

Abstract
Objective
The aim of the study was to report the proportion of homozygous and compound heterozygous variants in the survival motor neuron 1 (SMN1) gene in a large population of patients with spinal muscular atrophy (SMA) and to correlate the severity of the disease with the presence of specific intragenic variants in SMN1 and with the SMN2 copy number.
Methods
Four hundred fifty Brazilian patients with SMA were included in a retrospective study, and clinical data were analyzed compared with genetic data; the SMN2 copy number was obtained by multiplex ligation-dependent probe amplification and pathogenic variants in SMN1 by next-generation sequencing.
Results
Four hundred two patients (89.3%) presented homozygous exon 7-SMN1 deletion, and 48 (10.7%) were compound heterozygous for the common deletion in one allele and a point mutation in the other allele. Recurrent variants in exons 3 and 6 (c.460C>T, c.770_780dup and c.734_735insC) accounted for almost 80% of compound heterozygous patients. Another recurrent pathogenic variant was c.5C>G at exon 1. Patients with c.770_780dup and c.734_735insC had a clinical phenotype correlated with SMN2 copy number, whereas the variants c.460C>T and c.5C>G determined a milder phenotype independently of the SMN2 copies.
Conclusions
Patients with specific pathogenic variants (c.460C>T and c.5C>G) presented a milder phenotype, and the SMN2 copy number did not correlate with disease severity in this group.
Spinal muscular atrophy (SMA) is a neurodegenerative disease of lower motor neurons, leading to progressive weakness associated with ventilatory insufficiency. The most common form of SMA is caused by mutations in the survival motor neuron 1 (SMN1) gene located at 5q13.1 Exon 7 of SMN1 is not detectable in approximately 96% of patients with SMA, and approximately 4% of patients have a combination of the deletion and an intragenic mutation in the second allele.1,2 The centromeric homologous SMN2 gene cannot compensate for the SMN1 defect because a single nucleotide polymorphism located in exon 7 causes exon skipping in about 90% of SMN2 transcripts leading to a nonfunctional SMN protein.3
The SMN2 copy number varies, and several studies have demonstrated a strong correlation between the number of SMN2 copies and SMA severity.4–6 However, other genetic and environmental factors also influence the clinical severity of the disease. One example is the variant c.859G>C in exon 7 of SMN2, which acts as a positive modifier that results in an approximate 20% increase in full-length SMN RNA.7,8 In addition, intragenic mutations in SMN1 may contribute more significantly to clinical severity than SMN2 copy numbers in some patients with SMA.9
The aim of this study was to determine the proportion of homozygous and compound heterozygous deletions in the SMN1 gene in a Brazilian population of patients with SMA and to correlate the severity of the disease with the presence of specific intragenic variants in SMN1 and with the SMN2 copy number.
Methods
A national collaborative historical cohort study was conducted to describe the clinical and molecular features of consecutive patients with SMA referred to 12 neuromuscular centers in Brazil, from January 2016 to July 2019. The eligibility criterion was the presence of SMN1 mutations in patients with the SMA phenotype. General and clinical data of the patients were obtained through the records including age at birth, age at onset, SMA subtype, ventilatory status, use of gastrostomy, and sitting, standing, and walking capacity.
All patients fulfilled the diagnostic criteria of proximal SMA defined by the International SMA Consortium and were classified into the 5 main clinical phenotypes of SMA, from 0 to 4.10,11 All patients had a genetic confirmation of SMN1 gene mutation in both alleles. DNA used in the analysis was extracted from leukocytes from peripheral blood or from oral swab. Copy number variation in the SMN1 and SMN2 genes was detected by the multiplex ligation-dependent probe amplification (SALSA MLPA kit P060-SMA, MRC Holland) method for both SMN1 and SMN2. Patients who failed to show homozygous absence of SMN1 exon 7 by MLPA were submitted to sequencing of SMN1/SMN2 genes by next-generation sequencing (NGS). The methodology used was exon capture with Agilent Mendelics Custom Panel V3 followed by sequencing with Illumina HiSeq.
Sequence variations were compared with data available in the Human Gene Mutation Database and ClinVar with reference to the GRCh37 version of the human genome. Variants were classified according to the 2015 American College of Medical Genetics and Genomics criteria. PolyPhen-2, Sorting Intolerant From Tolerant, Combined Annotation Dependent Depletion, Mendelian Clinically Applicable Pathogenicity score, MutationTaster, Human Splicing Finder v3.0, and ESEfinder v.3.0 were used for in silico analysis. Phylogenetic conservation was estimated with Genomic Evolutionary Rate Profiling, and allele frequencies were searched on the Genome Aggregation Database and the 1000 genomes browser.
