

Summary
The pathophysiology, immune reaction, and differential vulnerability of different population groups and viral host immune system evasion strategies of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection are not yet well understood. Here, we reviewed the multitude of known strategies of coronaviruses and other viruses to usurp mitochondria-associated mechanisms involved in the host innate immune response and put them in context with the current knowledge on SARS-CoV-2. We argue that maintenance of mitochondrial integrity is essential for adequate innate immune system responses and to blunt mitochondrial modulation by SARS-CoV-2. Mitochondrial health thus may determine differential vulnerabilities to SARS-CoV-2 infection rendering markers of mitochondrial functions promising potential biomarkers for SARS-CoV-2 infection risk and severity of outcome. Current knowledge gaps on our understanding of mitochondrial involvement in SARS-CoV-2 infection, lifestyle, and pharmacological strategies to improve mitochondrial integrity and potential reciprocal interactions with chronic and age-related diseases, e.g., Parkinson disease, are pointed out.
Subject Areas: Immunology, Virology, Cell Biology
Graphical Abstract
Immunology; Virology; Cell Biology
SARS-CoV-2 Biology: an Equation with Many Unknowns
The variability of individual vulnerability and regional mortality rates to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which caused the ongoing coronavirus disease (COVID-19) pandemic, is poorly understood. Although it is clear that age and pre-existing morbidities (Wang et al., 2020), such as cardiovascular and respiratory diseases, represent major risk factors, mechanistic explanations are lacking. In particular, it remains enigmatic, why sporadic severe disease progression may occur after SARS-CoV-2 infection in young individuals apparently without risk factors. Elucidating the variability in vulnerability, derivation of appropriate biomarkers, and the identification of efficient treatment strategies require thorough understanding of the pathophysiology and immune responses following SARS-CoV-2 infection.
Here we discuss how the cellular innate immune system of an infected host organism fighting viral infection strongly relies on host mitochondria. Viruses commonly manipulate host cells by infiltrating cellular, including mitochondria-mediated, defense mechanisms, leading to an inadequate host immune response that may promote adverse disease outcomes. Given the fast development of the COVID-19 pandemic and thus the only recently launched research efforts on SARS-CoV-2, knowledge on SARS-CoV-2 biology and pathophysiology in humans is still limited. However, the SARS-CoV-2 genome shares 79% with the genome of SARS-CoV (Coronaviridae Study Group, 2020), which caused the SARS epidemic in 2002. Building on research on other RNA viruses, in particular SARS-CoV, we integrate emerging evidence on the roles of mitochondria in the innate immune reaction to SARS-CoV-2 infection; how SARS-CoV-2 may evade host immune defense mechanisms by hijacking mitochondrial pathways, as recently suggested (Singh et al., 2020); and how dysfunctional mitochondria could contribute to severe disease progression. For these comparisons it is important to stress the substantial differences in SARS versus COVID-19 epidemiology and symptomatology (Fani et al., 2020; Petersen et al., 2020). A direct comparison of disease pathogenesis after infection with Middle East respiratory syndrome (MERS) CoV and SARS-CoV-2 in a non-human primate model compared with historical SARS-CoV infections has been recently published (Rockx et al., 2020).
We will conclude by highlighting the importance and suggesting promising therapeutic avenues of enhancing mitochondrial functions for an efficient innate immune response.
SARS-CoV-2 Infection
Cells infected by SARS-CoV, and most likely also by SARS-CoV-2 (Gordon et al., 2020), are mainly epithelial cells, alveolar epithelial cells, vascular endothelial cells, and macrophages in the lung. In this review we focus on the cellular innate immune response in these primarily infected cells from a mitochondrial perspective. Infection of a number of other cells, including specialized immune cells, has also been described for SARS-CoV (Tiku et al., 2020).
Upon attaching of the virus to the cell surface via angiotensin-converting enzyme 2 (ACE2) and cleavage of the viral spike protein by proteases like transmembrane protease, serine 2 (TMPRSS2) that enables fusion of the virus with the cell membrane, SARS-CoV-2 enters cells by endocytosis (Azkur et al., 2020). The resulting activation of different viral detection systems and innate immune responses of the host cells (Azkur et al., 2020) and the immunopathology of SARS-CoV-2 infection (Vabret et al., 2020) have recently been summarized.
An incubation period of 4–5 days usually precedes symptoms of SARS-CoV-2 infection. The peak of viral load is reached after another 5–6 days (Tay et al., 2020). In the case of an efficient immune response rapid inactivation of SARS-CoV-2, clearance of infected cells, and the induction of an adequate immune response halt lung vascular damage and adverse disease progression (Tay et al., 2020). Conversely, infection of airway epithelial cells by SARS-CoVs may induce pronounced pyroptosis, a type of cell death associated with a strong inflammatory response (Cookson and Brennan, 2001). This has been demonstrated to be triggered in human alveolar epithelial cells (Yue et al., 2018) and macrophages by the SARS-CoV 3a protein (Chen et al., 2019) and likely also happens in SARS-CoV-2 (Tay et al., 2020). Pyroptosis may result in substantial vascular leakage and release of cytokines and chemokines, attracting monocytes and T cells to the site of injury or inflammation. Direct viral damage can thus be intensified by high levels of proteases and oxidative stress, an integral factor in COVID-19 progression (Delgado-Roche and Mesta, 2020), caused by the host defense. SARS-CoV-2 infection is characterized by an inappropriate cytokine host response: a relatively low induction of type I and III interferons blunt the anti-viral host defense and is paralleled by a high induction of chemokines and interleukin (IL)-6 (Blanco-Melo et al., 2020), similar to what has been reported for SARS-CoV infection. Damage to the airways and the resulting inflammatory host response may in turn contribute to the development of symptoms resembling acute respiratory distress syndrome (ARDS). Characteristic diffuse alveolar damage, pulmonary edema, and occasional hyaline membranes impair pulmonary gas exchange and may cause hypoxemia (reduced arterial oxygen pressure) and breathing difficulties. Direct respiratory failure or systemic cytokine-storm-mediated sepsis (including, for example, death from circulatory failure) may ensue, both conditions representing common causes of COVID-19-related death (Ruan et al., 2020).
The Role of Mitochondria in the Innate Immune System
Innate immune responses are triggered by the activation of pattern recognition receptors, which non-specifically recognize pathogens via pathogen-associated molecular patterns or signals from damaged cells via damage-associated molecular patterns (DAMPs). In response to tissue injury, affected cells drive the recruitment of specialized innate immune cells, which in turn activate the specific adaptive immune response and trigger the production of molecules needed for inflammation and regeneration/repair of injured tissue. It is becoming increasingly clear that mitochondria are fundamentally involved in the mammalian innate immune system (Banoth and Cassel, 2018; Weinberg et al., 2015). Mitochondria are most commonly associated with energy production by oxidative phosphorylation, but they are also involved in a myriad of other functions, such as ion homeostasis, cellular signaling, differentiation, and cell death/survival.
Beside mitochondrial reactive oxygen species (ROS) (mtROS)-based involvement in immunity, mitochondrial participation in antiviral defense is now well established (West et al., 2011). Mitochondria play integral roles in anti-viral host response signaling upon infection, and they can activate the innate immune system upon cellular damage via mitochondrial DAMPs. Importantly, SARS-CoV proteins manipulate mitochondrial mechanisms to evade the host immune response and may alter mitochondrial functions leading to enhanced ROS production, perturbed signaling, and blunted antiviral defenses of the host organism.
The following sections summarize the mitochondrial involvement in the innate immune system after infection with RNA viruses and specifically with SARS-CoVs.
Mitochondrial Antiviral Signaling: The RLR Signaling Pathway
The mitochondrial membrane-anchored mitochondrial anti-viral signaling protein MAVS is an essential component of the cellular anti-viral defense system (West et al., 2011). Viruses in the cytoplasm can be detected by retinoic acid-inducible gene 1 (RIG-I)-like receptors (RLRs), including RIG-I and melanoma differentiation-associated gene 5 (MDA5) (Belgnaoui et al., 2011). RLRs activate MAVS, which upon activation forms oligomers in a prion-like manner (Hou et al., 2011). MAVS then recruits a number of downstream signaling effectors, including the E3 ligases tumor necrosis factor receptor-associated factors 3 and 6 (TRAF3 and TRAF6) leading to antiviral defense by the induction of nuclear factor kappa light chain enhancer of activated B cell (NF-κB) and type I interferons via interferon regulatory factors (IRFs). Formation of the MAVS complex (“MAVS signalosome”) is mediated by translocases of the outer mitochondrial membrane 70 (TOM70) and 20 (TOM20) and leads to activation of TANK-binding kinase 1 (TBK1) and phosphorylation of IRF-3. IRF-3 then associates with the cytosolic chaperone heat shock protein 90 (HSP90) to robustly activate an anti-viral response. Dysfunctional MAVS or TOM70 (e.g., due to mislocalization or low protein levels) impair the antiviral host defense. On the other hand, overexpression of TOM70 (West et al., 2011), or increase in the activity of MAVS (Song et al., 2019), boosts the cellular antiviral defenses. MAVS is thought to be tethered to the inner mitochondrial membrane by the proteins prohibitin 1 and 2 (PHB1 and PHB2), which therefore also might play important roles in the regulation of mitochondrial anti-viral innate immunity (Yoshinaka et al., 2019).
The MAVS pathway is preferentially targeted by viruses. For example, the hepatitis C virus serine-protease NS3/4A and the 3ABC protease of the hepatitis A virus cleave MAVS to dissociate it from its outer mitochondrial membrane base, resulting in inhibition of the pathway and thus reducing the cellular capacity to induce antiviral responses (Li et al., 2005; Yang et al., 2007). Similarly, influenza virus protein PB1-F2 was shown to bind and impair MAVS (Varga et al., 2012), thereby blunting the interferon response.
Mitochondrial Dynamics in the Innate Immune System
Cell metabolism in general (Zasłona and O'Neill, 2020) and mitochondria in specific play integral roles in the host defense of many diseases, including viral infections.
