

Highlights
-
•
HIF-1α and HIF-2α promote cellular adaptation to acute hypoxia, but during prolonged activation, these isoforms exert mutually antagonistic effects on the redox state and on proinflammatory pathways.
-
•
Imbalances in HIF-1α and HIF-2α may contribute to the evolution and progression of chronic cardiac, vascular, and renal disorders.
-
•
Selective activation of HIF-2α can be achieved with drugs that inhibit isoform-selective PHDs or that promote the redox sensor, SIRT-1 (e.g., SGLT2 inhibitors).
Key Words: cobalt chloride, hypoxia-inducible factor-1α, hypoxia-inducible factor-2α, roxadustat, sodium-glucose cotransporter 2 inhibitors
Abbreviations and Acronyms: HIF, hypoxia inducible factor; PHD, prolyl hydroxylase; SGLT2, sodium-glucose cotransporter 2; SIRT1, sirtuin-1
Summary
Hypoxia-inducible factor (HIF)-1α and HIF-2α promote cellular adaptation to acute hypoxia, but during prolonged activation, these isoforms exert mutually antagonistic effects on the redox state and on proinflammatory pathways. Sustained HIF-1α signaling can increase oxidative stress, inflammation, and fibrosis, actions that are opposed by HIF-2α. Imbalances in the interplay between HIF-1α and HIF-2α may contribute to the progression of chronic heart failure, atherosclerotic and hypertensive vascular disorders, and chronic kidney disease. These disorders are characterized by activation of HIF-1α and suppression of HIF-2α, which are potentially related to mitochondrial and peroxisomal dysfunction and suppression of the redox sensor, sirtuin-1. Hypoxia mimetics can potentiate HIF-1α and/or HIF-2α; ideally, such agents should act preferentially to promote HIF-2α while exerting little effect on or acting to suppress HIF-1α. Selective activation of HIF-2α can be achieved with drugs that: 1) inhibit isoform-selective prolyl hydroxylases (e.g., cobalt chloride and roxadustat); or 2) promote the actions of the redox sensor, sirtuin-1 (e.g., sodium-glucose cotransporter 2 inhibitors). Selective HIF-2α signaling through sirtuin-1 activation may explain the effect of sodium-glucose cotransporter 2 inhibitors to simultaneously promote erythrocytosis and ameliorate the development of cardiomyopathy and nephropathy.
Central Illustration

Hypoxia-inducible factors (HIFs) enhance adaptation to oxygen-related stress by promoting oxygen delivery and reducing oxygen consumption. These transcription factors are heterodimers that consist of a constitutively expressed HIF-1β subunit and an inducible oxygen-sensitive subunit (HIF-1α or HIF-2α). HIF-1α and HIF-2α have a 48% amino acid sequence identity and possess similar protein structures and functional domains, but they have distinct mechanisms of regulation and spatial expression patterns and differ meaningfully in their cellular actions (1,2).
Despite an identical DNA consensus recognition sequence, DNA binding does not correspond to increased transcriptional activity, and each isoform loads at a distinct repertoire of cell type−specific sites across the genome (3). HIF-1α is a universal master regulator for hypoxia-inducible gene expression and is expressed in a wide range of cell types (2,4). In contrast, HIF-2α is expressed selectively, that is, primarily in alveolar epithelial cells in the lung, specialized peritubular interstitial cells in the kidney, in hepatic parenchymal cells, and in endothelial cells in the heart (4, 5, 6, 7). Furthermore, the expression of HIF-1α is highly sensitive to environmental oxygen, and inactivation of HIF-1α completely abolishes induction of HIF target genes (2). In contrast, the expression of HIF-2α is less influenced by acute changes in oxygen tension (4−6,8) but its activity is upregulated by cellular stress and hypoxia mimetics. Although both HIF-1α and HIF-2α can modulate the transcription of the same genes, the 2 isoforms often do so in an opposing manner (9,10). Thus, the ability of HIF-1α and HIF-2α to activate specific target genes is highly context-dependent, particularly with respect to the inciting stimulus and cell type (2).
The activities of HIF-1α and HIF-2α are upregulated both by hypoxia and by drugs that mimic hypoxia under normoxic conditions (e.g., cobalt chloride) (4,6,11). Oxygen influences the activity of these isoforms by directly modulating the activation of a family of 3 prolyl hydroxylases (PHD1, PHD2, and PHD3) that function to degrade the 2 transcription factors; the PHDs are sensitive to environmental oxygen, and thus, act as oxygen sensors (12, 13, 14). Inhibition of these PDHs by hypoxia leads to stabilization of one or both HIFs, which leads to enhanced HIF signaling. However, PHD1, PHD2, and PHD3 differ meaningfully with respect to their expression profiles, subcellular localization, and their effects on HIF-1α or HIF-2α (15,16).
Therefore, despite the structural homology of HIF-1α and HIF-2α, differences in cellular and subcellular expression patterns, upstream regulators, and sensitivity to degradative enzymes explain why HIF-1α and HIF-2α exert highly distinctive effects under a range of specific conditions. In general, HIF-1α promotes the transcription of proteins that decrease oxygen use and increase angiogenesis, whereas HIF-2α is the primary stimulus for erythropoietin synthesis (17,18). In addition, under conditions of prolonged cellular stress, HIF-1α and HIF-2α exert diametrically opposing actions on critical pathways that determine the balance between health and disease, especially in the development of chronic cardiac, vascular, and renal disorders (9,10). As a result, switching from the expression of one isoform to another appears to play a role in the pathogenesis of many diseases and may mediate the effects of treatment (19).
Oxygen-Related Activation of HIF-1α and HIF-2α and the Interplay of These Isoforms to Modulating the Cellular Redox State and the Inflammatory Set Point
During commonly encountered physiological states, a reduction in environmental oxygen enhances signaling through both HIF-1α and HIF-2α. However, under the pathological conditions that prevail in many chronic cardiac, renal, and vascular disorders, HIF signaling is also stimulated by abnormalities in mitochondria and peroxisomes, the most important oxygen-consuming organelles in cells. Derangements in these cellular constituents redirects the use of oxygen toward the generation of reactive oxygen species, which (like hypoxia) has a direct effect to inhibit PDHs (20). The activation of HIFs by oxidative stress is dependent on the presence of mitochondria that are capable of consuming oxygen and generating reactive oxygen species (20,21). Therefore, the organellar dysfunction that characterizes many chronic diseases appears to play a critical role in enhancing HIF signaling.
