

Abstract
Tissue factor (TF) is the primary initiator of blood coagulation in vivo and the only blood coagulation factor for which a human genetic defect has not been described. As there are no routine clinical assays that capture the contribution of endogenous TF to coagulation initiation, the extent to which reduced TF activity contributes to unexplained bleeding is unknown. Using whole genome sequencing, we identified a heterozygous frameshift variant (p.Ser117HisfsTer10) in F3, the gene encoding TF, causing premature termination of TF (TFshort) in a woman with unexplained bleeding. Routine hematological laboratory evaluation of the proposita was normal. CRISPR-edited human induced pluripotent stem cells recapitulating the variant were differentiated into vascular smooth muscle and endothelial cells that demonstrated haploinsufficiency of TF. The variant F3 transcript is eliminated by nonsense-mediated decay. Neither overexpression nor addition of exogenous recombinant TFshort inhibited factor Xa or thrombin generation, excluding a dominant-negative mechanism. F3+/– mice provide an animal model of TF haploinsufficiency and exhibited prolonged bleeding times, impaired thrombus formation, and reduced survival following major injury. Heterozygous TF deficiency is present in at least 1 in 25,000 individuals and could limit coagulation initiation in undiagnosed individuals with abnormal bleeding but a normal routine laboratory evaluation.
Keywords: Hematology, Vascular Biology
Keywords: Coagulation, Proteases, endothelial cells
Introduction
Tissue factor (TF) is an integral membrane protein of the cytokine receptor superfamily that binds factor VII/VIIa to catalyze proteolytic activation of factors IX and X to trigger the blood coagulation cascade (1–8). TF is constitutively expressed in a hemostatic envelope that surrounds the vessel wall to ensure rapid hemostasis if injury compromises the spatial separation between vessel wall TF and plasma (9). Conversely, aberrant intravascular TF procoagulant cofactor activity contributes to the pathophysiology of myocardial infarction (10), venous thromboembolism (11), cancer-associated thrombosis (12), and sickle cell disease (13). TF is the primary initiator of blood coagulation in humans (14) and remains the only blood coagulation factor for which a human deficiency has not been described (15). As there are no routine clinical assays that capture the contribution of endogenous cellular TF to coagulation initiation, the extent to which reduced TF contributes to unexplained bleeding is unknown.
TF is encoded by the gene F3 with the major splice variant spanning 6 exons. The translated protein comprises an N-terminal signal sequence, 2 extracellular fibronectin type III domains, a single transmembrane helix, and a short cytoplasmic tail. The extracellular domains of TF create an elongated surface for the binding of factor VII/VIIa (6), an interaction that allosterically enhances catalysis of the factor VIIa serine protease domain by more than 105 for its substrate factor X (16). Because factors VIIa, IX, and X all require calcium ions to enable binding to membrane surfaces, the anchoring of TF to the lipid bilayer is critical for TF procoagulant activity (17, 18).
TF is essential for embryonic development and hemostasis (19–21). F3–/– mice experienced embryonic lethality by E10.5 in most genetic backgrounds, with hemorrhage and a defect in vitelline vascular integrity. F3+/– animals exhibit no spontaneous bleeding (19–21) but have only undergone limited evaluation following hemostatic challenge (20, 21). A murine chemical mutagenesis screen demonstrated that heterozygous F3 null alleles can suppress a lethal thrombotic phenotype in mice homozygous for factor V Leiden and haploinsufficient for tissue factor pathway inhibitor (TFPI), the major negative regulator of TF (22). This suppressive effect was confirmed using F3+/– mice (22), highlighting that even a 50% reduction in TF can lead to a survival phenotype in vivo. F3–/– mice can be rescued with a transgene expressing human TF at low levels (23), but they demonstrate a variety of spontaneous hemostatic defects including placental, postpartum, pulmonary, gastrointestinal, intracranial, and intracardiac hemorrhage (24–26).
The clinical diagnosis of rare bleeding disorders can be challenging and is often unsuccessful (27). The introduction of targeted next generation sequencing panels to diagnose disorders of hemostasis greatly reduces diagnostic cost and efficiency, but only 3.2% of cases will achieve a molecular diagnosis when standard laboratory evaluation is unrevealing (28). This highlights the fact that important components of the coagulation system are not captured by routine clinical laboratory assays, including the contribution of cellular TF. In order to identify unexplained heritable defects that contribute to human bleeding, we have applied whole genome sequencing to individuals with undiagnosed clinical bleeding as part of the NIHR BioResource Rare Diseases study for the 100,000 Genomes Project (29).
We have identified a heterozygous frameshift variant in F3 resulting in premature truncation of TF in a woman with mild unexplained bleeding. Routine hematologic evaluation was normal. As complete family studies proved impossible, we studied this TF mutation using protein biochemistry, reverse genetics, CRISPR/Cas9-engineered induced pluripotent stem cells (iPSCs), and F3+/– mice. We demonstrated that haploinsufficiency of TF contributes to impaired initiation of blood coagulation and, in mice, increased bleeding risk. The data support heterozygous TF deficiency as a novel modifier of bleeding risk that should be considered in individuals with undiagnosed bleeding and normal clinical laboratory evaluation.
Results
Identification of a truncating mutation in F3 associated with a human bleeding diathesis.
To determine the genetic basis for novel bleeding disorders, WGS has been applied to 973 probands with unexplained inherited bleeding, thrombotic, or platelet disorders enrolled in the NIHR BioResource Rare Diseases program (29). We identified an F3 variant in a subject characterized by unexplained bleeding. She is an otherwise healthy female who had menorrhagia, epistaxis, easy bruising, and bleeding following a dental extraction. Her mother experienced menorrhagia and postpartum hemorrhages and her maternal aunt had mild bleeding symptoms, but these individuals could not be evaluated nor could clinical testing results be obtained. The father is estranged from the family and his clinical history and medical records could not be obtained. Clinical laboratory assessment of the proband was unremarkable (Table 1). WGS revealed a 2 nucleotide deletion in one allele of F3 (c.249delAG), resulting in a heterozygous frameshift (Figure 1A) and premature termination (p.Ser117HisfsTer10); the other TF allele was normal. Paternal transmission of the F3 c.249delAG variant was observed. The mother and aunt did not carry this F3 variant and the etiology of their bleeding remains unknown. This F3 variant was not observed in 141,456 exome or genome sequences present in the Genome Aggregation Database (gnomAD) (30). No pathogenic variants were observed in genes known to cause bleeding in humans (31). If the p.Ser117HisfsTer10 protein were expressed, the predicted truncated form, TFshort, would, after signal sequence cleavage, include native TF residues 37–116 followed by the frameshifted amino acids. TFshort would thus lack the second fibronectin type III domain, transmembrane segment, and cytoplasmic tail (Figure 1B).
Table 1. Summary of the proband’s clinical laboratory evaluation.