Standard protocol approvals, registrations, and patient consents
The study was approved by the institutional ethics committees of the local centers. Informed consent was obtained for genetic studies from the patients or their legal representatives.
Data availability
Data corresponding to the total number of patients, distribution in SMA types, distribution of the variants in SMN1, and SMN2 copy number are available in figures 1–4. Individual details of heterozygous patients are fully shown in table 1. Any data not published within the article are available in a public repository. Methods and the statistical plan are also fully reported in the Methods section. Details regarding the statistical analysis by logistic regression are shown in table 2.
Figure 1. Prevalence of types of SMA.

Prevalence of types of SMA in the studied population (left chart) compared with the prevalence of SMA types in heterozygous patients (right chart). Note the significant increase in the proportion of patients with mild phenotypes (types 3 and 4) in the heterozygote group. SMA = spinal muscular atrophy.
Figure 3. Correlation between clinical phenotype of homozygous SMA patients X SMN2 copy number.

Correlation between clinical phenotype of SMA X SMN2 copy number in patients (%) with homozygous deletion of SMN1 detected by MLPA (n = 402). Data from SMA type 0 are not shown, as all 3 patients carried only 1 SMN2 copy. SMA = spinal muscular atrophy.
Figure 4. Correlation between clinical phenotype of compound heterozygous SMA patients X SMN2 copy number.
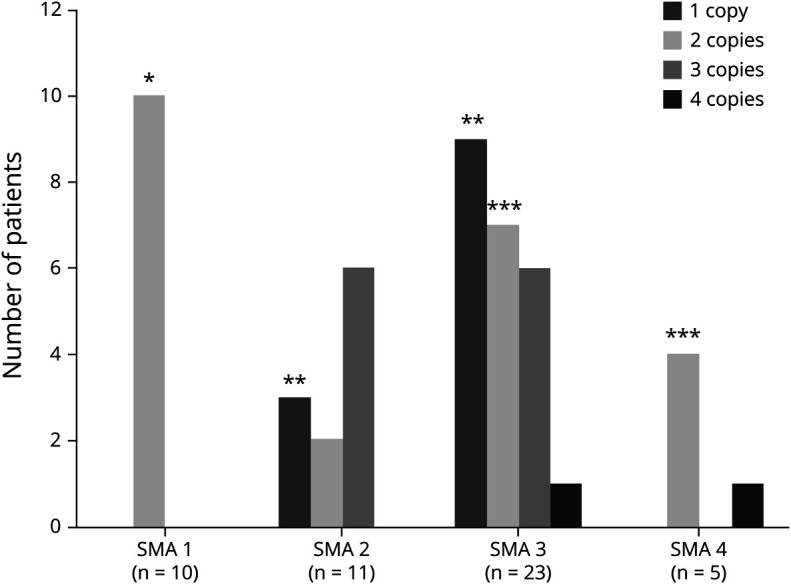
Correlation between clinical phenotype X SMN2 copy number in patients with specific intragenic variants (n = 48). *All patients with SMA type 1 presented with variants c. 770_780dup or c.734_745insC and carried 2 copies of SMN2. **Most patients with SMA types 2 and 3 carrying only 1 SMN2 copy presented the c.5>G variant. ***The variant c.460C>T was present in most patients with SMA types 3 or 4 carrying only 2 copies of SMN2. SMA = spinal muscular atrophy.
Table 1.
Genetic and clinical data of patients with SMA with compound heterozygous variants in SMN1


Table 2.
Multivariate analysis identifying factors associated with mild phenotype (SMA types 3 and 4) in heterozygous patients (n = 48)

Statistical analysis
For descriptive purposes, categorical variables were expressed as absolute and relative frequencies and then compared with Pearson χ2 in a univariate analysis. Continuous variables were expressed as the mean and SD and then compared with the Student t test. The mild phenotype was defined as SMA type 3 or 4. To identify factors associated with SMA phenotypes (sex, SMN2 copy number, and specific intragenic variants), Firth logistic regression analysis was performed for the heterozygous patients. The significant variables at the 0.10 level in the univariate analysis were included on the multivariate model. The results were expressed as odds ratios, coefficients, and 95% confidence intervals.