Once activated by oxygen-related organellar stresses, HIF-1α and HIF-2α act to mute these stresses by reducing the amount of oxygen consumed by mitochondria and peroxisomes, both directly and indirectly. HIF-1α directly inhibits both the biogenesis and oxidative functions of mitochondria (22). In addition, both HIF-1α and HIF-2α promote autophagy, a lysosome-dependent degradative pathway that mediates the clearance of dysfunctional organelles. HIF-1α enhances the autophagic clearance of damaged mitochondria (mitophagy) (23), whereas HIF-2α stimulates the autophagic disposal of injured peroxisomes (pexography) (24). Therefore, once activated by oxygen-related stress, HIF-1α and HIF-2α signaling can act to reduce this stress, particularly that caused by the organellar dysfunction characteristic of chronic heart and renal disease.
Mutual antagonism between HIF-1α and HIF-2α in the regulation of redox state and in the modulation of proinflammatory and profibrotic pathways
Although the actions of HIF-1α and HIF-2α can be concordant, these isoforms exert mutually antagonistic effects on many aspects of cellular homeostasis (Figure 1). In particular, the interplay of HIF-1α and HIF-2α helps to determine the set point for the redox state of cells. HIF-2α hypomorphic mice showed increased HIF-1α and an oxidized intracellular redox state, which was accompanied by exaggerated hypoxic sensitivity and was reversed by a HIF-1α inhibitor or a superoxide scavenger (25). Conversely, HIF-1α hypomorphic mice demonstrated increased levels of HIF-2α and a reduced intracellular redox state, which was accompanied by blunted oxygen sensing and was corrected by a HIF-2α inhibitor (25). HIF-2α may reduce oxidative stress not only by promoting pexophagy but also by transactivating genes that encode for antioxidant enzymes (26). Disorders characterized regional and transient derangements in oxygen delivery can cause striking imbalances between HIF-1α and HIF-2α signaling; for example, chronic intermittent hypoxia activates HIF-1α (27) but downregulates HIF-2α (28).
Figure 1.
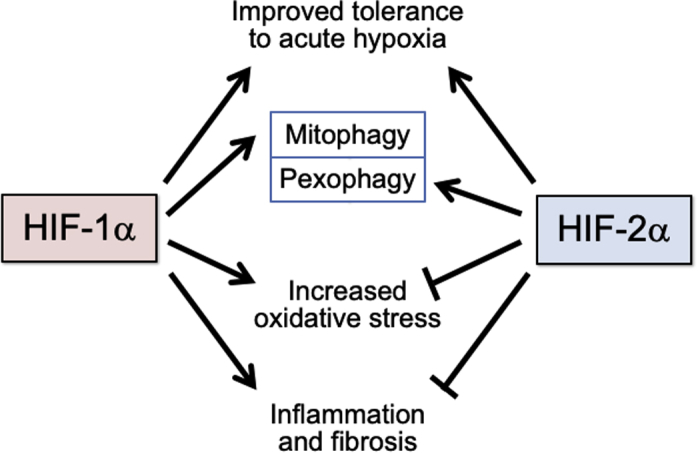
Mutually Reinforcing and Antagonistic Effects of the Actions of HIF Isoforms on the Determinants of Cellular Stress
The effects of hypoxia-inducible factor (HIF)-1α signaling to enhance adaption to acute hypoxia are immediate and short-lived, whereas the effects of HIF-1α signaling to promote oxidative stress, inflammation, and fibrosis are seen with sustained activation.
HIF-1α and HIF-2α also have mutual antagonistic effects on inflammation and fibrosis in diverse organs, including the heart, kidney, vasculature, and adipose tissue (29, 30, 31). HIF-1α promotes the activation of inducible nitric oxide synthase (a proinflammatory mediator) and enhances M1 macrophage polarization (a proinflammatory phenotype); both effects are opposed by the actions of HIF-2α (31). HIF-1α promotes (whereas HIF-2α inhibits) the actions of proinflammatory cytokines (32,33). The genes that encode for profibrotic chemokines and collagen deposition are activated by HIF-1α (34), whereas HIF-2α promotes collagen matrix degradation (35). Therefore, the balance between HIF-1α and HIF-2α may determine the set point for inflammation and fibrosis.
What influences the point of equilibrium between HIF-1α and HIF-2α? An important modulator of both isoforms is sirtuin-1 (SIRT1) (19), a nicotinamide adenine dinucleotide responsive deacetylase that serves as both as a redox rheostat and a nutrient and/or oxygen sensor. SIRT1 activation leads to downregulation of HIF-1α (36) but upregulation of HIF-2α (37); the latter effect is likely mediated by SIRT1-mediated deacetylation of HIF-2α. In many chronic disorders (e.g., obesity, diabetes, chronic heart failure, and chronic kidney disease), the activity of SIRT1 is suppressed (38,39), thus shifting the balance toward upregulation of HIF-1α and downregulation of HIF-2α. Conversely, activation of SIRT1 activation favors HIF-2α over HIF-1α, and SIRT1 has direct effects to preserve the integrity of mitochondria and peroxisomes similar to the actions of HIF-2α (40). For these reasons, SIRT1 is poised to act as a HIF-1α to HIF-2α switch (19).
Modulation of HIF-1α/HIF-2α Signaling in the Ischemic and Failing Heart
Following an abrupt decline in oxygen tension, activation of HIF-1α and HIF-2α promotes the adaptation of the myocardium to hypoxia; HIF upregulation ameliorates ischemia−reperfusion injury, whereas HIF downregulation exacerbates hypoxic dysfunction (41). Suppression of HIF-1α causes inadequate vascularization, and thus, decompensation of acute pressure overload or ischemic states (42).
However, enhanced HIF-1α signaling may not be beneficial in chronic heart failure, which is characterized by oxidative stress and SIRT1 suppression (Figure 2) (38). The action of HIF-1α to shift glucose metabolism from oxidative to glycolytic pathways limits cardiac performance because glycolysis is an inefficient mechanism of generating adenosine triphosphate; this shift also promotes lipid accumulation in cardiomyocytes, which leads to contractile dysfunction (43). Moreover, HIF-1α impairs mitochondrial biogenesis, further limiting the generation of adenosine triphosphate (22). HIF-1α can also promote hypertrophy and inflammation in cardiomyocytes (44, 45, 46) and may activate the sympathetic nervous system in chronic heart failure (47). For all of these reasons, marked, sustained, and isolated activation of HIF-1α acts to impair cardiac performance. Stabilization of HIF-1α at high levels leads to worsening myocardial function (48), and genetic overexpression of HIF-1α results in cardiomyopathy (49). Conversely, inhibition of HIF-1α slows the transition from cardiac hypertrophy to dilated cardiomyopathy (50). Therefore, it is interesting that expression of HIF-1α is increased in the failing heart, both experimentally and clinically (48,51). High circulating levels of HIF-1α are associated with a poor prognosis in patients with chronic heart failure (52), and drugs that are effective in treating heart failure can diminish the activation of HIF-1α (53).
