

Abstract
The correlation of HIV-specific antibody-dependent cellular cytotoxicity (ADCC) responses with protection from and delayed progression of HIV-1 infection provides a rationale to leverage ADCC-mediating antibodies for treatment purposes. We evaluated ADCC mediated by different combinations of 2 to 6 neutralizing and non-neutralizing anti–HIV-1 Envelope (Env) mAbs, using concentrations ≤ 1 μg/mL, to identify combinations effective at targeting latent reservoir HIV-1 viruses from 10 individuals. We found that within 2 hours, combinations of 3 mAbs mediated more than 30% killing of HIV-infected primary CD4+ T cells in the presence of autologous NK cells, with the combination of A32 (C1C2), DH511.2K3 (MPER), and PGT121 (V3) mAbs being the most effective. Increasing the incubation of target and effector cells in the presence of mAb combinations from 2 to 24 hours resulted in increased specific killing of infected cells, even with neutralization-resistant viruses. The same combination eliminated reactivated latently HIV-1–infected cells in an ex vivo quantitative viral outgrowth assay. Therefore, administration of a combination of 3 mAbs should be considered in planning in vivo studies seeking to eliminate persistently HIV-1–infected cells.
Keywords: AIDS/HIV
Keywords: Immunoglobulins, Immunotherapy, NK cells
Introduction
Although antiretroviral therapy (ART) has been effective in controlling virus replication, delaying disease progression, and reducing HIV-1 transmission (1), it has had limited effects in preventing the seeding of the latent reservoir that persists during ART (2, 3). The isolation of potent anti–HIV-1 Envelope (Env) antibodies from natural infection has allowed the development of novel therapeutic agents in treatment of HIV-1 infection (4–6). Passive immunization with a single broadly neutralizing antibody (bNAb) (VRC01, 10-1074, or 3BNC117) has mediated suppression of viremia in HIV-1–infected individuals (7–9), and only individuals with low plasma viral load demonstrated viral suppression to undetectable levels until systemic levels of antibody had decreased (9). In addition, passive immunization with VRC01 or 3BNC117 delayed viral rebound during analytical treatment interruption (10, 11). Unfortunately, bNAb monotherapy did not prevent viral rebound — either because of viral escape, indicating that bNAbs exerted selective pressure on the rebound virus (11), or because of outgrowth of preexisting resistant viral variants (8–10) — and, as recently reported, did not demonstrate a decline in the size of viral reservoir (12). The outcomes of these studies have prompted in silico and computational predictions of the requirements for successful use of bNAbs in the field of HIV-1 prevention. These studies suggest that treatment with single anti–HIV-1 monoclonal antibodies (mAbs) will lead to an emergence of viral resistance (13), and in vitro testing supported this prediction (14). Moreover, Wagh et al. created a mathematical model and demonstrated in vitro that combinations of 3 or 4 mAbs increased neutralization potency and breadth and are more effective than any single mAb or combination of 2 mAbs (15). These results were supported by an independent in vitro neutralization study of 125 HIV-1 pseudotyped strains from various clades by a combination of 2, 3, and 4 mAbs for additive or synergistic effects, which demonstrated that an optimal combination of 3 or more bNAbs improved neutralizing breadth against HIV-1 viruses (16).
Combinations of bNAbs have been administered to ART-naive humanized mouse and nonhuman primate preclinical models for treatment of established HIV-1 infection. Both animal models demonstrated that treatment with a combination of antibodies reduced viral load to undetectable levels, induced long-lasting immunity, and reduced measures of persistent viral infection; however, viral escape to at least one of the bNAbs in the combination was detected (17–23). Human clinical trials treating antiretroviral-suppressed participants with combinations of 2 bNAbs have also demonstrated a delay in viral rebound upon analytical treatment interruption (24–26). In both studies, more durable viral control was observed in individuals with viral sensitivity to all administered bNAbs. In these studies, the rebound virus either developed resistance to (25) or was linked to preexisting resistance to one of the bNAbs in the treatment combination but not to both bNAbs (24, 26), indicating that an optimal combination may achieve acceptable therapeutic outcome.
The effects of bNAbs can be mediated by both neutralization of virus and antibody-dependent cellular cytotoxicity (ADCC) through Fc receptor–mediated function, eliminating HIV-1–infected cells. It is proposed that ADCC contributes to the kinetics of viral load reduction seen in vivo in elite controllers (27) and following bNAb administration (28–33). ADCC, driven by bNAbs and non-neutralizing antibodies (non-NAbs), can also mediate killing of cells infected by the viruses that escaped neutralization and cells infected with neutralization-resistant viruses (34, 35).
Given the potential of mAbs in eradication of HIV-1 infection, we analyzed the potency and breadth of ADCC-mediating bNAb and non-NAb combinations against HIV-1–infected cells. In order to recapitulate the effects of mAb combinations in vivo, we used an autologous in vitro system in which primary activated and infected CD4+ T cells were used as targets and autologous purified natural killer (NK) cells were used as effectors. To ensure broad coverage of HIV-1 Env epitopes, we used 5 bNAbs that target nonoverlapping epitopes expressed on the viral Env spike as well as on the surface membrane of infected cells and 1 non-NAb that targets C1C2 epitope expressed on the Env upon engagement with the cell surface receptor CD4. To further validate our findings, we used latent reservoir HIV-1 viruses (LRVs) recovered from the resting CD4+ T cells of HIV-infected donors treated with fully, durably suppressive ART (36). Our results indicated that at least 3 mAbs with various epitope specificities were necessary for elimination of a diverse population of latently HIV-infected cells.
Results
The kinetics of ADCC in a primary in vitro autologous system.
We have previously demonstrated that the ADCC antibody responses detectable in the plasma of HIV-1–infected individuals were primarily dependent on the recruitment of NK cells present in the PBMC samples used as a source of effector cells in vitro (37). Therefore, we used an autologous primary in vitro system in which HIV-1NL-LucR.WITO.ecto–infected primary CD4+ T cells, isolated from healthy HIV-1–seronegative subjects, were used as target (T) cells. Autologous NK cells purified from the same donor were used as effector (E) cells. We first determined how soon after encountering the infected target cells mAbs were able to mediate NK-specific killing. We chose a panel of 6 mAbs targeting 6 distinct HIV-1 Env epitopes: C1C2 (A32) (38), CD4bs (CH557) (39), V2 glycan (PG9) (40), V3 glycan (PGT121) (41), gp120-gp41 interface (PGT151) (42), and MPER (DH511.2K3) (43). The neutralization sensitivity of WITO infectious molecular clone (IMC) to each mAb was tested and is shown in Supplemental Table 1 (supplemental material available online with this article; https://doi.org/10.1172/JCI135557DS1). WITO IMC was sensitive to neutralization by all bNAbs except PGT121 (IC50 > 5 μg/mL). HIV-infected CD4+ T cells were cocultured with mAbs and autologous NK cells for 2, 3, 4, or 6 hours or overnight, and then the percentage specific killing (or percentage ADCC) was measured (Figure 1). Percentage ADCC was observed to vary according to mAb specificity, concentration, and duration of incubation. A dose-dependent effect was often observed for ADCC mediated by the individual mAb. Moreover, we observed 2-fold increase at the concentration of peak ADCC when killing was measured overnight (green titration curve) compared with 2 hours after incubation (blue titration curve) for CH557 (10 μg/mL), PG9 (0.2–10 μg/mL), and PGT151 (0.2–10 μg/mL) mAbs. In comparison, for PGT121, an increase in killing was observed overnight compared with 2 hours after incubation at all concentrations (green and blue titration curves, respectively).
Figure 1. NK-specific killing of WITO-infected CD4+ T cells measured after culture with NK cells and mAbs for 2, 3, 4, or 6 hours or overnight.

NK-specific killing mediated by each antibody was tested using a primary in vitro autologous model, in which NL-LucR.WITO.ecto–infected primary CD4+ T cells were used as targets (T) and autologous purified and IL-15–stimulated NK cells as effectors (E). The percentage specific killing is reported on the y axes and the mAb dilutions on the x axes. Blue, red, purple, black, and green titration curves represent killing at 2, 3, 4, and 6 hours and overnight, respectively. CH65 (an anti-influenza monoclonal antibody) was used as negative control (83). The results show the average of 2 replicates from a single experiment.
While most of the neutralizing mAbs mediated greater levels of killing with higher concentrations and longer incubation times, A32 was unique: it was much more efficient at mediating ADCC at lower concentrations regardless of the duration of incubation.