Changes of metabolic or physiological conditions, such as infection (Banoth and Cassel, 2018), induce morphological changes (“mitochondrial dynamics”) of mitochondria with functional consequences importantly affecting the innate immune response. Mitochondria transition from shorter to longer organelles (fusion) and the other way around (fission) by means of factors regulating mitochondrial dynamics, including the fusion factors mitofusins 1 and 2 (MFN1 and MFN2), optic atrophy 1 (OPA1) and the fission factor dynamin-related protein-1 (DRP1). These mechanisms and consequences are described excellently and in detail elsewhere (Giacomello et al., 2020; Pernas and Scorrano, 2016), but due to the high relevance of the present work are briefly outlined also here. Mitochondria are enclosed by two membranes, the inner and outer mitochondrial membranes (IMM and OMM, respectively). The OMM enzymes MFN1 and MFN2 are effectors for the fusion of the OMM, which is followed by OPA1-mediated IMM fusion. OPA1 itself is regulated by the metalloproteases YME1L and OMA1.
Mitochondrial fission is mediated primarily by DRP1, which is recruited from the cytosol to the fission site. There it forms ring-like oligomers around the mitochondrion and induces mitochondria division by GTP hydrolysis-mediated DRP1 constriction. The recruitment of DRP1 to mitochondria is believed to be mediated by adaptor proteins, including mitochondrial fission factor (MFF), fission 1 (FIS1), and the mitochondrial dynamics proteins of 49 and 51 kDa (MiD49 and MiD51). Their functions are not completely understood, and in particular MiD49 and MiD51 may affect both fission and fusion depending on the cellular conditions (Pernas and Scorrano, 2016). DRP1-mediated fission is strongly dependent on posttranslational modifications and can be induced by elevated cytosolic Ca2+ levels that lead to the calcineurin-dependent de-phosphorylation of DRP1 Ser637 (Cribbs and Strack, 2007). Conversely, phosphorylation of DRP1 Ser616 (position reflecting the human DRP1) promotes fission (Taguchi et al., 2007).
Fusion is promoted in conditions of nutrient scarcity and is thought to boost mitochondrial bioenergetics efficiency, whereas fission is favored when nutrients are not limited and results in increased nutrient storage, but conversely reduced bioenergetics efficiency (Schrepfer and Scorrano, 2016). Mitochondrial fusion furthermore enables the exchange of mtDNA, metabolites, proteins, and lipids (Pernas and Scorrano, 2016), whereas fission is not only associated with increased ROS production but also facilitates mitochondrial quality control (mitophagy) and mitochondrial distribution, for example, to match local energy demands (Giacomello et al., 2020; Horn et al., 2020).
Mitochondrial dynamics are highly affected and in some cases compromised by viral infections, which affects host responses. These mechanisms have been reviewed specifically from that perspective (Khan et al., 2015; Kim et al., 2018). Viral infections may result in strongly elongated mitochondria potentially enhancing antiviral signaling (Castanier et al., 2010; Onoguchi et al., 2010). Several factors regulating mitochondrial dynamics appear to regulate mitochondrial antiviral signaling. Pro-fusion protein MFN2 levels have been shown to regulate MAVS efficiency, i.e., high levels of MFN2 impair MAVS by changing its active conformation (Sasaki et al., 2013), whereas MFN2 depletion enhances MAVS activity (Yasukawa et al., 2009). Conversely, reductions of MFN1 and OPA1 inhibited MAVS-mediated induction of NF-κB and IRF3 (Castanier et al., 2010). MFN2 furthermore has been reported to be involved in the Nod-like receptor family pyrin domain-containing 3 (NLRP3) inflammasome activation (Ichinohe et al., 2013).
The pro-fission factor DRP1 is commonly targeted by viruses. Human immunodeficiency virus (HIV) via its protein gp120, for example, has been shown to reduce overall DRP1 and DRP1 phosphorylated at Ser616 levels and thus impair mitochondrial fission (Fields et al., 2016). Mitochondrial fission is believed to be important for clearance of defective mitochondria (Pickles et al., 2018). Therefore, it is tempting to speculate that prevention of the clearance of infected mitochondria by mitophagy supports viral replication and survival.
Conversely, hepatitis B virus has been reported to induce mitochondrial fission by stimulating phosphorylation at Ser616. This facilitated mitophagy and enhanced viral survival by reducing virus-induced apoptosis (Kim et al., 2013). These examples demonstrate that viruses in principle can take advantage of both inhibition and enhancement of mitochondrial fission, in the described cases, primarily based on the modification of a single amino acid residue of DRP1, Ser616. The right balance of mitochondrial dynamics and mitophagy thus seems to be crucial in antiviral defense and in the regulation of inflammation (Gkikas et al., 2018).
The RING-between-RING (RBR) E3 ubiquitin protein ligase parkin, which is involved in the induction of mitophagy, has recently emerged as another important mitochondria-associated modulator of innate immune function (Manzanillo et al., 2013). It has been shown that hepatitis B virus modulates parkin function for immune evasion by recruitment of the linear ubiquitin assembly complex (LUBAC) to mitochondria to disrupt MAVS downstream signaling (Khan et al., 2016).
NLRP3 and cGAS-STING
Upon mitochondrial damage, mtDNA is released in the cytosol, where it can activate the NLRP3 inflammasome, a multi-protein complex that, mediated by caspase-1, promotes IL-1β and IL-18 secretion. NLRP3 is furthermore importantly regulated by mtROS and elevated mtROS activation of NLRP3 especially in bronchial epithelial cells may have detrimental consequences (Kim et al., 2014). Intriguingly, the loss of parkin has been recently associated with improved NLRP3-mediated viral clearance: parkin deficiency increased NLRP3 activation and anti-viral inflammation without affecting type I interferon production (Li et al., 2019).
mtDNA released in the cytosol also gets detected by DNA-sensing enzymes, such as toll-like receptor 9 (TLR9) or GMP-AMP synthase (cGAS). cGAS detection of mtDNA in turn via stimulator of interferon genes (STING)-IRF3 promotes type I interferon responses, which increases resistance to viruses (West et al., 2015). Detection by cGAS activates cGAS-STING and interferon-based host responses. RNA viruses, including coronaviruses, have also evolved strategies to evade cGAS-STING (Eaglesham and Kranzusch, 2020), comparable to the described MAVS evasion strategies. Strikingly, parkin also has been implicated in STING signaling, presumably regulating STING-induced inflammation (Sliter et al., 2018).
In conclusion, mitochondria are involved in many pathways related to innate immune responses and viruses commonly undermine these mechanisms. In the next section we will assess the existing knowledge on the role of mitochondria in the innate immune system with regard to SARS-CoVs. We are not aware of published research on the direct role of parkin in SARS-CoV infection. But the central roles of mitochondrial dysfunction and of parkin, also in relation to inflammation (Newman and Shadel, 2018), in sporadic and familial Parkinson disease, respectively, in combination with increasing evidence about COVID-19 as a potential risk factor for Parkinson disease (Antonini et al., 2020; Papa et al., 2020) are of great interest. Recent findings on the immediate relevance of influenza virus infection on Parkinson disease pathology (Marreiros et al., 2020) should additionally spur research on potential interdependencies of Parkinson disease and SARS-CoV-2 infection-related mechanisms.
Manipulation of Mitochondrial Processes by SARS- Coronaviruses
Many pathogens, including RNA viruses like CoVs, have developed targeted strategies to hijack mitochondria, damaging them or using them for their purposes, such as survival, propagation, or evading host immunity. Some viruses use mitochondrial membranes for genome replication (West et al., 2011). As shown for HIV infection, virus RNAs can also enter mitochondria, compromising mitochondrial integrity (Somasundaran et al., 1994). Recent computational work suggests that SARS-CoV-2 RNA preferentially localizes to mitochondria (Wu et al., 2020). Although no experimental data are yet available on whether this is really happening, SARS-CoV-2 RNA deposition in mitochondria could likewise impair mitochondrial function, potentially resulting in reduced energy and increased ROS generation (see part 5). Additional indirect support for a central role of mitochondria in SARS-CoV-2 infection comes from characteristic transcriptomic signatures of components of the oxidative phosphorylation system in peripheral mononuclear leukocytes and bronchoalveolar lavage fluid of patients with COVID-19 (Gardinassi et al., 2020).
Host Evasion Mechanisms
How SARS-CoV-2 evades the antiviral response is not well understood yet. But antagonistic mechanisms on cellular virus detection and interferon responses have been shown for SARS-CoV (Totura and Baric, 2012) and MERS-CoV (Shokri et al., 2019) proteins. These mechanisms impede host responses to infection, e.g., by blunting interferon induction; support viral replication; and increase pyroptotic events, and may be active as well for SARS-CoV-2 (Tay et al., 2020). From animal studies on MERS-CoV infection, we know that blocking the interferon response has detrimental consequences on virus clearance and further immune responses (Channappanavar et al., 2019).
A comprehensive overview on assumed host cell immunity evasion mechanisms of coronaviruses including SARS-CoV-2 has recently been published (Vabret et al., 2020). Some of these effects are afforded by the interaction of SARS-CoV proteins with cellular components affecting mitochondrial functions and thereby inhibiting immune responses. A systematically assembled list of such interactions for SARS-CoV is provided in Table 1. Many of these interactions result in the suppression of interferon signaling or in the modulation of apoptosis. Some of the most interesting interactions with regard to the role of mitochondria in the innate immune system are briefly discussed below and depicted in Figure 1. All viral proteins mentioned in Figure 1, except for ORF3b, are also expressed by SARS-CoV-2.
Table 1.