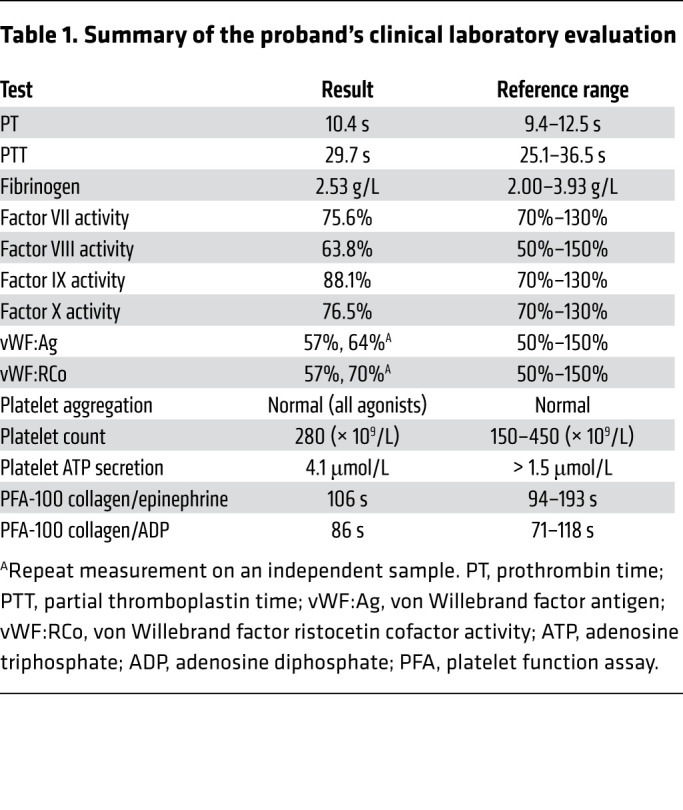
Figure 1. A truncating F3 variant in an individual with unexplained bleeding.

(A) Sanger sequencing confirmed a heterozygous 2 base pair deletion on chromosome 1:95,001,582 GCT>G. A red vertical arrow indicates the start of the deletion, after which competing sequences reflect the frameshift. (B) The frameshift encodes TF p.Ser117HisfsTer10. A schematic of the TF protein highlights features of the primary structure and domain organization. The mutation at position 117 is indicated by the vertical red arrow. SP, signal peptide; TM, transmembrane segment.
Truncated TF does not perturb bleeding via a dominant-negative mechanism.
We determined whether the truncated variant contributes to bleeding by inhibiting the function of the WT allele (dominant negative) or by inadequate production of functional TF (haploinsufficiency). The identical 2 base pair deletion was introduced into a plasmid encoding the extracellular region of human TF carrying an N-terminal His-Tag (32). TFshort was expressed in E. coli and purified using cobalt affinity chromatography (Figure 2A). We tested recombinant TFshort in a factor Xa generation assay in which primary human umbilical vein endothelial cells (HUVECs) provided functional TF on the membrane surface to support the factor VIIa–mediated conversion of recombinant factor X to factor Xa, as determined by cleavage of a chromogenic substrate. Quiescent HUVECs manifest negligible TF procoagulant activity, and no additional activity is conferred with exogenous TFshort at concentrations as high as 30 nM, a 50-fold molar excess over factor VIIa (Figure 2, B and C). When TF expression is induced on HUVECs by stimulation with tumor necrosis factor α (TNF-α), TFshort does not inhibit factor Xa generation. To further ensure that TFshort does not functionally interact with an unknown regulator of coagulation in plasma, recombinant TFshort was evaluated in a contact pathway-inhibited plasma-based thrombin generation assay initiated by limiting concentrations of lipidated recombinant TF. Titration of TFshort to concentrations up to 200 nM did not alter thrombin generation (Figure 2D).
Figure 2. Truncated TF neither enhances nor inhibits coagulant activity.

(A) Purified recombinant TFshort encoding p.Ser117HisfsTer10 visualized by Coomassie stain migrates as a single band following SDS-PAGE. (B) TFshort (30 nM) was evaluated for its influence on the TF/factor VIIa catalyzed conversion of factor X to factor Xa on the surface of TNF-α stimulated HUVECs, as determined by cleavage of a chromogenic substrate monitoring factor Xa activity. Controls include treatment with bovine serum albumin (BSA), a TF inhibitory antibody (anti-TF) or its isotype control, or omission of TNF-α or factor VIIa. A representative kinetic course is shown, with error bars depicting SEM for 3 replicates. (C) Quantification of B across n = 3 independent experiments; 1-way analysis of variance, ****P < 0.0001. (D) The kinetics of thrombin generation initiated by lipidated TF in the presence of ascending TFshort concentrations, as indicated. (E) HUVECs were transduced with lentivirus encoding TF, TFshort, or vector control and cells were analyzed for their ability to support factor Xa generation with or without TNF-α stimulation; n = 4, 1-way analysis of variance, ****P < 0.0001. (F) HUVECs were transduced as in E and F3 mRNA was analyzed via quantitative PCR; n = 3, 1-way analysis of variance, not significant. (G) As in F, but TF expression was determined by immunoblot with anti-TF and anti-GAPDH antibodies. (H) As in G, but cells were treated with 100 μM MG-132 or vehicle for 6 hours before analysis; asterisk highlights the stabilized fragment. See complete unedited blots in the supplemental material.
To exclude an intracellular inhibitory effect on TF presentation (33), HUVECs were transduced with lentiviral particles expressing TFshort. There was neither augmented nor inhibited cofactor activity (Figure 2E). While F3 mRNA was induced by 2 orders of magnitude in TF and TFshort transduced cells (Figure 2F), analysis of the same cells by SDS-PAGE followed by immunoblot detected full-length TF but only faintly detected TFshort (Figure 2G). TFshort accumulated in the presence of the proteasome inhibitor MG-132 (Figure 2H), indicating that the truncated form of TF is unstable and rapidly degraded by the proteasome via the endoplasmic reticulum–associated decay pathway. Taken together, these data eliminate the possibility that TFshort impairs coagulation through a dominant-negative mechanism.
Haploinsufficiency of TF impairs coagulation initiation.
To determine whether F3 c.249delAG is a null allele and results in TF haploinsufficiency, we engineered isogenic human iPSCs that recapitulate heterozygosity of the truncated F3 variant. iPSCs were edited using CRISPR/Cas9 nuclease activity guided by a specific sgRNA in the presence of a single-stranded DNA oligonucleotide template for homology-directed repair. Isolation of the heterozygous frameshift (HET) rather than the homozygote (HOM) required a pair of oligonucleotides, with one introducing synonymous blocking mutations (BLK) to prevent sgRNA homology and an unintended second round of genome editing (Figure 3A). Two independent iPSC clones for BLK, HET, HOM, and WT (WT) were isolated. These iPSC lines were differentiated into mesoderm using GSK3 inhibition and BMP4 treatment, followed by directed commitment to vascular smooth muscle cells (VSMCs) using PDGF-BB and activin A, or toward endothelial cells (ECs) by treatment with VEGF A and forskolin (34). Each lineage was confirmed by flow cytometry to detect expression of CD144 and CD140b. Whereas iPSCs do not express either CD144 or CD140b, progenitors differentiated into VSMCs were more than 90% positive for CD140b (Figure 3B). After directed differentiation into ECs, only about 75% of cells were CD144 positive (Figure 3B), so these cells were sorted using flow cytometry to isolate CD144-positive cells. The immunophenotype of ECs was validated by indirect immunofluorescence staining for von Willebrand factor and PECAM1 (Figure 3C). VSMCs were substantiated by indirect immunofluorescence staining for α-smooth muscle actin, myosin IIb, and smooth muscle protein 22-α (Figure 3C).
Figure 3. iPSCs heterozygous for the truncating F3 allele differentiated into vascular cells exhibit haploinsufficiency of coagulation initiation.

