Gastrointest Liver Physiol. 2020;318(5):G870–g88. doi: 10.1152/ajpgi.00045.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mileto SJ, Jarde T, Childress KO, Jensen JL, Rogers AP, Kerr G, Hutton ML, Sheedlo MJ, Bloch SC, Shupe JA, et al. Clostridioides difficile infection damages colonic stem cells via TcdB, impairing epithelial repair and recovery from disease. Proc Natl Acad Sci U S A. 2020;117(14):8064–8073. doi: 10.1073/pnas.1915255117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Engevik MA, Yacyshyn MB, Engevik KA, Wang J, Darien B, Hassett DJ, Yacyshyn BR, Worrell RT. Human Clostridium difficile infection: altered mucus production and composition. Am J Physiol Gastrointest Liver Physiol. 2015;308(6):G510–24. doi: 10.1152/ajpgi.00091.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Engevik MA, Engevik KA, Yacyshyn MB, Wang J, Hassett DJ, Darien B, Yacyshyn BR, Worrell RT. Human Clostridium difficile infection: inhibition of NHE3 and microbiota profile. Am J Physiol Gastrointest Liver Physiol. 2015;308:G497–509. doi: 10.1152/ajpgi.00090.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fischer S, Uckert AK, Landenberger M, Papatheodorou P, Hoffmann-Richter C, Mittler AK, Ziener U, Hagele M, Schwan C, Muller M, et al. Human peptide alpha-defensin-1 interferes with Clostridioides difficile toxins TcdA, TcdB, and CDT. Faseb J. 2020. doi: 10.1096/fj.201902816R. [DOI] [PubMed] [Google Scholar]
- 46.Zhu Z, Schnell L, Muller B, Muller M, Papatheodorou P, Barth H. The antibiotic bacitracin protects human intestinal Epithelial cells and stem cell-derived intestinal organoids from Clostridium difficile toxin TcdB. Stem Cells Int. 2019;2019:4149762. doi: 10.1155/2019/4149762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuhlmann FM, Santhanam S, Kumar P, Luo Q, Ciorba MA, Fleckenstein JM. Blood group O-dependent cellular responses to cholera toxin: parallel clinical and epidemiological links to severe cholera. Am J Trop Med Hyg. 2016;95:440–443. doi: 10.4269/ajtmh.16-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zomer-van Ommen DD, Pukin AV, Fu O, Quarles van Ufford LH, Janssens HM, Beekman JM, Pieters RJ. Functional characterization of cholera toxin inhibitors using human intestinal organoids. J Med Chem. 2016;59(14):6968–6972. doi: 10.1021/acs.jmedchem.6b00770. [DOI] [PubMed] [Google Scholar]
- 49.Haksar D, de Poel E, van Ufford LQ, Bhatia S, Haag R, Beekman J, Pieters RJ. Strong inhibition of cholera Toxin B subunit by affordable, polymer-based multivalent inhibitors. Bioconjug Chem. 2019;30(3):785–792. doi: 10.1021/acs.bioconjchem.8b00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cervin J, Boucher A, Youn G, Bjorklund P, Wallenius V, Mottram L, Sampson NS, Yrlid U. Fucose-Galactose polymers inhibit cholera toxin binding to fucosylated structures and galactose-dependent intoxication of human enteroids. ACS Infect Dis. 2020. doi: 10.1021/acsinfecdis.0c00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Poole NM, Green SI, Rajan A, Vela LE, Zeng XL, Estes MK, Maresso AW. Role for FimH in extraintestinal pathogenic Escherichia coli invasion and translocation through the intestinal epithelium. Infect Immun. 2017;85(11). doi: 10.1128/iai.00581-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Senger S, Ingano L, Freire R, Anselmo A, Zhu W, Sadreyev R, Walker WA, Fasano A. Human fetal-derived Enterospheres provide insights on intestinal development and a novel model to study Necrotizing Enterocolitis (NEC). Cell Mol Gastroenterol Hepatol. 2018;5(4):549–568. doi: 10.1016/j.jcmgh.2018.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meng D, Sommella E, Salviati E, Campiglia P, Ganguli K, Djebali K, Zhu W, Walker WA. Indole-3-lactic acid, a metabolite of tryptophan, secreted by Bifidobacterium longum subspecies infantis is anti-inflammatory in the immature intestine. Pediatr Res. 2020. doi: 10.1038/s41390-019-0740-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wu RY, Li B, Koike Y, Maattanen P, Miyake H, Cadete M, Johnson-Henry KC, Botts SR, Lee C, Abrahamsson TR, et al. Human Milk Oligosaccharides increase Mucin expression in experimental Necrotizing Enterocolitis. Mol Nutr Food Res. 2019;63:e1800658. [DOI] [PubMed] [Google Scholar]