Interaction of SARS-CoV Proteins with Mitochondrial Innate Immunity Components
| Viral Component | Target | Reference |
|---|---|---|
| Structural nucleocapsid protein | Suppression of IRF responses, interference of RIG-1 activation | (Kopecky-Bromberg et al., 2007), (Hu et al., 2017; Lu et al., 2011) |
| Structural membrane protein | Suppression of RIG-1/MAVS | (Siu et al., 2009) |
| S1 spike protein | Induces apoptosis | (Chen et al., 2018) |
| Envelop protein | Activates NLRP3 inflammasome | (Nieto-Torres et al., 2015) |
| Nsp1 | Suppression of IRF responses (downstream signaling) | (Jauregui et al., 2013), (Wathelet et al., 2007) |
| Nsp2 | Inhibits PHB1 and 2 (blocking mitochondrial biogenesis) | (Cornillez-Ty et al., 2009) |
| Nsp3 | Suppression of IRF responses (inhibits IRF3 phosphorylation) | (Mielech et al., 2014) |
| Nsp15 | Inhibits MAVS-induced apoptosis | (Lei et al., 2009) |
| ORF3a | p38 MAP kinase → modulates mitochondrial death pathways | (Padhan et al., 2008) |
| ORF3a | Activates NLRP3 inflammasome | (Chen et al., 2019; Siu et al., 2019) |
| ORF3a | Suppression of IRF responses (downstream signaling) | (Minakshi et al., 2009) |
| ORF3b | Suppression of IRF responses (inhibiting the phosphorylation and nuclear translocation of IRF3) | (Kopecky-Bromberg et al., 2007) |
| ORF3b | Inhibits RIG-1/MAVS | (Freundt et al., 2009) |
| ORF6 | Suppression of IRF responses (downstream signaling) | (Kopecky-Bromberg et al., 2007), (Frieman et al., 2007) |
| ORF7a | Induces apoptosis | (Tan et al., 2007) |
| ORF8a | Increased mitochondrial membrane potential, ROS, and apoptosis | (Chen et al., 2007) |
| ORF8b | Activates NLRP3 inflammasome | (Shi et al., 2019) |
| ORF9b | Inhibits MAVS, induces degradation of DRP-1 | (Shi et al., 2014) |
| SARS-CoV PLpro | Suppression of IRF responses (inhibits IRF3 phosphorylation) | (Devaraj et al., 2007), (Frieman et al., 2009) |
| SARS-CoV PLpro | Disruption of STING | (Sun et al., 2012) |
| SARS-CoV 3C-like protease | Overexpression changes expression of many mitochondrial proteins | (Lai et al., 2007) |
Please see text for available information on SARS-CoV-2 proteins.
Figure 1.

Putative Mechanisms of Mitochondria-Related Immune Functions Manipulated by SARS-CoV and SARS-CoV-2
Red rectangles represent viral proteins, whose association with mitochondrial proteins of the host cells have been shown in SARS-CoV (except for Nsp4 and ORF9c, bold, for which interaction with mitochondrial components has been suggested only for SARS-CoV-2. ORF3b is not expressed in SARS-CoV-2). ETS, electron transport system.
SARS-CoV nonstructural protein (Nsp) 2, for example, interacts with PHB1 and 2 and thereby impairs intracellular signaling and mitochondrial biogenesis (Cornillez-Ty et al., 2009; Tatsuta and Langer, 2017) as well as MAVS (Yoshinaka et al., 2019). Open reading frame 3b (ORF3b) inhibits type I interferon induction and thus antiviral defense via interaction with the mitochondrial virus-detecting protein RIG-1 (Freundt et al., 2009). ORF8b of SARS-CoV has been shown to robustly activate the NLRP3 inflammasome (Shi et al., 2019). The SARS-CoV protein expressed from ORF9b localizes to mitochondria, promotes mitochondrial fusion and modulates the host antiviral defense (Shi et al., 2014). By increasing ubiquitination and proteasomal degradation of the mitochondrial fission factor DRP1, it favors mitochondrial fusion, possibly promoting cell survival during viral replication. ORF9b furthermore triggers the degradation of MAVS, severely limiting host cell interferon responses (Shi et al., 2014).
Although experimental studies elucidating detailed mitochondrial modulation by SARS-CoV-2 are lacking, recent proteomic studies allow a preliminary assessment of the relevance of related findings in SARS-CoV. A recent SARS-CoV-2-human protein-protein interaction map derived from global affinity purification mass spectrometry analysis (Gordon et al., 2020) reveals putative interactions of SARS-CoV-2 proteins with human mitochondrial proteins, overlapping in many instances with available information on SARS-CoV. Beside a wide range of other human proteins, components of the innate immune system, including interferon signaling, were reported to possibly be regulated by viral proteins (Gordon et al., 2020). In line with the report on SARS-CoV ORF9b (Shi et al., 2014), Gordon et al. (2020) described potential modulation of MAVS by SARS-CoV-2 ORF9b via interaction with the mitochondrial import receptor TOM70. A very recent report also confirmed downregulation of MAVS in SARS-CoV-2-infected human colon epithelial carcinoma cells CaCo-2 (Bojkova et al., 2020).
Gordon and colleagues' study (Gordon et al., 2020) furthermore indicates the interaction of several SARS-CoV-2 proteins with mitochondrial components that have not been reported yet for SARS-CoV: SARS-CoV-2 Nsp4 may interact with the mitochondrial import machinery (TIM complexes); Nsp8, with mitochondrial ribosomes, and ORF9c, with several components of the mitochondrial electron transport system. Nsp13 and Nsp15 furthermore may also interfere with interferon activation (Gordon et al., 2020). Experimental confirmation of these results is needed to assess the impact of SARS-CoV-2 proteins on mitochondrial functions.
Manipulation of the cGAS-STING pathway by SARS-coronaviruses may also occur. SARS-CoV papain-like protease (PLpro), for example, which is also essential for CoV replication and spread, has been shown to interact with and inhibit STING (Chen et al., 2014; Sun et al., 2012). Similar to SARS-CoV PLpro, SARS-CoV-2 PLpro has recently been demonstrated to also impair type I interferon and NF-κB responses (Shin et al., 2020). Interestingly, SARS-CoV-2 PLpro exhibited a differential substrate specificity in this study, resulting in preferential cleavage of other host proteins than by SARS-CoV PLpro, highlighting mechanistic differences that may contribute to the different pathological outcomes of SARS and COVID-19.
The innate immune response is furthermore targeted by SARS-CoV-2 by inhibition of translation. SARS-CoV-2 Nsp1, for example, has the potential to strongly impair translation of proteins involved in the anti-viral response, such as interferons and RIG-1, and the structural characterization of this process has recently been published (Thoms et al., 2020).
Taken together, SARS-coronaviruses inhibit a number of mitochondrial antiviral defense mechanisms, including the reduction of interferon activation. It is known that pretreatment with interferon I/III is beneficial in SARS-CoV infection, but the protective effect may be even higher in SARS-CoV-2 (Vabret et al., 2020), suggesting similar or aggravated impairment of interferon activation in COVID-19.
A summary of how SARS-CoV and SARS-CoV-2 modulate mitochondrial functions related to the innate immune system is depicted in Figure 1.
Modulation of Mitochondria by SARS-CoV-2 and Oxygen Sensing
SARS-CoV-2 infection may furthermore be related to reduced oxygen sensing mediated by mitochondrial dysfunction, as hypothesized by Archer et al. (2020). It recently became apparent that ARDS related to SARS-CoV-2 infection when compared with traditional ARDS exhibits distinct manifestations: relatively preserved lung compliance, large intrapulmonary shunt, sometimes an absence of dyspnea (Archer et al., 2020; Gattinoni et al., 2020), pulmonary vessel abnormalities such as thrombosis, microangiopathy, and strongly increased angiogenesis (Ackermann et al., 2020). Mitochondrial oxidative stress may be directly involved in platelet dysfunction and coagulation pathways and contribute to elevated blood clotting and thrombosis (Saleh et al., 2020). Based on the observations of differential pathologies when compared with ARDS, Archer and colleagues speculated that hypoxemia in SARS-CoV-2 patients is caused by deficits in organismal oxygen sensing, which normally regulates oxygen uptake and systemic oxygen delivery. The response of the pulmonary circulation to pathological airway hypoxia is pulmonary artery constriction. This leads to changes in blood flow toward better ventilated alveoli. Reduced oxygen availability in the blood in turn is sensed by the carotid bodies, which induce increased respiration. Archer et al. (2020) argue that hypoxemia in patients infected with SARS-CoV-2 is most likely induced due to an impaired pulmonary artery constriction mechanism and carotid body dysfunction (see Figure 2) that sometimes may be combined with infection-induced ARDS. These authors propose that oxygen-sensing mechanisms for pulmonary artery constriction and in the carotid bodies, which depend on the mitochondrial complex I subunit NDUFS2 (Dunham-Snary et al., 2019), are impaired following SARS-CoV-2 infection. Carotid bodies furthermore express ACE2 and thus maybe directly infected by SARS-CoV-2 (Li and Schultz, 2006). Indeed, Gordon and colleagues identified several complex I subunits to interact with the SARS-CoV-2 proteins Nsp7 and ORF9c (although not NDUFS2), an interaction that the US Food and Drug Administration-approved diabetes drug metformin, which inhibits complex I and associated mtROS production and exerts a plethora of related and different molecular effects (Kulkarni et al., 2020), according to these authors' analysis could target (Gordon et al., 2020). Metformin's original purpose as an influenza drug (Amin et al., 2019) and other theoretical considerations, including regulation of ACE2 (Sharma et al., 2020), may suggest efficiency in COVID-19. Metformin furthermore reverses a typical inflammation phenotype of T cells, observed both in aging and diabetes, by enhancing autophagy and mitochondrial bioenergetics (Bharath et al., 2020). Finally, the reduction of IL-6 release mediated by the inhibition of mitochondrial complex I-linked ROS production by metformin (Soberanes et al., 2019) has recently been directly linked to COVID-19 (Menendez, 2020).
Figure 2.

Possible Role of Mitochondrial Dysfunction in Aggravation of Hypoxemia in SARS-CoV-2
Mitochondrial dysfunction due to SARS-CoV2 infection contributes to pulmonary tissue damage, deterioration of pulmonary function, and airway hypoxia. Impaired mitochondria in the carotid bodies may worsen hypoxemia due to impaired oxygen sensing and result in a compromised chemoreflex. Oxidative stress and impaired anti-viral signaling lead to an imbalanced innate immune response.
Taken together, modulation of mitochondria by viruses such as SARS-CoV-2 perturbs mitochondrial functions, possibly enabling host defense evasion strategies. SARS-CoV-2 induced oxygen-sensing deficiencies may aggravate systemic respirational deficits and promote severe infection outcome, but may be amenable to pharmacological treatments, necessitating intensified research.