Importantly, ADCC was detected as soon as 2 hours after incubation for each mAb (10%–40%), although the concentrations that allowed detectable ADCC varied for each mAb. It has been proposed that mAbs could be used in combination with latency-reversing agents (LRAs) to improve and accelerate the elimination of the HIV-1 latent reservoir (44, 45). LRAs have a very short half-life (46, 47), and mAbs are expected to be able rapidly recognize cells expressing HIV-1 Env epitopes. Therefore, we determined the concentration of each mAb sufficient to mediate specific killing individually and to capture any additive ADCC results when used in combination after 2 hours incubation. We chose low concentration of each broadly neutralizing antibody that individually shows low to minimal ADCC activity against WITO-infected cells and hypothesized that combinations of mAbs at low concentrations will lead to an increase in ADCC compared with individual mAbs. Therefore, we chose 1 μg/mL of bNAbs mediating 25% (CH557), 40% (PG9), 15% (PGT121 and PGT151 each), or 10% (DH511.2K3) specific killing against infected cells and 0.1 μg/mL of A32 mediating 40% specific killing at 2 hours after incubation of T, E, and mAb (Figure 1, blue curves).
Panel of latent reservoir HIV-1 viruses.
To study clearance of clinically relevant latent reservoir HIV-1 viruses (LRVs), we recruited 10 HIV-1–infected individuals with suppressed viremia (viral load <50 copies/mL) for at least 7 months; 1 individual, P500, had had uncontrolled viremia for over 18 years before full suppression (Table 1). Clinical characteristics of the donors are detailed in Table 1. The average duration of undetectable viral load for these 10 donors was 4 years, with the range of viral suppression from 0.77 to 8.7 years. Importantly, this cohort also represents the variability of current patients starting ART from as early as 15 days to 2.5 years after HIV-1 diagnosis. Viruses representing LRVs within each given donor were isolated using a quantitative viral outgrowth assay (QVOA). For each donor, between 9 and 12 p24-positive wells were pooled to generate LRVs. These viral pools are representative of the total viral population that exists within each participant. Sequencing of each LRV swarm yielded between 2 and 10 HIV env sequences (GenBank MT007547–MT007585). The observed viral sequences represent a fraction of the total diversity that may be observed within the participant. All LRV sequences were classified as clade B, as determined by the Los Alamos National Laboratory database (Supplemental Figure 1A). Sequences were aligned with 1916 filtered subtype B env sequences, and a maximum-likelihood tree was constructed to determine the genetic distribution of the isolates within clade B (Supplemental Figure 1B). Sequences from all 10 participants were distributed evenly throughout the phylogenetic tree and, consequently, widely represented clade B envs.
Table 1. Clinical characteristics of HIV-1–seropositive donors.

Combination of mAbs mediates improved ADCC activity against infected cells.
To identify whether combinations of mAbs, compared with individual mAbs, are more efficient at eliminating infected cells within 2 hours, we used 54 combinations of 2–6 mAbs to achieve broad and rapid NK-specific killing. We chose 0.1 μg/mL for A32 and submaximal ADCC concentrations of each bNAb (1 μg/mL) to reveal the presence of agonist or antagonist effects. As control, mAbs were also tested individually. Target cells were generated using primary human CD4+ T cells infected with LRVs from each of the 10 chronically infected individuals, and infectivity of the LRVs was confirmed by intracellular p24 staining (Supplemental Figure 2).
ADCC mediated by the 6 individual mAbs against LRV-infected cells after 2 hours of incubation was generally ≤20% (Figure 2A) with the median killing by individual mAbs at 11% (Figure 2B) with exceptions for the activity of DH511.2K3 (28%, 22%, 21.5%, and 21% specific killing against P500, P800, P725, and P749, respectively) and PGT121 (28%, 24%, 23.7%, and 23% specific killing against P500, P834, P1095, and P10785, respectively). Specific killing improved with an increase in the number of mAbs used in combinations: 2 mAbs demonstrated a median of 22%, 3- and 4-mAb combinations had a median of 27%, 5 mAbs had a median of 29%, and 6 mAbs had a median of 34% (Figure 2B). Groups 1 and 2 showed a difference in percentage killing (P = 0.0039), and similarly groups 2 and 3 showed a difference in percentage killing (P = 0.0020). Further addition of more mAbs, groups 4 and 5, did not show significant improvement compared with group 3 (Figure 2B). Group 2 when compared with groups 3, 4, and 5 showed a difference in percentage specific killing (P values < 0.05).
Figure 2. ADCC by individual and combination mAbs 2 hours after incubation of T+E+mAbs.

(A) NK-specific killing mediated by each mAb individually within 2 hours using 1 μg/mL of PGT151, PG9, CH577, DH511.2K3, or PGT121 and 0.1 μg/mL of A32. The anti-influenza mAb CH65 was used as a negative control at 1 μg/mL. (B) Average specific killing by the number of mAbs in combinations against LRVs. The thick line in the box plots denotes the median, and the ends of the box denote the 25th and 75th percentiles.
Comparison of mAb combinations by ADCC potency and breadth.
We sought to identify the combination of mAbs that would demonstrate the broadest and most potent ADCC activity against the majority of the LRVs tested. Therefore, we calculated an ADCC score using a principal component analysis method that combined 2 parameters: (a) a weighted average of the potency (or percentage specific killing) of a specific combination against cells infected with LRVs from each of the 10 individuals, and (b) the breadth (the ability of a combination to target LRVs from each of 10 individuals with percentage specific killing > 20%). Using this scoring method, we ranked combinations from the most potent to the weakest in ADCC score (Table 2 shows the top 6 combinations). The majority of combinations among these 6 consisted of 3 mAbs, while individual mAbs were among the weakest 11 out of a total 61 combinations tested (Figure 2B and Supplemental Table 2). This further indicates that we did not identify a single mAb that can efficiently target the variety of HIV-1 envelopes on the surface of infected cells, and sufficient breadth of ADCC activity requires a combination of at least 3 mAbs.
Table 2. Statistical analysis and significance of combinations.

The most effective combination according to ADCC score was A32+DH511.2K3+PGT121. This combination was able to mediate specific killing above 20% against LRVs from all individuals, except P725 (17.5%). We observed that DH511.2K3 was in all 6 top combinations, PGT121 was in 4 of the top 6 combinations, and PG9 was in 3 of the top 6 combinations against LRVs in our panel. Notably, A32 was in 5 of the top 6 combinations (Table 2).
We next analyzed whether the non-NAb A32 contributed to the increase in killing observed in groups f 3 mAbs (Figure 3A). We found that specific killing mediated by combinations of 3 mAbs that included A32 mAb was statistically higher in comparison with equivalent combinations of 2 bNAbs without A32 (P = 1.1 × 10–5) or combination of 3 bNAbs (P = 0.00096). Further analysis revealed that combinations of 3 bNAbs did not improve in specific killing compared with combinations of 2 bNAbs (P = 0.33; Figure 3A), indicating that an increase in amount of mAb in the assay was not responsible for the increase in killing, and A32 alone was responsible for the observed boost in specific killing by combinations of 3 mAbs.
Figure 3. Impact of A32 on specific killing mediated by bNAbs.

(A) Comparison of specific killing mediated by 2 bNAbs, equivalent combinations with A32 (2 bNAbs + A32), and 3 bNAbs. (B) Each group of mAb combinations was separated into bNAbs and equivalent combination with the addition of A32. Box plots with interquartile range represent median specific killing (y axis) by each group of bNAbs without A32 (pink) and with A32 (green). Statistical significance was calculated by paired Wilcoxon test.
To verify the direct contribution of A32 to each group, we compared killing mediated by each bNAb individually or combinations of bNAbs with the equivalent combination that included the non-NAb A32 (Figure 3B). We observed that the addition of A32 to any individual bNAb or combination of 2 or 3 bNAbs increased specific killing, while the same effect was not observed with addition of A32 to combinations of 4 bNAbs. These results further demonstrate the unique contribution of the non-neutralizing A32 mAb to potent and effective clearance of HIV-infected cells.
mAb neutralization and binding of LRVs.
Having determined the ADCC activity of each mAb in our panel, we sought to evaluate the ability of bNAbs in the top combination, A32+DH511.2K3+PGT121, to neutralize each of the LRVs in our panel. In addition, we chose another combination, A32+PG9+PGT121, as PG9 and PGT121 have shown complementary neutralization activity against the 200 subtype C HIV-1 representing those isolated during acute/early infection (15). Thus, we tested neutralization activity of individual bNAbs (A32, DH511.2K3, PGT121, and PG9), 2 bNAbs in each combination (DH511.2K3+PGT121 and PG9+PGT121), or triple combinations (A32+DH511.2K3+PGT121 and A32+PG9+PGT121; Table 3). We found that A32 was unable to neutralize LRVs from any of the individuals tested here. PGT121 was not able to neutralize LRVs from 3 of 10 individuals (P795, P673, P800), PG9 did not neutralize LRVs from 3 of 10 individuals (P834, P674, P800), and DH511.2K3 neutralized only 2 LRVs at ≤5 μg/mL (P834, IC50 = 4.3; P10785, IC50 = 4.4). The combination of PG9+PGT121 neutralized LRVs from P795 and P673 with an IC50 similar to that of PG9 alone, suggesting that neutralization was driven by PG9. We observed a similar pattern with PG9-resistant LRVs from individuals P834 and P674: LRVs from both individuals were neutralized by the combination of PG9+PGT121, which was driven by PGT121. Interestingly, while LRVs from both P795 and P673 were resistant to PGT121 and DH511.2K3 individually, the combination of PGT121+DH511.2K3 was able to neutralize these viruses with IC50 of 4.7 μg/mL and 2.6 μg/mL, respectively. Addition of the non-neutralizing mAb A32 to the combination of 2 bNAbs demonstrated similar IC50 values to the 2 bNAbs alone. Moreover, while the combination DH511.2K3+PGT121 was able to neutralize all LRVs at <5 μg/mL with the exception of LRVs from P800, addition of A32 reduced the neutralization potency of triple combination from IC50 of 0.57 μg/mL to 2.5 μg/mL against P674, and to IC50 > 5 μg/mL against P795 and P673 LRVs. The A32+PG9+PGT121 combination revealed levels of neutralization comparable to those seen with the combination of the 2 bNAbs (PG9+PGT121), indicating that no effects on neutralization were obtained by the addition of A32 to the combination. The reduction of neutralization potency was observed only against P674 LRVs, from IC50 of 2.2 μg/mL to IC50 > 5 μg/mL. Of the 10 individuals, we noticed that LRVs from P800 were resistant to neutralization by any individual bNAb or their combinations. On the basis of these results, we further grouped LRVs from the 10 donors based on their neutralization sensitivity (Table 3).