- 55.Afrazi A, Branca MF, Sodhi CP, Good M, Yamaguchi Y, Egan CE, Lu P, Jia H, Shaffiey S, Lin J, et al. Toll-like receptor 4-mediated endoplasmic reticulum stress in intestinal crypts induces necrotizing enterocolitis. J Biol Chem. 2014;289(14):9584–9599. doi: 10.1074/jbc.M113.526517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li B, Lee C, Cadete M, Zhu H, Koike Y, Hock A, Wu RY, Botts SR, Minich A, Alganabi M, et al. Impaired Wnt/beta-catenin pathway leads to dysfunction of intestinal regeneration during necrotizing enterocolitis. Cell Death Dis. 2019;10:743. doi: 10.1038/s41419-019-1987-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Livio S, Strockbine NA, Panchalingam S, Tennant SM, Barry EM, Marohn ME, Antonio M, Hossain A, Mandomando I, Ochieng JB, et al. Shigella isolates from the global enteric multicenter study inform vaccine development. Clin Infect Dis. 2014;59(7):933–941. doi: 10.1093/cid/ciu468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lampel KA, Formal SB, Maurelli AT. A brief history of Shigella. EcoSal Plus. 2018;8(1). doi: 10.1128/ecosalplus.ESP-0006-2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schroeder GN, Hilbi H. Molecular pathogenesis of Shigella spp.: controlling host cell signaling, invasion, and death by type III secretion. Clin Microbiol Rev. 2008;21:134–156. doi: 10.1128/CMR.00032-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Killackey SA, Sorbara MT, Girardin SE. Cellular aspects of Shigella pathogenesis: focus on the manipulation of host cell processes. Front Cell Infect Microbiol. 2016;6:38. doi: 10.3389/fcimb.2016.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schnupf P, Sansonetti PJ. Shigella pathogenesis: new insights through advanced methodologies. Microbiol Spectr. 2019;7(2). BAI-0023-2019. doi: 10.1128/microbiolspec. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sperandio B, Fischer N, Joncquel Chevalier-Curt M, Rossez Y, Roux P, Robbe Masselot C, Sansonetti PJ. Virulent Shigella flexneri affects secretion, expression, and glycosylation of gel-forming mucins in mucus-producing cells. Infect Immun. 2013;81(10):3632–3643. doi: 10.1128/IAI.00551-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sansonetti PJ, Arondel J, Cantey JR, Prevost MC, Huerre M. Infection of rabbit Peyer’s patches by Shigella flexneri: effect of adhesive or invasive bacterial phenotypes on follicle-associated epithelium. Infect Immun. 1996;64:2752–2764. doi: 10.1128/IAI.64.7.2752-2764.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wassef JS, Keren DF, Mailloux JL. Role of M cells in initial antigen uptake and in ulcer formation in the rabbit intestinal loop model of shigellosis. Infect Immun. 1989;57:858–863. doi: 10.1128/IAI.57.3.858-863.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mounier J, Vasselon T, Hellio R, Lesourd M, Sansonetti PJ. Shigella flexneri enters human colonic Caco-2 epithelial cells through the basolateral pole. Infect Immun. 1992;60:237–248. doi: 10.1128/IAI.60.1.237-248.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Parsot C. Shigella type III secretion effectors: how, where, when, for what purposes? Curr Opin Microbiol. 2009;12:110–116. doi: 10.1016/j.mib.2008.12.002. [DOI] [PubMed] [Google Scholar]
- 67.Zychlinsky A, Thirumalai K, Arondel J, Cantey JR, Aliprantis AO, Sansonetti PJ. In vivo apoptosis in Shigella flexneri infections. Infect Immun. 1996;64:5357–5365. doi: 10.1128/IAI.64.12.5357-5365.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zychlinsky A, Prevost MC, Sansonetti PJ. Shigella flexneri induces apoptosis in infected macrophages. Nature. 1992;358:167–169. doi: 10.1038/358167a0. [DOI] [PubMed] [Google Scholar]