(A) CRISPR was used to engineer iPSCs heterozygous (HET) or homozygous (HOM) for the patient’s deletion (red). Synonymous blocking mutations (yellow) prevented editing of the second allele. WT (WT) and blocking mutant (BLK) iPSC lines provided additional controls. Edited bases (red boxes) and the site of gRNA homology (gray) are highlighted. (B) Representative fluorescence activated cell sorting plots demonstrating differentiation of WT iPSCs (left) into VSMCs (middle) and ECs (right) by staining for CD140b (upper panel) and CD144 (lower panel). (C) Validation of endothelial cell differentiation by indirect immunofluorescence staining for PECAM1 and von Willebrand factor (VWF); VSMC differentiation was validated using myosin IIb, smooth muscle actin (SMA), and smooth muscle protein 22-α (SM22a). Nuclei are stained with Hoechst. Scale bars: 100 µm. (D) Engineered iPSCs differentiated into ECs were evaluated for their ability to support the factor VIIa catalyzed conversion of factor X to factor Xa (FXa), as determined by cleavage of a chromogenic substrate. A representative kinetic course reflects mean ± SEM for 3 replicates. (E) Quantification of D; n = 3 independent experiments for 2 independently isolated iPSC clones differentiated into ECs (n = 2 for a single clone), 1-way analysis of variance. (F) iPSC clones differentiated into VSMCs were tested for FXa generation as in D and E, n = 4. (G) TF expression in differentiated VSMCs was determined by immunoblot with anti-TF and anti-GAPDH antibodies. See complete unedited blots in the supplemental material. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Live cell monolayers of differentiated ECs and VSMCs were then tested for their ability to support TF-dependent factor Xa generation. When endothelial cells were tested, the heterozygous F3 variant yielded approximately 50% of the activity of WT, consistent with a haploinsufficiency mechanism (Figure 3, D and E). The homozygous clones expressed negligible coagulant activity, while the BLK was comparable to WT. VSMCs were similarly analyzed and also supported a mechanism of TF haploinsufficiency (Figure 3F) without apparent TFshort expression by immunoblot (Figure 3G).
F3 c.249delAG is a null allele degraded by nonsense-mediated decay.
The F3 c.249delAG allele introduces a premature stop codon in the third of 6 exons that contribute to the canonical F3 transcript (Figure 4A). We hypothesized that the presence of this premature stop codon before the final exon junction might target the transcript for degradation via the nonsense-mediated mRNA decay pathway. Because protein translation is required for the cell to recognize a premature termination codon, cells were treated with the ribosomal inhibitor cycloheximide to test whether the variant mRNA transcript could be rescued from degradation. We compared differentiated iPSCs that were homozygous for or lacked a single premature termination codon in F3. Cycloheximide treatment of both homozygous EC clones resulted in a 13-fold increase in the F3 mRNA level, whereas no significant increase was observed in control cells (Figure 4B). When VSMCs were tested, cycloheximide increased F3 transcript levels more than 100-fold (Figure 4C). These findings indicate that inhibition of protein translation suppresses nonsense-mediated decay of the prematurely terminated transcript and that F3 c.249delAG is a null allele.
Figure 4. Premature termination renders the TFshort mRNA a substrate for nonsense-mediated decay.

(A) Organization of the exons contributing to the canonical F3 transcript highlights the site of premature termination (Ter). Splicing schematic generated using Ensembl (47). (B and C) F3 mRNA expression was evaluated by quantitative PCR in ECs (B, n = 4) and VSMCs (C, n = 3) derived from each BLK or HOM clone following 3 hours treatment with 100 μM cycloheximide (CHX) or vehicle. Relative mRNA expression for F3 (CHX vs. vehicle treatment) is presented as mean ± SEM. Unpaired t test. *P < 0.05, **P < 0.01.
Prolonged bleeding and impaired survival following major injury in F3+/– mice replicating heterozygous TF deficiency.
In the absence of critical participation by this extended family to allow investigation of their medical history and to perform appropriate clinical laboratory measurements, we developed a mouse model with a parallel defect in TF. Our data provide a mechanism for how the inheritance of a single F3 null allele impairs initiation of blood coagulation, so we used F3+/– mice to directly demonstrate the consequences of heterozygous TF deficiency on hemostasis in vivo. Primary ear fibroblasts derived from F3+/– mice expressed half the TF cofactor activity of WT littermates in a factor Xa generation assay (Figure 5, A and B), indicating that the model recapitulates haploinsufficiency of F3 observed in humans heterozygous for the F3 c.249delAG allele. To evaluate the consequence of TF haploinsufficiency on hemostasis in vivo, we challenged a cohort of 8- to 10-week-old F3+/– and F3+/+ littermate control mice to an aggressive tail amputation bleeding model characterized by transection of the tail at 2 mm diameter. The bleeding times were prolonged in F3+/– as compared with F3+/+ animals (Figure 5C), with many animals failing to achieve hemostasis. Five of 14 F3+/– mice died of exsanguination before observation time concluded at 30 minutes, whereas no F3+/+ mice died, resulting in an overall survival advantage for WT animals following tail transection (Figure 5D).
Figure 5. Impaired coagulation initiation and hemostasis in F3+/– mice replicating heterozygous TF deficiency.

Primary ear fibroblasts were cultured from F3+/– and F3+/+ littermate controls and assayed for their ability to support the factor VIIa-mediated conversion of factor X to factor Xa, as determined using a chromogenic factor Xa substrate that absorbs at 405 nm. A negative control omits factor VIIa (-VIIa). (A) Representative kinetic course plots the mean ± SEM of 3 replicates. (B) Factor Xa generation (nM/min) across n = 5 independent experiments was quantified; 1-way analysis of variance. (C) Eight- to 10-week-old F3+/– and F3+/+ littermate controls were evaluated in an aggressive tail amputation model and time to initial cessation of bleeding was recorded over 30 minutes of observation for n ≥ 14 mice of each genotype. Unpaired t test. (D) Kaplan-Meier cumulative survival of mice in C following tail transection. Log rank test. *P < 0.05.
Impaired thrombus formation following vascular injury in F3+/– mice.
To directly observe the impact of a TF null allele in vivo, we used intravital microscopy to image the response to vessel wall injury in F3+/– and F3+/+ littermate control mice. The median kinetics of platelet accumulation (Figure 6A) and fibrin generation (Figure 6B) were visualized in live mice using anti-CD42b– and anti-fibrin–specific antibodies, respectively. Because our prior experiments demonstrated that haploinsufficiency of F3 caused prolonged bleeding following major injury, our analysis accounts for injury size (Figure 6C) (35). The kinetics of thrombus formation diverge when the large injuries for each genotype are compared. These differences in platelet accumulation and fibrin generation are statistically significant when the areas under the curve for each injury are compared (Figure 6, D and F), but no difference was observed for smaller injuries (Figure 6, E and G).
Figure 6. Impaired coagulation initiation and thrombus formation in F3+/– mice replicating heterozygous TF deficiency.

Thrombus formation was monitored for 180 seconds following laser-induced vessel wall injury in F3+/– and F3+/+ littermate control male mice. Platelet and fibrin accumulation were detected using anti-CD42b or anti-fibrin antibody conjugated to Dylight 488 and 647, respectively. Median integrated fluorescence over time in arbitrary units (AU) was determined for platelet (A) and fibrin (B) accumulation as stratified by injury size (C). The median area under the curve (AUC) was determined for larger injuries (n = 13) (D, platelets; F, fibrin) or smaller injuries (n = 15) (E, platelets; G, fibrin) than median (90 μm). Wilcoxon rank sum test. *P < 0.05.