Table 3. Neutralization by most effective single mAbs, dual and triple mAb combinations, and Env escape mutations.

To understand whether the lack of neutralization was due to the loss of epitopes, we isolated viral RNA from LRVs and used PacBio sequencing to sequence HIV envs. For each participant we obtained between 2 and 10 different env sequences from their LRVs, with little intra-participant diversity detected. Sequence analysis revealed that most LRVs resistant to PGT121/PG9 neutralization contained classical resistance mutations (Table 3): P674 LRVs had a proline at position 169, while absence of positively charged amino acid at this position has been associated with PG9 resistance (48). P795 LRVs lacked the N332 glycan, and P673 LRVs contained a tyrosine residue at position 330, which has been associated with PGT121 resistance (41, 49). P800 LRVs contained a tyrosine residue at position 330 and lacked glycan at position 160, which are associated with both PGT121 and PG9 resistance (50). Although DH511.2K3 has previously demonstrated broader neutralization potency of clade B viruses compared with other leading MPER mAbs (43), the LRVs tested here are swarms of viruses rather than a single T/F strain, which could reflect low neutralization potency of DH511.2K3 Despite this, DH511.2K3 alone was able to mediate ADCC against 3 LRVs above 20% (Figure 2A and Supplemental Table 2) and demonstrate potent ADCC when used in combination with other mAbs.
P725 LRVs demonstrated sensitivity to neutralization by PG9 and PGT121. However, neither A32+DH511.2K3+PGT121 nor A32+PG9+PGT121 was able to mediate ADCC ≥ 20% against P725. This may be because infected cells may display Env differently on the surface of infected cells, resulting in loss of mAb binding to the cellular membrane-bound Env and in a subsequent decrease in ADCC activity. Thus, we next examined the loss of epitopes on the surface of cells infected with LRVs using the 4 mAbs in our 2 combinations (A32, PG9, PGT121, and DH511.2K3) in an infected-cell antibody binding assay (Figure 4). LRVs from individuals with 4 different neutralization profiles were chosen: P725 (PGT121 and PG9 sensitive), P674 (PGT121 sensitive, PG9 resistant), P795 (PGT121 resistant, PG9 sensitive), and P800 (PGT121 and PG9 resistant). We observed that all 4 mAbs bound to P725 LRV–infected cells (Figure 4A); PGT121 but not PG9 bound to P674 LRV–infected cells (Figure 4B); PG9 but not PGT121 bound to P795 LRV–infected cells (Figure 4C); and only A32 bound to the cells infected with the resistant LRVs from P800 (Figure 4D).
Figure 4. Infected-cell antibody binding assay with LRVs.

Primary CD4+ T cells were infected with PGT121-sensitive, PG9-sensitive LRVs (P725) (A); PGT121-sensitive, PG9-resistant LRVs (P674) (B); PGT121-resistant, PG9-sensitive LRVs (P795) (C); and PGT121-resistant, PG9-resistant LRVs (P800) (D). The y axis indicates the frequency of infected cells (p24+) bound by the mAbs listed on the x axis. Asterisks represent binding to mock-infected cells.
Longer incubation time increases specific killing of infected cells.
The lack of ADCC activity of triple combinations against P725 LRVs was surprising, considering neutralization sensitivity to PG9 and PGT121 and expression of HIV-1 Env epitopes on virus-infected cells. While neutralization in general correlates with ADCC and antibody binding, some antibodies can show lack of polyfunctionality depending on antigen, epitope expression, and affinity of antibodies for Env on the surface of infected cells (35), which could be rescued by an increase of antibody concentration or prolong incubation of effector and target cells in the presence of antibody. Therefore, we sought to investigate whether an increase of incubation from 2 to 24 hours would allow detection of ADCC activity against P725 and improve specific killing against other viruses. LRVs from the same 4 individuals as above were used: P725, P674, P795, and P800 (Figure 5). After 24 hours, an increase in specific killing (>20%) with each mAb alone and with both combinations of 3 mAbs was observed against the dual-sensitive P725 LRVs. A32+DH511.2K3+PGT121 mediated 58% specific killing, and A32+PG9+PGT121 mediated 60% specific killing (Figure 5A, black bars). With 24 hours of incubation, mAbs alone and in the aforementioned combinations showed increased ADCC activity, except for PG9, which was not able to mediate ADCC against PG9 neutralization–resistant P674 LRVs (Figure 5B). We observed similar results with PGT121 neutralization–resistant P795 LRVs: there was a slight increase in ADCC by PGT121 from 10% at 2 hours to 21% at 24 hours, and an increase in ADCC to >20% with other mAbs and >40% with both combinations (Figure 5C). Notably, for triple bNAb-resistant P800 LRVs, only A32 of the individual mAbs increased specific killing (to 30%) with an increase in incubation period from 2 to 24 hours. Specific killing mediated by the most effective combination (A32+DH511.2K3+PGT121) increased from 34% at 2 hours to 55% at 24 hours (Figure 5D). In addition, the combination A32+PG9+PGT121 demonstrated specific killing below 20% against P800-infected cells at 2 hours, which increased to 60% at 24 hours (Figure 5D). These results indicate that the combinations of neutralizing and non-neutralizing mAbs are able to mediate ADCC and eliminate cells infected even with viruses that are resistant to neutralization by these mAbs, and that ADCC activity mediated by antibody combinations is increased over time.
Figure 5. Time-dependent elimination of infected cells.

ADCC of individual antibodies and combinations was tested using a primary in vitro autologous model, in which primary CD4+ T cells were used as targets and infected with PG9-resistant, PGT121-sensitive LRVs (P674) (A); PG9-sensitive, PGT121-sensitive LRVs (P725) (B); PGT121-resistant LRVs (P795) (C); and PG9-, PGT121-, DH511.2K3-resistant LRVs (P800) (D). The percentage specific killing is reported on the y axis and individual mAbs or combinations on the x axis. White bars represent killing after 2 hours; black bars represent killing after 24 hours; gray bars represent killing after 24 hours in the absence of effectors.
Latency clearance by combinations of mAbs.
To demonstrate the recognition and clearance of HIV-1–infected cells following latency reversal, we used a modified viral outgrowth assay (51) to assess the ability of mAb combinations to mediate clearance of latently infected cells once they were induced to emerge from latency and produce infectious virions. We focused our attention on donor P800, whose very unusual LRV isolates demonstrated resistance to the bNAbs of interest.
Briefly, resting CD4+ T cells (RCD4) from donor P800 were isolated, and latency was reversed either with phytohemagglutinin (PHA) as a positive control of maximal mitogen stimulation or with a clinically relevant dose of the histone deacetylase inhibitor vorinostat (VOR) (52). Reactivated cells were cultured alone or with autologous NK cells plus 1 mAb (A32), 2 mAbs (DH511.2K3+PGT121), or 3 mAbs (A32+DH511.2K3+PGT121), and plated in 12 wells per condition with PHA and 16 wells per condition with VOR. Viral production was measured at different time points, and the number of HIV-1–positive wells was compared between conditions by p24 ELISA. The combination of 3 mAbs with NK cells reduced the number of p24-positive wells in both the cultures of PHA- and VOR-reactivated cells, at all the time points assayed (Figure 6). PHA-reactivated cultures showed 3 positive wells (out of 12) at day 5 when the RCD4 cells were cultured in the absence of NK cells or mAbs. No reduction was observed when cells were cultured with NK cells and 1 or 2 antibodies, but, interestingly, in the condition with NK cells and the combination of 3 antibodies, no p24-positive wells were detected. The same trend was observed at days 8, 12, 15, and 19 of culture, with a gradual overall increase of p24-positive wells due to viral propagation (Figure 6A). In the cultures reactivated with VOR, wells did not become p24 positive until day 8, increasing to some extent over time. At day 25, 4 of 16 wells were positive in the cultures of only RCD4, and with A32, 5 wells were positive with DH511.2K3+PGT121 and only 1 was positive with the combination of 3 mAbs (Figure 6B). These results indicate that ADCC-mediated clearance of the RCD4 cells after latency reversal is augmented by 3 mAbs in this model system.
