

Abstract
Prodrugs (MRS7422, MRS7476) of highly selective A3 adenosine receptor (AR) agonists Cl-IB-MECA and MRS5698, respectively, were synthesized by succinylation of the 2′ and 3′ hydroxyl groups, and the parent, active drug was shown to be readily liberated upon incubation with liver esterases. The prodrug MRS7476 had greatly increased aqueous solubility compared with parent MRS5698 and was fully efficacious and with a longer duration than MRS7422 in reversing mouse neuropathic pain (chronic constriction injury model, 3 μmol/kg, p.o.), a known A3AR effect. MRS7476 (5 mg/kg, p.o., twice daily) was found to protect against non-alcoholic steatohepatitis (NASH) in the STAM mouse model, indicated by the NAFLD activity score. Hepatocyte ballooning, IL-10 production, and liver histology were significantly normalized in the MRS7476-treated mice, but not liver fibrosis (no change in ACTA2 levels) or inflammation. Hepatic expression of ADORA3 in human NAFLD patients was 1.9-fold lower compared to normal controls. Adora3 expression determined by qPCR in primary mouse liver was associated with the stellate cells, and its mouse full body A3AR knockout worsened liver markers of inflammation and steatosis. Thus, we have introduced a reversible prodrug strategy that enables water solubility and in vivo activity of masked A3AR agonists in models of two disease conditions.
Electronic supplementary material
The online version of this article (10.1007/s11302-020-09715-0) contains supplementary material, which is available to authorized users.
Keywords: Purinergic receptors, Nucleosides, Adenosine receptor, Prodrug, Pain, Steatohepatitis
Introduction
Adenosine acts as an anti-inflammatory mediator in many physiological systems and is considered a retaliatory metabolite; i.e., its production as a consequence of elevated ATP inside and outside the cell is a protective feedback signal [1, 2]. Extracellular adenosine acts through a family of four G protein-coupled receptors, two coupled principally to Gi (A1 and A3) [3, 4] and two coupled principally to Gs (A2A and A2B) [5, 6]. A3 adenosine receptor (A3AR) agonists IB-MECA 1 and Cl-IB-MECA 2 (Fig. 1) have demonstrated safety and efficacy in clinical trials for rheumatoid arthritis, psoriasis (1), and hepatocellular carcinoma (HCC) and other liver diseases (2) [7–11]. The A3AR is upregulated in pathological conditions and can serve as a predictive biomarker for clinical efficacy of A3AR agonists in inflammatory diseases [12–14]. A clinical trial of Cl-IB-MECA in non-alcoholic steatohepatitis (NASH) is underway, based on its modulation of Wnt and other signaling pathways in liver tissue mediating anti-inflammatory, anti-steatotic, and anti-fibrotic effects [15–17]. Recently, the utility of highly selective agonists chronic pain conditions was demonstrated [18–22]. These agonists include MRS5698 3 and MRS5663 4, having a rigid, extended substituent at the C2 position were shown to contribute to high A3AR selectivity (thousands of fold) across several species. The action of A3AR agonists in chronic neuropathic pain models has been shown to occur in peripheral, spinal, and supraspinal sites [18]. In a model of cancer chemotherapy-induced neuropathic pain, MRS5698 modulated the nucleotide-binding oligomerization domain-like receptor protein 3 (NLRP3) inflammasome, to attenuate proinflammatory IL-1β production and increase anti-inflammatory and neuroprotective IL-10 expression [20].
Fig. 1.

Structures of selective A3AR agonists in clinical trials (1, 2) and those used currently as pharmacological probes (3, 4)
Many of the highly selective A3AR agonists display low aqueous solubility, and we explore here, charged prodrug derivatives of several agonists as a means of increasing solubility while retaining in vivo activity. Drugs that have only sparing aqueous solubility are subject to variability in their in vivo responses [23]. Both peroral and parenteral drugs benefit from sufficient solubility to avoid the use of complicated vehicles for in vivo administration, which often drives the selection of the most promising drug molecules. There are well-explored approaches to derivatize nucleoside and nucleotide drugs as prodrugs [23]. Often such prodrugs are intended for intracellular action, for example, an O-acyl antiviral nucleoside prodrug that is orally bioavailable, unmasked after entering the cell, and phosphorylated for incorporation into viral RNA [24]. However, nucleosides that target cell surface G protein-coupled receptors (GPCRs) require a different prodrug approach. Ester derivatives of nucleosides, specifically AR agonists [25], have also been reported to be prodrugs for unmasking in vivo, typically in the intestinal epithelium, which is rich in esterases. Thus, we have chemically synthesized O-succinyl derivatives of nucleosides 2 and 3, in which the bioreversible esterification occurs at the free 2′- and 3′-hydroxyl groups (Fig. 2A).
