

Abstract
Human natural killer cell deficiency (NKD) arises from inborn errors of immunity that lead to impaired NK cell development, function, or both. Through the understanding of the biological perturbations in individuals with NKD, requirements for the generation of terminally mature functional innate effector cells can be elucidated. Here, we report a cause of NKD resulting from compound heterozygous mutations in minichromosomal maintenance complex member 10 (MCM10) that impaired NK cell maturation in a child with fatal susceptibility to CMV. MCM10 has not been previously associated with monogenic disease and plays a critical role in the activation and function of the eukaryotic DNA replisome. Through evaluation of patient primary fibroblasts, modeling patient mutations in fibroblast cell lines, and MCM10 knockdown in human NK cell lines, we have shown that loss of MCM10 function leads to impaired cell cycle progression and induction of DNA damage–response pathways. By modeling MCM10 deficiency in primary NK cell precursors, including patient-derived induced pluripotent stem cells, we further demonstrated that MCM10 is required for NK cell terminal maturation and acquisition of immunological system function. Together, these data define MCM10 as an NKD gene and provide biological insight into the requirement for the DNA replisome in human NK cell maturation and function.
Keywords: Immunology
Keywords: Genetic diseases, NK cells
Introduction
Human natural killer (NK) cells play a critical role in the control of viral infection and malignancy through contact-mediated killing of susceptible target cells and cytokine secretion. While rare, monogenic cases of human NK cell deficiency (NKD) lead to severe and life-threatening illness in which the abnormality of NK cells is the major clinically relevant immunological defect (reviewed in refs. 1–4). To date, 5 causes of classical or developmental NKD, in which loss of NK cells or aberrant NK cell development leads to absence or impaired maturation of peripheral blood NK cells, have been described (1). Genes implicated in developmental NKD include GATA binding protein 2 (GATA2), IFN regulatory factor 8 (IRF8), minichromosome maintenance 4 (MCM4), and go-ichi-ni-san complex subunit 1 (GINS1) (5–9). In addition, although only reported in one child, regulator of telomere elongation helicase (RTEL1) mutation has been described as a cause of NKD (10, 11). Each of these cases has revealed previously unidentified requirements for human NK cell differentiation and homeostasis and is distinct from a broader category of inborn defects of immunity that can include NK cell abnormalities as a minor immunological defect. The biology uncovered through discovery of NKD includes a requirement for the CDC45-MCM2-7-GINS (CMG) complex in NK cell terminal maturation, illustrated by patients with MCM4 and GINS1 deficiencies. Here, we describe mutations in another factor required for CMG function, minichromosomal maintenance complex member 10 (MCM10), which caused NKD in a child with a severe and ultimately fatal CMV infection.
Human NK cells develop from the common lymphoid progenitor and undergo increasingly restricted stages of lineage commitment, leading to terminal maturation. NK cells compose 5%–20% of the lymphocyte population in peripheral blood and can be further subdivided into 2 subsets, CD56bright and CD56dim, each with unique phenotypic and functional properties. CD56bright NK cells are potent producers of cytokines, particularly IFN-γ (12, 13). The majority of NK cells in peripheral blood are CD56dim, the subset specialized to mediate target cell lysis. As such, they express perforin and granzymes to lyse susceptible targets and the Fc receptor CD16 to rapidly recognize opsonized cells.
Multiple lines of evidence demonstrate that CD56bright NK cells are direct precursors of CD56dim NK cells, with this evidence including longer telomeres in CD56bright NK cells and the early appearance of CD56bright NK cells following hematopoietic stem cell (HSC) transplant (14). Similarly, in models of in vitro differentiation or humanized mouse reconstitution, the CD56bright subset is detected before the CD56dim subset, suggesting these are less mature precursors (15–17). Recent single-cell analyses of human NK cells include pseudotime projections of gene and protein expression in peripheral blood NK cell subsets that define a spectrum of development ranging from CD56bright to CD56dim (18). However, despite these data from a variety of model systems, the human NK cell developmental program is poorly understood, and identifying requirements for NK cell development is an ongoing challenge within the field.
Of the previously described classical NKD, most include an aspect of aberrant NK cell subset generation in addition to reduced NK cell frequencies within the lymphocyte population. A highly conserved feature of the NK cell phenotype in patients with GATA2 deficiency is the near or absolute absence of the CD56bright subset (8, 19–21). CD56dim NK cells may be present in these patients, while their intrinsic function of target cell lysis is impaired and likely contributes to severe herpesviral illness (8). In contrast, IRF8, MCM4, and GINS1 deficiency lead to reduction specifically of the CD56dim subset and concomitant overrepresentation of the CD56bright subset (5–7). In the case of biallelic IRF8 deficiency, the CD56dim NK cells that are present have impaired lytic function, and therefore it is likely that this impairment combined with reduced CD56dim NK cell frequency leads to unusual susceptibility to viral infections (7). Therefore, the deregulation of NK cell subset generation is frequently accompanied by impaired NK cell function and viral susceptibility.
First described in a cohort of endogamous Irish travelers with familial NKD, a hypomorphic MCM4 mutation causes susceptibility to viral infections, short stature, and adrenal insufficiency (5, 6, 22, 23). Further investigation into the NK cell phenotype in these patients revealed low NK cell numbers with significant overrepresentation of the CD56bright subset (5, 6). Subsequent to the report of MCM4-deficient patients was the description of individuals with GINS1 deficiency, which leads to a clinical and NK cell phenotype strikingly similar to that of MCM4 deficiency. In 5 patients from 4 kindreds, compound heterozygous mutations in GINS1 led to intrauterine growth retardation, neutropenia, and NKD (9, 22, 24). Specifically, low NK cell number in these patients is accompanied by a relative overrepresentation of the CD56bright subset that is suggestive of the NK cell phenotype in individuals with hypomorphic MCM4 mutations.
MCM4 and GINS1 are subunits of the CMG replicative helicase complex that binds to origins of replication and is required for DNA synthesis. The MCM2-7 complex binds to chromatin in a cell cycle–specific manner and is highly expressed in proliferating cells (25). Hypomorphic mutations of Mcm4 in Saccharomyces cerevisiae, or in the Mcm4Chaos3 mouse model, lead to genomic instability and increased tumor formation in the mouse (26). While Mcm4-knockout mice are not viable, mutation of F345I disrupts interactions between MCM family members and leads to cell cycle arrest (26). The reported NKD-causing variants in MCM4 and GINS1 are hypomorphic alleles, but their effect has not been defined using NK cell experimental systems. Fibroblasts from these individuals have increased genomic instability and impaired cell cycle progression, with increased induction of DNA damage–repair (DDR) pathways (5, 6, 9). The profound NK cell phenotype in these patients and accompanying viral susceptibility suggest that the CMG complex is an important regulator of human NK cell terminal maturation; however, the mechanism by which this effect is mediated is not well understood.
Here, we describe a single patient with unusual susceptibility to CMV infection having NKD. Genetic analyses identified a compound heterozygous mutation in MCM10. MCM10 is a highly conserved and nonredundant component of the eukaryotic replisome that binds directly to the MCM2-7 complex, CDC45, and both double- and single-stranded DNA (27). MCM10 is thought to play a critical role in CMG complex assembly and activation and replication fork processivity (28–30). Here, we sought to define the molecular nature of the MCM10 mutations discovered in a patient with NKD. We also sought to determine the role of MCM10 in human NK cell maturation and function using models of MCM10 knockdown (KD) in an NK cell line and primary NK cell precursors. Moreover, we recapitulated NK cell development in vitro and in vivo using patient-derived iPS cells. These studies demonstrate the importance of MCM10 function in human NK cell maturation, accentuate the importance of the CMG complex for NK cell development, and define MCM10 deficiency as a cause of classical NKD.
Results
Clinical history and variant allele confirmation.
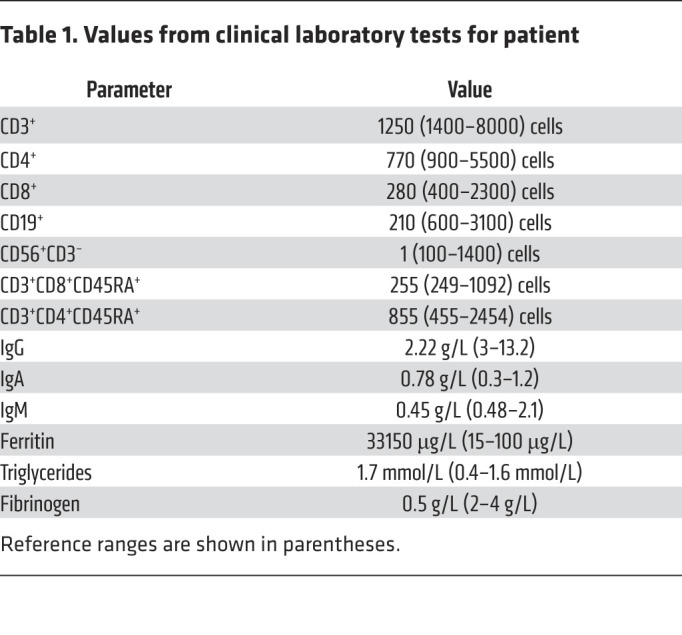
The male proband was born to healthy, nonconsanguineous parents, yet presented at 16 months of age with fever, organomegaly, diarrhea, and CMV infection (2 × 106 copies/mL). T and B cell numbers were slightly decreased with reduction in effector and memory T cells. Profoundly reduced NK cell numbers were noted, and further analysis suggested that 50% of these were in the CD56bright subset, although the severely reduced number of NK cells precluded precise quantification (Figure 1A and Table 1). T cell activation in response to phytohemagglutinin was reduced relative to control, but responses to phorbyl myristate acetate and CD3 activation were normal. While perforin expression was within normal range, elevated levels of ferritin and triglycerides and decreased fibrinogen concentration prompted consideration of hematophagocytic lymphohistiocytosis (HLH). Expression of SLAM-associated protein (SAP), X-linked inhibitor of apoptosis (XIAP), MHC I, and MHC II were normal, and no mutations in CD3ζ were detected. The patient underwent bone marrow transplantation for suspected primary immunodeficiency, but succumbed to overwhelming preexisting CMV at 24 months.