Mitochondrial Integrity and Antiviral Defense
Mitochondrial dysfunction, for example, due to aging or chronic disease, negatively affects immunity and inflammation (McGuire, 2019) and is a potential risk factor for SARS-CoV-2 infection. Mitochondrial integrity is paramount to mount adequate antiviral defense and prevent viral manipulation. Mitochondrial membrane potential, an important indicator of mitochondrial integrity, and oxidative phosphorylation have been shown to be necessary for the activation of the RLR-MAVS pathways (Koshiba et al., 2011; Yoshizumi et al., 2017). Moreover, the viral inhibition of MAVS by, e.g., the influenza virus protein PB1-F2 is achieved by a reduction of mitochondrial membrane potential (Varga et al., 2012; Yoshizumi et al., 2014). Supporting these results, diminished mitochondrial membrane potential is associated with a lower cytokine production after infection with various viruses (Ichinohe et al., 2013) and functional mitochondrial respiration is required to activate specialized immune cells (Sander and Garaude, 2018).
On the level of the lung, mitochondria not only provide energy for the regulation of airway and smooth muscles to regulate alveolar gas exchanges and to match ventilation and circulatory blood flow but also are integral for pulmonary cell and tissue protection (Cloonan and Choi, 2016; Piantadosi and Suliman, 2017). Acute lung injury is associated with mitochondrial dysfunction and oxidative stress (Piantadosi and Suliman, 2017), thus it is reasonable that maintaining mitochondrial integrity will improve the outcome. Indeed, in a lipopolysaccharide-induced lung injury model the observed severe mitochondrial dysfunction of pulmonary epithelia could be readily prevented by mitochondrial transfer of functional mitochondria from bone marrow-derived stromal cells, resulting in reduced leukocytosis, protein leak, and mortality in mice (Islam et al., 2012).
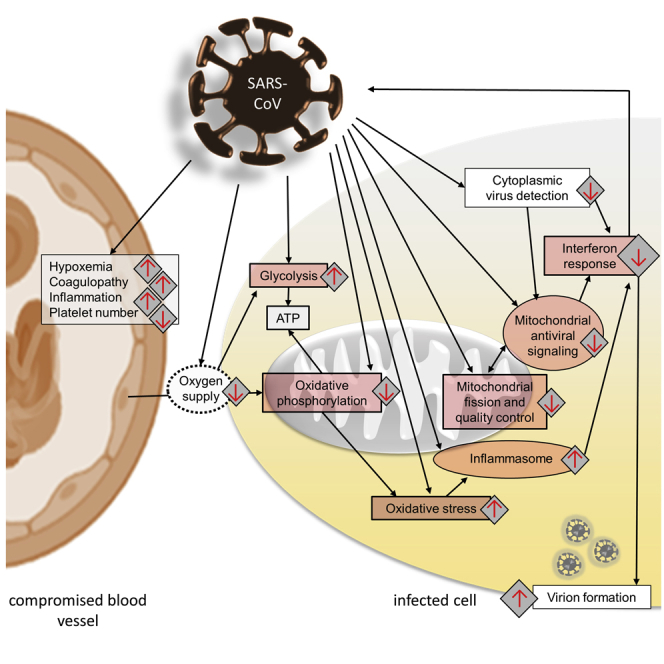
An overview of which mitochondrial functions related to the innate immune response are likely compromised by SARS-CoV-2 infection is depicted in Figure 3.
Figure 3.

Perturbance of Energy Metabolism, Mitochondrial Dynamics, and Mitochondrial Anti-viral Signaling due to SARS-Coronavirus Infection
SARS-coronavirus infection is believed to induce mitochondrial dysfunction, resulting in oxidative phosphorylation (OXPHOS) deficits, the production of mtROS, dysregulated mitochondrial dynamics, and compromised mitochondrial anti-viral signaling. In combination with hypoxemia it may change cellular bioenergetics, favoring glycolysis over OXPHOS. In summary the new cellular conditions promote viral replication.
An interesting aspect about COVID-19 epidemiology is the modulatory effect of genetic sex on immune response and disease progression, with men being at higher risk. Kloc et al. (2020) recently summarized the potential involvement of sexually dimorphic populations of mitochondria in differential immune responses in general and in COVID-19 specifically. They argue that, beside other factors, sex hormone-dependent higher variation of female intestinal microbiomes may support the immune function in COVID-19 by providing more adequate substrates to mitochondria. Due to the maternal inheritance of mitochondria and the associated female-biased quality control, mitochondrial DNA mutations that are harmful for males but not females may furthermore persist during the mammalian egg maturation (Kloc et al., 2020). Although the relevance of these effects in COVID-19 is speculative at this stage, they support the potential involvement of mitochondrial dysfunction in COVID-19.
How strongly mitochondrial integrity is involved in SARS-CoV-2 vulnerability remains to be tested experimentally. However, if compromised mitochondria are the reason for differential severity of COVID-19, this also warrants their assessment as biomarkers for disease progression as recently proposed (Shenoy, 2020). Identification of patients with increased likelihood of severe COVID-19 (or future epidemics) outcome is essential to select appropriate treatments and manage potentially limited health care capacities. Coagulopathy, circulating pro-inflammatory stimuli, and low platelet counts emerge as integral aspects of COVID-19 and have been associated with the severity of the disease outcome (Giannis et al., 2020; Merad and Martin, 2020). Oxidized phospholipids due to oxidative stress are linked to these pathologies, suggesting mitochondrial dysfunction or oxidative stress assessments to be candidates of peripherally measurable biomarkers. The recapitulation of mitochondrial dysfunction in blood samples of cardiac and skeletal muscles supports the investigation of the validity of such an approach for respiration (Tyrrell et al., 2016). Other blood-borne mitochondrial damage signals, like circulating dysfunctional, or functional (Al Amir Dache et al., 2020), mitochondria or mtDNA in peripheral blood cells may also hold promise (Fetterman et al., 2016). It has recently been argued that elevated oxidative and inflammatory states due to freely circulating mitochondria and mitochondrial components may result in dysfunction and apoptosis of platelets in COVID-19 and could thus be crucially involved in severe disease progression (Saleh et al., 2020).
In this review we focused on mitochondrial involvement in the innate immune system of primarily affected cells in COVID-19: epithelial cells, alveolar epithelial cells, vascular endothelial cells, which constitute an important factor of lung-specific immune response (Krausgruber et al., 2020), and resident macrophages in the lung. Clearly, specialized immune cell function also strongly depends on metabolic factors (immunometabolism), and mitochondria and may be affected by SARS-CoV-2 infection as they are by SARS-CoV (Tiku et al., 2020). A very informative review on the effect of SARS-CoV-2 on different immune cell types has been provided by Vabret et al. (2020); the knowledge on SARS-CoV-2-related immunometabolism is, however, still rudimentary. Activation of many immune cell types depends on mitochondrial function and oxidative phosphorylation (Sander and Garaude, 2018), although upon activation they often primarily rely on glycolysis (Banoth and Cassel, 2018). An excellent overview on these and related effects is provided in a recent review (Makowski et al., 2020). The exploration of the regulation and potential manipulation of mitochondria in specialized immune cells in SARS-CoV-2 will present another major scientific challenge.
In summary, enhanced mitochondrial integrity may be beneficial in lung injury following respiratory infection and improved mitochondrial function may be protective for SARS-CoV-2 infection. An overview on valuable therapeutic possibilities, aside from metformin (see above), to improve conditions for the mitochondria-mediated innate immune response or generally enhance mitochondrial functions with regard to immunity is provided in Boxes 1 and 2.
Box 1. Targeting Mitochondria to Enhance the Antiviral Immune Defense: Potential Applications for COVID-19.
a. Enhancing MAVS: Given its central role in the innate immune system, the major target MAVS seems to represent for SARS-CoVs, and the efficiency of interferon therapy in SARS-CoV infection, therapeutically targeting MAVS seems reasonable. Song et al. (2019) demonstrated the potential of D-glucosamine to upregulate MAVS efficiency by enhancing O-GlcNacylation of MAVS. This in turn promotes increased poly-ubiquitination of MAVS and thus induces more efficient induction of type I interferons via IRF3 signaling. The authors showed that a diet rich in D-glucosamine supplement indeed protected mice from RNA viruses such as human influenza virus.
b. Modulation of glycolysis in viral infection: Activation of the immune system induces changes in energy metabolism, mtROS production, and nutrient utilization and availability (Sander and Garaude, 2018). Cells infected by RNA viruses often upregulate glycolysis and downregulate oxidative phosphorylation for energy production (Ivashkiv, 2020), which might optimize viral replication conditions. The glycolysis product lactate inhibits RLR-mediated interferon activation, whereas glycolysis downregulation may be beneficial for the RLR-dependent anti-viral host defense (Yasukawa et al., 2020; Zhang et al., 2019). Activated specialized immune cells also rely more on glycolysis (Sander and Garaude, 2018). Although a potential metabolic shift upon innate immune system activation toward glycolysis in SARS-CoV-2 infection is insufficiently explored, the attenuation of glycolysis by pharmacological tools, such as the glycolysis inhibitor 2-deoxy-D-glucose (2DG) (Pålsson-McDermott and O'Neill, 2020), may represent a pharmacological avenue to explore.
Box 2. Boosting Mitochondria to Enhance the Innate Immunity: Potential Applications for COVID-19.
a. Boosting mitochondrial biogenesis and energy production: Mitochondria, most obviously and best studied in skeletal and cardiac muscles, respond rapidly to extracellular stimuli, such as exercise, by regulation of mitochondrial dynamics, mitochondrial membrane potential, and mitochondrial respiration (Hood et al., 2019). Moderate, regular exercise by ameliorating anti-oxidative capacities (Bouzid et al., 2018) is supposedly protective by preventing mitochondrial and tissue damage by exuberant ROS and can improve mitochondrial biogenesis, ATP production (Egan and Zierath, 2013), and immunity (Nieman and Wentz, 2019). Acute intensive bouts of exercise conversely reduce immune function transiently and thus are not to be recommended in the presence of infection risk (Nieman and Wentz, 2019).