Fig. 2.

A Synthesis of the 2′,3′-di-succinyl ester prodrugs using 8 equivalents of succinic anhydride (a) in dichloromethane in the presence of triethylamine (16 equivalents) and 4-dimethylaminopyridine (1 equivalent). The prodrugs were purified to homogeneity by HPLC and isolated as the partial Et3N salt forms (0.8 equivalents Et3N for MRS7422, 0.5 equivalents for MRS7476). B Enzymatic hydrolysis of MRS7422 using porcine liver esterase (PLE, b) at 37 °C in HEPES buffer, pH 7.3. Mean ± SD is shown. See Figure S3 for the corresponding data for MRS7476
Methods
Instruments and reagents used for the synthesis
Cl-IB-MECA (2) was purchased from Tocris Bioscience (Ellisville, MO). Compound 3 was synthesized in our laboratory as described.1,2 Other reagents and solvents were purchased from Sigma-Aldrich (St. Louis, MO). The prodrug synthesis and characterization are described in the Supporting information. 1H-NMR spectra were obtained with a Bruker 400 spectrometer using MeOH-d4 as a solvent. The chemical shifts are expressed as parts per million, and the coupling constants (J) are given in hertz. The RP-HPLC was performed using Phenomenex Luna 5 μm C18(2) 100 Å, AXIA, 250 × 21.2 mm column. The purity of final nucleoside derivatives was checked using a Hewlett−Packard 1100 HPLC equipped with an Agilent Eclipse 5 μm XDB-C18 analytical column (50 mm × 4.6 mm; Agilent Technologies Inc., Palo Alto, CA). Mobile phase: linear gradient solvent system, 10 mM TEAA (triethylammonium acetate):CH3CN from 95:5 to 0:100 in 20 min; the flow rate was 1.0 mL/min. Peaks were detected by UV absorption with a diode array detector at 230, 254, and 280 nm. All derivatives tested for biological activity showed > 95% purity in the HPLC systems. cLogP was calculated using ChemDraw 19.1 (PerkinElmer) for MAC.
Expression of adenosine receptors in human liver
Gene expression of ARs was assessed in baseline (pre-treatment) liver biopsies obtained in a clinical trial investigating the effects of vitamin E in patients with non-alcoholic fatty liver disease (ClinicalTrials.gov, NCT017921150) [reference currently under review]). The study was reviewed by the Institutional Review Board, and all subjects gave written informed consent. Liver samples were placed in RNAlater (Thermo Fisher Scientific, Waltham, MA) at the bedside and stored in − 80 °C. RNA was extracted using Trizol Reagent (Thermo Fisher Scientific) followed by purification with miRNAeasy kit (Qiagen, Hilden, Germany). The quality of extracted RNA was assessed using a NanoDrop spectrophotometer and Agilent 2100 Bioanalyzer. PolyA-enriched RNA was used to prepare a paired-end index library. Samples were pooled (2 pools) and run on an Illumina HiSeq4000 with 75 bp paired-end sequencing. Reads were aligned to the human genome (GRCh38) using STAR version 2.5.4a with default settings. Genes were counted with htseq-count using the mode intersection-nonempty. Counts were normalized and are shown as transcripts per million (TPM). Data were compared with normal liver tissue (n = 10) from the HPA data set obtained through the Human Protein Atlas (https://www.proteinatlas.org, PMID: 25613900).
Expression of Adora3 in mouse liver cells
Total RNA was extracted from primary hepatocytes and stellate cells isolated from C57BL/6 mice. First-strand cDNA was used as template for TaqMan probe-based quantitative polymerase chain reaction (qPCR) assay to quantify the gene expression level, which was normalized to the level of 18S rRNA. Primer sequences are in Table S1 (Supporting Information).
Mouse model of chronic neuropathic pain
The mouse experimental model of chronic neuropathic pain (CCI model [26]) has been published in various studies showing protection by highly selective A3AR agonists [19, 21]. Briefly, the drugs were administered p.o. on day 7 following constriction of the sciatic nerve, which is the time of peak pain. Following drug injection, mechano-allodynia was quantified at multiple time points according to the hind paw withdrawal response to von Frey filaments stimulation.
Mouse maintenance and diet
A3AR knockout (A3Δ/Δ) and WT (A3+/+) mice, on a C57BL/6 J background, used in this study were generated as described.27 Briefly, mice heterozygous for A3AR (A3Δ/+) were interbred to generate A3Δ/Δ and WT mice. Male littermates were used for all experiments.