All tests were 2 tailed, and final p values less than 0.05 were considered to be statistically significant. All analyses were performed with Statistical Package for Social Sciences software, version 20 (SPSS, IBM Statistics, Chicago, IL).
Results
A total of 450 patients with SMA with genetic confirmation were identified. Among them, 105 (23.3%) were classified as type 1 (3 of them were SMA type 0/1a), 153 (34%) as type 2, 183 (40.7%) as type 3, and 9 (2%) as type 4 (figure 1). Of the patients, homozygous deletion of SMN1 exon 7 was detected in 402 (89.3%), and 48 patients (10.7%) had 1 nondeleted allele of SMN1 exon 7 with an intragenic variant (figure 2). No patient was homozygous for intragenic variants. The quantification of the SMN2 copy number was possible in all 450 patients tested by MLPA.
Figure 2. Characterization of SMN1 mutations in the Brazilian population.

Among 450 patients with SMA, 402 (89.3%) had a homozygous deletion in exon 7, whereas 48 (10.7%) retained 1 copy of the SMN1 gene with point mutations, most of them localized at exons 1, 3, and 6. SMA = spinal muscular atrophy; SMN1 = survival motor neuron 1; YG = YG domain.
Within the group of 48 compound heterozygous patients, 10 (20.8%) were type 1, 11 (22.9%) type 2, 22 (45.8%) type 3, and 5 (10.5%) type 4 (figure 1).
As expected, the SMN2 copy number presented an inverse correlation with the severity of phenotype in the homozygous group. A greater proportion of patients with SMA type 1 carried 2 copies of SMN2 (72.5%), most patients with SMA type 2 had 3 copies (65%), and patients with type 3 presented 3 or 4 copies of SMN2 (69.6% and 30.5%, respectively) (figure 3).
A total of 7 different pathogenic variants were identified in SMN1/SMN2 (figure 2 and table 1), 4 of which were recurrent variants. The most frequent variant was the c.460C>T (p.Gln154*). This variant was identified in 16 patients and has not been previously reported in the literature; its allelic frequency in large databases (Genome Aggregation Database and Exome Aggregation Consortium) is zero, indicating that it is a pathogenic variant. The variant c.770_780dup (p.Gly261Leufs*) was identified in 13 patients, the variant c.5C>G (p.Ala2Gly) in 9 patients and the variant c.734_735insC (p.Pro246Thrfs*) in 7 patients. General clinical data of the compound heterozygous patients are presented in table 1.
In the group of compound heterozygous patients, we did not find the variants c.859G>C and A-44G that are considered positive modifiers.7,8,12 In these patients, it was possible to state that the variant c.859G>C was not detected because in our database, there are controls with this variant identified using the same NGS methodology. Because of methodological limitation, intragenic variants A-549G and C-1897T were not analyzed. All variables that were classically associated with disease severity (sex and SMN2 copy number), along with pathogenic variants that showed statistical significance (p < 0.05) in correlation with the phenotype in the univariate analysis, were included in the multivariate logistic regression analysis model (table 2). Although all patients carrying the variant c.5C>G presented as SMA type 2 or 3, we did not find a statistical significance association with the mild phenotype in the univariate analysis (p = 0.71), so these patients were not included in the multivariate analysis. In heterozygous patients, no clear correlation existed between the clinical phenotype and the SMN2 copy number (p = 0.498). However, as occurs in homozygous patients, the male sex showed an association with a more severe phenotype (p = 0.041). Several heterozygous patients with type 2 or 3 SMA carried only 1 SMN2 copy, and most patients with types 3 and 4 SMA carried 2 copies (figure 4). In these patients, we could demonstrate by logistic regression that the most important factor in determining the phenotype was the pathogenic variant that the patients presented, with variant c. 460C>T related to the mild phenotype (p = 0.014), whereas variants c.770_780dup and c.734_735insC were related to more severe phenotypes depending on SMN2 copy number (tables 1 and 2). Although the variant c.5C>G was not entered in the multivariate analysis, based on the model criteria, all patients with this variant had 1 SMN2 copy, but none of them had type 1 SMA, which one may expect.