Figure 1. Decreased frequency of peripheral blood NK cells with overrepresentation of the CD56bright subset in an individual with compound heterozygous mutations in MCM10.

Severe CMV infection in the male proband born to healthy parents led to evaluation of peripheral blood NK cells and whole exome sequencing of the proband and his immediate family. (A) Flow cytometric analysis of peripheral blood NK cells from a representative healthy donor (left) and the proband (right). NK cells are defined as CD56+CD3−. The relative frequency of CD56bright NK cells within the NK cell subset is defined by density of CD56 staining (histograms). (B) Whole exome sequencing identified compound heterozygous mutations that were rare and predicted to be damaging with familial segregation. (C) Location of identified variants relative to MCM10 domains. Dashed line indicates previously defined NLS (32). NTD, N-terminal domain; ID, internal domain; CTD, C-terminal domain.
Table 1. Values from clinical laboratory tests for patient.
Exome sequencing was performed on DNA derived from tissue from the affected individual and his parents as part of an effort to seek molecular diagnoses in a large cohort of primary immunodeficiency patients (31). A trio-based analysis of the whole-exome sequencing results was performed, comparing the affected individual’s results with those of his parents and filtering on low allelic frequency (31). Since the proband was affected and the parents apparently healthy, we focused on de novo occurring variants, hemizygous variants, and variants segregating in accordance with Mendelian expectations for a recessive disease trait. Noninherited variants with low quality reads and very few reads were excluded from the analysis and filtered out. Variants in genes associated with autosomal recessive disorders were excluded if they occurred in homozygous state in the normal variant databases, as were frequently occurring variants in genes for dominant disorders. X-linked variants observed in hemizygous states in the normal variant databases were also excluded. The remaining variants were interpreted via extensive literature and database review to consider potential relevance to disease phenotype, penetrance, segregation or inheritance, disease-causing mechanism, and potential pathogenicity of variants. Designated as potential novel gene 4 (patient 68.1 in Table E1) in our initial publication (31), the MCM10 variants were selected for further study at the time, as they were infrequent, changed a highly conserved nucleotide or amino acid site, and were located within a known disease gene or pathway gene that fitted with the immunological phenotype (5, 6, 22). These variant alleles segregated in accordance with Mendelian expectations for a recessive disease trait and were confirmed by Sanger sequencing (Supplemental Figure 1; supplemental material available online with this article; https://doi.org/10.1172/JCI134966DS1). Reanalysis of these variants with current databases underscored their pathogenicity and rarity (Supplemental Table 1 and Supplemental Methods).
A missense variant allele in exon 10 was inherited from the father ([NM_018518.5] c.1276C>T, p.R426C), and a nonsense variant introducing a premature stop codon at the end of exon 13 ([NM_018518.5] c.1744C>T, p.R582X) of this 19-exon gene was inherited from the mother (Figure 1B). This stop-gain variant is located N-terminally to the previously reported C-terminal domain required for nuclear localization in multicellular eukaryotes (ref. 32 and Figure 1C) and is predicted to be subject to degradation by nonsense-mediated decay (33). The missense variant was predicted to be disease causing by MutationTaster (score 0.99) (34) and likely damaging by PolyPhen2 (score 1.0) (35). The Combined Annotation Dependent Depletion score (CADD, http://cadd.gs.washington.edu/) was 24.3 (36), and the arginine in position 426 is conserved across all species. The nonsense variant was not found in ExAC (37) nor in GnomAD (38), while the missense variant was found at extremely low frequency (4.12 × 10–5 in a heterozygous state with no homozygotes in ExAC, 2.5 × 10–5 in GnomAD). The rarity and likely damaging effect of these 2 variants suggesting pathogenic alleles, combined with the clinical and NK cell phenotype of the patient, argued that MCM10 deficiency was responsible for his disease.
Mutations in MCM10 affect protein localization and function.
To determine the effect of the patient mutations on MCM10 protein expression and function, we performed immunoblotting of lysates from primary dermal fibroblasts obtained from the patient and a healthy, unrelated donor. Western blotting of patient fibroblasts showed the presence of protein at the predicted molecular weight for full-length MCM10, suggesting that the mRNA carrying the premature stop codon was degraded by nonsense-mediated decay and did not permit production of the truncated protein (Figure 2A and Supplemental Figure 2). While the nuclear localization signal (NLS) for human MCM10 has not been specifically defined, it has been localized to the metazoan-specific C-terminal region of the protein (32). To understand the effect of C-terminal MCM10 truncations, in the case in which they were expressed by the patient (even transiently), stable 293T cells were generated expressing N-terminally tagged full-length and truncation mutants. Western blot analyses demonstrated stable accumulation of all tagged versions of MCM10 (Figure 2B) Furthermore, immunofluorescence experiments showed that tagged MCM10 localizes to the nucleus when truncated C-terminally to the NLS, but remains cytoplasmic when truncated N-terminally to the NLS (Figure 2C). Given its position relative to the NLS, we hypothesized that if the truncated protein were expressed, the MCM10-R582X mutation would prevent its nuclear localization. To specifically test this hypothesis, we generated MCM10-turboGFP and turboGFP-MCM10-R582X constructs and transiently overexpressed these in 293T cells. Using confocal microscopy, we found that expression of the R582X nonsense mutation, but not the R426C variant, significantly impaired nuclear translocation of MCM10 (Figure 2D). Therefore, while endogenous protein in patient cells was not detected due to nonsense-mediated decay, any production of R582X variant demonstrates ineffective nuclear localization due to truncation of the NLS.
Figure 2. Expression and localization of MCM10 mutants.

(A) Primary fibroblasts from proband or healthy donor were lysed and probed for MCM10 (left). Intensity of MCM10 was normalized to loading control (actin, right). (B) Western blot of stable SFB-MCM10 expression in 293T whole cell extracts. Full-length SFB-MCM10 and truncation mutants were detected with anti-FLAG antibody (top) and with anti–α-tubulin antibody as a loading control (bottom). (C) Confocal imaging of SFB-MCM10 localization in stable 293T cell lines. Full-length SFB-MCM10 and truncation mutants were detected with anti-FLAG antibody (green) with DAPI staining (blue). Scale bars: 10 μm. (D) WT GFP-MCM10 or GFP-R582X MCM10 were transiently expressed in 293T cells and imaged by confocal microscopy with quantification of nuclear GFP (right). Mean ± 95% CI. ***P < 0.001, Kruskal-Wallis with Dunn’s multiple comparison test. n = 48 (WT); n = 55 (R426C); n = 40 (R582X). Scale bars: 10 μm. Data representative of 3 technical replicates.
The MCM10 c.1276C>T, p.R426C allele was predicted to be potentially damaging, but not to affect protein localization or stability, by PolyPhen2 prediction software (35). Expression and localization of WT and MCM10 R426C were comparable when examination was done with a bicistronic vector expressing full-length WT MCM10 with a GFP reporter in concert with MCM10 R426C with an mApple reporter (Supplemental Figure 3). The use of the bicistronic system also demonstrates stability of the R426C variant relative to WT MCM10, which we could not otherwise assume.
In budding yeast, Mcm10 plays a role in loading and stabilizing Polα at origins of replication (39–41). Furthermore, direct interaction of diubiquitinated MCM10 with PCNA is required for DNA elongation (42). Human MCM10 interacts directly with the CMG helicase through MCM2 and CDC45, supporting chromatin association of MCM10 and efficient firing and elongation during replication (28, 29). To further probe the effect of the R426C mutation on replisome formation, we immunoprecipitated WT MCM10 or MCM10 R426C using turboGFP and probed for POLA, MCM2, CDC45, and PCNA. The R426C variant did not affect the quantity of POLA, MCM2, and PCNA coimmunoprecipitated with MCM10, and we found a reproducible, but insignificant, increase of CDC45 binding to the R426C variant compared with WT MCM10 (Figure 3A). These data suggest that the R426C mutation does not impair formation of the replisome.
Figure 3. Independent damaging effects of R426C and R582X mutations.