Figure 2.
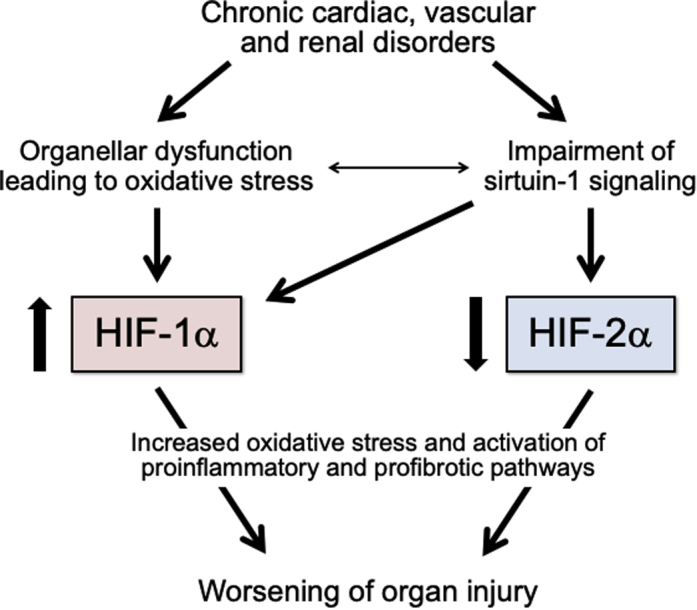
Upregulation of HIF-1α and Downregulation of HIF-2α in Chronic Cardiac, Vascular, and Renal Disorders
The effects of HIF-1α signaling to promote oxidative stress, inflammation, and fibrosis are seen with sustained activation. Abbreviation as in Figure 1.
In contradistinction to HIF-1α, activation of HIF-2α exerts protective effects in cardiomyocytes (54), and decreased HIF-2α expression is accompanied by inflammasome activation in the heart (55). Upregulation of HIF-2α occurs in myocardial regions most susceptible to ventricular remodeling, perhaps as an adaptive response (56). Despite these observations, little is known about the role of HIF-2α in chronic heart failure.
Modulation of HIF-1α/HIF-2α Signaling in Vascular Disorders
Hypertension and atherosclerosis involve increased sheer stress as well as hypoxia and inflammation in the arterial wall. Increased intravascular pressures and the medial infiltration by inflammatory cells promotes the activation of HIF-1α (Figure 2) (57). Upregulation of HIF-1α in macrophages, vascular smooth muscle, and endothelial cells has been implicated in the pathogenesis of neointimal proliferation, medial hypertrophy, and activation of proinflammatory pathways, and thus, the progression of atherosclerosis (58, 59, 60). Genetic silencing of HIF-1α in macrophages and vascular smooth muscle cells attenuates vascular inflammation, mitigating the deleterious actions of oxidized lipoproteins and the formation of foam cells, as well as slowing development of atherosclerotic plaque (58,61). HIF-1α hypomorphic mice showed decreased production of proinflammatory cytokines and a shift in the polarization of macrophages from the M1 to the M2 phenotype, leading to amelioration of vascular remodeling (62). The proinflammatory and mitogenic effects of inducible nitric oxide synthase may be mediated by HIF-1α (63). Vascular wall hypoxia can promote arterial thrombus formation through activation of HIF-1α (64).
In contrast, upregulation of HIF-2α suppresses the development of neointimal proliferation and atherosclerosis (65); however, oxidized lipoproteins abolish the hypoxic induction of HIF-2α (66), raising the possibility that atherosclerosis represents a HIF-2α−deficient state. Activation of HIF-2α in the endothelium and media of coronary and renal arteries reduces the magnitude of tissue injury following experimental ischemia (67,68).
Modulation of HIF-1α /HIF-2α signaling in the diabetic and nondiabetic kidney
During acute renal ischemia or hypoxia, the activation of HIF-1α mitigates renal injury, potentially by promoting angiogenesis and tissue repair (69,70). Silencing of HIF-1α during low-oxygen tension states exacerbates renal tubular epithelial cell necrosis, and potentiation of HIF-1α prevents this injurious effect (71). Upregulation of endothelial HIF-2α can exert a similar reno-protective action during acute ischemia (68).
However, prolonged HIF-1α activation exerts deleterious effects on the kidney, even if the causal mechanism is hypoxia or ischemia (Figure 2) (72). Sustained upregulation of HIF-1α promotes epithelial to mesenchymal transition in renal tubular epithelial cells (73). In chronic kidney disease, sustained signaling through HIF-1α promotes proinflammatory and profibrotic pathways in the glomerulus and renal tubules (72,74,75), which is characterized by activation of inflammation-related cytokines, profibrotic gene transcription, macrophage infiltration and collagen deposition, mesangial cell proliferation, and tubulo-interstitial inflammation (74, 75, 76, 77). Experimental suppression of HIF-1α attenuates mesangial matrix expansion and glomerulosclerosis and alleviates tubulointerstitial fibrosis (75,76,78).
Although HIF-1α promotes inflammation, activation of HIF-2α mutes inflammation and reduces injury in renal tissues (68,79). Chronic kidney disease is characterized by upregulation of HIF-1α but down-regulation of HIF-2α (30,79,80), and the deficiency in HIF-2α likely explains why chronic kidney disease is accompanied by blunted production of erythropoietin and anemia (81), and why impaired erythropoietin synthesis is accompanied by increased inflammatory and angiogenic markers (82). The interplay between HIF-1α and HIF-2α may be particularly important in diabetes (78), because hyperglycemia and advanced glycation end products directly promote the transcription of HIF-1α in glomerular and renal tubular cells (83,84). In addition, SIRT1 signaling (along with its ability to inhibit HIF-1α and activate HIF-2α) is impaired in the diabetic kidney (38,39).
Effect of Hypoxia Mimetics (PHD Inhibitors and SIRT1 Activators) on Chronic Cardiac, Renal, and Vascular Injury
Hypoxia mimetics enhance the activity of HIF-1α and/or HIF-2α, most commonly by inhibiting 1 or some of the 3 PHDs that are responsible for degradation of the isoforms. PHDs differ with respect to their expression profiles, subcellular localization, and effects on HIF-1α or HIF-2α. Inhibition of PHD2 acts primarily to boost the activity of HIF-1α (85), whereas PHD3 acts preferentially on HIF-2α (16,86,87). Hypoxia mimetics that increase the production of erythropoietin and that can treat anemia (e.g., cobalt chloride and roxadustat) inhibit PHD3 (3,85,88), although they may also suppress other PHDs (depending on dose). Drugs that selectively inhibit PHD1, PHD2, or PHD3 have been developed (Central Illustration), but studies of PHD inhibition have not often assessed the degree of selectivity. For example, most reports of the effects of cobalt chloride in experimental organ injury have been difficult to interpret, because they often failed to measure the effect of the drug on both HIF isoforms.