Discussion
We report on the largest cohort of patients with SMA in Latin America and bring new insights regarding the molecular diagnosis in SMA. The presence of compound heterozygous mutations occurred in approximately 10.7% of the patients with SMA, a frequency higher than that reported in the literature. This fact draws attention to our population, especially with the future advent of neonatal screening for the diagnosis and treatment of presymptomatic patients. Patients with milder phenotypes tend to have a compound heterozygous genotype more often than patients with type 1 SMA, and in our study, the frequency of type 1 SMA was lower than commonly reported in the literature10, most probably related to the higher mortality in this group of patients. More interesting is the finding that most of the patients with SMA type 4 (55.5%) were compound heterozygous, most of them carrying the same variant (p.Gln154*). These data reinforce the idea that diagnosis in SMA type 4 is more difficult even from the molecular point of view, and the average time from symptom onset to diagnosis in this group was 10 years, ranging from 2 to 18 years (data not shown in table).
Knowledge of the molecular portrait of the population is important to understand the clinical profile and natural history of patients and to assist in the therapeutic planning, considering that part of the currently available specific therapies modulate the SMN2 gene expression.13 The correlation between the copy number of the SMN2 gene and the phenotype followed a similar pattern to that reported in the literature,4–6 as patients with SMA type 1 usually carried 2 copies, whereas type 2 patients usually carried 3 copies and types 3 and 4 presented with 3 or 4 copies. As reported in the literature, such a correlation is not strict because to a lesser extent, patients with SMA type 1 had 3 copies and type 3 and 4 phenotypes had 2 copies.4,5 The lack of exact correlation must be related to other phenotypic modifiers, such as the presence of the variants c.859G>C in exon 7 and A-44G, A-549G, and C-1897T in intron 6 of SMN2 that act as positive modifier.7,8,12 Analysis of these variants in the SMN2 gene was not performed in our homozygous patients, and in the group of compound heterozygous patients, we did not find the variants c.859G>C and A-44G.
We identified 4 recurrent variants in our population: 3 nonsense and 1 missense variants. The most common variant identified was c.460C>T, located at exon 3, that creates a premature stop codon at residue 154 (p.Gln154*). Exon 3 is responsible for encoding the tudor domain of protein, which interacts with a complex of Sm proteins, kaolin, fibrillarin, and ribonucleoproteins.14 This information confirms that exon 3 is a hot spot for small mutations; in addition, it supports the importance of the tudor domain for SMN functioning.15,16 This variant was the most common variant in our population, suggesting a founder effect by a common ancestor. More detailed ancestral haplotype studies would be needed to confirm this hypothesis.
Among the 16 patients with this variant, 9 were still walking (table 1), and of those patients who lost the ability to walk, the mean age of gait loss was 19 years (11–36 years), compatible with an SMA type 3b phenotype.11 Of interest, all patients presented a milder phenotype (SMA types 3 and 4) even in the presence of 1 or 2 copies of SMN2. There is no clear explanation for the milder phenotype in this situation. Sossi et al.17 reported patients with mutations in exon 3 of SMN1 leading to premature termination codons and skipping of the entire exon generating in-frame transcripts and a better phenotype. However, many studies have shown severe missense mutations occurring in exon 3 that cause severe phenotypes and thus confirming that amino acids and the tudor domain located at exon 3 are essential to SMN function.15,16,18–20 A possibility to explain the milder phenotype associated with the c.460C>T is the presence of positive modifier variants in SMN2. The presence of the variants c.859G> C and A-44G was negative in these cases; however, we cannot exclude the possibility that other untested modifier variants are involved in the modulation of the clinical phenotype.
The second most common variant was c.770_780dup (p.Gly261Leufs*), located at exon 6, and has been previously reported to be associated with SMA.19,21,22 Parsons et al.21 showed through real-time PCR that the 11-bp duplication in exon 6 was associated with the SMN1 locus and not SMN2 and was sufficient to produce a severe type 1 SMA phenotype. In a Portuguese SMA population, this variant was the most frequent intragenic mutation in a total of 14 heterozygous unrelated patients, which leads to the hypothesis that this mutation migrated from the Portuguese population to Brazil22. In our study, as in an SMA Portuguese population study, all patients carrying this variant exhibited a strict correlation between the phenotype and the SMN2 copy number, with patients with 2 SMN2 copies presenting as SMA type 1, patients with 3 copies presenting as SMA type 2 or 3, and a patient with a very mild phenotype (SMA type 4) with the presence of 4 SMN2 copies (table 1).