(A) WT MCM10-GFP or R426C MCM10-GFP constructs transiently expressed in 293T cells. GFP was immunoprecipitated and blots probed for POLA, MCM2, PCNA, CDC45. (B) Whole cell extract (WCE) of parental (WT), R426C homozygous patient mutation (R426C/R426C), and R582X heterozygous patient mutation (R582X/+) hTERT RPE-1 cell lines probed for MCM10. Data are representative of 3 technical replicates. (C) Parental (WT), R426C homozygous patient mutation (R426C/R426C), and 3 R582X heterozygous mutation clones (R582X/+ 1, 2 and 3) counted after 72 hours to calculate doubling time. Data are represented as mean ± 95% CI. n = 3–6 technical replicates. Symbols directly over bars indicate significance of mutant compared with WT. (D) γH2AX imaged by confocal microscopy. Scale bar: 10 μm. (E) Mean number of γH2AX foci counted from cells treated with 20J UV or untreated. Data are represented as mean ± 95% CI; each point represents an independent technical replicate. n = 35–68 cells per condition. *P < 0.05; **P < 0.01; ****P < 0.0001, parametric 1-way ANOVA with multiple comparisons.
To further understand the effects of the patient mutations separately, we generated hTERT-RPE-1 cell lines homozygous for the R426C mutation or heterozygous for the R582X mutation. Homozygous lines for the R582X mutation could not be generated, as this is presumably a lethal null allele, given that MCM10 function is essential. Doubling time was assessed and compared with parental hTERT-RPE-1 cells. The R582X heterozygous mutation had a 50% reduction in MCM10 protein expression, as measured by Western blot, which was accompanied by 50% reduction in growth in 3 unique clones (Figure 3, B and C). The R426C homozygous cell line retained approximately 80% growth compared with WT, which is consistent with this mutation having a critical role in the patient’s phenotype, particularly when paired as a compound heterozygous allele with the more severe nonsense mutation (Figure 3C). This suggests that, in human cell lines, expression of the heterozygous R582X mutation leads to MCM10 haploinsufficiency. We also assessed the mutant cell lines for basal levels of DNA damage by immunostaining for γH2AX (Figure 3D). The parental and R426C homozygous cell lines had similar basal DNA damage, which increased upon treatment with 20J UV light (Figure 3E). The R582X heterozygous cell line exhibited slightly elevated basal DNA damage, but that was not statistically significant and did not increase following UV treatment (Figure 3E). Thus, the acute response to UV light exposure was not significantly altered in the individual mutant cell lines.
To specifically determine the effect of the patient MCM10 alleles in patient cells, SV-40 large T antigen transformation of primary dermal fibroblasts from the proband and an unrelated healthy donor was performed. Western blotting of fractionated cell lysates confirmed the presence of MCM10 in the nuclear fraction of patient-derived cells with slightly increased intensity relative to healthy controls (Figure 4A). In human cells, MCM10 is released from chromatin at the end of the S phase of the cell cycle (40). Given the increased intensity of MCM10 staining in the nucleus of patient-derived cells, we sought to further biochemically quantify the affinity of MCM10 binding to chromatin. We performed tight chromatin fractionation with differential salt extraction. At increased NaCl concentration (0.3M), we found MCM10 more tightly associated with chromatin in patient-derived cells when compared with healthy donor control cells (Figure 4B). Given that human MCM10 is normally released from DNA at the end of the S phase (40), increased chromatin association in patient cells is suggestive of aberrant cell cycle dynamics and therefore function of the complex containing variant MCM10.
Figure 4. Effect of compound heterozygous mutations.

Primary fibroblasts from the patient were generated by SV-40 large T antigen transformation. (A) Cells were fractionated as described in Methods, and nuclear and cytoplasmic fractions were probed for MCM10, lamin B1, and α-tubulin. (B) Chromatin fractionation was performed with increasing stringency of salt concentrations (0.15M, 0.3M) from patient fibroblasts immortalized by SV-40 large T antigen transduction. Lysates were probed for MCM10, lamin B1, or actin as a loading control. Intensity of bands from 0.3M condition is quantified relative to loading control. Data are represented as mean ± SD from 3 technical replicates performed on different days. MW, molecular weight marker; HD, healthy donor; Pt, patient.
Taken together, these data demonstrate the impact of each mutation in isolation, including undetectable expression of the R582X variant at endogenous levels and impaired release from chromatin of the R426C variant, which is accompanied by impaired growth in a nontransformed human cell line.
Patient cells have increased nuclear area and frequency of γH2AX foci.
Genomic integrity and repair of DNA damage require CMG helicase function. The previously reported hypomorphic mutations in MCM4 and GINS1 lead to abnormal nuclear morphology and increased genomic instability defined by metaphase breaks following aphidicolin treatment (5, 9). GINS1 mutations also lead to increased presence of DNA damage markers, including γH2AX and 53BP1 foci, in primary fibroblasts from patients (9). Using confocal microscopy, we visualized the presence of γH2AX foci in immortalized fibroblasts from the patient and a healthy donor (Figure 5A). We found a significantly higher frequency of γH2AX foci in patient cells, including increased intensity and area of γH2AX signal within the nucleus of patient cells (Figure 5, A and B). Counterstaining nuclei with DAPI also revealed increased nuclear area in patient cells (Figure 5, A and C), suggesting dysregulation of cell cycle and increased replication stress in patient cells, similar to that described for GINS1-deficient primary fibroblasts (9). Transient overexpression of the single mutants in 293T cells led to increased nuclear staining as a result of expression of the R426C variant, but not the R582X variant, further supporting the hypothesis that reduced CMG helicase function as a result of the R426C variant leads to replication stress and that this may be manifested as increased nuclear size (Figure 5D). Finally, we performed cell cycle analyses on immortalized patient fibroblasts with BrdU and 7-AAD. Examination of patient cells showed an increased number of cells found in the S phase, with an accompanying decrease of cells found in G2/M (Figure 5E). Taken together, these data suggest cell cycle defects and replication stress in patient-derived cells expressing both MCM10 variants.
Figure 5. R426C/R582X mutations lead to increased nuclear area and γH2AX staining in immortalized fibroblasts.

(A) Immortalized fibroblasts were fixed, permeabilized, and incubated with primary anti-MCM10 antibody followed by goat anti-mouse Alexa Fluor 488 secondary antibody and directly conjugated anti-γH2AX Alexa Fluor 647. Slides were mounted with ProLong Gold antifade media with DAPI and imaged by confocal microscopy. Scale bars: 10 μm. (B) MFI of γH2AX staining (left) and area of positive γH2AX signal (right) were measured in 28 to 35 cells per condition. ****P < 0.0001, unpaired t test (left) or Mann-Whitney U test (right). Data are represented as mean ± 95% CI. (C) Nuclear area was measured by positive DAPI staining in 31 (patient) and 46 (healthy donor) cells per condition. ****P < 0.0001, Mann-Whitney U test. Data are represented as mean ± 95% CI. (D) R426C or R582X variants were transiently overexpressed in 293T cells and prepared for microscopy, as described above. Nuclear area determined by DAPI staining was measured. *P < 0.05, Kruskal-Wallis with Dunn’s multiple comparison test. Data are represented as mean ± 95% CI. n = 69 (R426C); n = 95 (R582X, WT). (E) Healthy donor–derived (left) or patient-derived (right) immortalized fibroblast cells were labeled with BRDU and 7-AAD, and cell cycle was analyzed by FACS. All data shown are of 1 representative experiment of 3 technical replicates performed on different days.
Impaired cell cycle progression in MCM10-KD NK cells.
The effect of the R426C and R582X variants in patient cells was suggestive of impairment of CMG helicase function as a result of MCM10 mutation. To model MCM10 deficiency in an NK cell line using a complementary approach to patient cell analyses, we performed CRISPR/Cas9 gene editing of MCM10 in the NK92 human NK cell line. Intronic guides were designed to reduce, but not completely abolish, protein expression, as cells in which MCM10 was deleted were not viable (not shown). Single cell clones were isolated and expanded, and mRNA and protein expression were measured. One clone with approximately 10% protein expression of the parental line was primarily chosen for further study (clone 1), and 3 clones were verified with reduced protein expression accompanied by functional defects (Figure 6, A and B, and data not shown).
Figure 6. Cell cycle arrest in a cell line model of MCM10 KD.

MCM10 expression in NK92 cells was reduced by CRISPR/Cas9 gene editing, as described in Methods. Single clones were expanded and validated, and 1 was selected for further analysis. (A) RNA was isolated from WT NK92 or MCM10-KD cells and qPCR measurement of MCM10 mRNA was performed. Pooled data from 4 independent experiments done in quadruplicate. (B) WT and MCM10-KD NK92 cells were lysed and probed for MCM10 protein and actin as a loading control. Quantification of MCM10 protein normalized to loading control is shown below each lane. Data are representative of 3 technical replicates performed on different days. (C) WT and MCM10-KD NK92 cells were labeled with BRDU and 7-AAD, and cell cycle was analyzed by FACS. (D) Quantification of the frequency of cells in early S phase relative to the WT control is shown from 3 technical replicates performed on different days. *P < 0.05, 1-sample t test and Wilcoxon’s test. (E) Cell-doubling time was calculated by enumerating cells in culture. Data show 6 technical replicates performed on different days. **P < 0.01, Mann-Whitney U test. Data are represented as mean ± 95% CI.
To determine the functional effect of MCM10 KD, we performed cell cycle analyses in NK92 and NK92 MCM10-KD (MCM10-KD) cell lines using the verified clone described above (clone 1). These revealed a significant and consistent increase in the frequency of cells in early S phase with a decrease of cells in G2/M when compared with the control cell line (Figure 6, C and D). In addition, MCM10-KD cells had significantly increased doubling time (39.7 hours vs. 28.7 hours) (Figure 6E). Thus, the cell cycle profile of MCM10-KD NK92 cell lines was reminiscent of that seen in patient cells and suggested impairment in cell cycle progression, with a specific accumulation of cells detected at early S phase owing to a reduction in MCM10 function.