Central Illustration.

Mechanisms of Selective HIF-2α Activation by Drugs That Enhance Sirtuin-1 Signaling or Exert Preferential Inhibitory Effects on Certain Prolyl Hydroxlyases
The 2 HIF isoforms exert opposing effects on cellular stress and proinflammatory pathways, and thus, on the development of chronic cardiac, vascular and renal diseases. HIF = hypoxia-inducible factor isoform; SGLT2= sodium-glucose cotransporter 2.
Selective changes in the relative activities of HIF-1α and HIF-2α can also be achieved by modulating the activity of SIRT(1,19) because SIRT1 upregulation enhances signaling through HIF-2α, but suppresses the activity of HIF-1α (Central Illustration) (36,37). Resveratrol, a SIRT1 activator, inhibits HIF-1α in diverse tissues (89,90). Suppression of HIF-1α in the kidney by resveratrol diminishes inflammation and fibrosis in glomerular mesangial cells and ameliorates tubulointerstitial injury (90,91). Furthermore, the action of resveratrol to downregulate HIF-1α in cardiomyocytes reduces hypoxic injury (92). Resveratrol also increases erythropoietin (93), which is presumably related to an action on HIF-2α, although the effect of resveratrol on HIF-2α signaling has been directly studied.
Similarly, sodium-glucose cotransporter 2 (SGLT2) inhibitors that are used to treat type 2 diabetes have important cardioprotective and reno-protective effects, both experimentally and clinically (38,39). These drugs appear to activate SIRT1 and its downstream effectors by virtue of their ability to induce a fasting-like transcriptional paradigm (94, 95, 96, 97); the effects of SGLT2 inhibitors on SIRT1 signaling have been proposed to mediate their favorable effects on the heart and kidneys (38,39). SIRT1 upregulation may explain the effect of SGLT2 inhibitors to suppress HIF-1α in the kidney (98,99), as well as their effect to enhance the production of erythropoietin and promote erythrocytosis (100) (which is potentially related to SIRT1-mediated activation of HIF-2α37). The importance of this latter effect is reinforced by the results of statistical mediation analyses, which showed that changes in hemoglobin produced by SGLT2 inhibitors are a powerful predictor of their ability to reduce the risk of cardiovascular death and hospitalizations for heart failure (101,102).
Conclusions
HIF-1α and HIF-2α exert mutually antagonistic effects on the redox state and on proinflammatory pathways. Although the short-term actions of HIF-1α can reduce hypoxia-related injury, prolonged signaling through HIF-1α leads to increases in oxidative stress, inflammation, and fibrosis, but these deleterious effects are opposed by the actions of HIF-2α. The interplay between HIF-1α and HIF-2α may contribute to the evolution and progression of chronic heart failure, atherosclerotic and hypertensive vascular disorders, and chronic kidney disease. These disorders are characterized by activation of HIF-1α and suppression of HIF-2α; the latter effect explains why many chronic inflammatory diseases decrease the production of erythropoietin and cause anemia. Hypoxia mimetics are capable of potentiating both HIF-1α and HIF-2α; ideally, such agents should act preferentially to promote HIF-2α while exerting little effect on or acting to suppress HIF-1α. Selective upregulation of HIF-2α can be achieved with drugs that inhibit isoform-selective PHDs or that promote the actions of SIRT1 (i.e., SGLT2 inhibitors). Augmentation of SIRT1 and HIF-2α signaling may explain the link between the proerythrocytic action of SGLT2 inhibitors and their cardioprotective effects.
Footnotes
Dr. Packer has consulted for Abbvie, Actavis, Akcea, Amarin, Amgen, AstraZeneca, Boehringer Ingelheim, Cardiorentis, Daiichi Sankyo, Johnson & Johnson, Novartis, NovoNordisk, Pfizer, Relypsa, Sanofi, Synthetic Biologics, and Theravance.
The author attests they are in compliance with human studies committees and animal welfare regulations of the authors’ institutions and Food and Drug Administration guidelines, including patient consent where appropriate. For more information, visit the JACC: Basic to Translational Scienceauthor instructions page.
References
- 1.Semenza G.L. Hypoxia-inducible factors in physiology and medicine. Cell. 2012;148:399–408. doi: 10.1016/j.cell.2012.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Loboda A., Jozkowicz A., Dulak J. HIF-1 and HIF-2 transcription factors--similar but not identical. Mol Cells. 2010;29:435–442. doi: 10.1007/s10059-010-0067-2. [DOI] [PubMed] [Google Scholar]
- 3.Smythies J.A., Sun M., Masson N. Inherent DNA-binding specificities of the HIF-1α and HIF-2α transcription factors in chromatin. EMBO Rep. 2019;20 doi: 10.15252/embr.201846401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park S.K., Dadak A.M., Haase V.H., Fontana L., Giaccia A.J., Johnson R.S. Hypoxia-induced gene expression occurs solely through the action of hypoxia-inducible factor 1alpha (HIF-1alpha): role of cytoplasmic trapping of HIF-2alpha. Mol Cell Biol. 2003;23:4959–4971. doi: 10.1128/MCB.23.14.4959-4971.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mole D.R., Blancher C., Copley R.R. Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J Biol Chem. 2009;284:16767–16775. doi: 10.1074/jbc.M901790200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wiesener M.S., Jürgensen J.S., Rosenberger C. Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 2003;17:271–273. doi: 10.1096/fj.02-0445fje. [DOI] [PubMed] [Google Scholar]
- 7.Bernhardt W.M., Schmitt R., Rosenberger C. Expression of hypoxia-inducible transcription factors in developing human and rat kidneys. Kidney Int. 2006;69:114–122. doi: 10.1038/sj.ki.5000062. [DOI] [PubMed] [Google Scholar]
- 8.Uchida T., Rossignol F., Matthay M.A. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression in lung epithelial cells: implication of natural antisense HIF-1alpha. J Biol Chem. 2004;279:14871–14878. doi: 10.1074/jbc.M400461200. [DOI] [PubMed] [Google Scholar]