One prominent example to boost mitochondrial function pharmacologically is the modulation of the nicotinamide dinucleotide (NAD+) synthesis (Katsyuba et al., 2020), the disruption of which has been demonstrated to be deleterious for the immune system (Minhas et al., 2019). Recently, Heer et al. (2020) indeed presented preliminary data that SARS-CoV-2 might dysregulate the NAD gene set, compromising immune responses.
b. Modulation of mitochondrial dynamics: SARS-coronaviruses are thought to modulate mitochondrial dynamics by degrading DRP1, thus favoring mitochondrial fusion. This may impair clearance of dysfunctional, for example, viral RNA or viral protein-containing, mitochondria and support survival of the infected cell during viral replication. Another consequence of impaired fission may be reduced cell membrane repair capacities of mitochondria for which fission is essential (Horn et al., 2020) and thereby could aggravate cell and tissue damage.
Inhibition of fusion to restore homeostasis of fusion and fission thus may be beneficial for SARS-CoV-2 infection. Recently, a new pharmacological tool for this purpose was presented in the form of an OPA1 inhibitor. Notably, OPA1 in the study of Herkenne et al. (2020) also was shown to promote angiogenesis by limiting NF-κB. Angiogenesis is commonly controlled by viruses to establish microenvironments favorable for their own pathogenesis (Alkharsah, 2018). OPA1 inhibition thus potentially has the capacity to also target another occasional pathological feature of SARS-CoV-2 infection, intussusceptive angiogenesis (Ackermann et al., 2020).
On the other hand, mitochondrial fusion is thought to be generally beneficial for mitochondrial antiviral signaling, for example, by enhancing mitochondrial interaction with the endoplasmic reticulum by bringing MAVS and STING components closer together, and thus increasing the efficiency of type I interferon induction (Pernas and Scorrano, 2016; West et al., 2011). Whether the benefits of fusion inhibition outweigh the expected negative effects on cellular anti-viral defense specifically by reduction of OPA1 (Castanier et al., 2010) and mitochondrial fusion in general remains to be investigated.
Future Perspectives
Despite immense scientific efforts to further our understanding of SARS-CoV-2, the SARS-CoV-2 biology and COVID-19 pathogenesis are still poorly understood. Due to this reason, many of the here-presented interactions between mitochondria and SARS-CoV-2 remain speculative, with unclear clinical relevance warranting further research. Specifically, experimental evidences regarding the effects of SARS-CoV-2-induced mitochondrial dysfunctions in infected cells and in those affected secondarily by compromised oxygen supply are important. We hope with this perspective to motivate scientific investigation in the presented topics that in our opinion have great potential to better understand the biological basis of the ongoing pandemic and thus may contribute to improved management of COVID-19.
In particular, the elucidation of the interactions between chronic diseases (both preconditions and as potential long-term effects of SARS-CoV-2) and COVID-19, specifically for metabolic and age-related diseases (e.g. Parkinson disease) is desirable. Epidemiological assessment of the correlation of chronic diseases involving mitochondrial dysfunctions is necessary to confirm the hypothesis that pre-existing mitochondrial deficits might predict infection and disease severity.
Furthermore, if prominent mitochondrial dysfunction occurs in COVID-19, evaluation of its use as a biomarker for infection susceptibility and severity of disease progression will be of interest, thanks to accessible mitochondria-related biomarkers (e.g., in the blood), such as oxidative stress markers, respirational capacities of blood cells, or circulating mitochondrial DNA.
Several strategies to target mitochondria, both pharmacologically and lifestyle interventions, hold promise to prevent SARS-CoV-2 infection or mitigate disease progression (see Box 1). Up to date, any experimental evidence on their potential is lacking. As mitochondria are often compromised following different types of viral infections, boosting their function might be an approach with potential not only in COVID-19 but also to fight other viruses. Targeting shared features of a broad array of viruses may be an important approach for future epidemics, especially as long as effective antibody treatments are lacking. Better understanding of mitochondrial involvement in immune functions eventually will also contribute to our understanding of the role of infections and inflammation in chronic diseases and aging.
In conclusion, coronavirus infection may cause mitochondrial dysfunctions and functional mitochondria are integral for cell and tissue protection and for initiation and maintenance of immune responses. Here the manifold ways, how RNA viruses, and specifically SARS-coronaviruses, modulate mitochondrial function to evade the innate immune response of host cells, were summarized and put into context with the current understanding of SARS-CoV-2. We argue that enhancing or preserving mitochondrial function may prevent mitochondrial manipulation and dysfunction following viral infection. Regular physical activity or targeted pharmacological interventions to boost mitochondrial capacities are likely efficient to increase mitochondrial function and may enhance innate immunity in face of SARS-CoV-2 infection. In addition, the exploration of mitochondrial functions or mitochondrial damage markers in accessible tissues (e.g., blood) in response to SARS-CoV-2-infection may pave the way for the development of mitochondrial biomarkers for vulnerability and expected disease severity. The many knowledge gaps we currently have on SARS-CoV-2 biology and host pathophysiology give room for much future research.
In summary, mitochondrial involvement in the innate immune response to SARS-CoV-2-infection represents a mechanism targetable by lifestyle and pharmacological strategies. Its thorough investigation is warranted for a better understanding of the pathogen-host biology and the development of adequate treatment strategies. Pre-existing mitochondrial dysfunction, for example, due to age or disease (e.g., cardiovascular disease, a prominent risk-factor of COVID-19), may represent factors contributing to differential vulnerabilities to SARS-CoV-2-infection and thus should be considered as a biomarker for the risk of infection and severity of outcome.
Acknowledgments
This work was partly supported by the JSPS KAKENHI Grant 20H04914 (to T.K.).
Author Contributions
Conceptualization, J.B.; Methodology, J.B., G.C., A.O., T.K., and G.P.M.; Writing – Original Draft, J.B.; Writing – Review and Editing, J.B., G.C., A.O., T.K., and G.P.M.
References
- Ackermann M., Verleden S.E., Kuehnel M., Haverich A., Welte T., Laenger F., Vanstapel A., Werlein C., Stark H., Tzankov A. Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. New Engl. J. Med. 2020;383:120–128. doi: 10.1056/NEJMoa2015432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Amir Dache Z., Otandault A., Tanos R., Pastor B., Meddeb R., Sanchez C., Arena G., Lasorsa L., Bennett A., Grange T. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J. 2020;34:3616–3630. doi: 10.1096/fj.201901917RR. [DOI] [PubMed] [Google Scholar]
- Alkharsah K.R. VEGF upregulation in viral infections and its possible therapeutic implications. Int. J. Mol. Sci. 2018;19:1642. doi: 10.3390/ijms19061642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin S., Lux A., O'callaghan F. The journey of metformin from glycaemic control to mTOR inhibition and the suppression of tumour growth. Br. J. Clin. Pharmacol. 2019;85:37–46. doi: 10.1111/bcp.13780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonini A., Leta V., Teo J., Chaudhuri K.R. Outcome of Parkinson's Disease patients affected by COVID-19. Movement Disord. 2020;35:905–908. doi: 10.1002/mds.28104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer S.L., Sharp W.W., Weir E.K. Differentiating COVID-19 Pneumonia from acute respiratory distress syndrome (ARDS) and high altitude pulmonary edema (HAPE): therapeutic implications. Circulation. 2020;142:101–104. doi: 10.1161/CIRCULATIONAHA.120.047915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azkur A.K., Akdis M., Azkur D., Sokolowska M., van de Veen W., Bruggen M.C., O'Mahony L., Gao Y., Nadeau K., Akdis C.A. Immune response to SARS-CoV-2 and mechanisms of immunopathological changes in COVID-19. Allergy. 2020;75:1564–1581. doi: 10.1111/all.14364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banoth B., Cassel S.L. Mitochondria in innate immune signaling. Transl Res. 2018;202:52–68. doi: 10.1016/j.trsl.2018.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgnaoui S.M., Paz S., Hiscott J. Orchestrating the interferon antiviral response through the mitochondrial antiviral signaling (MAVS) adapter. Curr. Opin. Immunol. 2011;23:564–572. doi: 10.1016/j.coi.2011.08.001. [DOI] [PubMed] [Google Scholar]
- Bharath L.P., Agrawal M., McCambridge G., Nicholas D.A., Hasturk H., Liu J., Jiang K., Liu R., Guo Z., Deeney J. Metformin enhances autophagy and normalizes mitochondrial function to alleviate aging-associated inflammation. Cell Metab. 2020;32:44–55.e6. doi: 10.1016/j.cmet.2020.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Melo D., Nilsson-Payant B.E., Liu W.-C., Uhl S., Hoagland D., Møller R., Jordan T.X., Oishi K., Panis M., Sachs D. Imbalanced host response to SARS-CoV-2 drives development of COVID-19. Cell. 2020;181:1036–1045.e9. doi: 10.1016/j.cell.2020.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojkova D., Klann K., Koch B., Widera M., Krause D., Ciesek S., Cinatl J., Münch C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature. 2020;583:469–472. doi: 10.1038/s41586-020-2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzid M.A., Filaire E., Matran R., Robin S., Fabre C. Lifelong voluntary exercise modulates age-related changes in oxidative stress. Int. J. Sports Med. 2018;39:21–28. doi: 10.1055/s-0043-119882. [DOI] [PubMed] [Google Scholar]