MCM10 KD leads to replication stress, but does not impair cytotoxicity.
To further define the role of MCM10 in NK cell genomic integrity, we determined the effect of MCM10 KD on replication stress in the NK92 cell line. NK92 MCM10-KD cells had greater number and intensity of γH2AX foci at baseline, as measured by confocal microscopy (Figure 7A). While the frequency of gH2AX-positive MCM10-KD cells was slightly increased relative to those of WT NK92 (Figure 7B), enumeration of γH2AX foci showed a significant increase in the number of γH2AX foci at baseline in MCM10-KD cells (Figure 7C). To further test responses to genotoxic stress, cells were irradiated and then rested for 24 hours before quantitative analysis of γH2AX, which demonstrated that the frequency of gH2AX foci was also significantly greater in response to irradiation in the MCM10-KD line (Figure 7D).
Figure 7. Increased replication stress in an MCM10-KD cell line.

WT or MCM10-KD NK92 cells were fixed, permeabilized, and incubated with anti-γH2AX Alexa Fluor 647. (A) Images were acquired by confocal microscopy. Scale bar: 5 μm. (B) The frequency of cells per field of view that were positive for γH2AX by microscopy was scored by manual counting. Shown are the means from 3 independent replicates performed on different days and normalized to WT NK92. NS, 1-sample t test and Wilcoxon’s test. Data are represented as mean ± 95% CI. (C) The number of γH2AX foci per cell were determined by manual counting of microscopy images. Shown are the means from 3 independent replicates performed on different days and normalized to WT NK92. n = 18–60 cells per condition. **P < 0.01, 1-sample t test and Wilcoxon’s test. Data are represented as mean ± 95% CI. (D) WT NK92 or NK92 MCM10-KD were irradiated with 2 Gy and allowed to recover for 24 hours before fixing and immunostaining for γH2AX. Images were acquired by confocal microscopy, and foci were enumerated by manual counting. Data shown are means from 3 technical replicates performed on different days normalized to WT NK92. n = 10–29 cells per condition. Data are represented as mean ± 95% CI. *P < 0.05, 1-sample t test and Wilcoxon’s test. (E) Cytotoxic function of WT NK92 or MCM10-KD cells against K562 targets was performed by 51Cr release assay. Representative data shown from 3 technical replicates performed on different days. E:T ratio, effector/target ratio. (F) CD56 expression on NK92 (solid line) or MCM10-KD (dashed line) NK92 cells was measured by FACS analysis. Data are representative of 3 technical replicates performed on different days.
The severe CMV infection in this patient suggested impaired NK cell cytotoxic function; however, this was not tested due to the limited availability of peripheral blood from the deceased patient. To determine whether NK92 lytic function was directly affected by MCM10 KD in the NK92 cell line, we performed 4-hour 51Cr release assays and found no difference between WT and MCM10-KD cells when cells were seeded at equal densities overnight before the assay to approximate cell cycle synchronicity (Figure 7E). Furthermore, expression of NK cell receptors, including CD56, were unaffected (Figure 7F). Thus, the roles of MCM10 in promoting effective replication and cell cycle progression defined in other cells can be extended to NK cells specifically. Further, the impact on primary NK cell function in the patient arose from aberrant development and not from a direct role for MCM10 in regulating cytotoxic function.
In vitro differentiation of MCM10-KD NK cells from CD34+ precursors.
While there was limited biological material available from our patient, flow cytometry of peripheral blood lymphocytes demonstrated profoundly reduced frequency of NK cells, with increased representation of the CD56bright subset. This was analogous to the NK cell phenotype seen in patients with MCM4 and GINS1 mutations (5, 9); however, the mechanism leading to this phenotype in any of these disorders is unknown. CD56bright NK cells are thought to be the precursors of the CD56dim subset, and previous studies suggest that hyperproliferation of the CD56bright subset is required for their terminal maturation (5, 9, 43). However, whether this defect occurs over time and reflects impaired NK cell homeostasis in the periphery or occurs during differentiation has been untested.
To determine the effect of MCM10 KD on NK cell terminal maturation, we performed CRISPR/Cas9 gene editing of CD34+ HSCs from a healthy donor to generate MCM10-KD precursors, which were used for the generation of mature NK cells by in vitro differentiation (7, 8, 44). CD34+ HSCs were isolated by FACS from apheresis product, and the CRISPR/Cas9 guide found to be most effective for the NK92 cell line or an empty vector containing GFP was nucleofected into purified HSCs. These were expanded for 3 days in TPO, IL-3, IL-7, and SCF, resorted to select GFP+ cells, and were then cultured in vitro in the presence of EL08.1D2 stromal cells to generate mature NK cells. Following 30 days of culture, cells were harvested and NK cell developmental stages were assessed by flow cytometry. NK cells derived from CD34+ HSCs transfected with empty vector underwent terminal maturation with increased frequency when compared with those transfected with MCM10 CRISPR guide constructs. Specifically, control NK cells upregulated CD16, whereas MCM10-deficient NK cells showed decreased CD16 signal (Figure 8A). Developmental subsets were quantified using previously described phenotypic markers that mark progression from stage 1 (CD34+ CD117− CD94−CD16−) through stage 2 (CD34+CD117+CD94−CD16−), then stage 3 (CD34−CD117+CD94−CD16−), stage 4 (CD34−CD117+/−CD94+CD16−), and finally stage 5 (CD34−CD117−CD94+/−CD16+) (45). There was an increase in stage 3/early stage 4 cells in MCM10-KD cells when compared with mock transfected cells. As suggested by the reduced CD16 expression on MCM10-KD cells, the increase in stage 3 cells was accompanied by a decrease in more mature stage 4 cells and stage 5 NK cells when compared with control cells (Figure 8B). In addition, a subset of cells remained in stage 2 under MCM10-KD conditions, whereas there were no detectable stage 2 cells under control conditions. Taken together, these data demonstrate that decreased expression of MCM10 leads to the NK cell phenotype observed in the peripheral blood of a patient with MCM10 mutations. The effect is therefore specific to MCM10 and not a feature of any other genetic influences in the patient, as the developmental abnormality can be recreated in healthy donor cells. Importantly, this distinctive phenotype can be recapitulated in vitro and suggests that progression through maturation is impaired at multiple stages of NK cell development and that intact MCM10 function is needed for NK cell maturation.
Figure 8. MCM10-KD in primary cells impairs NK cell maturation from CD34+ HSCs.

CD34+ HSC precursors were isolated from apheresed peripheral blood and transfected with MCM10 CRISPR/Cas9-GFP. After 3 days of expansion, GFP+ or GFP– cells were sorted and then cocultured with EL08.1D2 stromal cells in the presence of cytokines, including IL-15, as described in Methods. Cells were harvested at 28 days, and NK cell maturation was analyzed by FACS. (A) Representative histograms of CD16 expression as a marker of NK cell terminal maturation. (B) Relative frequency of cells according to defined stages of NK cell maturation: stage 1 (CD34+CD117−CD94−CD16−); stage 2 (CD34+CD117+CD94−CD16−); stage 3 (CD34−CD117+CD94−CD16−); stage 4 (CD34−CD117+/−CD94+CD16−); and stage 5 (CD34−CD117−CD94+/−CD16+) (45). Data are representative of 2 biological replicates.
Generation of patient-derived NK cells in a humanized mouse model.
Finally, we sought to definitively link patient mutations with the NK cell developmental phenotype generated in vitro and observed in the limited ex vivo patient samples. While there were no remaining preserved peripheral blood cells from the deceased patient, we reprogrammed primary patient fibroblasts using a footprint-free modified RNA method to generate iPS cell lines. An iPS cell line from a healthy donor was generated in parallel. We found both patient and control iPS cell lines to express pluripotency markers and undergo differentiation into 3-germ layers.
To test the effect of patient mutations on NK cell development in an in vivo model, we reconstituted humanized mice with CD34+ precursors generated from patient or healthy donor iPS cells. For production of CD34+ cells, teratomas were generated, harvested, and purified. These were injected into NSG mice along with OP9w3a feeder cells and were allowed to develop for 21 days. Tissues were harvested and evaluated by flow cytometry using a human NK cell developmental marker phenotype panel. While the frequency of total human CD56+CD3− NK cells was comparable to or higher than that of mice reconstituted with patient-derived cells (Supplemental Figure 4), there was a consistent enrichment of the CD56bright subset within engrafted human NK cells in the blood and spleen from all 4 patient-generated mice (Figure 9, A and B). Further, confocal microscopy showed an increase in the number of γH2AX foci in enriched human NK cells from mice reconstituted with patient-derived iPS cells (Figure 9C), reminiscent of the increase in foci seen in NK92 MCM10-KD and MCM10 variant-expressing cell lines. This was significantly greater when all 4 mice were taken into account (Figure 9D). Therefore, using complementary mechanisms of modeling patient mutations and MCM10 insufficiency, we demonstrate that MCM10 function is required for the generation of terminally mature NK cell subsets both in vitro and in vivo and that biallelic variants can result in a human NKD.
Figure 9. iPS cell–derived NK cells from the proband have impaired terminal maturation and increased replication stress.