Population level analysis of F3 null alleles.
We examined 141,456 aggregated exome or whole genome sequences present in gnomAD (30) to estimate the incidence of previously undiagnosed heterozygous TF deficiency in the general human population. While the F3 c.249delAG allele was unique, we identified 4 additional F3 null alleles (one allele was observed in 2 individuals) with a combined allele frequency of 2 × 10–5 (Table 2). The combined allele frequency of these high confidence null alleles is 2 × 10–5, meaning that the frequency of any individual in the population inheriting one such allele is 4 × 10–5. Heterozygous TF deficiency is thus present in at least 1 in every 25,000 persons. gnomAD constraint metrics also help to estimate the extent to which loss of F3 is tolerated in humans, indicating an observed (5)/expected (13.1) ratio of predicted loss-of-function single nucleotide variants of 0.38 (90% confidence interval: 0.2–0.8) and a probability of being loss-of-function intolerant (pLI) of 0.02 (30). These data demonstrate that heterozygous TF deficiency is not exceedingly rare and can be tolerated in humans.
Table 2. Summary of F3 null alleles identified in 141,456 human exome or whole genome sequences.

Discussion
TF is the primary initiator of blood coagulation in vivo and the only major blood coagulation factor in which a human genetic defect has never previously been described. Here, we applied whole genome sequencing to assist in the diagnosis of an unexplained bleeding disorder in a patient with a normal clinical laboratory evaluation. We found a heterozygous null mutation in F3 characterized by a 2 base pair deletion that resulted in a premature nonsense codon, accelerated F3 mRNA degradation, and, when translated, an unstable truncated TF fragment. An analysis of null mutations in F3 exons from 141,456 human genomes or exomes indicates that, while the variant observed in this kindred is unique, 4 similar F3 null alleles were observed. Indeed, heterozygous TF deficiency is tolerated in humans but remains undiagnosed and exists in at least 1 in 25,000 individuals.
Although WGS of the proposita did not identify a pathogenic variant in known genes that cause bleeding in humans (36), it is probable that she carries unidentified variants that modify the penetrance of this F3 null allele. The discovery that the F3 null allele was inherited paternally supports the possibility that bleeding in association with heterozygous TF deficiency has incomplete penetrance that is modified by the genetic background and unmasked by variable hemostatic challenge. The clinical history of the proposita’s father is not available, so he may or may not have experienced abnormal bleeding. However, a history of bleeding on the maternal side suggests a polygenic inheritance pattern with additional maternal variants modifying the bleeding phenotype. Though one could hypothesize, a priori, that bleeding in the proposita is unrelated to the truncating variant in F3, our data using CRISPR-edited iPSCs and multiple mouse models highlight the significance of heterozygous TF deficiency independent of the genetic background.
Because relatives of the proposita were unavailable for evaluation, isogenic human iPSC lines and backcrossed mice were used to deconvolute the variant from genetic background. The penetrance of complete F3 deficiency (F3–/–) in mice is dependent upon genetic background. Whereas nearly all F3–/– embryos are dead by E10.5 and none survive gestation in the 129/SvJ × 129/SvEv background, 14% of the expected number of F3–/– embryos survived gestation in the 129/SvJ × C57BL/6 background and 2% even survived 1 to 2 days postpartum (37). An ENU (N-ethyl-N-nitrosourea) mutagenesis screen in mice found that a mutation mapping to F3 could suppress lethal thrombosis in mice homozygous for factor V Leiden and heterozygous for TFPI deficiency, a survival phenotype that could be replicated using F3+/– mice (22). It is probable that F3 null and other hypomorphic alleles modify thrombotic as well as bleeding risk in diverse genetic backgrounds across human populations.
Studies in mice and humans suggest that heterozygous TF deficiency can be tolerated. When F3+/– mice are crossed, F3+/– and F3+/+ progeny result at a 2:1 ratio in most genetic backgrounds without an overt phenotype, but no F3–/– pups were observed. This is consistent with the requirement for at least low levels of TF expression (19–21). F3–/– mice rescued with a low expressing human F3 transgene demonstrate cardiac, pulmonary, and other hemorrhagic pathologies not seen in unchallenged F3+/– animals (24–26). Our biochemical and differentiated iPSC experiments demonstrate that the F3 c.249delAG variant identified in the proposita is a true null allele, indicating that haploinsufficiency of TF is compatible with life in humans. Although we identify a specific human mutation that leads to heterozygous TF deficiency, other types of genetic defects could lead to a similar phenotype.
Analysis of population-level human genomic data generalizes this conclusion. Our examination of 141,456 human genome or exome sequences present in gnomAD identified 4 additional unique F3 null alleles (one was observed twice) with a combined allele frequency of 2 × 10–5. The accuracy of these estimates will be enhanced as additional genomes become available, but predict that heterozygous TF deficiency has an incidence of at least 1 in 25,000 individuals, comparable to the incidence of hemophilia B. These figures are a lower limit because they neglect other classes of predicted null alleles, including those due to impaired F3 splicing and nonsynonymous SNPs predicted to be hypomorphic. The probability of having a loss-of-function intolerant score (0.02) together with the observed/expected (0.38) metric, calculated in gnomAD, suggests that although heterozygous TF deficiency may be under weak negative selection, it is viable, consistent with an otherwise healthy adult carrying the F3 p.Ser117HisfsTer10 null allele (30). A human carrying 2 F3 null alleles has never been observed (30, 38).
F3+/– mice lack a spontaneous bleeding phenotype (19–21), but our studies of mice replicating haploinsufficiency of TF demonstrate prolonged bleeding and impaired thrombus formation after substantial injury. Caged mice rarely demonstrate spontaneous bleeding in general and especially mucosal bleeding, and that the F3+/– mice don’t exhibit uterine bleeding may be related to physiological differences, as female mice experience an estrous rather than menstrual cycle. Bleeding in F3+/– as compared with F3+/+ animals has previously been observed following tail biopsy (21) and nail bed injury (20). While no difference was apparent, these data were not presented in either study and it is unclear whether these experiments were powered with sufficient animals to distinguish more nuanced phenotypes, particularly given the inherent variability of bleeding assays. Nevertheless, these findings are consistent with our own experience that heterozygous TF deficiency is unmasked in an injury-dependent context. We observed prolonged time to bleeding cessation and inferior overall survival in F3+/– mice as compared with WT after tail amputation. This is a more severe injury than the routine tail bleeding assays generally used, and it suggests that full TF activity is necessary to protect against traumatic selective pressures such as catastrophic bleeding. Similarly, we observed via intravital microscopy that injury size determines the extent to which heterozygous TF deficiency influences platelet accumulation and fibrin deposition following vascular injury. Larger injuries may result in enhanced exposure of TF and could account for the greater impairment to thrombus formation observed following large as opposed to small vessel injury in F3+/– animals (39).
We have demonstrated a novel hereditary defect in the F3 gene and how 1 deleterious F3 allele can contribute to bleeding. Routine clinical laboratory evaluations are not sensitive to the effect of endogenous TF on coagulation initiation, so patients with a bleeding phenotype will not be diagnosed with abnormalities of TF. Identification of an expanded cohort of individuals with heterozygous TF deficiency will further clarify the natural history of these variants in humans and establish diverse mechanisms that can lead to heterozygous TF deficiency. Our findings demonstrate that haploinsufficiency of TF is a modifier of coagulation initiation not captured by routine clinical laboratory testing that should be considered in individuals with undiagnosed bleeding disorders. A simple genetic test for the integrity of F3 may prove effective in identifying bleeding risk.