- 9.Chae K.S., Kang M.J., Lee J.H. Opposite functions of HIF-α isoforms in VEGF induction by TGF-β1 under non-hypoxic conditions. Oncogene. 2011;30:1213–1228. doi: 10.1038/onc.2010.498. [DOI] [PubMed] [Google Scholar]
- 10.Ahmad A., Ahmad S., Malcolm K.C. Differential regulation of pulmonary vascular cell growth by hypoxia-inducible transcription factor-1α and hypoxia-inducible transcription factor-2α. Am J Respir Cell Mol Biol. 2013;49:78–85. doi: 10.1165/rcmb.2012-0107OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yuan Y., Hilliard G., Ferguson T., Millhorn D.E. Cobalt inhibits the interaction between hypoxia-inducible factor-alpha and von Hippel-Lindau protein by direct binding to hypoxia-inducible factor-alpha. J Biol Chem. 2003;278:15911–15916. doi: 10.1074/jbc.M300463200. [DOI] [PubMed] [Google Scholar]
- 12.Sridharan V., Guichard J., Bailey R.M., Kasiganesan H., Beeson C., Wright G.L. The prolyl hydroxylase oxygen-sensing pathway is cytoprotective and allows maintenance of mitochondrial membrane potential during metabolic inhibition. Am J Physiol Cell Physiol. 2007;292:C719–C728. doi: 10.1152/ajpcell.00100.2006. [DOI] [PubMed] [Google Scholar]
- 13.Tarhonskaya H., Hardy A.P., Howe E.A. Kinetic investigations of the role of factor inhibiting hypoxia-inducible factor (FIH) as an oxygen sensor. J Biol Chem. 2015;290:19726–19742. doi: 10.1074/jbc.M115.653014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berchner-Pfannschmidt U., Tug S., Trinidad B. Nuclear oxygen sensing: induction of endogenous prolyl-hydroxylase 2 activity by hypoxia and nitric oxide. J Biol Chem. 2008;283:31745–31753. doi: 10.1074/jbc.M804390200. [DOI] [PubMed] [Google Scholar]
- 15.Vengellur A., Woods B.G., Ryan H.E., Johnson R.S., LaPres J.J. Gene expression profiling of the hypoxia signaling pathway in hypoxia-inducible factor 1alpha null mouse embryonic fibroblasts. Gene Expr. 2003;11:181–197. doi: 10.3727/000000003108749062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Appelhoff R.J., Tian Y.M., Raval R.R. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 17.Kapitsinou P.P., Liu Q., Unger T.L. Hepatic HIF-2 regulates erythropoietic responses to hypoxia in renal anemia. Blood. 2010;116:3039–3048. doi: 10.1182/blood-2010-02-270322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Warnecke C., Zaborowska Z., Kurreck J. Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J. 2004;18:1462–1464. doi: 10.1096/fj.04-1640fje. [DOI] [PubMed] [Google Scholar]
- 19.Koh M.Y., Powis G. Passing the baton: the HIF switch. Trends Biochem Sci. 2012;37:364–372. doi: 10.1016/j.tibs.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chandel N.S., McClintock D.S., Feliciano C.E. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem. 2000;275:25130–25138. doi: 10.1074/jbc.M001914200. [DOI] [PubMed] [Google Scholar]
- 21.Pan Y., Mansfield K.D., Bertozzi C.C. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol Cell Biol. 2007;27:912–925. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang H., Gao P., Fukuda R. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407–420. doi: 10.1016/j.ccr.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Schönenberger M.J., Kovacs W.J. Hypoxia signaling pathways: modulators of oxygen-related organelles. Front Cell Dev Biol. 2015;3:42. doi: 10.3389/fcell.2015.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walter K.M., Schönenberger M.J., Trötzmüller M. Hif-2α promotes degradation of mammalian peroxisomes by selective autophagy. Cell Metab. 2014;20:882–897. doi: 10.1016/j.cmet.2014.09.017. [DOI] [PubMed] [Google Scholar]
- 25.Yuan G., Peng Y.J., Reddy V.D. Mutual antagonism between hypoxia-inducible factors 1α and 2α regulates oxygen sensing and cardio-respiratory homeostasis. Proc Natl Acad Sci U S A. 2013;110:E1788−96. doi: 10.1073/pnas.1305961110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26.Scortegagna M., Ding K., Oktay Y. Multiple organ pathology, metabolic abnormalities and impaired homeostasis of reactive oxygen species in Epas1-/- mice. Nat Genet. 2003;35:331–340. doi: 10.1038/ng1266. [DOI] [PubMed] [Google Scholar]
- 27.Yuan G., Nanduri J., Khan S., Semenza G.L., Prabhakar N.R. Induction of HIF-1alpha expression by intermittent hypoxia: involvement of NADPH oxidase, Ca2+ signaling, prolyl hydroxylases, and mTOR. J Cell Physiol. 2008;217:67–85. doi: 10.1002/jcp.21537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nanduri J., Wang N., Yuan G. Intermittent hypoxia degrades HIF-2alpha via calpains resulting in oxidative stress: implications for recurrent apnea-induced morbidities. Proc Natl Acad Sci U S A. 2009;106:1199–1204. doi: 10.1073/pnas.0811018106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ban J.J., Ruthenborg R.J., Cho K.W., Kim J.W. Regulation of obesity and insulin resistance by hypoxia-inducible factors. Hypoxia (Auckl) 2014;2:171–183. doi: 10.2147/HP.S68771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yu X., Fang Y., Liu H. The balance of beneficial and deleterious effects of hypoxia-inducible factor activation by prolyl hydroxylase inhibitor in rat remnant kidney depends on the timing of administration. Nephrol Dial Transplant. 2012;27:3110–3119. doi: 10.1093/ndt/gfr754. [DOI] [PubMed] [Google Scholar]
- 31.Takeda N., O'Dea E.L., Doedens A. Differential activation and antagonistic function of HIF-{alpha} isoforms in macrophages are essential for NO homeostasis. Genes Dev. 2010;24:491–501. doi: 10.1101/gad.1881410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tashiro N., Segawa R., Tobita R. Hypoxia inhibits TNF-α-induced TSLP expression in keratinocytes. PLoS One. 2019;14 doi: 10.1371/journal.pone.0224705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Charron C.E., Chou P.C., Coutts D.J. Hypoxia-inducible factor 1alpha induces corticosteroid-insensitive inflammation via reduction of histone deacetylase-2 transcription. J Biol Chem. 2009;284:36047–36054. doi: 10.1074/jbc.M109.025387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang Z., Tang L., Zhu Q. Hypoxia-inducible factor-1α contributes to the profibrotic action of angiotensin II in renal medullary interstitial cells. Kidney Int. 2011;79:300–310. doi: 10.1038/ki.2010.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ishizuka S., Sakai T., Hiraiwa H. Hypoxia-inducible factor-2α induces expression of type X collagen and matrix metalloproteinases 13 in osteoarthritic meniscal cells. Inflamm Res. 2016;65:439–448. doi: 10.1007/s00011-016-0926-1. [DOI] [PubMed] [Google Scholar]