iPS cells reprogrammed from patient primary fibroblasts were differentiated by teratoma formation to CD34+ HSCs, purified, and transplanted into NSG mice as described in Methods. Organs were harvested 21 days following transplantation, and human CD45+CD56+CD3− cells were analyzed for density of CD56 expression. n = 4 mice per genotype (patient and healthy donor–derived iPS cells). (A) Representative FACS histograms of NK cells from bone marrow of mice reconstituted with human NK cells from healthy donor– or patient-derived CD34+ cells generated from iPS cells. (B) Frequency of CD56bright NK cells from 4 mice per genotype from blood, spleen, and bone marrow, as indicated. NK cells were identified as human CD45+CD56+CD3− (bone marrow: 93–979 NK cells; blood: 20–405 NK cells; spleen: 60–880 NK cells, all from >106 cells from each organ per mouse), and the frequency of CD56bright NK cells based on CD56 density within the human NK cell population is shown. *P < 0.05, Mann-Whitney U test. (C) Splenocytes from mice transplanted with HD or patient-specific iPS cell–derived CD34+ cells were fixed, permeabilized, and incubated with anti-γH2AX antibody. Images were acquired by confocal microscopy. Scale bar: 5 μm. (D) Frequency of γH2AX foci per cell were enumerated by manual counting of 109 (HD) and 119 (patient) cells. ****P < 0.0001, Mann-Whitney U test. Data were pooled from 4 mice per genotype and are represented as mean ± 95% CI.
Discussion
The study of human NKD provides important insights into the requirements for NK cell differentiation and maturation, particularly when NK cells are the primarily affected lymphocyte subset. To date, 5 monogenic causes of NKD affecting development have been described in the literature. Notably, 2 of these, MCM4 and GINS1 deficiencies, are a result of mutations that destabilize the CMG complex and lead to reduced numbers of peripheral blood NK cells with relative overrepresentation of the CD56bright and reduced frequency of CD56dim NK cells (5, 6, 9). While other immune subsets, including neutrophils, innate lymphoid cells (ILCs), and T cells, may also be affected to varying degrees (9), the profound NK cell–intrinsic defects observed in these rare cases and their association with the patient’s clinical phenotype define these monogenic discorders as NKD.
Here, we report what we believe to be a novel classical NKD as a result of compound heterozygous mutations in MCM10. By modeling MCM10 deficiency in a human NK cell line, we demonstrate that MCM10 function is required for fidelity of cell cycle progression and that decreased MCM10 function leads to increased replication stress. In addition, we recapitulate the NK cell phenotype observed in our proband using CRISPR/Cas9-mediated modeling of MCM10 deficiency, followed by NK cell in vitro differentiation, and a humanized mouse model of NK cell differentiation from patient iPS cells. These approaches show for what we believe is the first time that impaired function of a eukaryotic replisome complex member can directly lead to the NK cell phenotype observed in peripheral blood of patients with CMG complex mutations, namely, reduced NK cell number and increased representation of the CD56bright subset. While the extremely limited access to peripheral blood cells from this patient made it difficult to perform extended analyses, we demonstrate that the initial observation of nearly absent NK cells in the periphery with a relative overrepresentation of CD56bright NK cells is recapitulated by our modeling of MCM10 deficiency.
The clinical hallmark of NKD is susceptibility to viral infections, particularly those of the herpesviral family, and the remarkable susceptibility of the proband to CMV prompted investigation into NK cell phenotype and function. This evaluation revealed a profoundly reduced frequency of NK cells in peripheral blood (<1%). In addition, the phenotype of NK cells in peripheral blood was reflective of impaired terminal maturation or survival of terminally mature subsets, defined by substantive overrepresentation of the CD56bright subset relative to CD56dim NK cells. Exome sequencing of the patient and his immediate family identified variants in MCM10 that segregated appropriately and were predicted to be highly damaging. The novelty and predicted damage of these mutations, taken together with the consistency of the NK cell phenotype reported previously for MCM4 and GINS1 deficiencies (5, 6, 9), made MCM10 the strongest gene candidate in this patient.
MCM10 is a critical functional regulator of the CMG complex, and here, we demonstrate that both the R426C and R582X mutations in concert are damaging (albeit in different ways) and lead to replication stress and impaired cell cycle progression. Specifically, we show that the R582X variant, which introduces a premature stop codon, presumably leads to nonsense-mediated decay. While the mechanistic impact of the R426C mutation is less clear, our data suggest that it affects cell proliferation, possibly by impairing origin firing and/or replication elongation and the resulting inefficiency. This is manifested by increased retention of MCM10 on chromatin and impaired cell growth. The effects of both mutations are increased accumulation of cells in early S phase and replication stress indicated by increased phosphorylation of γH2AX. The slightly increased retention of MCM10 on chromatin under high-salt conditions when isolated from patient cells is compatible with altered replication dynamics despite intact interactions with the MCM2-7 complex and Polα. While overexpression of the R582X mutant allele demonstrated its nuclear exclusion, we failed to detect truncated protein in lysates from patient cells by Western blotting. This suggests that MCM10 R582X protein is not expressed at endogenous levels. While quantitative measurement of protein levels did not identify differences in expression of full-length protein between the patient and healthy donors, the cell cycle–dependent changes in expression of MCM10 also make it difficult to draw quantitative conclusions based on this measurement. With relative expression of MCM10 highest in S phase (29), it is likely that cell cycle–dependent protein regulation affects the overall expression of MCM10 in patient cells, which we have shown are more frequently detected in S phase than healthy donor control cells.
The addition of MCM10 deficiency as an NKD strengthens the importance of the CMG complex and DNA replicative machinery in human NK cell functional maturation. With highly conserved NK cell and clinical phenotypes, patients with MCM4, GINS1, and MCM10 mutations have illustrated the importance of the eukaryotic DNA helicase in NK cell development. Despite the effect of these mutations on cell cycle progression and DNA damage response in extra-immune cell types, the most substantive clinical phenotype in these patients is unusually severe viral susceptibility with accompanying NKD. CD56bright NK cells from patients with GINS1 or MCM4 mutations fail to proliferate in response to IL-2, whereas the CD56dim subset undergoes proliferation at rates similar to those of controls from healthy donors (5). In addition, while less well described, the presence of NK cell impairment in patients with POLE1 (46) and POLE2 mutations (47) and a single patient with RTEL1 mutations (10, 11) suggests that these other regulators of DNA replication and damage repair may similarly function to regulate NK cell homeostasis.
It is remarkable that NK cells are seemingly profoundly sensitive to loss-of-function mutations in these proteins, and the outstanding question remains as to why this is the case. Mouse models demonstrate that complete deletion of MCM genes, including Mcm4 (26) or Mcm10 (48), leads to embryonic lethality. Hypomorphic alleles of Mcm4 (Mcm4chaos3/chaos3), however, lead to defects in cell proliferation and cancer susceptibility (26), and while milder than what exists in MCM4 patient phenotypes, decreased frequencies of NK cells are found in these mice (5). Relatively decreased CMG complex expression in stem cells, which occurs with aging, leads to increased replicative stress and decreased ribosomal function (49). Overexpression or haploinsufficiency causes similar defects (50), demonstrating the importance of tuned levels of protein expression in CMG helicase function.
Tightly controlled CMG complex protein expression is linked to the importance of replicative helicase components to both initiate replication and repair stalled forks if necessary (30). Therefore, it may be that cells with a critical requirement for rapid proliferation, including T cells, have higher production of these proteins and thus are more tolerant of loss of function than NK cells. In support of this, both effector T cells and germinal center B cells have significantly higher expression of MCM10 than mature NK cells in peripheral blood (51). The potential for varying availability of functional CMG protein to differentially affect different lymphocyte subsets makes it conceivable that there are more damaging mutations that lead to combined immune deficiency and have not been detected; similarly, these may not be supportive of viability. In addition, the transient T cell lymphopenia observed in GINS1-deficient patients and the impaired T cell differentiation noted among our patient’s PBMCs further suggest that, depending on the severity of the mutation, there may be variable effects on other lymphocyte subsets. However, we propose that the particular mutations that cause NKD fall within a window that primarily affects NK cells, rendering the NK cell–mediated defense inadequate, thus leading to susceptibility to viral infections, particularly of the herpesvirus family. As such, the NK cell abnormality would be the main clinically relevant immune deficiency. This is supported by the finding that the clinical severity of disease correlates with the relative levels of GINS complex expression in GINS1-deficient individuals (9). Finally, while the immune compartment in these patients is highly affected, there is an impact of these mutations on cell cycle and DDR pathways in fibroblasts. While some MCM4 patients report malignancies (5), unfortunately, many of these patients succumb to viral infections at an unusually young age, perhaps obscuring unusual cancer susceptibility.
As in the case of MCM4 mutations, unlike in GINS1-deficient patients, neutrophil numbers were normal in our patient. As such, there are subtle differing effects of CMG mutations on both the clinical manifestations and immune cell phenotypes in patients with MCM4, MCM10, and GINS1 deficiencies. Adrenal insufficiency was reported in MCM4 patients, but has not been detected in GINS1 patients or in the patient reported here with MCM10 deficiency (and our patient did not have intrauterine growth abnormalities). It is unclear whether these differences reflect nonredundant roles for CMG complex proteins or whether they are indicative of differing severity of mutations leading to varying effects on different cell lineages. While ILC subsets were not examined in our patient due to limited availability of patient material, GINS1, but not MCM4, patients had loss of ILC subsets in peripheral blood, again reflecting either a more conserved requirement for GINS1 in neutrophil and ILC development in addition to that of NK cells or reflecting a more severe mutational burden that led to this phenotype.