Figure 6. Elimination of latently HIV-infected cells from a chronically infected individual after latency reversal.

ADCC activity of each antibody and antibody combination was tested using reactivated latently infected cells isolated from an HIV-seropositive donor, P800, as targets and autologous purified NK cells as effectors, in the presence of 1, 2, or 3 mAbs. Resting CD4+ T cells (RCD4) were reactivated with phytohemagglutinin (PHA) (A) or vorinostat (VOR) (B) and cultured alone or in the presence of NK and mAbs at an effector/target ratio of 1:20. The number of p24-positive wells is reported on the y axis and the duration of mAb treatment on the x axis.
The effect of cell surface CD4 downregulation on mAb binding.
HIV has developed an escape mechanism to common humoral immune responses targeting epitopes expressed on Env after CD4 engagement (CD4-induced [CD4i] epitopes) via Nef-mediated downregulation of surface CD4 expression. This prevents the cis association of surface CD4 with Env on the membrane of infected cells, reducing the expression of CD4i epitopes (53–55). We determined whether CD4 downregulation affects antibody binding to LRV-infected cells. First, we analyzed the level of CD4 downregulation after infection with each of the LRVs. We observed a range of CD4 downregulation from 40% (P674, P500, and P725 LRVs) to 70% (P795 and P1095 LRVs; Figure 7, A and B). We then analyzed the binding of mAbs to both subsets of infected cells. We chose the P1095 LRV, which demonstrated neutralization sensitivity to PG9 and PGT121 (Table 3) and expressed cell surface HIV-1 Env epitopes recognized by all mAbs in our panel. We observed that A32 (C1C2) and DH511.2K3 (MPER) mAbs preferentially bound to infected cells with CD4 molecules still present on the surface (CD4+p24+), while CH557 (CD4bs), PG9 (V2 glycan), PGT151 (gp120-gp41 interface), and PGT121 (V3 glycan) preferentially bound infected cells that had downregulated CD4 (CD4–p24+) (Figure 7C). These results indicate that the binding of mAbs depends on the accessibility of the relevant epitope, which is affected by cell surface CD4 expression.
Figure 7. CD4 downregulation and its effect on antibody binding.

(A) Frequency of infected cells that express surface CD4 (CD4+p24+) and infected cells in which CD4 is downregulated (CD4–p24+) following infection with 10 LRVs. (B) Example of the gating strategy to determine p24+CD4+/– mock- or P1095 LRV–infected primary CD4+ T cells 72 hours after infection. CD4 expression is shown on the y axis and p24 on the x axis. The blue quadrant represents CD4+ infected cells (CD4+p24+), and the red quadrant represents CD4– infected cells (CD4–p24+). (C) The frequency of cells with antibody bound to their surface among CD4+p24+ (blue) and CD4–p24+ (red) P1095 LVR–infected cells.
Influence of CD4 downregulation on ADCC activity of mAbs.
Since mAbs demonstrated a binding preference to Env on infected cells based on CD4 expression, we investigated whether this would result in the preferential killing of double-positive cells (CD4+p24+) or infected cells with downregulated CD4 (CD4–p24+). Cells were infected with the P1095 LRVs, and NK-specific killing was measured 6 hours after incubation of T+E+mAbs. We detected increased ADCC activity of A32 and DH511.2K3 mAbs against CD4+ infected cells compared with infected cells with downregulated CD4 (Supplemental Figure 3, blue line for CD4+ and red line for CD4–). Conversely, the titration curves of CH557 and PGT151 indicated increased specific killing against infected cells with downregulated surface CD4 (Supplemental Figure 3). PGT121 and PG9 demonstrated similar ADCC activity against both subsets of infected cells. Overall, we observed that the ADCC activity of each mAb was affected by the CD4 expression on target cells.
We next analyzed whether mAb combinations were able to target both CD4-positive and CD4-downregulated infected cells. ADCC scoring was carried out as previously described. This revealed that the 6 most effective combinations against CD4-positive infected cells (CD4+p24+) were combinations of 3 mAbs, with the best combination being the same as against total infected cells: A32+DH511.2K3+PGT121 (Table 4). In addition, 4 of the 6 best combinations were groups of 3 mAbs. The top 6 combinations against CD4-downregulated infected cells (CD4–p24+) (Table 4) ranged from combinations of 2 to 6 mAbs with A32+PG9+PGT121 ranking ninth and A32+DH511.2K3+PGT121 ranking tenth. Importantly, most of the combinations targeting the CD4+p24+ population and CD4–p24+ cells included A32, indicating the importance of a potent mAb targeting CD4i epitopes. Moreover, there is evidence revealing a population of HIV-1–infected cells that display CD4i epitopes due to incomplete CD4 downregulation (56), and responses to these epitopes correlate with control of virus replication (57). Since de novo infection with HIV-1 viruses or reactivation of latently infected cells may lead to various levels of CD4 expression, it is important to target HIV-1 Env epitopes coexpressed on those infected cell populations to efficiently eliminate pools of infected cells. The best combination for this purpose is A32+DH511.2K3+PGT121, covering epitopes that may be expressed in both CD4-positive and CD4-downregulated cell populations.
Table 4. Statistical analysis of all combinations against CD4+p24+ and CD4–p24+ cells, showing top 6 combinations.

Discussion
Several studies using broadly neutralizing antibodies (bNAbs) to treat HIV-1 infection in clinical trials have demonstrated significant delay in viral rebound after ART interruption, but failed to eliminate the viral reservoir (31–33). Potent ADCC-mediating antibodies have the ability to target infected cells and assist their clearance, potentially depleting the latent reservoir. In this study, we have shown that within 2 hours a triple combination of 1 non-neutralizing and 2 neutralizing ADCC-mediating mAbs at concentrations ≤ 1 μg/mL eliminated cells infected with 9 of 10 viruses representing the latent reservoir in 10 aviremic, ART-treated participants. Notably, the combinations of 3 mAbs were able to mediate NK-specific killing of LRVs that are resistant to individual or multiple bNAbs. Even in the face of virally induced CD4 downmodulation, most of the mAb combinations with the highest ADCC activity were able to target infected cell populations, illustrating the importance of combining mAbs with different specificities and complementary function.
ART can suppress the HIV-1 replication cycle, but it cannot eradicate existing viral reservoirs. Passively administered potent anti–HIV-1 bNAbs can serve as potential agents for treatment and cure of HIV-1 infection. bNAbs are able to neutralize virus and engage host adaptive and innate immune responses, and can be engineered to have long half-life (17, 58–60). There is growing interest in the use of such antibodies in combination with latency-reversing agents (LRAs) to seek the eradication of all latently infected cells (61, 62). Upon reactivation, it is important to eliminate infected cells as soon as Env is expressed on the cell surface and, possibly, before the release of newly infectious virions. Therefore, Fc receptor functions of antibodies, such as ADCC, may play a crucial role in eliminating these cells (34, 63, 64). Several studies have demonstrated the importance of Fc receptor function of non-NAbs and bNAbs, although some controversy has recently been raised on the importance of the Fc-mediated function in one study of the mAb PGT121 (65). Recent results from a phase IIa clinical trial of coadministration of romidepsin with 3BNC117 demonstrated no obvious impact on the reduction of viral reservoir or the delay in viral rebound (12), thus supporting the importance of combination of mAbs with various function to achieve significant elimination or eradication of viral infection. In this study, we have demonstrated that low concentrations (≤1 μg/mL) of 3 mAbs targeting neutralizing and non-neutralizing epitopes led to an increase in ADCC of infected cells within only 2 hours of incubation in the presence of effector cells; ADCC also increased in a time-dependent manner.
While there is wide consensus that bNAbs will play an important role in cure strategies as evidenced by animal models (17–23), there has been debate about the potential role of non-NAbs. This is particularly true for antibodies targeting CD4-induced (CD4i) epitopes on the Env, which are presented to antibodies when both Env and CD4 are present on the surface of infected cells, leaving Env in a state 2A conformation (66). Non-NAbs targeting C1C2 HIV-1 Env epitopes tend to recognize a broad number of HIV-1 strains and mediate potent ADCC (67, 68). The relatively broad breadth of non-NAbs may ultimately prove important for cure strategies. In this study, we have demonstrated that non-NAb A32 significantly boosted ADCC mediated by individual bNAbs or combination of 2 bNAbs. Interestingly, while A32 alone did not mediate specific killing above 20% against LRVs from any of 10 participants tested in this study, the addition of A32 to other antibodies or antibody combination boosted specific killing. This may be explained by the binding of multiple mAbs with different epitopes to the same Env spike, resulting in increased potency for infected cells to be recognized and eliminated by FcR-bearing NK cells. In general, it seems that a single category of mAbs will not be optimal for viral clearance.