- Castanier C., Garcin D., Vazquez A., Arnoult D. Mitochondrial dynamics regulate the RIG-I-like receptor antiviral pathway. EMBO Rep. 2010;11:133–138. doi: 10.1038/embor.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Channappanavar R., Fehr A.R., Zheng J., Wohlford-Lenane C., Abrahante J.E., Mack M., Sompallae R., McCray P.B., Jr., Meyerholz D.K., Perlman S. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. J. Clin. Invest. 2019;130:3625–3639. doi: 10.1172/JCI126363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.Y., Ping Y.H., Lee H.C., Chen K.H., Lee Y.M., Chan Y.J., Lien T.C., Jap T.S., Lin C.H., Kao L.S. Open reading frame 8a of the human severe acute respiratory syndrome coronavirus not only promotes viral replication but also induces apoptosis. J. Infect Dis. 2007;196:405–415. doi: 10.1086/519166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I.Y., Moriyama M., Chang M.F., Ichinohe T. Severe acute respiratory syndrome coronavirus viroporin 3a activates the NLRP3 inflammasome. Front. Microbiol. 2019;10:50. doi: 10.3389/fmicb.2019.00050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X., Yang X., Zheng Y., Yang Y., Xing Y., Chen Z. SARS coronavirus papain-like protease inhibits the type I interferon signaling pathway through interaction with the STING-TRAF3-TBK1 complex. Protein & Cell. 2014;5:369–381. doi: 10.1007/s13238-014-0026-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Zhang Z., Li J., Gao Y., Zhou L., Ge X., Han J., Guo X., Yang H. Porcine epidemic diarrhea virus S1 protein is the critical inducer of apoptosis. Virol. J. 2018;15:170. doi: 10.1186/s12985-018-1078-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloonan S.M., Choi A.M. Mitochondria in lung disease. J. Clin. Invest. 2016;126:809–820. doi: 10.1172/JCI81113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson B.T., Brennan M.A. Pro-inflammatory programmed cell death. Trends Microbiol. 2001;9:113–114. doi: 10.1016/s0966-842x(00)01936-3. [DOI] [PubMed] [Google Scholar]
- Cornillez-Ty C.T., Liao L., Yates J.R., Kuhn P., Buchmeier M.J. Severe acute respiratory syndrome coronavirus nonstructural protein 2 interacts with a host protein complex involved in mitochondrial biogenesis and intracellular signaling. J. Virol. 2009;83:10314–10318. doi: 10.1128/JVI.00842-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coronaviridae Study Group, I The species Severe acute respiratory syndrome-related coronavirus: classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020;5:536–544. doi: 10.1038/s41564-020-0695-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cribbs J.T., Strack S. Reversible phosphorylation of Drp1 by cyclic AMP-dependent protein kinase and calcineurin regulates mitochondrial fission and cell death. EMBO Rep. 2007;8:939–944. doi: 10.1038/sj.embor.7401062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Roche L., Mesta F. Oxidative stress as key player in severe acute respiratory syndrome coronavirus (SARS-CoV) infection. Arch. Med. Res. 2020;51:384–387. doi: 10.1016/j.arcmed.2020.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devaraj S.G., Wang N., Chen Z., Chen Z., Tseng M., Barretto N., Lin R., Peters C.J., Tseng C.-T.K., Baker S.C. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007;282:32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunham-Snary K.J., Wu D., Potus F., Sykes E.A., Mewburn J.D., Charles R.L., Eaton P., Sultanian R.A., Archer S.L. Ndufs2, a core subunit of mitochondrial complex I, is essential for acute oxygen-sensing and hypoxic pulmonary vasoconstriction. Circ. Res. 2019;124:1727–1746. doi: 10.1161/CIRCRESAHA.118.314284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaglesham J.B., Kranzusch P.J. Conserved strategies for pathogen evasion of cGAS-STING immunity. Curr. Opin. Immunol. 2020;66:27–34. doi: 10.1016/j.coi.2020.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B., Zierath Juleen R. Exercise metabolism and the molecular regulation of skeletal muscle adaptation. Cell Metab. 2013;17:162–184. doi: 10.1016/j.cmet.2012.12.012. [DOI] [PubMed] [Google Scholar]
- Fani M., Teimoori A., Ghafari S. Comparison of the COVID-2019 (SARS-CoV-2) pathogenesis with SARS-CoV and MERS-CoV infections. Future Virol. 2020 doi: 10.2217/fvl-2020-0050. [DOI] [Google Scholar]
- Fetterman J.L., Holbrook M., Westbrook D.G., Brown J.A., Feeley K.P., Bretón-Romero R., Linder E.A., Berk B.D., Weisbrod R.M., Widlansky M.E. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovasc. Diabetology. 2016;15:53. doi: 10.1186/s12933-016-0372-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields J.A., Serger E., Campos S., Divakaruni A.S., Kim C., Smith K., Trejo M., Adame A., Spencer B., Rockenstein E. HIV alters neuronal mitochondrial fission/fusion in the brain during HIV-associated neurocognitive disorders. Neurobiol. Dis. 2016;86:154–169. doi: 10.1016/j.nbd.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freundt E.C., Yu L., Park E., Lenardo M.J., Xu X.-N. Molecular determinants for subcellular localization of the severe acute respiratory syndrome coronavirus open reading frame 3b protein. J. Virol. 2009;83:6631–6640. doi: 10.1128/JVI.00367-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Ratia K., Johnston R.E., Mesecar A.D., Baric R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009;83:6689–6705. doi: 10.1128/JVI.02220-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieman M., Yount B., Heise M., Kopecky-Bromberg S.A., Palese P., Baric R.S. Severe acute respiratory syndrome coronavirus ORF6 antagonizes STAT1 function by sequestering nuclear import factors on the rough endoplasmic reticulum/Golgi membrane. J. Virol. 2007;81:9812–9824. doi: 10.1128/JVI.01012-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardinassi L.G., Souza C.O.S., Sales-Campos H., Fonseca S.G. Immune and metabolic signatures of COVID-19 revealed by transcriptomics data reuse. Front. Immunol. 2020;11:1636. doi: 10.3389/fimmu.2020.01636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L., Coppola S., Cressoni M., Busana M., Rossi S., Chiumello D. COVID-19 does not lead to a "typical" acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 2020;201:1299–1300. doi: 10.1164/rccm.202003-0817LE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giacomello M., Pyakurel A., Glytsou C., Scorrano L. The cell biology of mitochondrial membrane dynamics. Nat. Rev. Mol. Cell Biol. 2020;21:204–224. doi: 10.1038/s41580-020-0210-7. [DOI] [PubMed] [Google Scholar]
- Giannis D., Ziogas I.A., Gianni P. Coagulation disorders in coronavirus infected patients: COVID-19, SARS-CoV-1, MERS-CoV and lessons from the past. J. Clin. Virol. 2020;127:104362. doi: 10.1016/j.jcv.2020.104362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gkikas I., Palikaras K., Tavernarakis N. The role of mitophagy in innate immunity. Front. Immunol. 2018;9:1283. doi: 10.3389/fimmu.2018.01283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D.E., Jang G.M., Bouhaddou M., Xu J., Obernier K., White K.M., O’Meara M.J., Rezelj V.V., Guo J.Z., Swaney D.L. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heer C.D., Sanderson D.J., Alhammad Y.M.O., Schmidt M.S., Trammell S.A.J., Perlman S., Cohen M.S., Fehr A.R., Brenner C. Coronavirus infection and PARP expression dysregulate the NAD metabolome: a potentially actionable component of innate immunity. bioRxiv. 2020 doi: 10.1101/2020.04.17.047480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herkenne S., Ek O., Zamberlan M., Pellattiero A., Chergova M., Chivite I., Novotná E., Rigoni G., Fonseca T.B., Samardzic D. Developmental and tumor angiogenesis requires the mitochondria-shaping protein Opa1. Cell Metab. 2020;31:987–1003.e8. doi: 10.1016/j.cmet.2020.04.007. [DOI] [PubMed] [Google Scholar]
- Hood D.A., Memme J.M., Oliveira A.N., Triolo M. Maintenance of skeletal muscle mitochondria in health, exercise, and aging. Annu. Rev. Physiol. 2019;81:19–41. doi: 10.1146/annurev-physiol-020518-114310. [DOI] [PubMed] [Google Scholar]
- Horn A., Raavicharla S., Shah S., Cox D., Jaiswal J.K. Mitochondrial fragmentation enables localized signaling required for cell repair. J. Cell Biol. 2020;219:e201909154. doi: 10.1083/jcb.201909154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou F., Sun L., Zheng H., Skaug B., Jiang Q.-X., Chen Z.J. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y., Li W., Gao T., Cui Y., Jin Y., Li P., Ma Q., Liu X., Cao C. The severe acute respiratory syndrome coronavirus nucleocapsid inhibits type I interferon production by interfering with TRIM25-mediated RIG-I ubiquitination. J. Virol. 2017;91 doi: 10.1128/JVI.02143-16. e02143–02116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinohe T., Yamazaki T., Koshiba T., Yanagi Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. 2013;110:17963–17968. doi: 10.1073/pnas.1312571110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam M.N., Das S.R., Emin M.T., Wei M., Sun L., Westphalen K., Rowlands D.J., Quadri S.K., Bhattacharya S., Bhattacharya J. Mitochondrial transfer from bone-marrow–derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012;18:759. doi: 10.1038/nm.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivashkiv L.B. The hypoxia–lactate axis tempers inflammation. Nat. Rev. Immunol. 2020;20:85–86. doi: 10.1038/s41577-019-0259-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jauregui A.R., Savalia D., Lowry V.K., Farrell C.M., Wathelet M.G. Identification of residues of SARS-CoV nsp1 that differentially affect inhibition of gene expression and antiviral signaling. PLoS One. 2013;8:e62416. doi: 10.1371/journal.pone.0062416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsyuba E., Romani M., Hofer D., Auwerx J. NAD+ homeostasis in health and disease. Nat. Metab. 2020;2:9–31. doi: 10.1038/s42255-019-0161-5. [DOI] [PubMed] [Google Scholar]