Regardless of their differences, the conserved impact on NK cells is a notable feature of CMG helicase mutations, especially in light of this being the third independent association of NKD with the CMG complex. Given the impaired proliferation and increased apoptosis of the CD56bright subset, it seems that impaired generation of the CD56dim NK cell subset in particular is a feature of the NK cells found in peripheral blood of MCM4- and GINS1-deficient patients (5, 9). Here, we have described an additional patient fitting this paradigm, but with biallelic MCM10 mutations associated with reduced NK cell numbers in the periphery and overrepresentation of the CD56bright subset. Notably, we show that this particular NK cell phenotype can be recapitulated using in vitro NK cell differentiation from MCM10 KD CD34+ precursors or the generation of patient NK cells from iPS cells in a humanized mouse model. There does not appear to be a specific deficit in the numbers of NK cells present in the blood, bone marrow, or spleen of mice reconstituted with CD34+ cells derived from patient iPS cells relative to healthy donors. These data suggest that either a survival defect of the CD56dim subset is manifested within the relatively short time frame of these experiments or there is impaired generation of these cells within these systems. Interestingly, the highest expression of MCM complex components is in stem cells, and CD34+ HSCs, as well as other progenitors, have significantly increased expression of MCM10 relative to other cells of hematopoietic and immune origin (51). Our data using in vitro differentiation show that progression through early stages of NK cell development is also impaired, suggesting that the requirement for proliferation is not restricted to later stages of maturation, but limits access to it.
Finally, there may be additional functions mediated by the CMG helicase that contribute to the NK cell phenotype in these patients. MCM5 binds directly to the STAT1α promoter and mediates direct transcriptional control of IFN-γ response genes (52). Alternatively, activation of DDR pathway–associated genes in murine NK cells, including Chk2 and Atm, is mediated by RAG recombinase expression. NK cells from RAG-deficient mice have perturbed induction of DDR pathway genes, increased apoptosis, and impaired NK cell proliferative response to viral infection, suggesting that there may be a direct role for the DDR pathway in the generation or survival of mature effector NK cells (53). Careful genetic and functional analyses of the effect of these mutations on gene expression throughout NK cell development will be important for definitively elucidating the nature of these deficiencies on NK cell maturation and function. In addition, fully understanding the effect of such mutations on adaptive NK cells may shed additional light on the requirement for proliferation in their generation and the role that the adaptive subset plays in viral control. Unfortunately, we were extremely limited in our access to cells from the proband, and no peripheral blood cells were cryopreserved. We were therefore limited in our ability to directly test patient NK cell phenotype or function, although we were fortunate to have access to tissue for the generation of the primary fibroblast line and, subsequently, the iPS cells from the patient. Given the substantial expansion of adaptive NK cells during viral infection (54), we would predict that this response would be specifically impaired in the proband; however, we were unable to directly test this hypothesis.
In summary, here we define a classical NKD as a result of biallelic mutations in MCM10 leading to impaired generation of terminally mature NK cells and loss of NK cell function. While a full mechanistic understanding of the role of the CMG complex in human NK cell development and function remains to be elucidated, the biology demonstrated by these rare patients is extremely compelling. Although they are rare, NKDs provide invaluable insight into requirements for human NK cell differentiation and function. The discovery of MCM10 as an NKD gene strengthens the importance of intact function of the eukaryotic helicase complex in human NK cell functional maturation, with this representing the third such association. In addition, through our modeling of MCM10 insufficiency in NK cell development, we provide important mechanistic insight into the underlying immunological and biological cause of these defects.
Methods
Animal studies.
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ mice were purchased from The Jackson Laboratory and housed in pathogen-free conditions in the vivarium at Texas Children’s Hospital.
Exome sequencing and analysis.
Exome sequencing was performed on genomic DNA as part of the primary immunodeficiency cohort study at Baylor-Hopkins Center for Mendelian Genomics (BHCMG, https://mendeliangenomics.org/) (31, 55). Further details of sequencing and analysis are provided in Supplemental Methods.
Cell isolation, cell lines, and transfection.
Primary PBMCs were isolated by Ficoll-Paque density centrifugation. CD34+ cells for in vitro differentiation were isolated from discarded apheresis product from routine red cell exchange at Texas Children’s Hospital. NK92 cells (ATCC) were maintained in MyeloCult medium supplemented with penicillin/streptomycin and 100 U/mL IL-2 (Roche). EL08.1D2 stromal cell lines were a gift from J.S. Miller (University of Minnesota, Minneapolis, Minnesota, USA) and were maintained as previously described (44). K562 target cells were a gift from J.L. Strominger (Harvard University, Boston, Massachusetts, USA) and were maintained in complete RPMI medium with 10% fetal calf serum. 293T and hTERT RPE-1 lines were from ATCC. OP9w3a cells were a gift from P. Kincade (University of Oklahoma Health Sciences Center, Oklahoma City, Oklahoma, USA). NK92 and CD34+ cells were gene edited, as described in Supplemental Methods.
Plasmids.
For generation of 805X, 771X, 705X, and 607X truncation mutations in 293T cells, the MCM10 coding sequence in the pDONR201 (Invitrogen) was used to generate truncation mutants using the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent). The full-length and truncation sequences were transferred into a Gateway compatible destination vector that included an N-terminal triple-epitope tag (S protein tag, FLAG epitope tag, and streptavidin-binding peptide tag) using the LR Clonase Enzyme Kit (Thermo Fisher). Plasmids were transfected into 293T cells using FuGENE Transfection Reagent (Promega), and stable cell lines were selected using puromycin.
For generation of patient MCM10 variants and expression in 293T cells full-length MCM10 (NM_018518.5) was subcloned from pCMV6-X6 (Origene) into pLENTI-N-GFP. Site-directed mutagenesis was performed to generate missense (c.1276C>T, p.[Arg426Cys]) or truncation (c.1744C>T, p.[Arg582*]) mutations. 293T cells were transiently transfected using FUGENE6, and microscopy was performed 48 hours after transfection.
qPCR.
RNA was extracted from WT or NK92 MCM10-KD cells using the RNeasy Mini Kit (QIAGEN). First-strand cDNA was generated using Superscript VILO MasterMix (Thermo Fisher). Quantitative PCR (qPCR) for MCM10 was performed in quadruplicate on a Roche LightCycler with 25 ng of cDNA using TaqMan assay probe Hs00218560_m1 (Thermo Fisher) or GAPDH as a housekeeping gene (Hs02786624_m1).
Western blots.
Western blots of patient cells and cell lines were performed as described in Supplemental Methods. All uncut gels are available in the supplemental material.
Doubling-time assay.
Cell doubling time was determined by plating 3 × 104 cells per condition in a 6-well plate and counting 72 hours later. These conditions were established to not allow cells to become greater than 90% confluent during this assay to prevent contact inhibition from affecting doubling-time analysis. Statistical analysis and graphical representation were completed using Prism 8.4.1.
Cell cycle analyses.
For analysis of cell cycle, NK92 cells or primary patient fibroblasts were incubated with BrdU for 12 hours before fixation and staining with 7-AAD (BD Biosciences). Cells were analyzed via FACS on a BD Fortessa with 18-color configuration, and the resulting data were evaluated using Kaluza (Beckman Coulter) or FlowJo (BD Biosciences) software. Quantification of cells found in different cell cycle stages was performed by gating analyses using the flow cytometric data.
Cytotoxicity assays.
NK92 or MCM10-KD cell lines were seeded at equal density before overnight culture to approximate cell cycle synchronicity, then were counted and incubated with 104 K562 target cells that had been preincubated with 100 μCi 51Cr for 4 hours at 37°C 5% CO2; 1% IGEPAL (v/v) (MilliporeSigma) was used to lyse maximal release control wells, and plates were centrifuged. Supernatant was transferred to a LUMA plate (PerkinElmer) and dried overnight. Plates were read with a TopCount NXT and percentage of specific lysis was calculated as follows: (sample – average spontaneous release)/(average total release – average spontaneous release) × 100.
Microscopy and image analysis.
Confocal microscopy of patient fibroblasts, transiently infected and stable cell lines, and iPS cell–derived NK cells was performed as described in Supplemental Methods.
In vitro NK cell differentiation.
CD34+ precursors were isolated by FACS sorting from discarded apheresis collections from Texas Children’s Hospital. Initial lineage depletion was performed using RosetteSep enrichment (Stem Cell Technologies), followed by incubation with anti-CD34 PE (4H11, eBioscience), anti-CD56 Brilliant Violet 605 (HCD56, BioLegend), and anti-CD3 Brilliant Violet 711 (SK7, BioLegend) antibodies. Cells were gated on the CD56−CD3−CD34+ population and collected. Following collection, CD34+ cells were nucleofected with the MCM10 CRISPR guide with the GFP tag described above or a GFP-expressing negative control, then maintained in IL-3, thrombopoietin, IL-6, and IL-7 for 3 days (44). Cells were sorted for GFP positivity, and 2000 cells were cocultured with EL08.1D2 stromal cells for 28 days in the presence of IL-3 (first week only), IL-6, IL-7, Flt3L, SCF, and IL-15 (44). Following coculture, lymphocytes were analyzed by FACS for cell surface receptors associated with NK cell development, as described in Supplemental Methods.