The A32+DH511.2K3+PGT121 and A32+PG9+PGT121 mAb combinations were able to direct ADCC to target LRVs in a cohort of 10 chronically infected individuals within 2–24 hours in vitro. Addition of further antibodies to combinations of 3 mAbs did not offer significant benefit in this system. It is worth noting that the LRVs widely represented clade B HIV-1 env sequences; however, there was limited intra-participant diversity. Among these 10 LRVs, each containing between 2 and 10 different sequences, several were partially resistant to either PG9 or PGT121, or both. This insensitivity was likely due to mutations within the binding epitope for these mAbs, resulting in neutralization resistance and loss of mAb binding. The majority of LRVs also demonstrated neutralization resistance to DH511.2K3. Although DH511.2K3 has demonstrated broader neutralization potency of clade B viruses compared with other leading MPER mAbs, neutralization is not always predictive of ADCC (35). Using a primary in vitro assay system, we demonstrated that increased time of coculture with autologous NK cells and mAbs markedly augmented killing of infected cells. Three mAbs optimally enhanced this effect, even with neutralization-resistant LRVs.
Lastly, combinations of mAbs were effective in a novel ex vivo platform to measure clearance of autologous viruses emerging from the latent reservoir after exposure to LRAs that reveal env sequences not identified from the pooled outgrowth viruses used in earlier experiments. Clearance was observed when infected cells were fully activated or after pharmacologically relevant exposures to LRA. In fact, the best combination of 3 mAbs, A32+DH511.2K3+PGT121, was able to eliminate reactivated latently HIV-1–infected cells by ADCC ex vivo in the patient whose P800 LRVs were resistant to the individual bNAbs (DH511.2K3 and PGT121) and had little sensitivity to the combination during the initial 2-hour incubation. Despite these caveats, the mAb combination A32+DH511.2K3+PGT121 induced clearance after latency reversal with either PHA or VOR. These results suggest the important concept that mAb combinations may increase the activity of ADCC against cells infected with neutralization-resistant HIV isolates. This observation should be tested in future in vivo studies.
It has been reported that HIV-1 Env–specific mAbs vary in antigen-binding affinity (69, 70), neutralization potency (71, 72), and ADCC function (29, 73). Further caveats in the use of mAbs for cure strategies are likely to include the differential expression of viral epitopes on infected cells in various states of quiescence or activation, in distinct contrast to epitopes seen on free virions. Therefore, it may be difficult to find a single broad and potent mAb that will be suitable as passive immunotherapy to mediate robust clearance of residual infection. The administration of a combination of mAbs with selected epitope specificities, neutralizing activity, and Fc receptor functions can potentially overcome preexisting resistance, and increase the efficacy in clearance of persistently infected cells, particularly if appropriately paired with latency reversal strategies.
Our results demonstrated that combinations of the CD4i non-NAb A32, the V3 glycan bNAb PGT121, and either the V1V2 glycan bNAb PG9 or the MPER bNAb DH511.2K3 can eliminate cells infected with LRVs despite the varying neutralization and mAb-binding profiles of individual antibodies. It is important to note that upon latency reversal, Env epitope exposure on infected cells is highly variable across viral isolates (63). The potency of mAb combinations for ADCC in our study parallels the proposed potency of a combination of bNAbs for passive protection (24–26). Therefore, we propose that administration of appropriate combinations of mAbs with a breadth of specificities will ensure the broadest and most potent recognition of infected cells, and will allow more complete elimination of infected cells.
The coadministration of combinations of mAbs to ART-treated patients in combination with safe and effective latency reversal agents must now be tested in appropriate animal models, and ultimately in human studies. Moreover, the results from preclinical studies will provide some guidance in the design of novel mAb-based molecules such as bispecific mAbs (74) and other variations (75) and bispecific dual affinity retargeting (DART) molecules (45) that are currently being evaluated in phase I clinical trials and animal models.
Methods
Study samples.
Samples were derived either from HIV-negative donors or from HIV-infected donors on ART (P673, P674, P10785, P749, P795, P800, P500, P725, P1095, P834) with sustained plasma viremia suppression (<50 copies/mL) for at least 6 months. PBMCs from HIV-seronegative and HIV-seropositive donors were obtained by Ficoll gradient from buffy coats. Immediately after isolation, PBMCs were counted, resuspended in 10% DMSO, 20% FCS, 70% RPMI medium, and cooled to –80°C at a rate of –1°C/h overnight. The next day, PBMCs were stored in liquid nitrogen.
Generation of IMCs and LRVs.
Infectious molecular clones (IMCs) were constructed in an isogenic proviral backbone as previously described (76), encoding the full-length transmitted/founder sequence of subtype B virus WITO, and expressing the Renilla luciferase (LucR) reporter gene under the control of the HIV-1 Tat protein. Latent reservoir HIV-1 viruses (LRVs) were isolated from the supernatants of quantitative viral outgrowth assay from resting CD4+ T cells from HIV-1–seropositive individuals. For each donor, supernatants from all HIV-positive wells were pooled together, generating a swarm virus that represented the virus of the latent reservoir of the donor. HIV-1 NL-LucR.WITO.ecto plasmid was provided by Christina Ochsenbauer (Department of Medicine, University of Alabama at Birmingham, Birmingham, Alabama, USA).
Sequencing of env and alignment.
Viral RNA was isolated from the pooled LRVs and reverse-transcribed into cDNA using SuperScript III Reverse Transcriptase and an oligo-dT primer as described previously (77). For each participant, 3′ half genomes (HXB2 positions 4924–9604) were sequenced from the LRV swarm by isolating RNA, converting it to cDNA using an oligo-dT primer, and performing PCR using barcoded primers. The SMRTBell Template Prep Kit (PacBio) was used to add adaptors to amplicons, and the library was submitted for PacBio sequencing with a movie time of 10 hours. Sequences were demultiplexed by barcode using the PacBio LIMA package, and analyzed using the PacBio Long Amplicon Analysis (LAA) package. The Env amino acid sequences were produced using Los Alamos Genecutter. The PacBio env sequences were deposited in the GenBank repository (MT007547–MT007585).
A phylogenetic tree of all LRVs was generated with Muscle v3.8.31 using the 39 envelope (env) sequences from 10 individuals that were added to 1916 filtered subtype B env sequences available on the Los Alamos National Laboratory database and aligned using SynchAlign (www.hiv.lanl.gov/content/sequence/SYNCH_ALIGNS/SynchAligns.html). An unrooted phylogenetic tree was generated using FastTree 2.1 (78) and visualized using Figtree v1.4.4 (tree.bio.ed.ac.uk/software/tree).
Infection of primary cells.
The generation of infected cells was described previously (79). Here, 3 × 106 cells were infected with the appropriate dose of IMC or latent reservoir virus supernatant.
Monoclonal antibodies.
Six human mAbs targeting different regions of the Env, the non-neutralizing C1C2 (A32) (38), CD4bs (CH557) (39), V2 glycan (PG9) (40), V3 glycan (PGT121) (41), gp120-gp41 interface (PGT151) (42), and MPER (DH511.2K3) (43), were isolated from HIV-1–infected individuals, expressed as IgG1, and optimized for Fcγ receptor binding on the effector cells (80).
Luciferase ADCC assay.
ADCC was determined as previously described (81, 82). Briefly, primary CD4+ T cells were used as targets after infection with the HIV-1 IMCs. NK cells, purified from HIV-seronegative cryopreserved PBMCs, were used as effector cells, at an effector/target ratio of 5:1. The effector and target cells were plated with 5-fold serial dilutions of mAbs starting at 50 μg/mL in opaque 96-well half-area plates and incubated for 2, 3, 4, or 6 hours or overnight at 37°C in 5% CO2. The final readout was the luminescence intensity (relative light units [RLU]) generated by the presence of residual intact target cells that had not been lysed by the effectors in the presence of ADCC-mediating mAbs (ViviRen substrate, Promega). The percentage of specific killing was calculated using the formula:
(Equation 1)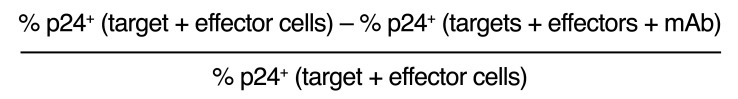
The RLU of the target plus effector wells represents spontaneous lysis in the absence of any antibodies. The anti–Flu HA CH65 mAb (provided by M. Anthony Moody, Department of Pediatrics, Division of Infectious Diseases and the Department of Immunology, Duke University Medical Center) was used as a negative control (83).
Infected-cell elimination assay.