- 36.Yoon H., Shin S.H., Shin D.H., Chun Y.S., Park J.W. Differential roles of Sirt1 in HIF-1α and HIF-2α mediated hypoxic responses. Biochem Biophys Res Commun. 2014;444:36–43. doi: 10.1016/j.bbrc.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 37.Chen R., Xu M., Hogg R.T. The acetylase/deacetylase couple CREB-binding protein/sirtuin 1 controls hypoxia-inducible factor 2 signaling. J Biol Chem. 2012;287:30800–30811. doi: 10.1074/jbc.M111.244780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Packer M. Role of deranged energy deprivation signaling in the pathogenesis of cardiac and renal disease in states of perceived nutrient overabundance. Circulation. 2020;141:2095–2105. doi: 10.1161/CIRCULATIONAHA.119.045561. [DOI] [PubMed] [Google Scholar]
- 39.Packer M. Role of impaired nutrient and oxygen deprivation signaling and deficient autophagic flux in diabetic CKD development: implications for understanding the effects of sodium-glucose cotransporter 2-inhibitors. J Am Soc Nephrol. 2020;31:907–919. doi: 10.1681/ASN.2020010010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tang B.L. Sirt1 and the mitochondria. Mol Cells. 2016;39:87–95. doi: 10.14348/molcells.2016.2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hyvärinen J., Hassinen I.E., Sormunen R. Hearts of hypoxia-inducible factor prolyl 4-hydroxylase-2 hypomorphic mice show protection against acute ischemia-reperfusion injury. J Biol Chem. 2010;285:13646–13657. doi: 10.1074/jbc.M109.084855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wei H., Bedja D., Koitabashi N. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of TGF-β signaling. Proc Natl Acad Sci U S A. 2012;109 doi: 10.1073/pnas.1202081109. E841−50. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 43.Krishnan J., Suter M., Windak R. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 2009;9:512–524. doi: 10.1016/j.cmet.2009.05.005. [DOI] [PubMed] [Google Scholar]
- 44.Wei Q., Bian Y., Yu F. Chronic intermittent hypoxia induces cardiac inflammation and dysfunction in a rat obstructive sleep apnea model. J Biomed Res. 2016;30:490–495. doi: 10.7555/JBR.30.20160110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pu J., Zhu S., Zhou D. Propofol alleviates apoptosis induced by chronic high glucose exposure via regulation of HIF-1α in H9c2 cells. Oxid Med Cell Longev. 2019;2019:4824035. doi: 10.1155/2019/4824035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar S., Wang G., Liu W. Hypoxia-induced mitogenic factor promotes cardiac hypertrophy via calcium-dependent and hypoxia-inducible factor-1α mechanisms. Hypertension. 2018;72:331–342. doi: 10.1161/HYPERTENSIONAHA.118.10845. [DOI] [PubMed] [Google Scholar]
- 47.Sharma N.M., Cunningham C.J., Zheng H., Liu X., Patel K.P. Hypoxia-inducible factor-1a mediates increased sympathoexcitation via glutamatergic N-Methyl-d-aspartate receptors in the paraventricular nucleus of rats with chronic heart failure. Circ Heart Fail. 2016;9 doi: 10.1161/CIRCHEARTFAILURE.116.003423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hölscher M., Schäfer K., Krull S. Unfavourable consequences of chronic cardiac HIF-1α stabilization. Cardiovasc Res. 2012;94:77–86. doi: 10.1093/cvr/cvs014. [DOI] [PubMed] [Google Scholar]
- 49.Bekeredjian R., Walton C.B., MacCannell K.A. Conditional HIF-1alpha expression produces a reversible cardiomyopathy. PLoS One. 2010;5 doi: 10.1371/journal.pone.0011693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guo J., Mihic A., Wu J. Canopy 2 attenuates the transition from compensatory hypertrophy to dilated heart failure in hypertrophic cardiomyopathy. Eur Heart J. 2015;36:2530–2540. doi: 10.1093/eurheartj/ehv294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pohl J., Hendgen-Cotta U.B., Stock P. Myocardial expression of macrophage migration inhibitory factor in patients with heart failure. J Clin Med. 2017;6:95. doi: 10.3390/jcm6100095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li G., Lu W.H., Wu X.W. Admission hypoxia-inducible factor 1α levels and in-hospital mortality in patients with acute decompensated heart failure. BMC Cardiovasc Disord. 2015;15:79. doi: 10.1186/s12872-015-0073-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shyu K.G., Lu M.J., Chang H., Sun H.Y., Wang B.W., Kuan P. Carvedilol modulates the expression of hypoxia-inducible factor-1alpha and vascular endothelial growth factor in a rat model of volume-overload heart failure. J Card Fail. 2005;11:152–159. doi: 10.1016/j.cardfail.2004.06.433. [DOI] [PubMed] [Google Scholar]
- 54.Martin C.M., Ferdous A., Gallardo T. Hypoxia-inducible factor-2alpha transactivates Abcg2 and promotes cytoprotection in cardiac side population cells. Circ Res. 2008;102:1075–1081. doi: 10.1161/CIRCRESAHA.107.161729. [DOI] [PubMed] [Google Scholar]
- 55.Mastrocola R., Collino M., Penna C. Maladaptive modulations of NLRP3 inflammasome and cardioprotective pathways are involved in diet-induced exacerbation of myocardial ischemia/reperfusion injury in mice. Oxid Med Cell Longev. 2016;2016:3480637. doi: 10.1155/2016/3480637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jürgensen J.S., Rosenberger C., Wiesener M.S. Persistent induction of HIF-1alpha and -2alpha in cardiomyocytes and stromal cells of ischemic myocardium. FASEB J. 2004;18:1415–1417. doi: 10.1096/fj.04-1605fje. [DOI] [PubMed] [Google Scholar]