- Khan M., Syed G.H., Kim S.-J., Siddiqui A. Mitochondrial dynamics and viral infections: a close nexus. Biochim. Biophys. Acta (Bba) - Mol. Cell Res. 2015;1853:2822–2833. doi: 10.1016/j.bbamcr.2014.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan M., Syed G.H., Kim S.-J., Siddiqui A. Hepatitis B virus-induced parkin-dependent recruitment of linear ubiquitin assembly complex (LUBAC) to mitochondria and attenuation of innate immunity. PLoS Pathog. 2016;12:e1005693. doi: 10.1371/journal.ppat.1005693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.-J., Ahn D.-G., Syed G.H., Siddiqui A. The essential role of mitochondrial dynamics in antiviral immunity. Mitochondrion. 2018;41:21–27. doi: 10.1016/j.mito.2017.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.J., Khan M., Quan J., Till A., Subramani S., Siddiqui A. Hepatitis B virus disrupts mitochondrial dynamics: induces fission and mitophagy to attenuate apoptosis. Plos Pathog. 2013;9:e1003722. doi: 10.1371/journal.ppat.1003722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S.R., Kim D., Kim S., Lee H., Lee K.S., Cho S.H., Lee Y.C. NLRP3 inflammasome activation by mitochondrial ROS in bronchial epithelial cells is required for allergic inflammation. Cell Death Dis. 2014;5:e1498. doi: 10.1038/cddis.2014.460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloc M., Ghobrial R.M., Kubiak J.Z. The role of genetic sex and mitochondria in response to COVID-19 infection. Int. Arch. Allergy Immunol. 2020;181:629–634. doi: 10.1159/000508560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopecky-Bromberg S.A., Martínez-Sobrido L., Frieman M., Baric R.A., Palese P. Severe acute respiratory syndrome coronavirus open reading frame (ORF) 3b, ORF 6, and nucleocapsid proteins function as interferon antagonists. J. Virol. 2007;81:548. doi: 10.1128/JVI.01782-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koshiba T., Yasukawa K., Yanagi Y., Kawabata S.-i. Mitochondrial membrane potential is required for MAVS-mediated antiviral signaling. Sci. Signal. 2011;4:ra7. doi: 10.1126/scisignal.2001147. [DOI] [PubMed] [Google Scholar]
- Krausgruber T., Fortelny N., Fife-Gernedl V., Senekowitsch M., Schuster L.C., Lercher A., Nemc A., Schmidl C., Rendeiro A.F., Bergthaler A. Structural cells are key regulators of organ-specific immune responses. Nature. 2020;583:296–302. doi: 10.1038/s41586-020-2424-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni A.S., Gubbi S., Barzilai N. Benefits of metformin in attenuating the hallmarks of aging. Cell Metab. 2020;32:15–30. doi: 10.1016/j.cmet.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai C.C., Jou M.J., Huang S.Y., Li S.W., Wan L., Tsai F.J., Lin C.W. Proteomic analysis of up-regulated proteins in human promonocyte cells expressing severe acute respiratory syndrome coronavirus 3C-like protease. Proteomics. 2007;7:1446–1460. doi: 10.1002/pmic.200600459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Y., Moore C.B., Liesman R.M., O'Connor B.P., Bergstralh D.T., Chen Z.J., Pickles R.J., Ting J.P. MAVS-mediated apoptosis and its inhibition by viral proteins. PLoS One. 2009;4:e5466. doi: 10.1371/journal.pone.0005466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Ma C., Long F., Yang D., Liu X., Hu Y., Wu C., Wang B., Wang M., Chen Y. Parkin impairs antiviral immunity by suppressing the mitochondrial reactive oxygen species-Nlrp3 axis and antiviral inflammation. iScience. 2019;16:468–484. doi: 10.1016/j.isci.2019.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.-D., Sun L., Seth R.B., Pineda G., Chen Z.J. Hepatitis C virus protease NS3/4A cleaves mitochondrial antiviral signaling protein off the mitochondria to evade innate immunity. Proc. Natl. Acad. Sci. 2005;102:17717–17722. doi: 10.1073/pnas.0508531102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.L., Schultz H.D. Enhanced sensitivity of Kv channels to hypoxia in the rabbit carotid body in heart failure: role of angiotensin II. J. Physiol. 2006;575:215–227. doi: 10.1113/jphysiol.2006.110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X., Pan J.a., Tao J., Guo D. SARS-CoV nucleocapsid protein antagonizes IFN-β response by targeting initial step of IFN-β induction pathway, and its C-terminal region is critical for the antagonism. Virus genes. 2011;42:37–45. doi: 10.1007/s11262-010-0544-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makowski L., Chaib M., Rathmell J.C. Immunometabolism: from basic mechanisms to translation. Immunological Rev. 2020;295:5–14. doi: 10.1111/imr.12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzanillo P.S., Ayres J.S., Watson R.O., Collins A.C., Souza G., Rae C.S., Schneider D.S., Nakamura K., Shiloh M.U., Cox J.S. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature. 2013;501:512–516. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marreiros R., Müller-Schiffmann A., Trossbach S.V., Prikulis I., Hänsch S., Weidtkamp-Peters S., Moreira A.R., Sahu S., Soloviev I., Selvarajah S. Disruption of cellular proteostasis by H1N1 influenza A virus causes α-synuclein aggregation. Proc. Natl. Acad. Sci. 2020;117:6741–6751. doi: 10.1073/pnas.1906466117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire P.J. Mitochondrial dysfunction and the aging immune system. Biology. 2019;8:26. doi: 10.3390/biology8020026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez J. Metformin and SARS-CoV-2: mechanistic lessons on air pollution to weather the cytokine/thrombotic storm in COVID-19. Aging. 2020;12:8760–8765. doi: 10.18632/aging.103347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merad M., Martin J.C. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat. Rev. Immunol. 2020;20:355–362. doi: 10.1038/s41577-020-0331-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielech A.M., Kilianski A., Baez-Santos Y.M., Mesecar A.D., Baker S.C. MERS-CoV papain-like protease has deISGylating and deubiquitinating activities. Virology. 2014;450-451:64–70. doi: 10.1016/j.virol.2013.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minakshi R., Padhan K., Rani M., Khan N., Ahmad F., Jameel S. The SARS Coronavirus 3a protein causes endoplasmic reticulum stress and induces ligand-independent downregulation of the type 1 interferon receptor. PLoS One. 2009;4:e8342. doi: 10.1371/journal.pone.0008342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minhas P.S., Liu L., Moon P.K., Joshi A.U., Dove C., Mhatre S., Contrepois K., Wang Q., Lee B.A., Coronado M. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019;20:50–63. doi: 10.1038/s41590-018-0255-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman L.E., Shadel G.S. Pink1/Parkin link inflammation, mitochondrial stress, and neurodegeneration. J. Cell. Biol. 2018;217:3327–3329. doi: 10.1083/jcb.201808118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieman D.C., Wentz L.M. The compelling link between physical activity and the body's defense system. J. Sport Health Sci. 2019;8:201–217. doi: 10.1016/j.jshs.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto-Torres J.L., Verdiá-Báguena C., Jimenez-Guardeño J.M., Regla-Nava J.A., Castaño-Rodriguez C., Fernandez-Delgado R., Torres J., Aguilella V.M., Enjuanes L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology. 2015;485:330–339. doi: 10.1016/j.virol.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onoguchi K., Onomoto K., Takamatsu S., Jogi M., Takemura A., Morimoto S., Julkunen I., Namiki H., Yoneyama M., Fujita T. Virus-infection or 5′ ppp-RNA activates antiviral signal through redistribution of IPS-1 mediated by MFN1. PLoS Pathog. 2010;6:e1001012. doi: 10.1371/journal.ppat.1001012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padhan K., Minakshi R., Towheed M.A.B., Jameel S. Severe acute respiratory syndrome coronavirus 3a protein activates the mitochondrial death pathway through p38 MAP kinase activation. J. Gen. Virol. 2008;89:1960–1969. doi: 10.1099/vir.0.83665-0. [DOI] [PubMed] [Google Scholar]
- Pålsson-McDermott E.M., O’Neill L.A.J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020;30:300–314. doi: 10.1038/s41422-020-0291-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa S.M., Brundin P., Fung V.S., Kang U.J., Burn D.J., Colosimo C., Chiang H.L., Alcalay R.N., Trenkwalder C., Committee M.-S.I. Impact of the COVID-19 pandemic on Parkinson’s disease and movement disorders. Mov. Disord. 2020;35:711–715. doi: 10.1002/mds.28067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pernas L., Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu. Rev. Physiol. 2016;78:505–531. doi: 10.1146/annurev-physiol-021115-105011. [DOI] [PubMed] [Google Scholar]
- Petersen E., Koopmans M., Go U., Hamer D.H., Petrosillo N., Castelli F., Storgaard M., Al Khalili S., Simonsen L. Comparing SARS-CoV-2 with SARS-CoV and influenza pandemics. Lancet Infect. Dis. 2020;20:e238–e244. doi: 10.1016/S1473-3099(20)30484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piantadosi C.A., Suliman H.B. Mitochondrial dysfunction in lung pathogenesis. Annu. Rev. Physiol. 2017;79:495–515. doi: 10.1146/annurev-physiol-022516-034322. [DOI] [PubMed] [Google Scholar]
- Pickles S., Vigié P., Youle R.J. Mitophagy and quality control mechanisms in mitochondrial maintenance. Curr. Biol. 2018;28:R170–R185. doi: 10.1016/j.cub.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rockx B., Kuiken T., Herfst S., Bestebroer T., Lamers M.M., Oude Munnink B.B., de Meulder D., van Amerongen G., van den Brand J., Okba N.M.A. Comparative pathogenesis of COVID-19, MERS, and SARS in a nonhuman primate model. Science. 2020;368:1012. doi: 10.1126/science.abb7314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan Q., Yang K., Wang W., Jiang L., Song J. Clinical predictors of mortality due to COVID-19 based on an analysis of data of 150 patients from Wuhan, China. Intensive Care Med. 2020;46:846–848. doi: 10.1007/s00134-020-05991-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh J., Peyssonnaux C., Singh K.K., Edeas M. Mitochondria and microbiota dysfunction in COVID-19 pathogenesis. Mitochondrion. 2020;54:1–7. doi: 10.1016/j.mito.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander L.E., Garaude J. The mitochondrial respiratory chain: a metabolic rheostat of innate immune cell-mediated antibacterial responses. Mitochondrion. 2018;41:28–36. doi: 10.1016/j.mito.2017.10.008. [DOI] [PubMed] [Google Scholar]
- Sasaki O., Yoshizumi T., Kuboyama M., Ishihara T., Suzuki E., Kawabata S.-i., Koshiba T. A structural perspective of the MAVS-regulatory mechanism on the mitochondrial outer membrane using bioluminescence resonance energy transfer. Biochim. Biophys. Acta (Bba) - Mol. Cell Res. 2013;1833:1017–1027. doi: 10.1016/j.bbamcr.2013.01.010. [DOI] [PubMed] [Google Scholar]