The third most common variant was the missense c.5C>G (p.Ala2Gly), located at exon 1. This variant was first reported in 3 patients with SMA, all of whom had only 1 SMN2 copy, and 2 of whom had a mild phenotype (type 3 SMA).18 More recently, 3 additional patients with this variant, a mild phenotype and only 1 SMN2 copy were also reported.23 Another mutation in the same location, the p.Ala2Val variant, has already been described, and all patients had only 1 SMN2 copy with a mild phenotype (type 3 SMA).9 Thus, in the presence of this variant, the SMN2 copy number did not correlate with the phenotype. The mutation effect of p.Ala2Val, as well as p.Ala2Gly, appears to be much less deleterious.
A deleterious effect of the c.5C>G variant has been demonstrated in an SMA mouse model, although the mutation did not produce a total loss of protein function.24 The authors reported that in the absence of the SMN2 gene, the transgenic SMN c.5C>G mutant is unable to rescue the embryonic lethality, but in its presence, the c.5C>G transgene delays the onset of motor neuron loss, resulting in mice with mild SMA. They concluded that only in the presence of low levels of full-length SMN is the c.5C>G transgene able to form partially functional higher-order SMN complexes essential for its functions, demonstrating the importance of SMN levels in SMA, even if the protein is expressed from a mutant allele.24 Similarly, in Brazilian patients carrying this variant, none presented a severe phenotype of the disease, such as type 0 or 1, even in the presence of only 1 SMN2 copy. Patients presented with type 2 or 3 SMA with a typical age at disease onset (table 1). Of interest, 3 patients with this variant were in need of continuous invasive ventilation before age 10 years, which would not be expected due to the disease duration presented by patients. This may represent a phenotypically characteristic effect of the c.5C>G variant on this population, although deficient respiratory care in our population cannot be ruled out.
The last recurrent variant identified in our population was c.734_735insC (p.Pro246Thrfs*). This variant was not previously reported in the literature as a recurrent variant in patients with SMA. As occurs with the c.770_780dup variant, this mutation is located at exon 6, and all patients carrying this variant also exhibited a strict correlation between the phenotype and the SMN2 copy number. These data lead us to suppose that this mutation severely compromises protein function at the YG domain of interaction with protein Gemin3.25 Accordingly, the clinical phenotype of those patients appears to be dependent exclusively on the SMN2 copy number.
Our study has some limitations as it was a retrospective study, and there was a bias of survival because the most severe cases (SMA type 1) had a low prevalence in our cohort. Another limitation is that NGS cannot distinguish whether pathogenic variants are located in SMN1 or SMN2, although it is a reliable test that can be used in real life. The large number of patients with intragenic small variants identified in our population could be explained by the current access to the large-scale sequencing of SMN genes, especially for patients with clinical features of SMA but negative in the MLPA assays.
We report a large cohort with important findings, such as a higher frequency of compound heterozygous patients with SMA in the Brazilian population than reported in the literature. Most of these patients have a milder phenotype, and the SMN2 copy number did not correlate well with the severity of disease, as observed in patients with homozygous deletion in SMN1 exon 7. The result of our study indicates a specific point mutation spectrum in the Brazilian population and may help in the search for an early diagnosis in the era of therapies, especially in newborn screening. Variants in exons 3 and 6 (c.460C>T, c.770_780dup, c.734_735insC, c806T>C, and c.346A>T) accounted for almost 80% of our patients. In patients with exon 7 deletion in 1 SMN1 allele, analysis of exons 3 and 6 should be prompt, as it seems to be a specific hot spot for small mutations and should be sequenced first. If the patient presents with mild phenotype and only 1 copy of SMN2, a search for the c.5C>G variant at exon 1 should be performed.
Glossary
- MLPA
multiplex ligation-dependent probe amplification
- NGS
next-generation sequencing
- SMA
spinal muscular atrophy
- SMN1
survival motor neuron 1
Appendix. Authors



Study funding
No targeted funding reported.