- 57.Wu D., Huang R.T., Hamanaka R.B. HIF-1α is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. Elife. 2017;6 doi: 10.7554/eLife.25217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu D., Lei L., Desir M. Smooth muscle hypoxia-inducible factor 1α links intravascular pressure and atherosclerosis--brief report. Arterioscler Thromb Vasc Biol. 2016;36:442–445. doi: 10.1161/ATVBAHA.115.306861. [DOI] [PubMed] [Google Scholar]
- 59.Aarup A., Pedersen T.X., Junker N. Hypoxia-inducible factor-1α expression in macrophages promotes development of atherosclerosis. Arterioscler Thromb Vasc Biol. 2016;36:1782–1790. doi: 10.1161/ATVBAHA.116.307830. [DOI] [PubMed] [Google Scholar]
- 60.Schultz K., Fanburg B.L., Beasley D. Hypoxia and hypoxia-inducible factor-1alpha promote growth factor-induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 2006;290:H2528–H2534. doi: 10.1152/ajpheart.01077.2005. [DOI] [PubMed] [Google Scholar]
- 61.Na T.Y., Lee H.J., Oh H.J., Huh S., Lee I.K., Lee M.O. Positive cross-talk between hypoxia inducible factor-1α and liver X receptor α induces formation of triglyceride-loaded foam cells. Arterioscler Thromb Vasc Biol. 2011;31:2949–2956. doi: 10.1161/ATVBAHA.111.235788. [DOI] [PubMed] [Google Scholar]
- 62.Nakayama T., Kurobe H., Sugasawa N. Role of macrophage-derived hypoxia-inducible factor (HIF)-1α as a mediator of vascular remodelling. Cardiovasc Res. 2013;99:705–715. doi: 10.1093/cvr/cvt146. [DOI] [PubMed] [Google Scholar]
- 63.Natarajan R., Jones D.G., Fisher B.J., Wallace T.J., Ghosh S., Fowler A.A., 3rd Hypoxia inducible factor-1: regulation by nitric oxide in posthypoxic microvascular endothelium. Biochem Cell Biol. 2005;83:597–607. doi: 10.1139/o05-047. [DOI] [PubMed] [Google Scholar]
- 64.Matsuura Y., Yamashita A., Iwakiri T. Vascular wall hypoxia promotes arterial thrombus formation via augmentation of vascular thrombogenicity. Thromb Haemost. 2015;114:158–172. doi: 10.1160/TH14-09-0794. [DOI] [PubMed] [Google Scholar]
- 65.Zhang X., Zhang Y., Wang P. Adipocyte hypoxia-inducible factor 2α suppresses atherosclerosis by promoting adipose ceramide catabolism. Cell Metab. 2019;30:937–951. doi: 10.1016/j.cmet.2019.09.016. [DOI] [PubMed] [Google Scholar]
- 66.Poitz D.M., Augstein A., Weinert S., Braun-Dullaeus R.C., Strasser R.H., Schmeisser A. OxLDL and macrophage survival: essential and oxygen-independent involvement of the Hif-pathway. Basic Res Cardiol. 2011;106:761–772. doi: 10.1007/s00395-011-0186-8. [DOI] [PubMed] [Google Scholar]
- 67.Zhang S., Zhao L., Wang J., Chen N., Yan J., Pan X. HIF-2α and Oct4 have synergistic effects on survival and myocardial repair of very small embryonic-like mesenchymal stem cells in infarcted hearts. Cell Death Dis. 2017;8:e2548. doi: 10.1038/cddis.2016.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kapitsinou P.P., Sano H., Michael M. Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J Clin Invest. 2014;124:2396–2409. doi: 10.1172/JCI69073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Xu X., Song N., Zhang X. Renal protection mediated by hypoxia inducible factor-1α depends on proangiogenesis function of miR-21 by targeting thrombospondin 1. Transplantation. 2017;101:1811–1819. doi: 10.1097/TP.0000000000001501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conde E., Giménez-Moyano S., Martín-Gómez L. HIF-1α induction during reperfusion avoids maladaptive repair after renal ischemia/reperfusion involving miR127-3p. Sci Rep. 2017;7:41099. doi: 10.1038/srep41099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fang Y., Zhang H., Zhong Y., Ding X. Prolyl hydroxylase 2 (PHD2) inhibition protects human renal epithelial cells and mice kidney from hypoxia injury. Oncotarget. 2016;7:54317–54328. doi: 10.18632/oncotarget.11104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lourenço B.N., Coleman A.E., Schmiedt C.W. Profibrotic gene transcription in renal tissues from cats with ischemia-induced chronic kidney disease. Am J Vet Res. 2020;81:180–189. doi: 10.2460/ajvr.81.2.180. [DOI] [PubMed] [Google Scholar]
- 73.Luo L., Luo G., Fang Q., Sun Z. Stable expression of hypoxia-inducible factor-1α in human renal proximal tubular epithelial cells promotes epithelial to mesenchymal transition. Transplant Proc. 2014;46:130–134. doi: 10.1016/j.transproceed.2013.06.024. [DOI] [PubMed] [Google Scholar]
- 74.Li Z.L., Lv L.L., Tang T.T. HIF-1α inducing exosomal microRNA-23a expression mediates the cross-talk between tubular epithelial cells and macrophages in tubulointerstitial inflammation. Kidney Int. 2019;95:388–404. doi: 10.1016/j.kint.2018.09.013. [DOI] [PubMed] [Google Scholar]
- 75.Deng W., Ren Y., Feng X. Hypoxia inducible factor-1 alpha promotes mesangial cell proliferation in lupus nephritis. Am J Nephrol. 2014;40:507–515. doi: 10.1159/000369564. [DOI] [PubMed] [Google Scholar]
- 76.Baumann B., Hayashida T., Liang X., Schnaper H.W. Hypoxia-inducible factor-1α promotes glomerulosclerosis and regulates COL1A2 expression through interactions with Smad3. Kidney Int. 2016;90:797–808. doi: 10.1016/j.kint.2016.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kimura K., Iwano M., Higgins D.F. Stable expression of HIF-1alpha in tubular epithelial cells promotes interstitial fibrosis. Am J Physiol Renal Physiol. 2008;295:F1023−9. doi: 10.1152/ajprenal.90209.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Nayak B.K., Shanmugasundaram K., Friedrichs W.E. HIF-1 mediates renal fibrosis in OVE26 type 1 diabetic mice. Diabetes. 2016;65:1387–1397. doi: 10.2337/db15-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kong K.H., Oh H.J., Lim B.J. Selective tubular activation of hypoxia-inducible factor-2α has dual effects on renal fibrosis. Sci Rep. 2017;7:11351. doi: 10.1038/s41598-017-11829-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kojima I., Tanaka T., Inagi R. Protective role of hypoxia-inducible factor-2alpha against ischemic damage and oxidative stress in the kidney. J Am Soc Nephrol. 2007;18:1218–1226. doi: 10.1681/ASN.2006060639. [DOI] [PubMed] [Google Scholar]