- 69.Guichon A, Zychlinsky A. Apoptosis as a trigger of inflammation in Shigella-induced cell death. Biochem Soc Trans. 1996;24:1051–1054. doi: 10.1042/bst0241051. [DOI] [PubMed] [Google Scholar]
- 70.Clerc P, Ryter A, Mounier J, Sansonetti PJ. Plasmid-mediated intracellular multiplication of Shigella flexneri. Ann Inst Pasteur Microbiol. 1985;137A 1986:315–320. [DOI] [PubMed] [Google Scholar]
- 71.Sansonetti PJ, Ryter A, Clerc P, Maurelli AT, Mounier J. Multiplication of Shigella flexneri within HeLa cells: lysis of the phagocytic vacuole and plasmid-mediated contact hemolysis. Infect Immun. 1986;51:461–469. doi: 10.1128/IAI.51.2.461-469.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bernardini ML, Mounier J, d’Hauteville H, Coquis-Rondon M, Sansonetti PJ. Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin. Proc Natl Acad Sci U S A. 1989;86:3867–3871. doi: 10.1073/pnas.86.10.3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Monack DM, Theriot JA. Actin-based motility is sufficient for bacterial membrane protrusion formation and host cell uptake. Cell Microbiol. 2001;3:633–647. doi: 10.1046/j.1462-5822.2001.00143.x. [DOI] [PubMed] [Google Scholar]
- 74.Singer M, Sansonetti PJ. IL-8 is a key chemokine regulating neutrophil recruitment in a new mouse model of Shigella-induced colitis. J Immunol. 2004;173:4197–4206. doi: 10.4049/jimmunol.173.6.4197. [DOI] [PubMed] [Google Scholar]
- 75.Sansonetti PJ, Arondel J, Huerre M, Harada A, Matsushima K. Interleukin-8 controls bacterial transepithelial translocation at the cost of epithelial destruction in experimental shigellosis. Infect Immun. 1999;67:1471–1480. doi: 10.1128/IAI.67.3.1471-1480.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 77.Perdomo JJ, Gounon P, Sansonetti PJ. Polymorphonuclear leukocyte transmigration promotes invasion of colonic epithelial monolayer by Shigella flexneri. J Clin Invest. 1994;93:633–643. doi: 10.1172/JCI117015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Perdomo OJ, Cavaillon JM, Huerre M, Ohayon H, Gounon P, Sansonetti PJ. Acute inflammation causes epithelial invasion and mucosal destruction in experimental shigellosis. J Exp Med. 1994;180:1307–1319. doi: 10.1084/jem.180.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Philpott DJ, Yamaoka S, Israel A, Sansonetti PJ. Invasive Shigella flexneri activates NF-kappa B through a lipopolysaccharide-dependent innate intracellular response and leads to IL-8 expression in epithelial cells. J Immunol. 2000;165:903–914. doi: 10.4049/jimmunol.165.2.903. [DOI] [PubMed] [Google Scholar]
- 80.Zhang Z, Jin L, Champion G, Seydel KB, Stanley SL Jr., Kaufmann SHE. Shigella infection in a SCID mouse-human intestinal xenograft model: role for neutrophils in containing bacterial dissemination in human intestine. Infect Immun. 2001;69(5):3240–3247. doi: 10.1128/IAI.69.5.3240-3247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Sansonetti PJ, Arondel J, Fontaine A, d’Hauteville H, Bernardini ML. OmpB (osmo-regulation) and icsA (cell-to-cell spread) mutants of Shigella flexneri: vaccine candidates and probes to study the pathogenesis of shigellosis. Vaccine. 1991;9:416–422. doi: 10.1016/0264-410X(91)90128-S. [DOI] [PubMed] [Google Scholar]
- 82.Ahmad T, Gogarty M, Walsh EG, Brayden DJ. A comparison of three Peyer’s patch “M-like” cell culture models: particle uptake, bacterial interaction, and epithelial histology. Eur J Pharm Biopharm. 2017;119:426–436. doi: 10.1016/j.ejpb.2017.07.013. [DOI] [PubMed] [Google Scholar]