Disclosure
E. Zanoteli and A.P.Q.C. Araújo have received grant support to conduct clinical trials on SMA from Hoffmann-La Roche and serve as consultants to AveXis, Biogen, and Hoffmann-La Roche. R.H. Mendonça, C. Matsui Jr, G.J. Polido, A.M.S. Silva, L. Kulikowski, A.T. Dias, E.A. Zanardo, D.J.F. Solla, J. Gurgel-Giannetti, A.C.M.L. Moura, G.P.C. Sampaio, A.S.B. Oliveira, P.V.S. Souza, W.B.V.R. Pinto, E.A. Gonçalves, I.B. Farias, F. Nardes, W. Marques Jr., P.J. Tomaselli, M.D.O. Ribeiro, J.P. Kitajima, F.P. Monteiro, J.A.M. Saute, M.M. Becker, M.L. Saraiva-Pereira, A.C. Brusius-Facchin, V. van der Linden, R.N. Florêncio, A.V.S. Barbosa, M.C. Machado-Costa, A.L.S. Pessoa, L.S. Souza, M.C. Franca Jr., F. Kok, and U.C. Reed report no disclosures relevant to this manuscript. Go to Neurology.org/NG for full disclosures.
References
- 1.Lefebvre S, Burglen L, Reboullet S, et al. . Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995;80:155–165. [DOI] [PubMed] [Google Scholar]
- 2.Burghes AH. When is a deletion not a deletion? When it is converted. Am J Hum Genet 1997;61:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cartegni L, Krainer AR. Disruption of an SF2/ASF-dependent exonic splicing enhancer in SMN2 causes spinal muscular atrophy in the absence of SMN1. Nat Genet 2002;30:377–384. [DOI] [PubMed] [Google Scholar]
- 4.McAndrew PE, Parsons DW, Simard LR, et al. . Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet 1997;60:1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wirth B, Herz M, Wetter A, et al. . Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype-phenotype correlation, and implications for genetic counseling. Am J Hum Genet 1999;64:1340–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Prior TW, Swoboda KJ, Scott HD, Hejmanowski AQ. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am J Med Genet A 2004;130A:307–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vezain M, Saugier-Veber P, Goina E, et al. . A rare SMN2 variant in a previously unrecognized composite splicing regulatory element induces exon 7 inclusion and reduces the clinical severity of spinal muscular atrophy. Hum Mutat 2010;31:E1110–E1125. [DOI] [PubMed] [Google Scholar]
- 8.Prior TW, Krainer AR, Hua Y, et al. . A positive modifier of spinal muscular atrophy in the SMN2 gene. Am J Hum Genet 2009;85:408–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamamoto T, Sato H, Lai PS, et al. . Intragenic mutations in SMN1 may contribute more significantly to clinical severity than SMN2 copy numbers in some spinal muscular atrophy (SMA) patients. Brain Dev 2014;36:914–920. [DOI] [PubMed] [Google Scholar]
- 10.Munsat TL, Davies KE. International SMA consortium meeting. (26-28 June 1992, Bonn, Germany). Neuromuscul Disord 1992;2:423–428. [DOI] [PubMed] [Google Scholar]
- 11.Finkel R, Bertini E, Muntoni F, et al. . 209th ENMC International Workshop: outcome measures and clinical trial readiness in spinal muscular atrophy 7-9 November 2014, Heemskerk, The Netherlands. Neuromuscul Disord 2015;25:593–602. [DOI] [PubMed] [Google Scholar]
- 12.Ruhno C, McGovern VL, Avenarius MR, et al. . Complete sequencing of the SMN2 gene in SMA patients detects SMN gene deletion junctions and variants in SMN2 that modify the SMA phenotype. Hum Genet 2019;138:241–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reed UC, Zanoteli E. Therapeutic advances in 5q-linked spinal muscular atrophy. Arq Neuropsiquiatr 2018;76:265–272. [DOI] [PubMed] [Google Scholar]
- 14.Pellizoni L, Kataoka N, Charroux B, Dreyfuss G. A novel function for SMN, the spinal muscular atrophy disease gene product, in pre-mRNA splicing. Cell 1998;95:615–624. [DOI] [PubMed] [Google Scholar]
- 15.Cusco I, Barcelo MJ, del Rio E, et al. . Detection of novel mutations in the SMN tudor domain in type I SMA patients. Neurology 2004;63:146–149. [DOI] [PubMed] [Google Scholar]
- 16.Jędrzejowska M, Gos M, Zimowski JG, Kostera-Pruszczyk A, Ryniewicz B, Hausmanowa-Petrusewicz I. Novel point mutations in survival motor neuron 1 gene expand the spectrum of phenotypes observed in spinal muscular atrophy patients. Neuromuscul Disord 2014;24:617–623. [DOI] [PubMed] [Google Scholar]