As previously described (79), HIV-1–infected or mock-infected CD8-depleted PBMCs were used as targets. Autologous cryopreserved PBMCs rested overnight in R10 supplemented with 10 ng/mL of IL-15 (Miltenyi Biotec) were used as a source of effector cells. The day of the assay, NK cells were isolated using a human NK cell isolation kit (130-092-657, Miltenyi Biotec). Infected and uninfected target cells were labeled with a fluorescent target-cell marker (TFL4, OncoImmunin) and a viability marker (NFL1, OncoImmunin) for 15 minutes at 37°C, as specified by the manufacturer. Target cells (0.4 × 106 cells/mL) were mixed with autologous NK cells (2 × 106 cells/mL) at an effector/target ratio of 5:1. The target/effector cell suspension was plated in V-bottom 96-well plates and cocultured with each individual antibody or antibody combinations (0.1 μg/mL of A32 and 1 μg/mL of CH557, PG9, DH511.2K3, PGT151). Cocultures were incubated for 2 or 24 hours (as indicated) at 37°C in 5% CO2. Specific killing was determined as described above. The results are the average of 2 independent experiments tested in 2 replicates each.
ADCC assay in the absence of effectors.
The assay was performed as described above in the absence of effector cells. The mixture of target cells with mAbs was incubated for 24 hours. Mock-infected cells were used to appropriately position live-cell p24+/– and CD4+/– gates.
Infected-cell antibody binding assay.
The binding of mAbs to the cells was measured as described previously (84). Briefly, 2 × 105 cells per well were incubated with 1 μg/mL mAb for 2 hours at 37°C followed by surface staining with anti-CD4–PerCP–Cy5.5 for 20 minutes at room temperature. Cells were then resuspended in 100 μL/well Cytofix/Cytoperm and incubated for 20 minutes at 4°C, followed by staining with anti-p24 antibody and a secondary FITC-conjugated antibody [goat anti-human IgG(H+L)–FITC, KPL] for 25 minutes at 4°C. Cells were washed and resuspended in 125 μL PBS–1% paraformaldehyde. The samples were acquired within 24 hours using a BD Fortessa cytometer. The results show the average of 2 independent experiments.
Latency clearance assay.
A modified viral outgrowth assay was optimized to assess the ability of NK cells to clear latently infected cells after reactivation (36, 51). Resting CD4+ T cells were negatively isolated (Stemcell Technologies) from HIV-infected, ART-treated virally suppressed donor P800, and either were reactivated with 2 μg/mL phytohemagglutinin (PHA) or underwent reversal of latency with 335 nM vorinostat (VOR). Cells were washed after 16 hours, and NK cells were added to selected cultures at an effector/target ratio of 1:20, along with 1 (A32), 2 (DH511.2K3+PGT121), or 3 (A32+DH511.2K3+PGT121) antibodies. Cocultures were left for 24 hours and then washed to eliminate free antibodies and plated in 12 or 16 replicates for reactivation with PHA and VOR, respectively. Allogeneic stimulated CD8-depleted cells were added at the amount of 1.6 × 106 cells per well to all cultures 24 hours after coculture to allow infection propagation. At day 8, cultures were split and additional feeder cells (1.6 × 106 cells per well) were added, and medium was refreshed every 3–4 days during the 25 days’ duration of the culture. Supernatants were harvested at different time points to measure viral production by p24 ELISA (ABL Inc.) and record the number of HIV-positive wells under each condition.
Neutralization assay.
Neutralization assays were performed with the assay validated according to Good Clinical Laboratory Practice guidelines using HIV-1 WITO.IMC and LRVs to infect TZM-bl cells as previously described (85). A 4-parameter hill slope equation was used to fit neutralization curves by nonlinear regression and for determination of maximum percentage inhibition values. Titers were calculated as IC50 and reported as the concentration of antibody causing a 50% reduction in relative light units (RLU) in comparison with virus control wells after subtraction of background RLU in cell control wells.
ADCC score.
Specific killing above 20% was considered to be a positive response. Antibodies and their combination do not target all the LRVs with equal affinity. Hence, we calculated a weighted average for ADCC score, where weights are a measure of the variation in affinity of antibodies toward LRVs. Weighted average was calculated using principal component analysis (PCA) (86). In one of the earlier studies published by Moody et al. in 2016 (87), PCA was used to calculate breadth score of neutralizing antibodies. PCA uses eigenvector decomposition of the correlation matrix of the variables, where each variable is represented by LRVs in our study. ADCC scores were obtained from principal component 1 (PC1). PC1 is a vector that explains maximum variance of the percentage killing induced by different antibodies on LRVs. Scores obtained from PC1 account for 70% variability in the affinity of the antibodies toward the LRVs. This method of calculation of ADCC score was described by Easterhoff et al. (79).
Statistics.
Data were classified into different groups according to the number of antibodies used. Group 1 refers to all observations for which only 1 antibody was used. Groups 2 and 3 refer to observations for which combination of any 2 and 3 different antibodies, respectively, was used; similarly, in groups 4 and 5, combinations of 4 and 5 different antibodies, respectively, were used. Group 6 refers to the combination of all different antibodies used in the study. The thick line in the box plots denotes the median, and the ends of the box denote the 25th and 75th percentiles. The whiskers denote the most extreme data points that were no more than 1.5 times the interquartile range. Kruskal-Wallis test was used to compare percentage of specific killing between groups. In order to assess whether 2 groups had different percentage of killing, pairwise comparisons between groups were conducted using Wilcoxon rank sum test (2-sided). A P value of less than 0.05 was considered to be statistically significant. Statistical analysis was performed using SAS software (SAS Institute Inc.).
Study approval.
Human samples were acquired via a protocol approved by the University of North Carolina at Chapel Hill Biomedical IRB and a protocol approved by the Duke University Biomedical IRB.
Author contributions
MT, CG, GF, and DMM designed the experimental procedures. MT and CG conducted the experiments. D Mielke, S Jha, MM, S Joseph, CL, and D Montefiori analyzed the data. BFH generated some of the mAbs. MT, GF, and DMM wrote the manuscript. All the authors provided critical review of the manuscript.
Supplementary Material
Acknowledgments
We thank Nancie Archin, Brigitte Allard, Erin Stuelke, Katherine Sholtis, and Jennifer Kirchherr for the processing of HIV-positive donor cell samples; Amanda Eaton for neutralization assay; and Justin Pollara for insightful comments and discussion. We also thank the HIV-positive participants for their essential assistance with this study. The research was supported, in part, by the Collaboratory of AIDS Researchers for Eradication (CARE), a Martin Delaney Collaboratory program; the National Institute of Allergy and Infectious Diseases (NIAID), National Institute of Neurological Disorders and Stroke, National Institute on Drug Abuse, and National Institute of Mental Health of the NIH (grant 1UM1AI126619); the NIAID (P01-AI120756-01A1); the Immunology Core of the Duke University Center for AIDS Research (5P30 AI064518); and the External Quality Assurance Program Oversight Laboratory (HHSN272201000045C and HHSN272201700061C). MT was supported by the NIH Ruth L. Kirschstein National Research Service Award (5T32AI007392). The content is solely the responsibility of the authors. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Version 1. 06/25/2020
In-Press Preview
Version 2. 08/24/2020
Electronic publication
Version 3. 10/01/2020
Print issue publication
Footnotes
Conflict of interest: GF and BFH have submitted a patent application (15/514,420) on HIV antibodies described in this article.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(10):5157–5170.https://doi.org/10.1172/JCI135557.
Contributor Information
Marina Tuyishime, Email: mt286@duke.edu.
Carolina Garrido, Email: carolina_garrido@med.unc.edu.
Shalini Jha, Email: shalini.jha@duke.edu.
Dieter Mielke, Email: dieter.mielke@duke.edu.
Celia LaBranche, Email: celia.labranche@duke.edu.
David Montefiori, Email: monte@duke.edu.
Barton F. Haynes, Email: barton.haynes@duke.edu.
David M. Margolis, Email: dmargo@med.unc.edu.
Guido Ferrari, Email: gflmp@dm.duke.edu.