- 83.Rouch JD, Scott A, Lei NY, Solorzano-Vargas RS, Wang J, Hanson EM, Kobayashi M, Lewis M, Stelzner MG, Dunn JCY, et al. Development of functional microfold (M) cells from intestinal stem cells in primary human Enteroids. PLoS One. 2016;11(1):e0148216. doi: 10.1371/journal.pone.0148216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wood MB, Rios D, Williams IR. TNF-alpha augments RANKL-dependent intestinal M cell differentiation in enteroid cultures. Am J Physiol Cell Physiol. 2016;311:C498–507. doi: 10.1152/ajpcell.00108.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. Bidirectional transport of amino acids regulates mTOR and autophagy. Cell. 2009;136(3):521–534. doi: 10.1016/j.cell.2008.11.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tattoli I, Sorbara MT, Vuckovic D, Ling A, Soares F, Carneiro LA, Yang C, Emili A, Philpott DJ, Girardin SE. Amino acid starvation induced by invasive bacterial pathogens triggers an innate host defense program. Cell Host Microbe. 2012;11:563–575. doi: 10.1016/j.chom.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 87.Crump JA, Mintz ED. Global trends in typhoid and paratyphoid fever. Clin Infect Dis. 2010;50:241–246. doi: 10.1086/649541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Majowicz SE, Musto J, Scallan E, Angulo FJ, Kirk M, O'Brien SJ, Jones TF, Fazil A, Hoekstra RM. The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis. 2010;50:882–889. doi: 10.1086/650733. [DOI] [PubMed] [Google Scholar]
- 89.Higginson EE, Simon R, Tennant SM. Animal models for Salmonellosis: applications in vaccine research. Clin Vaccine Immunol. 2016;23:746–756. doi: 10.1128/CVI.00258-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Galan JE, Ginocchio C, Costeas P. Molecular and functional characterization of the Salmonella invasion gene invA: homology of InvA to members of a new protein family. J Bacteriol. 1992;174:4338–4349. doi: 10.1128/JB.174.13.4338-4349.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Smith AC, Heo WD, Braun V, Jiang X, Macrae C, Casanova JE, Scidmore MA, Grinstein S, Meyer T, Brumell JH, et al. A network of Rab GTPases controls phagosome maturation and is modulated by Salmonella enterica serovar Typhimurium. J Cell Biol. 2007;176(3):263–268. doi: 10.1083/jcb.200611056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang AZ, Ojakian GK, Nelson WJ. Steps in the morphogenesis of a polarized epithelium. II. Disassembly and assembly of plasma membrane domains during reversal of epithelial cell polarity in multicellular epithelial (MDCK) cysts. J Cell Sci. 1990;95:153–165. [DOI] [PubMed] [Google Scholar]
- 93.Fleckenstein JM, Hardwidge PR, Munson GP, Rasko DA, Sommerfelt H, Steinsland H. Molecular mechanisms of enterotoxigenic Escherichia coli infection. Microbes Infect. 2010;12:89–98. doi: 10.1016/j.micinf.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Schulz S, Green CK, Yuen PS, Garbers DL. Guanylyl cyclase is a heat-stable enterotoxin receptor. Cell. 1990;63:941–948. doi: 10.1016/0092-8674(90)90497-3. [DOI] [PubMed] [Google Scholar]
- 95.Duan Q, Xia P, Nandre R, Zhang W, Zhu G. Review of newly identified functions associated with the Heat-Labile toxin of Enterotoxigenic Escherichia coli. Front Cell Infect Microbiol. 2019;9:292. doi: 10.3389/fcimb.2019.00292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhu Y, Luo Q, Davis SM, Westra C, Vickers TJ, Fleckenstein JM. Molecular determinants of Enterotoxigenic Escherichia coli Heat-Stable toxin secretion and delivery. Infect Immun. 2018;86(11). doi: 10.1128/IAI.00526-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Heiman KE, Mody RK, Johnson SD, Griffin PM, Gould LH. Escherichia coli O157 outbreaks in the United States, 2003-2012. Emerg Infect Dis. 2015;21:1293–1301. doi: 10.3201/eid2108.141364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lai Y, Rosenshine I, Leong JM, Frankel G. Intimate host attachment: enteropathogenic and enterohaemorrhagic Escherichia coli. Cell Microbiol. 2013;15:1796–1808. doi: 10.1111/cmi.12179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Johannes L, Römer W. Shiga toxins – from cell biology to biomedical applications. Nat Rev Microbiol. 2010;8:105–116. doi: 10.1038/nrmicro2279. [DOI] [PubMed] [Google Scholar]