Methods
Cell culture.
HUVECs (Lonza) were maintained for up to 5 passages in complete EGM-2 Endothelial Cell Growth Medium (Lonza) on gelatin-coated culture flasks. HEK293T cells (ATCC) were cultured in DMEM (Life Technologies) supplemented with 10% fetal bovine serum (HyClone). Cells were passaged using trypsin EDTA (0.05%, Thermo Fisher Scientific). Cells were cultured at 37°C and 5% carbon dioxide. iPSC methods are detailed below (see “iPSC culture” and “Differentiation of iPSCs”).
Factor Xa and thrombin generation assays.
Equal cell numbers were seeded in 96-well (20,000 HUVECs, 2500 differentiated VSMCs) or 384-well (3000 differentiated ECs) plate format and cultured in the appropriate medium (see “Cell culture”). Where indicated, endothelial cells were stimulated with tumor necrosis factor-α (TNF-α; 10 ng/mL; Calbiochem) for 3.5 hours at 37°C. Cells were washed with HBS-BSA (20 mM HEPES, pH 7.4, 150 mM NaCl, 5 mM KCl, 5 mM CaCl2 1 mg/mL bovine serum albumin) that was then replaced with HBS-BSA containing factor VIIa (0.3–0.6 nM, Haematologic Technologies), factor X (125 nM; Haematologic Technologies), and the chromogenic factor X substrate BIOPHEN CS-11 (22) (150 μM; HYPHEN BioMed). A SpectraMax spectrophotometer was used to monitor absorbance at 405 nm every minute for 2 hours. Where exogenous TF or TF inhibitory antibody (4509, BioMedica Diagnostics) was evaluated, cells were washed in HBS-BSA and then incubated in HBS-BSA containing the protein of interest at the indicated concentration. After 10 minutes, the reaction was triggered by the addition of factors VIIa, X, and the Xa substrate. Thrombin generation experiments were performed using human pooled normal plasma and the Technothrombin TGA assay (Diapharma). The assay was performed according to the manufacturer’s instruction but was adapted by reducing the amount of plasma in the assay by 10 μL to enable addition of vehicle or recombinant protein in phosphate buffered saline (PBS) and supplementation with corn trypsin inhibitor (25 μg/mL, Haematologic Technologies) to prevent aberrant contact activation.
Protein expression and purification.
Site-directed mutagenesis was used to introduce the 2 base pair deletion encoding Ser117HisfsTer10 into the pTRCHISC plasmid encoding the soluble TF ectodomain (amino acids 1-218) fused with an N-terminal His-tag (32). Mutagenesis was performed using oligonucleotide 5′-cagggaatgtggagcaccggttctgctg-3′ and its reverse complement. Mutant TF was expressed in E. coli after induction with 1 mM isopropyl β-D-1-thiogalactopyranoside and purified from a clarified lysate by cobalt affinity chromatography.
Lentiviral gene transfer.
Site-directed mutagenesis using the same primer as was previously used to introduce the 2 base pair deletion encoding Ser117HisfsTer10 into the lentiviral transfer vector pLX304 encoding human F3 (clone HsCD00413770, Harvard PlasmID). 293T cells (ATCC) were cotransfected with the indicated pLX304 lentiviral transfer vector and packaging vectors (psPAX2, pMD2.G, REV; Addgene) using Lipofectamine 2000 (Life Technologies). Beginning after 24 hours, the viral supernatant was collected every 12 hours and replaced with fresh DMEM supplemented with 10% fetal bovine serum.
Genome engineering of iPSCs.
The parent cell line for gene editing was the diploid 46,XY iPSC line BJFF.6 (Genome Engineering and iPSC Center [GEiC], Washington University in St. Louis). Cells were engineered using a CRISPR/Cas9-based strategy (40) and the gRNA, 5′-uggagagcaccgguucugcu-3′. Single-stranded oligodeoxynucleotides (ssODNs) were introduced together with the gRNA via nucleofection (Amaxa) to provide a template for homology-directed repair (HDR). An ssODN encoding the intended 2 base pair deletion and adjacent synonymous blocking substitutions (indicated by capital letters) 5′-gaaggatgtgaagcagacgtacttggcacgggtcttctcctacccggcagggaatgtggaATaccggttctgctggggagcctctgtatgagaactccccagagttcacaccttacctggagagtaagtgg-3′ as well as an ssODN encoding only the silent blocking variant 5′- gaaggatgtgaagcagacgtacttggcacgggtcttctcctacccggcagggaatgtggaAagTaccggttctgctggggagcctctgtatgagaactccccagagttcacaccttacctggagagtaagtg-3′ were required to prevent editing of the second F3 allele and enable selection of heterozygous clones. Single-cell clones were isolated, expanded, and analyzed by next generation sequencing, as previously described (41). Two independent iPSC clones reflecting the WT, HET, HOM, and BLK genotypes were validated and used for subsequent differentiation and phenotyping. All clones tested free of mycoplasma contamination using MycoAlert (Lonza).
iPSC culture.
Tissue culture–treated dishes were coated for 1 hour at room temperature with Geltrex (Life Technologies) reconstituted in DMEM medium (Life Technologies) supplemented with 4.5 g/L glucose, l-glutamine, and sodium pyruvate (Corning). iPSC stocks were thawed in complete StemFlex medium (Life Technologies) containing 10 μM Rho-associated, coiled-coil–containing protein kinase (ROCK) inhibitor Y-27632 (Calbiochem) and cultured on Geltrex-coated dishes. After 24 hours, media was replaced with complete StemFlex free of ROCK inhibitor. Cells were detached using ReLeSR (StemCell Technologies) and split 1:5 to 1:10 before reaching confluence; cells were cultured in complete StemFlex media supplemented with ROCK for the first 24 hours. All culture media were supplemented with penicillin-streptomycin (Life Technologies) and Plasmocin (Invivogen), and media was changed every other day unless otherwise specified.
Differentiation of iPSCs into vascular smooth muscle and endothelial cells.
iPSCs were differentiated as previously described (34). Briefly, iPSC clones were split to 50% confluence on Geltrex-coated plates and cultured overnight in StemFlex medium (Life Technologies). The next day (day 1), differentiation was initiated by replacing media with N2B27 prepared by combining equal parts of DMEM/F12 GlutaMAX (Life Technologies) and Neurobasal medium (Life Technologies) supplemented with concentrates of B-27 supplement without vitamin A (Life Technologies), N-2 supplement (Life Technologies), 6 μM CHIR-99021 (StemCell Technologies), and 25 ng/mL BMP4 (R&D Systems) for 3 days with daily media replacement. To generate VSMCs, media were replaced on day 4 with N2B27 supplemented with 10 ng/mL PDGF-BB (Pepro-Tech) and 2 ng/mL Activin A (Pepro-Tech). On day 6, VSMCs were split with trypsin-EDTA (Life Technologies) into dishes coated with human fibronectin (MilliporeSigma) and maintained in Medium 231 (Life Technologies) supplemented with Smooth Muscle Growth Supplement (Life Technologies). To generate ECs, the identical protocol was followed for 3 days to achieve the mesodermal lineage, but on day 4 media was instead replaced with complete N2B27 media supplemented with 200 ng/mL VEGF (Pepro-Tech) and 2 μM forskolin (MilliporeSigma). On day 6, cells were split using Accutase (StemCell Technologies) and differentiated ECs were either MACS-separated using CD144 MicroBeads (Miltenyi Biotec) or flow cytometry–sorted for CD144-positive cells to isolate the population expressing VE-cadherin. Differentiated ECs were maintained in EGM-2 Endothelial Cell Growth Medium (Lonza) replaced daily. All culture media were supplemented with penicillin-streptomycin (Life Technologies) and Plasmocin (Invivogen), and changed every other day in EGM-2 unless otherwise specified.