- 81.Landau D., London L., Bandach I., Segev Y. The hypoxia inducible factor/erythropoietin (EPO)/EPO receptor pathway is disturbed in a rat model of chronic kidney disease related anemia. PLoS One. 2018;13 doi: 10.1371/journal.pone.0196684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Olmos G., Muñoz-Félix J.M., Mora I. Impaired erythropoietin synthesis in chronic kidney disease is caused by alterations in extracellular matrix composition. J Cell Mol Med. 2018;22:302–314. doi: 10.1111/jcmm.13319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bondeva T., Heinzig J., Ruhe C., Wolf G. Advanced glycated end-products affect HIF-transcriptional activity in renal cells. Mol Endocrinol. 2013;27:1918–1933. doi: 10.1210/me.2013-1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Isoe T., Makino Y., Mizumoto K. High glucose activates HIF-1-mediated signal transduction in glomerular mesangial cells through a carbohydrate response element binding protein. Kidney Int. 2010;78:48–59. doi: 10.1038/ki.2010.99. [DOI] [PubMed] [Google Scholar]
- 85.Cioffi C.L., Liu X.Q., Kosinski P.A., Garay M., Bowen B.R. Differential regulation of HIF-1 alpha prolyl-4-hydroxylase genes by hypoxia in human cardiovascular cells. Biochem Biophys Res Commun. 2003;303:947–953. doi: 10.1016/s0006-291x(03)00453-4. [DOI] [PubMed] [Google Scholar]
- 86.Aprelikova O., Chandramouli G.V., Wood M. Regulation of HIF prolyl hydroxylases by hypoxia-inducible factors. J Cell Biochem. 2004;92:491–501. doi: 10.1002/jcb.20067. [DOI] [PubMed] [Google Scholar]
- 87.Miikkulainen P., Högel H., Seyednasrollah F., Rantanen K., Elo L.L., Jaakkola P.M. Hypoxia-inducible factor (HIF)-prolyl hydroxylase 3 (PHD3) maintains high HIF2A mRNA levels in clear cell renal cell carcinoma. J Biol Chem. 2019;294:3760–3771. doi: 10.1074/jbc.RA118.004902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fukui K., Shinozaki Y., Kobayashi H. JTZ-951 (enarodustat), a hypoxia-inducible factor prolyl hydroxylase inhibitor, stabilizes HIF-α protein and induces erythropoiesis without effects on the function of vascular endothelial growth factor. Eur J Pharmacol. 2019;859:172532. doi: 10.1016/j.ejphar.2019.172532. [DOI] [PubMed] [Google Scholar]
- 89.Li X., Li J., Wang L. The role of metformin and resveratrol in the prevention of hypoxia-inducible factor 1α accumulation and fibrosis in hypoxic adipose tissue. Br J Pharmacol. 2016;173:2001–2015. doi: 10.1111/bph.13493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ryu D.R., Yu M.R., Kong K.H. Sirt1-hypoxia-inducible factor-1α interaction is a key mediator of tubulointerstitial damage in the aged kidney. Aging Cell. 2019;18 doi: 10.1111/acel.12904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Shao Y., Lv C., Wu C., Zhou Y., Wang Q. Mir-217 promotes inflammation and fibrosis in high glucose cultured rat glomerular mesangial cells via Sirt1/HIF-1α signaling pathway. Diabetes Metab Res Rev. 2016;32:534–543. doi: 10.1002/dmrr.2788. [DOI] [PubMed] [Google Scholar]
- 92.Wang S., Qian Y., Gong D., Zhang Y., Fan Y. Resveratrol attenuates acute hypoxic injury in cardiomyocytes: correlation with inhibition of iNOS-NO signaling pathway. Eur J Pharm Sci. 2011;44:416–421. doi: 10.1016/j.ejps.2011.08.029. [DOI] [PubMed] [Google Scholar]
- 93.Pektaş M.B., Sadi G., Koca H.B. Resveratrol ameliorates the components of hepatic inflammation and apoptosis in a rat model of streptozotocin-induced diabetes. Drug Dev Res. 2016;77:12–19. doi: 10.1002/ddr.21287. [DOI] [PubMed] [Google Scholar]
- 94.Osataphan S., Macchi C., Singhal G. SGLT2 inhibition reprograms systemic metabolism via FGF21-dependent and -independent mechanisms. JCI Insight. 2019;4 doi: 10.1172/jci.insight.123130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Swe M.T., Thongnak L., Jaikumkao K., Pongchaidecha A., Chatsudthipong V., Lungkaphin A. Dapagliflozin not only improves hepatic injury and pancreatic endoplasmic reticulum stress, but also induces hepatic gluconeogenic enzymes expression in obese rats. Clin Sci (Lond) 2019;133:2415–2430. doi: 10.1042/CS20190863. [DOI] [PubMed] [Google Scholar]
- 96.Packer M. SGLT2 inhibitors produce cardiorenal benefits by promoting adaptive cellular reprogramming to induce a state of fasting mimicry: a paradigm shift in understanding their mechanism of action. Diabetes Care. 2020;43:508–511. doi: 10.2337/dci19-0074. [DOI] [PubMed] [Google Scholar]
- 97.Mohamed H.E., Asker M.E., Keshawy M.M., Hasan R.A., Mahmoud Y.K. Inhibition of tumor necrosis factor-α enhanced the antifibrotic effect of empagliflozin in an animal model with renal insulin resistance. Mol Cell Biochem. 2020;466:45–54. doi: 10.1007/s11010-020-03686-x. [DOI] [PubMed] [Google Scholar]
- 98.Li J., Liu H., Takagi S., Nitta K. Renal protective effects of empagliflozin via inhibition of EMT and aberrant glycolysis in proximal tubules. JCI Insight. 2020;5 doi: 10.1172/jci.insight.129034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bessho R., Takiyama Y., Takiyama T. Hypoxia-inducible factor-1α is the therapeutic target of the SGLT2 inhibitor for diabetic nephropathy. Sci Rep. 2019;9:14754. doi: 10.1038/s41598-019-51343-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mazer C.D., Hare G.M.T., Connelly P.W. Effect of empagliflozin on erythropoietin levels, iron stores and red blood cell morphology in patients with type 2 diabetes and coronary artery disease. Circulation. 2020;141:704–707. doi: 10.1161/CIRCULATIONAHA.119.044235. [DOI] [PubMed] [Google Scholar]
- 101.Inzucchi S.E., Zinman B., Fitchett D. How does empagliflozin reduce cardiovascular mortality? Insights from a mediation analysis of the EMPA-REG OUTCOME trial. Diabetes Care. 2018;41:356–363. doi: 10.2337/dc17-1096. [DOI] [PubMed] [Google Scholar]
- 102.Li J.W., Woodward M., Perkovic V. Mediators of the effects of canagliflozin on heart failure in patients with type 2 diabetes. J Am Coll Cardiol HF. 2020;8:57–66. doi: 10.1016/j.jchf.2019.08.004. [DOI] [PubMed] [Google Scholar]