- 17.Sossi V, Giuli A, Vitali T, et al. . Premature termination mutations in exon 3 of the SMN1 gene are associated with exon skipping and a relatively mild SMA phenotype. Eur J Hum Genet 2001;9:113–120. [DOI] [PubMed] [Google Scholar]
- 18.Parsons DW, McAndrew PE, Iannaccone ST, Mendell JR, Burghes AH, Prior TW. Intragenic telSMN mutations: frequency, distribution, evidence of a founder effect, and modification of the spinal muscular atrophy phenotype by cenSMN copy number. Am J Hum Genet 1998;63:1712–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alías L, Bernal S, Fuentes-Prior P, et al. . Mutation update of spinal muscular atrophy in Spain: molecular characterization of 745 unrelated patients and identification of four novel mutations in the SMN1 gene. Hum Genet 2009;125:29–39. [DOI] [PubMed] [Google Scholar]
- 20.Bussaglia E, Clermont O, Tizzano E, et al. . A frame-shift deletion in the survival motor neuron gene in Spanish spinal muscular atrophy patients. Nat Genet 1995;11:335–337. [DOI] [PubMed] [Google Scholar]
- 21.Parsons DW, McAndrew PE, Monani UR, et al. . An 11 base pair duplication in exon 6 of the SMN gene produces a type I spinal muscular atrophy (SMA) phenotype: further evidence for SMN as the primary SMA-determining gene. Hum Mol Genet 1996;5:1727–1732. [DOI] [PubMed] [Google Scholar]
- 22.Gonçalves-Rocha M, Oliveira J, Rodrigues L, Santos R. New approaches in molecular diagnosis and population carrier screening for spinal muscular atrophy. Genet Test Mol Biomarkers 2011;15:319–326. [DOI] [PubMed] [Google Scholar]
- 23.Kubo Y, Nishio H, Saito K. A new method for SMN1 and hybrid SMN gene analysis in spinal muscular atrophy using long-range PCR followed by sequencing. J Hum Genet 2015;60:233–239. [DOI] [PubMed] [Google Scholar]
- 24.Monani UR, Pastore MT, Gavrilina TO, et al. . A transgene carrying an A2G missense mutation in the SMN gene modulates phenotypic severity in mice with severe (type I) spinal muscular atrophy. J Cell Biol 2003;160:41–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.BuÈhler D, Raker V, LuÈhrmann R, Fischer U. Essential role for the entire domain of SMN in spliceosomal U snRNP assembly: implications for spinal muscular atrophy. Hum Mol Genet 1999;8:2351–2357. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data corresponding to the total number of patients, distribution in SMA types, distribution of the variants in SMN1, and SMN2 copy number are available in figures 1–4. Individual details of heterozygous patients are fully shown in table 1. Any data not published within the article are available in a public repository. Methods and the statistical plan are also fully reported in the Methods section. Details regarding the statistical analysis by logistic regression are shown in table 2.
Figure 1. Prevalence of types of SMA.
Prevalence of types of SMA in the studied population (left chart) compared with the prevalence of SMA types in heterozygous patients (right chart). Note the significant increase in the proportion of patients with mild phenotypes (types 3 and 4) in the heterozygote group. SMA = spinal muscular atrophy.
Figure 3. Correlation between clinical phenotype of homozygous SMA patients X SMN2 copy number.
Correlation between clinical phenotype of SMA X SMN2 copy number in patients (%) with homozygous deletion of SMN1 detected by MLPA (n = 402). Data from SMA type 0 are not shown, as all 3 patients carried only 1 SMN2 copy. SMA = spinal muscular atrophy.
Figure 4. Correlation between clinical phenotype of compound heterozygous SMA patients X SMN2 copy number.
Correlation between clinical phenotype X SMN2 copy number in patients with specific intragenic variants (n = 48). *All patients with SMA type 1 presented with variants c. 770_780dup or c.734_745insC and carried 2 copies of SMN2. **Most patients with SMA types 2 and 3 carrying only 1 SMN2 copy presented the c.5>G variant. ***The variant c.460C>T was present in most patients with SMA types 3 or 4 carrying only 2 copies of SMN2. SMA = spinal muscular atrophy.
Table 1.
Genetic and clinical data of patients with SMA with compound heterozygous variants in SMN1
Table 2.
Multivariate analysis identifying factors associated with mild phenotype (SMA types 3 and 4) in heterozygous patients (n = 48)