Quantitative PCR.
F3 mRNA expression was determined by quantitative PCR for ECs cultured in 96-well plate format using the Cells-to-CT 2-Step TaqMan Kit (Life Technologies) and TaqMan gene expression assays for F3 (Hs00175225_m1; Life Technologies) and GAPDH (Hs03929097_g1; Life Technologies) as an endogenous control. For VSMCs, RNA was isolated using the Purelink RNA Mini Kit (Thermo Fisher Scientific) before reverse transcription using the iScript cDNA synthesis kit (Bio-Rad) and analysis with the identical TaqMan gene expression assays for F3 and GAPDH. After reverse transcription, quantitative PCR was performed using the Applied Biosystems 7300 Real-time PCR System. HUVECs expressing WT or mutant TF constructs were analyzed 48 hours after transduction to confirm gene expression. To evaluate whether the mutant F3 mRNA is subject to nonsense-mediated decay, iPSCs differentiated into ECs and VSMCs were treated with 100 μM cycloheximide (Roche) or ethanol vehicle for 3 hours before evaluation.
Flow cytometry.
The cell-surface immunophenotype of iPSCs, ECs, and VSMCs was established by FACS. Cells were dissociated using Accutase (MilliporeSigma), pelleted at 300g for 5 minutes, incubated in complete media for 30 minutes at room temperature, washed once in PBS, and resuspended in PBS supplemented with 0.5% BSA. Phycoerythrin-conjugated antibodies against CD140b (1:10, BD Pharmingen) or CD144 (1:10, BD Pharmingen) were added as indicated and incubated for 30 minutes at 4°C. Cells were washed twice and ultimately resuspended in PBS containing 0.5% BSA.
Immunofluorescence microscopy.
The immunophenotype of forward programmed vascular cells was confirmed via indirect immunofluorescence microscopy. Cells were cultured on coverslips and washed in PBS, fixed for 10 minutes in PBS/4% paraformaldehyde, permeabilized in PBS/0.05% Triton X-100 for 10 minutes, and washed again 3 times in PBS. Cells were blocked in PBS containing 1% BSA and 10% goat serum for 1.5 hours. Cells were stained overnight at 4°C with the indicated primary antibodies diluted into blocking solution: von Willebrand Factor (1:400, A0082, Dako) and PECAM1 (1:100, AF806, R&D Systems) for ECs, SMA (1:250, sc-130616, Santa Cruz Biotechnology), myosin IIb (1:200, clone D8H8, Cell Signaling Technologies), and SM22α (1:200, AF7886, R&D Systems) for VSMCs. Cells were washed 3 times in PBS containing 1% Tween-20 and species-specific secondary antibody conjugated to Alexa 488 (1:1000, Thermo Fisher Scientific) in blocking solution. Hoechst (1 μg/mL, catalog 3342, Thermo Fisher Scientific) was added for the final 15 minutes to stain nuclei. Cells were mounted with Aqua Poly (Polysciences) on glass coverslips and imaged on an Olympus BX62 fluorescence microscope.
Mice.
Heterozygous TF (F3+/–) mice on the C57BL/6J background were a gift from George Broze, Washington University School of Medicine , St. Louis, Missouri (21). These mice were mated with C57BL/6J (stock number 000664, Jackson Laboratory) to generate the F3+/– and F3+/+ littermate control animals used in these experiments.
Intravital microscopy and laser-induced vessel wall injury model.
Thrombus formation was visualized via intravital microscopy following laser-induced injury to the cremasteric arteriole in male mice as previously described (42). Platelets were detected using anti-CD42b antibody (0.1 mg/g; Emfret Analytics) conjugated to Dylight-488 (Thermo Fisher Scientific) and fibrin was detected with anti-fibrin antibody (0.5 mg/g; clone 59D8) conjugated to Dylight-647 (Thermo Fisher Scientific) infused via internal jugular vein catheter. The cremasteric microcirculation was surgically exposed and injury to the cremasteric arteriole was provoked using a MicroPoint Laser System (Photonics Instruments). Data were digitally captured via an Orca Flash 4.0v2 CMOS camera in both the 488/520 nm and 647/670 nm fluorescence channels at a rate of 2 frames per second beginning before and extending for 180 seconds after laser injury. Images were analyzed using Slidebook version 6.0 (Intelligent Imaging Innovations). Injury size was determined using calipers one frame after laser injury (our unpublished observations). At least n = 13 injuries greater (large) or less than (small) median (90 μm) across at least 3 mice were used to enable statistical analysis (see “Statistics” below). The operator was blinded to genotype in all mouse experiments.
Tail amputation bleeding assay.
Tail transection was assayed as described (43), but the tail was inserted into a 2 mm diameter sizer and marked for transection. Animals were tested at 8–10 weeks of age and anesthetized via weight-based i.p. ketamine/xylazine injection. The tail was incubated in saline at 37°C for 2 minutes before downward transection with a new 10 blade scalpel at the marked site corresponding to 2 mm tail diameter. The tail was returned to a 15 mL conical of warmed saline and the time to cessation of bleeding was recorded over a 30-minute period of observation. Vital status was monitored. Animals were euthanized after 30 minutes.
Isolation and characterization of primary murine fibroblasts.
After euthanasia of mice in the aforementioned studies, a segment of ear tissue was harvested and soaked in 70% ethanol for 2 minutes. The ear was rinsed in DMEM and minced with a scalpel before transfer to a tube containing 2 mL trypsin EDTA and vortexed every 10 minutes during a 1 hour incubation at 37°C. Complete DMEM was then added to inactivate trypsin before centrifugation at 200g for 5 minutes. Cellular debris was resuspended in DMEM supplemented with 10% FBS and cultured in 6-well plates. After outgrowth of fibroblasts, cells were passaged and 2000 cells/well were seeded in 96-well plate format. Factor Xa generation was assayed as described above (see “Factor Xa and thrombin generation assays”) except the factor VIIa concentration was 0.3 nM.
Statistics.
Statistics were computed using the GraphPad Prism version 7.0 software package. Comparison between 2 normally distributed groups was performed using the Student’s t test. One-way analysis of variance was used for comparison of more than 2 groups with application of the Tukey’s method to adjust for multiple comparisons. Sample size was determined in rodent experiments using historical measurements of experimental variation and estimated effect size. Intravital data were not normally distributed and binary comparisons were made using the Wilcoxon rank sum test as previously described (44–46). Statistically significant P values of less than 0.05 were indicated by a single asterisk (*), less than 0.01 by a double asterisk (**), less than 0.001 by a triple asterisk (***), and less than 0.0001 by a quadruple asterisk (****). Comparisons that failed to achieve statistical significance were highlighted as not significant where relevant. Error bars represent the standard error of the mean except where otherwise noted.
Study approval.