In vitro iPS cell generation.
Dermal fibroblasts were expanded and reprogrammed into iPS cells using a nonintegration method based on modified mRNA encoding Oct4, Klf4, Sox2 c-myc, Lin28, and control GFP (56, 57) using StemRNA repogramming (Stemgent). Reprogrammed cells were validated as described in Supplemental Methods.
In vivo HSC generation.
To generate human HSCs, 2 × 106 iPS cells were mixed with 106 OP9w3a that expressed WNT3a, and the cell mixture was injected into female 4- to 8-week-old NSG mice for teratoma formation (58). In order to create niche space for transplanted human HSCs, 1-day-old NSG or Sirp1a pups received a sublethal dose of 100 cGy of x-ray irradiation in a biological irradiator. iPS cells and Op9W3a cells were detached from culture flasks using 0.25% trypsin-EDTA when they reached 80% confluence. Cells were washed with PBS and resuspended in PBS + 2% FBS. Cells were counted, and iPS cells were mixed with Op9W3a cells at a 2:1 ratio and resuspended in PBS + 2% FBS so that the final volume was 100 μL per mouse to be injected. NSG mice were injected with 2 million iPS cells and 1 million Op9W3a cells (3 million total cells in 100 μL solution) subcutaneously into the dorsal neck region.
Human CD34+ HSC magnetic column purification and reconstitution of NSG mice.
From 8 to 12 weeks after injection of OP9w3a-secreting and iPS cells, CD34+ HSCs were isolated from teratomas of recipient NSG mice as described in Supplemental Methods. Reconstituted mice were sacrificed 21 days after injection of CD34+ cells, and cells were harvested and prepared for flow cytometry as described in Supplemental Methods.
Analysis of NK cells from humanized mice by flow cytometry.
Cells were isolated as described above and kept on ice for all immunostaining. Human NK cells were detected by flow cytometry as described in Supplemental Methods.
Statistics.
See Supplemental Methods.
Study approval.
All mice were cared for in the animal facilities of the Center for Comparative Medicine at Baylor College of Medicine and Texas Children’s Hospital, and all protocols were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee. All human studies were performed in accordance with the Declaration of Helsinki with the written and informed consent of all participants under the guidance of the Children’s Hospital of Philadelphia (Philadelphia, Pennsylvania, USA), Baylor College of Medicine, Columbia University, and University Hospital of Wales.
Author contributions
EMM, JSO, AKB, SP, and JRL designed experiments and interpreted data. EMM, JSO, AKB, and SP wrote the manuscript. MIC, RMB, MMS, ST, AEP, NCG, M Mukherjee, SP, PPB, and SLP performed experiments and analyzed data. Exome sequencing and data analysis were performed as part of immunodeficiency cohort testing at BHCMG, and the proband in the current publication is individual 68.1 in ref. 31, in which MCM10 was designated potential novel gene 4. IKC, ZCA, SNJ, ASP, RAG, and DMM performed exome sequencing and exome data analyses. SJ, PPC, AGH, M Moody, CS, and REB guided clinical evaluation of the patient and supervised clinical care. SP conceptualized, designed, and performed experiments for reconstitution of NSG mice with patient iPS cells. MB and JC designed, performed, and validated reprogramming of patient cells to iPS cells.
Supplementary Material
Acknowledgments
This work was supported by NIH R01AI120989 (to JSO), NIH R01AI37275 (to EMM), NIH GM074917 (to AKB), NIH T32-CA009138 (to RMB), NIH National Center for Advancing Translational Sciences TL1R002493 and UL1TR002494 (MMS) and the University of Minnesota Imaging Centers, NIH Medical Scientist Training Program (MSTP) grant T32 GM008244 (to MMS), and National Science Center POLONEZ grant 2015/19/P/NZ3/03452 (to MB). JRL is supported in part by the NIH National Institute of Neurological Disorders and Stroke (R35NS105078) and the National Human Genome Research Institute/National Heart, Lung, and Blood Institute (UM1 HG006542 to the BHCMG). The authors wish to acknowledge Blake Heath, Jansen Smith, Laura Angelo, and Emily Haines for technical assistance with the development and experimentation on the humanized mouse model and Diane Yang for technical assistance with iPS cell generation and validation.
Version 1. 08/31/2020
Electronic publication
Version 2. 10/01/2020
Print issue publication
Footnotes
M Mukherjee’s present affiliation is: Merck, Philadelphia, Pennsylvania, USA.
AEP’s present affiliation is: Department of Dermatology, Baylor College of Medicine, Houston, Texas, USA.
ST’s present affiliation is: University of Chicago Pritzker School of Medicine, Chicago Illinois, USA.
PPB’s present affiliation is: MD Anderson Cancer Center, Houston, Texas, USA.
Conflict of interest: Baylor College of Medicine and Miraca Holdings Inc. have formed a joint venture with shared ownership and governance of Baylor Genetics (BG), which performs clinical microarray analysis and clinical exome sequencing. JRL serves on the scientific advisory board of BG. JRL has stock ownership in 23andMe, is a paid consultant for Regeneron Pharmaceuticals, and is a coinventor on multiple US and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting (US-8129353-B2, JP-2016025868-A, WO-02051981-A3, EP-0610396-B1, AU-633495-B2, US-5294533-A, AU-6538698-A, US-5306616-A, US-6132954-A, AU-2747802-A, AU-2226592-A, WO-9000624-A1).
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(10):5272–5286.https://doi.org/10.1172/JCI134966.
Contributor Information
Emily M. Mace, Email: em3375@cumc.columbia.edu.
Silke Paust, Email: paust@scripps.edu.
Ryan M. Baxley, Email: baxle002@umn.edu.
Sagar L. Patil, Email: sagarlpatil1@gmail.com.
Nicole C. Guilz, Email: ncm2146@cumc.columbia.edu.
Malini Mukherjee, Email: mmukherj@bcm.edu.
Ashley E. Pezzi, Email: aepezzi@gmail.com.
Jolanta Chmielowiec, Email: jolanta.chmielowiec@student.uwm.edu.pl.
Swetha Tatineni, Email: Swetha.Tatineni@uchospitals.edu.
Ivan K. Chinn, Email: ivan.chinn@bcm.edu.
Zeynep Coban Akdemir, Email: Zeynep.CobanAkdemir@bcm.edu.
Mo Moody, Email: mo.moody77@gmail.com.
Richard A. Gibbs, Email: agibbs@bcm.edu.
Malgorzata Borowiak, Email: borowiak@bcm.edu.
Stephen Jolles, Email: jollessr@cardiff.ac.uk.