- 100.Griffin PM, Tauxe RV. The epidemiology of infections caused by Escherichia coli O157: H7, other enterohemorrhagic E. coli, and the associated hemolytic uremic syndrome. Epidemiol Rev. 1991;13:60–98. doi: 10.1093/oxfordjournals.epirev.a036079. [DOI] [PubMed] [Google Scholar]
- 101.In JG, Foulke-Abel J, Clarke E, Kovbasnjuk O. Human Colonoid monolayers to study interactions between Pathogens, Commensals, and host Intestinal Epithelium. Journal of Visualized Experiments: JoVE. 2019. doi: 10.3791/59357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Das S, Jayaratne R, Barrett KE. The role of ion transporters in the pathophysiology of infectious diarrhea. Cell Mol Gastroenterol Hepatol. 2018;6:33–45. doi: 10.1016/j.jcmgh.2018.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Dundas S, Todd WT, Stewart AI, Murdoch PS, Chaudhuri AK, Hutchinson SJ. The central Scotland Escherichia coli O157: h7outbreak: risk factors for the hemolytic uremic syndrome and death among hospitalized patients. Clin Infect Dis. 2001;33:923–931. doi: 10.1086/322598. [DOI] [PubMed] [Google Scholar]
- 104.Lima AAM, Medeiros P, Havt A. Enteroaggregative Escherichia coli subclinical and clinical infections. Curr Opin Infect Dis. 2018;31:433–439. doi: 10.1097/QCO.0000000000000477. [DOI] [PubMed] [Google Scholar]
- 105.Nataro JP. Enteroaggregative Escherichia coli pathogenesis. Curr Opin Gastroenterol. 2005;21:4–8. [PubMed] [Google Scholar]
- 106.Henderson IR, Czeczulin J, Eslava C, Noriega F, Nataro JP. Characterization of pic, a secreted protease of Shigella flexneri and enteroaggregative Escherichia coli. Infect Immun. 1999;67:5587–5596. doi: 10.1128/IAI.67.11.5587-5596.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hebbelstrup Jensen B, Olsen KE, Struve C, Krogfelt KA, Petersen AM. Epidemiology and clinical manifestations of enteroaggregative Escherichia coli. Clin Microbiol Rev. 2014;27:614–630. doi: 10.1128/CMR.00112-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Navarro-Garcia F, Gutierrez-Jimenez J, Garcia-Tovar C, Castro LA, Salazar-Gonzalez H, Cordova V. Pic, an autotransporter protein secreted by different pathogens in the Enterobacteriaceae family, is a potent mucus secretagogue. Infect Immun. 2010;78:4101–4109. doi: 10.1128/IAI.00523-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wong AR, Pearson JS, Bright MD, Munera D, Robinson KS, Lee SF, Frankel G, Hartland EL. Enteropathogenic and enterohaemorrhagic Escherichia coli: even more subversive elements. Mol Microbiol. 2011;80(6):1420–1438. doi: 10.1111/j.1365-2958.2011.07661.x. [DOI] [PubMed] [Google Scholar]
- 110.Mellies JL, Navarro-Garcia F, Okeke I, Frederickson J, Nataro JP, Kaper JB, DiRita VJ. espC pathogenicity island of enteropathogenic Escherichia coli encodes an enterotoxin. Infect Immun. 2001;69(1):315–324. doi: 10.1128/IAI.69.1.315-324.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Croxen MA, Finlay BB. Molecular mechanisms of Escherichia coli pathogenicity. Nat Rev Microbiol. 2010;8:26–38. doi: 10.1038/nrmicro2265. [DOI] [PubMed] [Google Scholar]
- 112.Hackam DJ, Sodhi CP, Good M. New insights into necrotizing enterocolitis: from laboratory observation to personalized prevention and treatment. J Pediatr Surg. 2019;54:398–404. doi: 10.1016/j.jpedsurg.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Chandrasekaran R, Lacy DB. The role of toxins in Clostridium difficile infection. FEMS Microbiol Rev. 2017;41:723–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shen A. Clostridium difficile toxins: mediators of inflammation. J Innate Immun. 2012;4:149–158. doi: 10.1159/000332946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Orrell KE, Zhang Z, Sugiman-Marangos SN, Melnyk RA. Clostridium difficile toxins A and B: receptors, pores, and translocation into cells. Crit Rev Biochem Mol Biol. 2017;52:461–473. doi: 10.1080/10409238.2017.1325831. [DOI] [PubMed] [Google Scholar]