The NIHR BioResource Rare Disease Study was approved by the East of England – Cambridge South UK Research Ethics Committee. After providing written informed consent, patients were enrolled in the Bleeding, Thrombotic, and Platelet Disorders (BPD) project of the NIHR BioResource Rare Disease program. The Institutional Review Board of the University Hospital Gasthuisberg Leuven approved the genetic study (ML3580). All mouse care and experimentation was approved by the Beth Israel Deaconess Medical Center Institutional Animal Care and Use Committee.
Author contributions
SS designed, conducted, supervised, and analyzed experiments. BF introduced the study of TF and arranged for participation in the NIHR BioResource study. EED conducted and analyzed all experiments. LL conducted recombinant TF experiments. KP enrolled the patient and collected the clinical data. KF, SS, ET, and WHO designed, conducted, and interpreted human genetic experiments. MAB, RJW, GMS, CRS, and RF conducted and analyzed rodent experiments. MF, MHCF, and CAC conducted and analyzed iPSC experiments. The manuscript was written by SS and BF with contributions from all authors.
Supplementary Material
Acknowledgments
We thank NIHR BioResource volunteers for their participation, and gratefully acknowledge the NIHR BioResource centers and NHS trusts and staff for their contributions. We thank the National Institute for Health Research and NHS Blood and Transplant. The views expressed are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research (NIHR), or the Department of Health and Social Care. See Supplemental Acknowledgments for a complete list of participants in NIHR BioResource (Supplemental material available online with this article; https://doi.org/10.1172/JCI133780DS1). We thank Monica Sentmanat and Yong Miao at the Washington University Genome Engineering and iPSC Center for their expertise in genome editing iPSCs. We thank David Ginsburg, Nigel Mackman, and Oskar Eriksson for helpful insight and discussion. This work was supported by the National Hemophilia Foundation (Judith Graham Pool Fellowship to SS), the Hemostasis and Thrombosis Research Society (Mentored Research Award to SS supported by an educational grant from CSL Behring), CSL Behring (Professor Heimburger Award to SS, grants to KP and KF), Bayer (grants to KP and KF), SOBI (grants to KP and KF), KULeuven BOF (grant C14/19/096 to KP and KF), the NIHR (RG65966 to WHO and ET), and the NIH (K08HL146797 and DP5OD028129 to SS, R01HL136394 to BF, R01HL135035 to RJW, R35HL135775 and R01HL125275 to RF).
Version 1. 07/14/2020
In-Press Preview
Version 2. 08/31/2020
Electronic publication
Version 3. 10/01/2020
Print issue publication
Funding Statement
Not related to this manuscript
Not related to this manuscript
This is a an independently sponsored research grant awarded by an independent academic peer review body and CSL Behring had no role whatsoever in the planning, design, execution, or preparation of this manuscript.
Footnotes
Conflict of interest: RF is a founder of and consultant for Platelet Diagnostics and a consultant for Relay Therapeutics. ET is a partner and scientific director with Tachyon Ventures LLC. CC has ownership in and receives income from Sana Biotechnology. SS is the recipient of a research grant from CSL Behring.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(10):5302–5312.https://doi.org/10.1172/JCI133780.
Contributor Information
Sol Schulman, Email: sschulm1@bidmc.harvard.edu.
Emale El-Darzi, Email: emaledarzi@gmail.com.
Mary H.C. Florido, Email: kiwiflorido@g.harvard.edu.
Max Friesen, Email: maxfriesen@gmail.com.
Glenn Merrill-Skoloff, Email: gmerrill@bidmc.harvard.edu.
Marisa A. Brake, Email: mabrake@oakland.edu.
Calvin R. Schuster, Email: cschuste@bidmc.harvard.edu.
Lin Lin, Email: llin2@bidmc.harvard.edu.
Randal J. Westrick, Email: rjwestrick@oakland.edu.
Chad A. Cowan, Email: ccowan@bidmc.harvard.edu.
Robert Flaumenhaft, Email: rflaumen@bidmc.harvard.edu.
NIHR BioResource, Email: sp605@medschl.cam.ac.uk.
Willem H. Ouwehand, Email: Ouwehand-pa@medschl.cam.ac.uk.
Kathelijne Peerlinck, Email: kathelijne.peerlinck@med.kuleuven.be.
Kathleen Freson, Email: kathleen.freson@kuleuven.be.
Ernest Turro, Email: et341@cam.ac.uk.
Bruce Furie, Email: bfurie@bidmc.harvard.edu.
References
- 1.Morrissey JH, Fakhrai H, Edgington TS. Molecular cloning of the cDNA for tissue factor, the cellular receptor for the initiation of the coagulation protease cascade. Cell. 1987;50(1):129–135. doi: 10.1016/0092-8674(87)90669-6. [DOI] [PubMed] [Google Scholar]
- 2.Fisher KL, Gorman CM, Vehar GA, O’Brien DP, Lawn RM. Cloning and expression of human tissue factor cDNA. Thromb Res. 1987;48(1):89–99. doi: 10.1016/0049-3848(87)90349-5. [DOI] [PubMed] [Google Scholar]
- 3.Scarpati EM, et al. Human tissue factor: cDNA sequence and chromosome localization of the gene. Biochemistry. 1987;26(17):5234–5238. doi: 10.1021/bi00391a004. [DOI] [PubMed] [Google Scholar]
- 4.Spicer EK, et al. Isolation of cDNA clones coding for human tissue factor: primary structure of the protein and cDNA. Proc Natl Acad Sci USA. 1987;84(15):5148–5152. doi: 10.1073/pnas.84.15.5148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruf W, Rehemtulla A, Morrissey JH, Edgington TS. Phospholipid-independent and -dependent interactions required for tissue factor receptor and cofactor function. J Biol Chem. 1991;266(24):16256. [PubMed] [Google Scholar]
- 6.Banner DW, et al. The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature. 1996;380(6569):41–46. doi: 10.1038/380041a0. [DOI] [PubMed] [Google Scholar]
- 7.Furie B, Furie BC. The molecular basis of blood coagulation. Cell. 1988;53(4):505–518. doi: 10.1016/0092-8674(88)90567-3. [DOI] [PubMed] [Google Scholar]
- 8.Konigsberg WH, Nemerson Y. Molecular cloning of the cDNA for human tissue factor. Cell. 1988;52(5):639–640. doi: 10.1016/0092-8674(88)90399-6. [DOI] [PubMed] [Google Scholar]
- 9.Drake TA, Morrissey JH, Edgington TS. Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am J Pathol. 1989;134(5):1087–1097. [PMC free article] [PubMed] [Google Scholar]
- 10.Owens AP, Mackman N. Role of tissue factor in atherothrombosis. Curr Atheroscler Rep. 2012;14(5):394–401. doi: 10.1007/s11883-012-0269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wolberg AS, et al. Venous thrombosis. Nat Rev Dis Primers. 2015;1:15006. doi: 10.1038/nrdp.2015.6. [DOI] [PubMed] [Google Scholar]
- 12.Zwicker JI, et al. Tumor-derived tissue factor-bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res. 2009;15(22):6830–6840. doi: 10.1158/1078-0432.CCR-09-0371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sparkenbaugh EM, Chantrathammachart P, Chandarajoti K, Mackman N, Key NS, Pawlinski R. Thrombin-independent contribution of tissue factor to inflammation and cardiac hypertrophy in a mouse model of sickle cell disease. Blood. 2016;127(10):1371–1373. doi: 10.1182/blood-2015-11-681114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bauer KA, Kass BL, ten Cate H, Hawiger JJ, Rosenberg RD. Factor IX is activated in vivo by the tissue factor mechanism. Blood. 1990;76(4):731–736. [PubMed] [Google Scholar]