References
- 1.Orange JS. Human natural killer cell deficiencies. Curr Opin Allergy Clin Immunol. 2006;6(6):399–409. doi: 10.1097/ACI.0b013e3280106b65. [DOI] [PubMed] [Google Scholar]
- 2.Orange JS. Natural killer cell deficiency. J Allergy Clin Immunol. 2013;132(3):515–525. doi: 10.1016/j.jaci.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ham H, Billadeau DD. Human immunodeficiency syndromes affecting human natural killer cell cytolytic activity. Front Immunol. 2014;5:2. doi: 10.3389/fimmu.2014.00002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Voss M, Bryceson YT. Natural killer cell biology illuminated by primary immunodeficiency syndromes in humans. Clin Immunol. 2017;177:29–42. doi: 10.1016/j.clim.2015.11.004. [DOI] [PubMed] [Google Scholar]
- 5.Gineau L, et al. Partial MCM4 deficiency in patients with growth retardation, adrenal insufficiency, and natural killer cell deficiency. J Clin Invest. 2012;122(3):821–832. doi: 10.1172/JCI61014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hughes CR, et al. MCM4 mutation causes adrenal failure, short stature, and natural killer cell deficiency in humans. J Clin Invest. 2012;122(3):814–820. doi: 10.1172/JCI60224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mace EM, et al. Biallelic mutations in IRF8 impair human NK cell maturation and function. J Clin Invest. 2017;127(1):306–320. doi: 10.1172/JCI86276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mace EM, et al. Mutations in GATA2 cause human NK cell deficiency with specific loss of the CD56(bright) subset. Blood. 2013;121(14):2669–2677. doi: 10.1182/blood-2012-09-453969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cottineau J, et al. Inherited GINS1 deficiency underlies growth retardation along with neutropenia and NK cell deficiency. J Clin Invest. 2017;127(5):1991–2006. doi: 10.1172/JCI90727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Etzioni A, Eidenschenk C, Katz R, Beck R, Casanova JL, Pollack S. Fatal varicella associated with selective natural killer cell deficiency. J Pediatr. 2005;146(3):423–425. doi: 10.1016/j.jpeds.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 11.Hanna S, Béziat V, Jouanguy E, Casanova JL, Etzioni A. A homozygous mutation of RTEL1 in a child presenting with an apparently isolated natural killer cell deficiency. J Allergy Clin Immunol. 2015;136(4):1113–1114. doi: 10.1016/j.jaci.2015.04.021. [DOI] [PubMed] [Google Scholar]
- 12.Cooper MA, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood. 2001;97(10):3146–3151. doi: 10.1182/blood.V97.10.3146. [DOI] [PubMed] [Google Scholar]
- 13.Angelo LS, et al. Practical NK cell phenotyping and variability in healthy adults. Immunol Res. 2015;62(3):341–356. doi: 10.1007/s12026-015-8664-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romagnani C, et al. CD56brightCD16- killer Ig-like receptor- NK cells display longer telomeres and acquire features of CD56dim NK cells upon activation. J Immunol. 2007;178(8):4947–4955. doi: 10.4049/jimmunol.178.8.4947. [DOI] [PubMed] [Google Scholar]
- 15.Chan A, et al. CD56bright human NK cells differentiate into CD56dim cells: role of contact with peripheral fibroblasts. J Immunol. 2007;179(1):89–94. doi: 10.4049/jimmunol.179.1.89. [DOI] [PubMed] [Google Scholar]
- 16.Huntington ND, et al. IL-15 trans-presentation promotes human NK cell development and differentiation in vivo. J Exp Med. 2009;206(1):25–34. doi: 10.1084/jem.20082013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCullar V, et al. Mouse fetal and embryonic liver cells differentiate human umbilical cord blood progenitors into CD56-negative natural killer cell precursors in the absence of interleukin-15. Exp Hematol. 2008;36(5):598–608. doi: 10.1016/j.exphem.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Collins PL, et al. Gene regulatory programs conferring phenotypic identities to human NK cells. Cell. 2019;176(1-2):348–360.e12. doi: 10.1016/j.cell.2018.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickinson RE, et al. The evolution of cellular deficiency in GATA2 mutation. Blood. 2014;123(6):863–874. doi: 10.1182/blood-2013-07-517151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maciejewski-Duval A, et al. Altered chemotactic response to CXCL12 in patients carrying GATA2 mutations. J Leukoc Biol. 2016;99(6):1065–1076. doi: 10.1189/jlb.5MA0815-388R. [DOI] [PubMed] [Google Scholar]
- 21.Schlums H, et al. Adaptive NK cells can persist in patients with GATA2 mutation depleted of stem and progenitor cells. Blood. 2017;129(14):1927–1939. doi: 10.1182/blood-2016-08-734236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eidenschenk C, et al. A novel primary immunodeficiency with specific natural-killer cell deficiency maps to the centromeric region of chromosome 8. Am J Hum Genet. 2006;78(4):721–727. doi: 10.1086/503269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Riordan SM, Lynch SA, Hindmarsh PC, Chan LF, Clark AJ, Costigan C. A novel variant of familial glucocorticoid deficiency prevalent among the Irish Traveler population. J Clin Endocrinol Metab. 2008;93(7):2896–2899. doi: 10.1210/jc.2008-0034. [DOI] [PubMed] [Google Scholar]
- 24.Bernard F, et al. A novel developmental and immunodeficiency syndrome associated with intrauterine growth retardation and a lack of natural killer cells. Pediatrics. 2004;113(1 Pt 1):136–141. doi: 10.1542/peds.113.1.136. [DOI] [PubMed] [Google Scholar]
- 25.Tye BK. MCM proteins in DNA replication. Annu Rev Biochem. 1999;68:649–686. doi: 10.1146/annurev.biochem.68.1.649. [DOI] [PubMed] [Google Scholar]
- 26.Shima N, et al. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat Genet. 2007;39(1):93–98. doi: 10.1038/ng1936. [DOI] [PubMed] [Google Scholar]
- 27.Thu YM, Bielinsky AK. Enigmatic roles of Mcm10 in DNA replication. Trends Biochem Sci. 2013;38(4):184–194. doi: 10.1016/j.tibs.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Perna R, et al. The physical interaction of Mcm10 with Cdc45 modulates their DNA-binding properties. Biochem J. 2013;454(2):333–343. doi: 10.1042/BJ20130059. [DOI] [PubMed] [Google Scholar]
- 29.Izumi M, et al. The Mcm2-7-interacting domain of human mini-chromosome maintenance 10 (Mcm10) protein is important for stable chromatin association and origin firing. J Biol Chem. 2017;292(31):13008–13021. doi: 10.1074/jbc.M117.779371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baxley RM, Bielinsky AK. Mcm10: a dynamic scaffold at eukaryotic replication forks. Genes (Basel) 2017;8(2):E73. doi: 10.3390/genes8020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Stray-Pedersen A, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol. 2017;139(1):232–245. doi: 10.1016/j.jaci.2016.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharma A, Kaur M, Kar A, Ranade SM, Saxena S. Ultraviolet radiation stress triggers the down-regulation of essential replication factor Mcm10. J Biol Chem. 2010;285(11):8352–8362. doi: 10.1074/jbc.M109.041129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coban-Akdemir Z, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain-of-function alleles. Am J Hum Genet. 2018;103(2):171–187. doi: 10.1016/j.ajhg.2018.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schwarz JM, Cooper DN, Schuelke M, Seelow D. MutationTaster2: mutation prediction for the deep-sequencing age. Nat Methods. 2014;11(4):361–362. doi: 10.1038/nmeth.2890. [DOI] [PubMed] [Google Scholar]
- 35.Adzhubei IA, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46(3):310–315. doi: 10.1038/ng.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karczewski KJ, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581(7809):434–443. doi: 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heller RC, Kang S, Lam WM, Chen S, Chan CS, Bell SP. Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell. 2011;146(1):80–91. doi: 10.1016/j.cell.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ricke RM, Bielinsky AK. Mcm10 regulates the stability and chromatin association of DNA polymerase-alpha. Mol Cell. 2004;16(2):173–185. doi: 10.1016/j.molcel.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 41.Ricke RM, Bielinsky AK. A conserved Hsp10-like domain in Mcm10 is required to stabilize the catalytic subunit of DNA polymerase-alpha in budding yeast. J Biol Chem. 2006;281(27):18414–18425. doi: 10.1074/jbc.M513551200. [DOI] [PubMed] [Google Scholar]
- 42.Das-Bradoo S, Ricke RM, Bielinsky AK. Interaction between PCNA and diubiquitinated Mcm10 is essential for cell growth in budding yeast. Mol Cell Biol. 2006;26(13):4806–4817. doi: 10.1128/MCB.02062-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lutz CT, et al. Human NK cells proliferate and die in vivo more rapidly than T cells in healthy young and elderly adults. J Immunol. 2011;186(8):4590–4598. doi: 10.4049/jimmunol.1002732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cichocki F, Miller JS. In vitro development of human killer-immunoglobulin receptor-positive NK cells. Methods Mol Biol. 2010;612:15–26. doi: 10.1007/978-1-60761-362-6_2. [DOI] [PubMed] [Google Scholar]
- 45.Freud AG, Caligiuri MA. Human natural killer cell development. Immunol Rev. 2006;214:56–72. doi: 10.1111/j.1600-065X.2006.00451.x. [DOI] [PubMed] [Google Scholar]
- 46.Logan CV, et al. DNA polymerase epsilon deficiency causes IMAGe syndrome with variable immunodeficiency. Am J Hum Genet. 2018;103(6):1038–1044. doi: 10.1016/j.ajhg.2018.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Frugoni F, et al. A novel mutation in the POLE2 gene causing combined immunodeficiency. J Allergy Clin Immunol. 2016;137(2):635–638.e1. doi: 10.1016/j.jaci.2015.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lim HJ, Jeon Y, Jeon CH, Kim JH, Lee H. Targeted disruption of Mcm10 causes defective embryonic cell proliferation and early embryo lethality. Biochim Biophys Acta. 2011;1813(10):1777–1783. doi: 10.1016/j.bbamcr.2011.05.012. [DOI] [PubMed] [Google Scholar]
- 49.Flach J, et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature. 2014;512(7513):198–202. doi: 10.1038/nature13619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chuang CH, Wallace MD, Abratte C, Southard T, Schimenti JC. Incremental genetic perturbations to MCM2-7 expression and subcellular distribution reveal exquisite sensitivity of mice to DNA replication stress. PLoS Genet. 2010;6(9):e1001110. doi: 10.1371/journal.pgen.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mabbott NA, Baillie JK, Brown H, Freeman TC, Hume DA. An expression atlas of human primary cells: inference of gene function from coexpression networks. BMC Genomics. 2013;14:632. doi: 10.1186/1471-2164-14-632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zhang JJ, et al. Ser727-dependent recruitment of MCM5 by Stat1alpha in IFN-gamma-induced transcriptional activation. EMBO J. 1998;17(23):6963–6971. doi: 10.1093/emboj/17.23.6963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Karo JM, Schatz DG, Sun JC. The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell. 2014;159(1):94–107. doi: 10.1016/j.cell.2014.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lopez-Vergès S, et al. Expansion of a unique CD57+NKG2Chi natural killer cell subset during acute human cytomegalovirus infection. Proc Natl Acad Sci USA. 2011;108(36):14725–14732. doi: 10.1073/pnas.1110900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lupski JR, et al. Exome sequencing resolves apparent incidental findings and reveals further complexity of SH3TC2 variant alleles causing Charcot-Marie-Tooth neuropathy. Genome Med. 2013;5(6):57. doi: 10.1186/gm461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 57.Warren L, et al. Highly efficient reprogramming to pluripotency and directed differentiation of human cells with synthetic modified mRNA. Cell Stem Cell. 2010;7(5):618–630. doi: 10.1016/j.stem.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amabile G, et al. In vivo generation of transplantable human hematopoietic cells from induced pluripotent stem cells. Blood. 2013;121(8):1255–1264. doi: 10.1182/blood-2012-06-434407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.