- 116.Hayashi H, Szászi K, Coady-Osberg N, Furuya W, Bretscher AP, Orlowski J, Grinstein S. Inhibition and redistribution of NHE3, the apical Na+/H+ exchanger, by Clostridium difficile toxin B. J Gen Physiol. 2004;123(5):491–504. doi: 10.1085/jgp.200308979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Di Masi A, Leboffe L, Polticelli F, Tonon F, Zennaro C, Caterino M, Stano P, Fischer S, Hägele M, Müller M, et al. Human serum albumin is an essential component of the host defense mechanism against clostridium difficile intoxication. J Infect Dis. 2018;218(9):1424–1435. doi: 10.1093/infdis/jiy338. [DOI] [PubMed] [Google Scholar]
- 118.Schnell L, Felix I, Muller B, Sadi M, von Bank F, Papatheodorou P, Popoff MR, Aktories K, Waltenberger E, Benz R, et al. Revisiting an old antibiotic: bacitracin neutralizes binary bacterial toxins and protects cells from intoxication. Faseb J. 2019;33:5755–5771. doi: 10.1096/fj.201802453R. [DOI] [PubMed] [Google Scholar]
- 119.Kaper JB, Morris JG Jr., Levine MM. Cholera. Clin Microbiol Rev. 1995;8:48–86. doi: 10.1128/CMR.8.1.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Branson TR, Turnbull WB. Bacterial toxin inhibitors based on multivalent scaffolds. Chem Soc Rev. 2013;42:4613–4622. doi: 10.1039/C2CS35430F. [DOI] [PubMed] [Google Scholar]
- 121.Grassart A, Malarde V, Gobaa S, Sartori-Rupp A, Kerns J, Karalis K, Marteyn B, Sansonetti P, Sauvonnet N. Bioengineered human organ-on-chip reveals intestinal microenvironment and mechanical forces impacting Shigella infection. Cell Host Microbe. 2019;26:435–44 e4. doi: 10.1016/j.chom.2019.08.007. [DOI] [PubMed] [Google Scholar]
- 122.Jalili-Firoozinezhad S, Gazzaniga FS, Calamari EL, Camacho DM, Fadel CW, Bein A, Swenor B, Nestor B, Cronce MJ, Tovaglieri A, et al. A complex human gut microbiome cultured in an anaerobic intestine-on-a-chip. Nat Biomed Eng. 2019;3(7):520–531. doi: 10.1038/s41551-019-0397-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kasendra M, Tovaglieri A, Sontheimer-Phelps A, Jalili-Firoozinezhad S, Bein A, Chalkiadaki A, Scholl W, Zhang C, Rickner H, Richmond CA, et al. Development of a primary human small intestine-on-a-chip using biopsy-derived organoids. Sci Rep. 2018;8(1):2871. doi: 10.1038/s41598-018-21201-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tovaglieri A, Sontheimer-Phelps A, Geirnaert A, Prantil-Baun R, Camacho DM, Chou DB, Jalili-Firoozinezhad S, de Wouters T, Kasendra M, Super M, et al. Species-specific enhancement of enterohemorrhagic E. coli pathogenesis mediated by microbiome metabolites. Microbiome. 2019;7(1):43. doi: 10.1186/s40168-019-0650-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Sontheimer-Phelps A, Chou DB, Tovaglieri A, Ferrante TC, Duckworth T, Fadel C, Frismantas V, Sutherland AD, Jalili-Firoozinezhad S, Kasendra M, et al. Human colon-on-a-chip enables continuous in vitro analysis of colon mucus layer accumulation and physiology. Cell Mol Gastroenterol Hepatol. 2020;9(3):507–526. doi: 10.1016/j.jcmgh.2019.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cardenas D, Bhalchandra S, Lamisere H, Chen Y, Zeng XL, Ramani S, Karandikar UC, Kaplan DL, Estes MK, Ward HD. Two- and three-dimensional bioengineered human intestinal tissue models for Cryptosporidium. Methods Mol Biol (Clifton, NJ). 2020;2052:373–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Knight E, Murray B, Carnachan R, Przyborski S. Alvetex(R): polystyrene scaffold technology for routine three dimensional cell culture. Methods Mol Biol (Clifton, NJ). 2011;695:323–340. [DOI] [PubMed] [Google Scholar]
- 128.Mondrinos MJ, Yi YS, Wu NK, Ding X, Huh D. Native extracellular matrix-derived semipermeable, optically transparent, and inexpensive membrane inserts for microfluidic cell culture. Lab Chip. 2017;17:3146–3158. doi: 10.1039/C7LC00317J. [DOI] [PMC free article] [PubMed] [Google Scholar]