- Schrepfer E., Scorrano L. Mitofusins, from mitochondria to metabolism. Mol. Cell. 2016;61:683–694. doi: 10.1016/j.molcel.2016.02.022. [DOI] [PubMed] [Google Scholar]
- Sharma S., Ray A., Sadasivam B. Metformin in COVID-19: a possible role beyond diabetes. Diabetes Res. Clin. Pract. 2020;164:108183. doi: 10.1016/j.diabres.2020.108183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy S. Coronavirus (Covid-19) sepsis: revisiting mitochondrial dysfunction in pathogenesis, aging, inflammation, and mortality. Inflamm. Res. 2020;69:1077–1085. doi: 10.1007/s00011-020-01389-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C.-S., Qi H.-Y., Boularan C., Huang N.-N., Abu-Asab M., Shelhamer J.H., Kehrl J.H. SARS-coronavirus open reading frame-9b suppresses innate immunity by targeting mitochondria and the MAVS/TRAF3/TRAF6 signalosome. J. Immunol. 2014;193:3080–3089. doi: 10.4049/jimmunol.1303196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi C.S., Nabar N.R., Huang N.N., Kehrl J.H. SARS-Coronavirus Open Reading Frame-8b triggers intracellular stress pathways and activates NLRP3 inflammasomes. Cell Death Discov. 2019;5:101. doi: 10.1038/s41420-019-0181-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D., Mukherjee R., Grewe D., Bojkova D., Baek K., Bhattacharya A., Schulz L., Widera M., Mehdipour A.R., Tascher G. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature. 2020 doi: 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shokri S., Mahmoudvand S., Taherkhani R., Farshadpour F. Modulation of the immune response by Middle East respiratory syndrome coronavirus. J. Cell. Physiol. 2019;234:2143–2151. doi: 10.1002/jcp.27155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K.K., Chaubey G., Chen J.Y., Suravajhala P. American Physiological Society; 2020. Decoding SARS-CoV-2 Hijacking of Host Mitochondria in Pathogenesis of COVID-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu K.L., Kok K.H., Ng M.H., Poon V.K., Yuen K.Y., Zheng B.J., Jin D.Y. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKepsilon complex. J. Biol. Chem. 2009;284:16202–16209. doi: 10.1074/jbc.M109.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu K.L., Yuen K.S., Castaño-Rodriguez C., Ye Z.W., Yeung M.L., Fung S.Y., Yuan S., Chan C.P., Yuen K.Y., Enjuanes L. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019;33:8865–8877. doi: 10.1096/fj.201802418R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sliter D.A., Martinez J., Hao L., Chen X., Sun N., Fischer T.D., Burman J.L., Li Y., Zhang Z., Narendra D.P. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–262. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Soberanes S., Misharin A.V., Jairaman A., Morales-Nebreda L., McQuattie-Pimentel A.C., Cho T., Hamanaka R.B., Meliton A.Y., Reyfman P.A., Walter J.M. Metformin targets mitochondrial electron transport to reduce air-pollution-induced thrombosis. Cell Metab. 2019;29:335–347.e5. doi: 10.1016/j.cmet.2018.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somasundaran M., Zapp M.L., Beattie L.K., Pang L., Byron K.S., Bassell G.J., Sullivan J.L., Singer R.H. Localization of HIV RNA in mitochondria of infected cells: potential role in cytopathogenicity. J. Cell. Biol. 1994;126:1353–1360. doi: 10.1083/jcb.126.6.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song N., Qi Q., Cao R., Qin B., Wang B., Wang Y., Zhao L., Li W., Du X., Liu F. MAVS O-GlcNAcylation is essential for host antiviral immunity against lethal RNA viruses. Cell Rep. 2019;28:2386–2396.e5. doi: 10.1016/j.celrep.2019.07.085. [DOI] [PubMed] [Google Scholar]
- Sun L., Xing Y., Chen X., Zheng Y., Yang Y., Nichols D.B., Clementz M.A., Banach B.S., Li K., Baker S.C. Coronavirus papain-like proteases negatively regulate antiviral innate immune response through disruption of STING-mediated signaling. PLoS One. 2012;7:e30802. doi: 10.1371/journal.pone.0030802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi N., Ishihara N., Jofuku A., Oka T., Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J. Biol. Chem. 2007;282:11521–11529. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- Tan Y.X., Tan T.H., Lee M.J., Tham P.Y., Gunalan V., Druce J., Birch C., Catton M., Fu N.Y., Yu V.C. Induction of apoptosis by the severe acute respiratory syndrome coronavirus 7a protein is dependent on its interaction with the Bcl-XL protein. J. Virol. 2007;81:6346–6355. doi: 10.1128/JVI.00090-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatsuta T., Langer T. Prohibitins. Curr. Biol. 2017;27:R629–R631. doi: 10.1016/j.cub.2017.04.030. [DOI] [PubMed] [Google Scholar]
- Tay M.Z., Poh C.M., Rénia L., MacAry P.A., Ng L.F.P. The trinity of COVID-19: immunity, inflammation and intervention. Nat. Rev. Immunol. 2020;20:363–374. doi: 10.1038/s41577-020-0311-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoms M., Buschauer R., Ameismeier M., Koepke L., Denk T., Hirschenberger M., Kratzat H., Hayn M., Mackens-Kiani T., Cheng J. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science. 2020;369:1249–1255. doi: 10.1126/science.abc8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiku V., Tan M.-W., Dikic I. Mitochondrial functions in infection and immunity. Trends Cell Biol. 2020;30:263–275. doi: 10.1016/j.tcb.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totura A.L., Baric R.S. SARS coronavirus pathogenesis: host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012;2:264–275. doi: 10.1016/j.coviro.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyrrell D.J., Bharadwaj M.S., Jorgensen M.J., Register T.C., Molina A.J.A. Blood cell respirometry is associated with skeletal and cardiac muscle bioenergetics: implications for a minimally invasive biomarker of mitochondrial health. Redox Biol. 2016;10:65–77. doi: 10.1016/j.redox.2016.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vabret N., Britton G.J., Gruber C., Hegde S., Kim J., Kuksin M., Levantovsky R., Malle L., Moreira A., Park M.D. Immunology of COVID-19: current state of the science. Immunity. 2020;52:910–941. doi: 10.1016/j.immuni.2020.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varga Z.T., Grant A., Manicassamy B., Palese P. Influenza virus protein PB1-F2 inhibits the induction of type I interferon by binding to MAVS and decreasing mitochondrial membrane potential. J. Virol. 2012;86:8359–8366. doi: 10.1128/JVI.01122-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B., Li R., Lu Z., Huang Y. Does comorbidity increase the risk of patients with COVID-19: evidence from meta-analysis. Aging (Albany NY) 2020;12:6049. doi: 10.18632/aging.103000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wathelet M.G., Orr M., Frieman M.B., Baric R.S. Severe acute respiratory syndrome coronavirus evades antiviral signaling: role of nsp1 and rational design of an attenuated strain. J. Virol. 2007;81:11620–11633. doi: 10.1128/JVI.00702-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg S.E., Sena L.A., Chandel N.S. Mitochondria in the regulation of innate and adaptive immunity. Immunity. 2015;42:406–417. doi: 10.1016/j.immuni.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A.P., Khoury-Hanold W., Staron M., Tal M.C., Pineda C.M., Lang S.M., Bestwick M., Duguay B.A., Raimundo N., MacDuff D.A. Mitochondrial DNA stress primes the antiviral innate immune response. Nature. 2015;520:553–557. doi: 10.1038/nature14156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West A.P., Shadel G.S., Ghosh S. Mitochondria in innate immune responses. Nat. Rev. Immunol. 2011;11:389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu K.E., Fazal F.M., Parker K.R., Zou J., Chang H.Y. RNA-GPS predicts SARS-CoV-2 RNA residency to host mitochondria and nucleolus. Cell Syst. 2020;11:102–108.e3. doi: 10.1016/j.cels.2020.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y., Liang Y., Qu L., Chen Z., Yi M., Li K., Lemon S.M. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc. Natl. Acad. Sci. U S A. 2007;104:7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa K., Kinoshita D., Yaku K., Nakagawa T., Koshiba T. The microRNAs miR-302b and miR-372 regulate mitochondrial metabolism via the SLC25A12 transporter, which controls MAVS-mediated antiviral innate immunity. J. Biol. Chem. 2020;295:444–457. doi: 10.1074/jbc.RA119.010511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasukawa K., Oshiumi H., Takeda M., Ishihara N., Yanagi Y., Seya T., Kawabata S.-i., Koshiba T. Mitofusin 2 inhibits mitochondrial antiviral signaling. Sci. Signal. 2009;2:ra47. doi: 10.1126/scisignal.2000287. [DOI] [PubMed] [Google Scholar]
- Yoshinaka T., Kosako H., Yoshizumi T., Furukawa R., Hirano Y., Kuge O., Tamada T., Koshiba T. Structural basis of mitochondrial scaffolds by prohibitin complexes: Insight into a role of the coiled-coil region. iScience. 2019;19:1065–1078. doi: 10.1016/j.isci.2019.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizumi T., Ichinohe T., Sasaki O., Otera H., Kawabata S.-i., Mihara K., Koshiba T. Influenza A virus protein PB1-F2 translocates into mitochondria via Tom40 channels and impairs innate immunity. Nat. Commun. 2014;5:4713. doi: 10.1038/ncomms5713. [DOI] [PubMed] [Google Scholar]
- Yoshizumi T., Imamura H., Taku T., Kuroki T., Kawaguchi A., Ishikawa K., Nakada K., Koshiba T. RLR-mediated antiviral innate immunity requires oxidative phosphorylation activity. Scientific Rep. 2017;7:5379. doi: 10.1038/s41598-017-05808-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y., Nabar N.R., Shi C.-S., Kamenyeva O., Xiao X., Hwang I.-Y., Wang M., Kehrl J.H. SARS-Coronavirus Open Reading Frame-3a drives multimodal necrotic cell death. Cell Death Dis. 2018;9:904. doi: 10.1038/s41419-018-0917-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zasłona Z., O’Neill L.A. Cytokine-like roles for metabolites in immunity. Mol. Cell. 2020;78:814–823. doi: 10.1016/j.molcel.2020.04.002. [DOI] [PubMed] [Google Scholar]
- Zhang W., Wang G., Xu Z.-G., Tu H., Hu F., Dai J., Chang Y., Chen Y., Lu Y., Zeng H. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell. 2019;178:176–189.e15. doi: 10.1016/j.cell.2019.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]