- 15.Grover SP, Mackman N. Tissue Factor: An essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. 2018;38(4):709–725. doi: 10.1161/ATVBAHA.117.309846. [DOI] [PubMed] [Google Scholar]
- 16.Ruf W, Rehemtulla A, Morrissey JH, Edgington TS. Phospholipid-independent and -dependent interactions required for tissue factor receptor and cofactor function. J Biol Chem. 1991;266(24):16256. [PubMed] [Google Scholar]
- 17.Paborsky LR, Caras IW, Fisher KL, Gorman CM. Lipid association, but not the transmembrane domain, is required for tissue factor activity. Substitution of the transmembrane domain with a phosphatidylinositol anchor. J Biol Chem. 1991;266(32):21911–21916. [PubMed] [Google Scholar]
- 18.Waters EK, Morrissey JH. Restoring full biological activity to the isolated ectodomain of an integral membrane protein. Biochemistry. 2006;45(11):3769–3774. doi: 10.1021/bi052600m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carmeliet P, et al. Role of tissue factor in embryonic blood vessel development. Nature. 1996;383(6595):73–75. doi: 10.1038/383073a0. [DOI] [PubMed] [Google Scholar]
- 20.Bugge TH, et al. Fatal embryonic bleeding events in mice lacking tissue factor, the cell-associated initiator of blood coagulation. Proc Natl Acad Sci USA. 1996;93(13):6258–6263. doi: 10.1073/pnas.93.13.6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Toomey JR, Kratzer KE, Lasky NM, Stanton JJ, Broze GJ. Targeted disruption of the murine tissue factor gene results in embryonic lethality. Blood. 1996;88(5):1583–1587. [PubMed] [Google Scholar]
- 22.Westrick RJ, et al. Sensitized mutagenesis screen in Factor V Leiden mice identifies thrombosis suppressor loci. Proc Natl Acad Sci USA. 2017;114(36):9659–9664. doi: 10.1073/pnas.1705762114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parry GC, Erlich JH, Carmeliet P, Luther T, Mackman N. Low levels of tissue factor are compatible with development and hemostasis in mice. J Clin Invest. 1998;101(3):560–569. doi: 10.1172/JCI814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Erlich J, et al. Tissue factor is required for uterine hemostasis and maintenance of the placental labyrinth during gestation. Proc Natl Acad Sci USA. 1999;96(14):8138–8143. doi: 10.1073/pnas.96.14.8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pawlinski R, et al. Tissue factor deficiency causes cardiac fibrosis and left ventricular dysfunction. Proc Natl Acad Sci USA. 2002;99(24):15333–15338. doi: 10.1073/pnas.242501899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bode MF, Mackman N. A combined deficiency of tissue factor and PAR-4 is associated with fatal pulmonary hemorrhage in mice. Thromb Res. 2016;146:46–50. doi: 10.1016/j.thromres.2016.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quiroga T, et al. High prevalence of bleeders of unknown cause among patients with inherited mucocutaneous bleeding. A prospective study of 280 patients and 299 controls. Haematologica. 2007;92(3):357–365. doi: 10.3324/haematol.10816. [DOI] [PubMed] [Google Scholar]
- 28.Downes K, et al. Diagnostic high-throughput sequencing of 2396 patients with bleeding, thrombotic, and platelet disorders. Blood. 2019;134(23):2082–2091. doi: 10.1182/blood.2018891192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Turro E, et al. Whole-genome sequencing of patients with rare diseases in a national health system. Nature. 2020;583(7814):96–102. doi: 10.1038/s41586-020-2434-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Megy K, et al. Curated disease-causing genes for bleeding, thrombotic, and platelet disorders: Communication from the SSC of the ISTH. J Thromb Haemost. 2019;17(8):1253–1260. doi: 10.1111/jth.14479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stone MJ, Ruf W, Miles DJ, Edgington TS, Wright PE. Recombinant soluble human tissue factor secreted by Saccharomyces cerevisiae and refolded from Escherichia coli inclusion bodies: glycosylation of mutants, activity and physical characterization. Biochem J. 1995;310(Pt 2):605–614. doi: 10.1042/bj3100605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandal SK, Pendurthi UR, Rao LV. Cellular localization and trafficking of tissue factor. Blood. 2006;107(12):4746–4753. doi: 10.1182/blood-2005-11-4674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Patsch C, et al. Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat Cell Biol. 2015;17(8):994–1003. doi: 10.1038/ncb3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Grover SP, et al. Injury measurements improve interpretation of thrombus formation data in the cremaster arteriole laser-induced injury model of thrombosis: communication from the SSC of the ISTH. J Thromb Haemost. doi: 10.1111/jth.15059. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Simeoni I, et al. A high-throughput sequencing test for diagnosing inherited bleeding, thrombotic, and platelet disorders. Blood. 2016;127(23):2791–2803. doi: 10.1182/blood-2015-12-688267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Toomey JR, Kratzer KE, Lasky NM, Broze GJ. Effect of tissue factor deficiency on mouse and tumor development. Proc Natl Acad Sci USA. 1997;94(13):6922–6926. doi: 10.1073/pnas.94.13.6922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saleheen D, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544(7649):235–239. doi: 10.1038/nature22034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Taubman MB, Giesen PL, Schecter AD, Nemerson Y. Regulation of the procoagulant response to arterial injury. Thromb Haemost. 1999;82(2):801–805. [PubMed] [Google Scholar]
- 40.Paquet D, et al. Efficient introduction of specific homozygous and heterozygous mutations using CRISPR/Cas9. Nature. 2016;533(7601):125–129. doi: 10.1038/nature17664. [DOI] [PubMed] [Google Scholar]
- 41.Sentmanat MF, Peters ST, Florian CP, Connelly JP, Pruett-Miller SM. A survey of validation strategies for CRISPR-Cas9 editing. Sci Rep. 2018;8(1):888. doi: 10.1038/s41598-018-19441-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8(10):1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 43.Brake MA, Ivanciu L, Maroney SA, Martinez ND, Mast AE, Westrick RJ. Assessing blood clotting and coagulation factors in mice. Curr Protoc Mouse Biol. 2019;9(2):e61. doi: 10.1002/cpmo.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dubois C, Panicot-Dubois L, Gainor JF, Furie BC, Furie B. Thrombin-initiated platelet activation in vivo is vWF independent during thrombus formation in a laser injury model. J Clin Invest. 2007;117(4):953–960. doi: 10.1172/JCI30537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vandendries ER, Hamilton JR, Coughlin SR, Furie B, Furie BC. Par4 is required for platelet thrombus propagation but not fibrin generation in a mouse model of thrombosis. Proc Natl Acad Sci USA. 2007;104(1):288–292. doi: 10.1073/pnas.0610188104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ivanciu L, Krishnaswamy S, Camire RM. New insights into the spatiotemporal localization of prothrombinase in vivo. Blood. 2014;124(11):1705–1714. doi: 10.1182/blood-2014-03-565010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zerbino DR, et al. Ensembl 2018. Nucleic Acids Res. 2018;46(D1):D754–D761. doi: 10.1093/nar/gkx1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.