References
- 1.Cohen MS. Successful treatment of HIV eliminates sexual transmission. Lancet. 2019;393(10189):2366–2367. doi: 10.1016/S0140-6736(19)30701-9. [DOI] [PubMed] [Google Scholar]
- 2.Finzi D, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278(5341):1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 3.Chun TW, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 1997;94(24):13193–13197. doi: 10.1073/pnas.94.24.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Euler Z, Alter G. Exploring the potential of monoclonal antibody therapeutics for HIV-1 eradication. AIDS Res Hum Retroviruses. 2015;31(1):13–24. doi: 10.1089/aid.2014.0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Caskey M, Klein F, Nussenzweig MC. Broadly neutralizing antibodies for HIV-1 prevention or immunotherapy. N Engl J Med. 2016;375(21):2019–2021. doi: 10.1056/NEJMp1613362. [DOI] [PubMed] [Google Scholar]
- 6.Klein F, Mouquet H, Dosenovic P, Scheid JF, Scharf L, Nussenzweig MC. Antibodies in HIV-1 vaccine development and therapy. Science. 2013;341(6151):1199–1204. doi: 10.1126/science.1241144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caskey M, et al. Viraemia suppressed in HIV-1-infected humans by broadly neutralizing antibody 3BNC117. Nature. 2015;522(7557):487–491. doi: 10.1038/nature14411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Caskey M, et al. Antibody 10-1074 suppresses viremia in HIV-1-infected individuals. Nat Med. 2017;23(2):185–191. doi: 10.1038/nm.4268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lynch RM, et al. Virologic effects of broadly neutralizing antibody VRC01 administration during chronic HIV-1 infection. Sci Transl Med. 2015;7(319):319ra206. doi: 10.1126/scitranslmed.aad5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bar KJ, et al. Effect of HIV antibody VRC01 on viral rebound after treatment interruption. N Engl J Med. 2016;375(21):2037–2050. doi: 10.1056/NEJMoa1608243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scheid JF, et al. HIV-1 antibody 3BNC117 suppresses viral rebound in humans during treatment interruption. Nature. 2016;535(7613):556–560. doi: 10.1038/nature18929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gruell H, et al. A randomized trial of the impact of 3BNC117 and romidepsin on the HIV-1 reservoir. Paper presented at: Conference on Retroviruses and Opportunistic Infections (CROI); March 8–11, 2020; Boston, Massachusetts, USA. http://www.natap.org/2020/CROI/croi_05.htm.
- 13.Hake A, Pfeifer N. Prediction of HIV-1 sensitivity to broadly neutralizing antibodies shows a trend towards resistance over time. PLoS Comput Biol. 2017;13(10):e1005789. doi: 10.1371/journal.pcbi.1005789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouvin-Pley M, et al. Drift of the HIV-1 envelope glycoprotein gp120 toward increased neutralization resistance over the course of the epidemic: a comprehensive study using the most potent and broadly neutralizing monoclonal antibodies. J Virol. 2014;88(23):13910–13917. doi: 10.1128/JVI.02083-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagh K, et al. Optimal combinations of broadly neutralizing antibodies for prevention and treatment of HIV-1 clade C infection. PLoS Pathog. 2016;12(3):e1005520. doi: 10.1371/journal.ppat.1005520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kong R, et al. Improving neutralization potency and breadth by combining broadly reactive HIV-1 antibodies targeting major neutralization epitopes. J Virol. 2015;89(5):2659–2671. doi: 10.1128/JVI.03136-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nishimura Y, et al. Early antibody therapy can induce long-lasting immunity to SHIV. Nature. 2017;543(7646):559–563. doi: 10.1038/nature21435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klein F, et al. HIV therapy by a combination of broadly neutralizing antibodies in humanized mice. Nature. 2012;492(7427):118–122. doi: 10.1038/nature11604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diskin R, et al. Restricting HIV-1 pathways for escape using rationally designed anti-HIV-1 antibodies. J Exp Med. 2013;210(6):1235–1249. doi: 10.1084/jem.20130221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horwitz JA, et al. HIV-1 suppression and durable control by combining single broadly neutralizing antibodies and antiretroviral drugs in humanized mice. Proc Natl Acad Sci U S A. 2013;110(41):16538–16543. doi: 10.1073/pnas.1315295110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shingai M, et al. Antibody-mediated immunotherapy of macaques chronically infected with SHIV suppresses viraemia. Nature. 2013;503(7475):277–280. doi: 10.1038/nature12746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bolton DL, et al. Human immunodeficiency virus type 1 monoclonal antibodies suppress acute simian-human immunodeficiency virus viremia and limit seeding of cell-associated viral reservoirs. J Virol. 2016;90(3):1321–1332. doi: 10.1128/JVI.02454-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Barouch DH, et al. Therapeutic efficacy of potent neutralizing HIV-1-specific monoclonal antibodies in SHIV-infected rhesus monkeys. Nature. 2013;503(7475):224–228. doi: 10.1038/nature12744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trkola A, et al. Delay of HIV-1 rebound after cessation of antiretroviral therapy through passive transfer of human neutralizing antibodies. Nat Med. 2005;11(6):615–622. doi: 10.1038/nm1244. [DOI] [PubMed] [Google Scholar]
- 25.Mehandru S, et al. Adjunctive passive immunotherapy in human immunodeficiency virus type 1-infected individuals treated with antiviral therapy during acute and early infection. J Virol. 2007;81(20):11016–11031. doi: 10.1128/JVI.01340-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mendoza P, et al. Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature. 2018;561(7724):479–484. doi: 10.1038/s41586-018-0531-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ackerman ME, et al. Polyfunctional HIV-Specific Antibody Responses Are Associated with Spontaneous HIV Control. PLoS Pathog. 2016;12(1):e1005315. doi: 10.1371/journal.ppat.1005315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lu CL, et al. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science. 2016;352(6288):1001–1004. doi: 10.1126/science.aaf1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bruel T, et al. Elimination of HIV-1-infected cells by broadly neutralizing antibodies. Nat Commun. 2016;7:10844. doi: 10.1038/ncomms10844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bournazos S, Klein F, Pietzsch J, Seaman MS, Nussenzweig MC, Ravetch JV. Broadly neutralizing anti-HIV-1 antibodies require Fc effector functions for in vivo activity. Cell. 2014;158(6):1243–1253. doi: 10.1016/j.cell.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bournazos S, Ravetch JV. Fcγ receptor pathways during active and passive immunization. Immunol Rev. 2015;268(1):88–103. doi: 10.1111/imr.12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parsons MS, Chung AW, Kent SJ. Importance of Fc-mediated functions of anti-HIV-1 broadly neutralizing antibodies. Retrovirology. 2018;15(1):58. doi: 10.1186/s12977-018-0438-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hessell AJ, et al. Early short-term treatment with neutralizing human monoclonal antibodies halts SHIV infection in infant macaques. Nat Med. 2016;22(4):362–368. doi: 10.1038/nm.4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis GK. Role of Fc-mediated antibody function in protective immunity against HIV-1. Immunology. 2014;142(1):46–57. doi: 10.1111/imm.12232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.von Bredow B, et al. Comparison of antibody-dependent cell-mediated cytotoxicity and virus neutralization by HIV-1 Env-specific monoclonal antibodies. J Virol. 2016;90(13):6127–6139. doi: 10.1128/JVI.00347-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sung JA, et al. Expanded cytotoxic T-cell lymphocytes target the latent HIV reservoir. J Infect Dis. 2015;212(2):258–263. doi: 10.1093/infdis/jiv022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pollara J, et al. High-throughput quantitative analysis of HIV-1 and SIV-specific ADCC-mediating antibody responses. Cytometry A. 2011;79(8):603–612. doi: 10.1002/cyto.a.21084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wyatt R, Moore J, Accola M, Desjardin E, Robinson J, Sodroski J. Involvement of the V1/V2 variable loop structure in the exposure of human immunodeficiency virus type 1 gp120 epitopes induced by receptor binding. J Virol. 1995;69(9):5723–5733. doi: 10.1128/JVI.69.9.5723-5733.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bonsignori M, et al. Maturation pathway from germline to broad HIV-1 neutralizer of a CD4-mimic antibody. Cell. 2016;165(2):449–463. doi: 10.1016/j.cell.2016.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Walker LM, et al. Broad and potent neutralizing antibodies from an African donor reveal a new HIV-1 vaccine target. Science. 2009;326(5950):285–289. doi: 10.1126/science.1178746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walker LM, et al. Broad neutralization coverage of HIV by multiple highly potent antibodies. Nature. 2011;477(7365):466–470. doi: 10.1038/nature10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Falkowska E, et al. Broadly neutralizing HIV antibodies define a glycan-dependent epitope on the prefusion conformation of gp41 on cleaved envelope trimers. Immunity. 2014;40(5):657–668. doi: 10.1016/j.immuni.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams LD, et al. Potent and broad HIV-neutralizing antibodies in memory B cells and plasma. Sci Immunol. 2017;2(7):eaal2200. doi: 10.1126/sciimmunol.aal2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee WS, Parsons MS, Kent SJ, Lichtfuss M. Can HIV-1-specific ADCC assist the clearance of reactivated latently infected cells? Front Immunol. 2015;6:265. doi: 10.3389/fimmu.2015.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferrari G, Haynes BF, Koenig S, Nordstrom JL, Margolis DM, Tomaras GD. Envelope-specific antibodies and antibody-derived molecules for treating and curing HIV infection. Nat Rev Drug Discov. 2016;15(12):823–834. doi: 10.1038/nrd.2016.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rubin EH, et al. A study to determine the effects of food and multiple dosing on the pharmacokinetics of vorinostat given orally to patients with advanced cancer. Clin Cancer Res. 2006;12(23):7039–7045. doi: 10.1158/1078-0432.CCR-06-1802. [DOI] [PubMed] [Google Scholar]
- 47.Mu S, et al. Panobinostat PK/PD profile in combination with bortezomib and dexamethasone in patients with relapsed and relapsed/refractory multiple myeloma. Eur J Clin Pharmacol. 2016;72(2):153–161. doi: 10.1007/s00228-015-1967-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Andrabi R, et al. Identification of common features in prototype broadly neutralizing antibodies to HIV envelope V2 apex to facilitate vaccine design. Immunity. 2015;43(5):959–973. doi: 10.1016/j.immuni.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ferguson AL, Falkowska E, Walker LM, Seaman MS, Burton DR, Chakraborty AK. Computational prediction of broadly neutralizing HIV-1 antibody epitopes from neutralization activity data. PLoS One. 2013;8(12):e80562. doi: 10.1371/journal.pone.0080562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Doria-Rose NA, et al. New member of the V1V2-directed CAP256-VRC26 lineage that shows increased breadth and exceptional potency. J Virol. 2016;90(1):76–91. doi: 10.1128/JVI.01791-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garrido C, et al. Interleukin-15-stimulated natural killer cells clear HIV-1-infected cells following latency reversal ex vivo. J Virol. 2018;92(12):e00235-18. doi: 10.1128/JVI.00235-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Archin NM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487(7408):482–485. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Garcia JV, Miller AD. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature. 1991;350(6318):508–511. doi: 10.1038/350508a0. [DOI] [PubMed] [Google Scholar]
- 54.Bell I, Ashman C, Maughan J, Hooker E, Cook F, Reinhart TA. Association of simian immunodeficiency virus Nef with the T-cell receptor (TCR) zeta chain leads to TCR down-modulation. J Gen Virol. 1998;79(pt 11):2717–2727. doi: 10.1099/0022-1317-79-11-2717. [DOI] [PubMed] [Google Scholar]
- 55.Veillette M, et al. The HIV-1 gp120 CD4-bound conformation is preferentially targeted by antibody-dependent cellular cytotoxicity-mediating antibodies in sera from HIV-1-infected individuals. J Virol. 2015;89(1):545–551. doi: 10.1128/JVI.02868-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Grau-Expósito J, et al. A novel single-cell FISH-flow assay identifies effector memory CD4+ T cells as a major niche for HIV-1 transcription in HIV-infected patients. mBio. 2017;8(4):e00876-17. doi: 10.1128/mBio.00876-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tuen M, et al. Immune correlates of disease progression in linked HIV-1 infection. Front Immunol. 2019;10:1062. doi: 10.3389/fimmu.2019.01062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gilbert PB, et al. Basis and statistical design of the passive HIV-1 antibody mediated prevention (AMP) test-of-concept efficacy trials. Stat Commun Infect Dis. 2017;9(1):20160001. doi: 10.1515/scid-2016-0001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lu CL, et al. Enhanced clearance of HIV-1-infected cells by broadly neutralizing antibodies against HIV-1 in vivo. Science. 2016;352(6288):1001–1004. doi: 10.1126/science.aaf1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schoofs T, et al. HIV-1 therapy with monoclonal antibody 3BNC117 elicits host immune responses against HIV-1. Science. 2016;352(6288):997–1001. doi: 10.1126/science.aaf0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee WS, et al. Anti-HIV-1 ADCC antibodies following latency reversal and treatment interruption. J Virol. 2017;91(15):e00603-17. doi: 10.1128/JVI.00603-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rasmussen TA, et al. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. Lancet HIV. 2014;1(1):e13–e21. doi: 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- 63.Lee WS, et al. Antibody-dependent cellular cytotoxicity against reactivated HIV-1-infected cells. J Virol. 2016;90(4):2021–2030. doi: 10.1128/JVI.02717-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bournazos S, Ravetch JV. Anti-retroviral antibody FcγR-mediated effector functions. Immunol Rev. 2017;275(1):285–295. doi: 10.1111/imr.12482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parsons MS, et al. Fc-dependent functions are redundant to efficacy of anti-HIV antibody PGT121 in macaques. J Clin Invest. 2019;129(1):182–191. doi: 10.1172/JCI122466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Alsahafi N, et al. An asymmetric opening of HIV-1 envelope mediates antibody-dependent cellular cytotoxicity. Cell Host Microbe. 2019;25(4):578–587.e5. doi: 10.1016/j.chom.2019.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ferrari G, et al. An HIV-1 gp120 envelope human monoclonal antibody that recognizes a C1 conformational epitope mediates potent antibody-dependent cellular cytotoxicity (ADCC) activity and defines a common ADCC epitope in human HIV-1 serum. J Virol. 2011;85(14):7029–7036. doi: 10.1128/JVI.00171-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lewis GK, Finzi A, DeVico AL, Pazgier M. Conformational masking and receptor-dependent unmasking of highly conserved Env epitopes recognized by non-neutralizing antibodies that mediate potent ADCC against HIV-1. Viruses. 2015;7(9):5115–5132. doi: 10.3390/v7092856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mouquet H, et al. Polyreactivity increases the apparent affinity of anti-HIV antibodies by heteroligation. Nature. 2010;467(7315):591–595. doi: 10.1038/nature09385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Prigent J, et al. Conformational plasticity in broadly neutralizing HIV-1 antibodies triggers polyreactivity. Cell Rep. 2018;23(9):2568–2581. doi: 10.1016/j.celrep.2018.04.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Caskey M, Klein F, Nussenzweig MC. Broadly neutralizing anti-HIV-1 monoclonal antibodies in the clinic. Nat Med. 2019;25(4):547–553. doi: 10.1038/s41591-019-0412-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jaworski JP, Vendrell A, Chiavenna SM. Neutralizing monoclonal antibodies to fight HIV-1: on the threshold of success. Front Immunol. 2016;7:661. doi: 10.3389/fimmu.2016.00661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pollara J, Bonsignori M, Moody MA, Pazgier M, Haynes BF, Ferrari G. Epitope specificity of human immunodeficiency virus-1 antibody dependent cellular cytotoxicity [ADCC] responses. Curr HIV Res. 2013;11(5):378–387. doi: 10.2174/1570162X113116660059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sun M, Li Y, Zheng H, Shao Y. Recent progress toward engineering HIV-1-specific neutralizing monoclonal antibodies. Front Immunol. 2016;7:391. doi: 10.3389/fimmu.2016.00391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Xu L, et al. Trispecific broadly neutralizing HIV antibodies mediate potent SHIV protection in macaques. Science. 2017;358(6359):85–90. doi: 10.1126/science.aan8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Edmonds TG, et al. Replication competent molecular clones of HIV-1 expressing Renilla luciferase facilitate the analysis of antibody inhibition in PBMC. Virology. 2010;408(1):1–13. doi: 10.1016/j.virol.2010.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Abrahams MR, et al. The replication-competent HIV-1 latent reservoir is primarily established near the time of therapy initiation. Sci Transl Med. 2019;11(513):eaaw5589. doi: 10.1126/scitranslmed.aaw5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Price MN, Dehal PS, Arkin AP. FastTree 2 — approximately maximum-likelihood trees for large alignments. PLoS One. 2010;5(3):e9490. doi: 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Easterhoff D, et al. Boosting with AIDSVAX B/E enhances Env constant region 1 and 2 antibody-dependent cellular cytotoxicity breadth and potency. J Virol. 2020;94(4):e01120-19. doi: 10.1128/JVI.01120-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shields RL, et al. High resolution mapping of the binding site on human IgG1 for Fc gamma RI, Fc gamma RII, Fc gamma RIII, and FcRn and design of IgG1 variants with improved binding to the Fc gamma R. J Biol Chem. 2001;276(9):6591–6604. doi: 10.1074/jbc.M009483200. [DOI] [PubMed] [Google Scholar]
- 81.Liao HX, et al. Vaccine induction of antibodies against a structurally heterogeneous site of immune pressure within HIV-1 envelope protein variable regions 1 and 2. Immunity. 2013;38(1):176–186. doi: 10.1016/j.immuni.2012.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pollara J, et al. HIV-1 vaccine-induced C1 and V2 Env-specific antibodies synergize for increased antiviral activities. J Virol. 2014;88(14):7715–7726. doi: 10.1128/JVI.00156-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Whittle JR, et al. Broadly neutralizing human antibody that recognizes the receptor-binding pocket of influenza virus hemagglutinin. Proc Natl Acad Sci U S A. 2011;108(34):14216–14221. doi: 10.1073/pnas.1111497108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mielke D, et al. Antibody-dependent cellular cytotoxicity (ADCC)-mediating antibodies constrain neutralizing antibody escape pathway. Front Immunol. 2019;10:2875. doi: 10.3389/fimmu.2019.02875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Montefiori DC. Measuring HIV neutralization in a luciferase reporter gene assay. Methods Mol Biol. 2009;485:395–405. doi: 10.1007/978-1-59745-170-3_26. [DOI] [PubMed] [Google Scholar]
- 86. Hotelling H. Analysis of a complex of statistical variables into principal components. Warwick & York; 1933. [Google Scholar]
- 87.Moody MA, et al. Immune perturbations in HIV-1-infected individuals who make broadly neutralizing antibodies. Sci Immunol. 2016;1(1):aag0851. doi: 10.1126/sciimmunol.aag0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.