Mice were housed on a 12-h light/12-h dark cycle in a pathogen-free barrier facility at room temperature (23 °C). The mice had ad libitum access to water. The mice were maintained on standard mouse chow (7022 NIH-07 diet, 15% kcal fat, energy density 3.1 kcal/g, Envigo, Inc., Huntingdon, UK) or a high fat diet (HFD, 36% fat, equivalent to 60% kcal; 36% carbohydrate; F3282; Bio-Serv, Flemington, NJ). Mice maintained on chow diet (CD) were transferred to HFD at 13 weeks of age, and mice were compared after 32 weeks on HFD. The study and its procedures were approved by the Animal Care and Use Committee of National Institute of Diabetes and Digestive and Kidney Diseases, animal protocol K083-LBC-17.
Gene expression analysis by quantitative real time-PCR
Gene expression analysis was carried out as described [27]. Liver tissues were dissected and were frozen in liquid nitrogen. RNAeasy mini kit (Qiagen, Germantown, MD) was used for the extraction of total liver mRNA. After synthesizing cDNA using Superscript III First Strand synthesis Super Mix (Invitrogen, Thermo Fisher, Rockville, MD), qRT-PCR was performed using SYBR green reagent (Applied Biosystems, Beverly, MA) in triplicates. 18s rRNA was used as the normalization control for relative gene expression analysis using ΔΔCt method. Primer sequences are provided in Table S2.
Histological assessment of liver sections from HFD A3Δ/Δ mice
Liver tissues were dissected out of A3Δ/Δ and WT mice after 32 weeks on HFD and were frozen in liquid nitrogen. Liver tissues were sectioned and stained using standard techniques. Hematoxylin-eosin (HE) stained liver sections were visualized using Keyence Microscope BZ-9000.
Statistics
All data are expressed as the mean ± SEM. Data were tested for statistical significance by two-tailed, unpaired Student’s t test, as appropriate. A P value of less than 0.05 was considered significant.
Results
Prodrug design and synthesis
The recent generation of highly specific A3AR agonists, such as MRS5698, generally has low aqueous solubility. The maximal solubility of MRS5698 was determined to be only 1.6 μg/ml at pH 7.4, consistent with its bearing hydrophobic substituents at C2 and N6 positions [19]. In comparing prodrug strategies used with nucleosides, we did not want to use a derivatization scheme that would increase diffusion into the cell because of the cell surface location of the A3AR. Rather, we targeted liberation of the free nucleoside by ester formation, that is subject to hydrolysis by widespread esterases, including in the intestinal epithelium. Simple alkyl ester prodrugs of various A3AR nucleoside antagonists and agonists, including Cl-IB-MECA, were previously reported to require hydrolysis in order to liberate significant pharmacological activity associated with the parent drugs [25]. Small 2′ and 3′ alkyl diesters were found to be more readily cleaved enzymatically than similar diesters with large alkyl groups, such as 2′,3′-dihexanoate esters. Since the free ribosyl hydroxyl groups of Cl-IB-MECA and other nucleoside agonists are generally needed for AR activation [4], we again planned to block these groups in prodrug derivatives. None of the previously reported ester prodrugs of A3AR ligands [25] utilized water-solubilizing succinyl esters, although this has been attempted for other prodrugs, linkers, and nanocarrier assemblies [28–31]. The approach of adding a water-solubilizing group to form prodrugs of poorly soluble parent drugs is often accomplished with a phosphate ester [32]. However, we were unable to synthesize and purify analogous 2′ or 3′-phosphate esters of Cl-IB-MECA.
These two A3AR agonists, Cl-IB-MECA (already in clinical trials for liver conditions) and MRS5698 (a pharmacological probe compound), were derivatized as 2′,3′-di-succinyl monoesters, which were isolated as triethylammonium salt forms. The synthetic method is straightforward (Fig. 2A, with details in the Supporting information): treatment of the parent nucleoside with excess succinic anhydride in dichloromethane in the presence of triethylamine and acylation catalyst 4-(dimethylamino)pyridine. Each of the prodrug molecules, MRS7422 5 and MRS7476 6, was purified by HPLC and lyophilized as a salt form. The structures were confirmed by NMR and mass spectrometry (Figure S2, Supporting information), and partial equivalents of triethylamine were associated with each. As anticipated, the positions of derivatization were shown by NMR to be at the 2′- and 3′-hydroxyl groups.
The reported A3AR affinity (Ki, nM) is 1.5 (human) and 0.80 (mouse) for Cl-IB-MECA and 3.49 (human) and 3.08 (mouse) for MRS5698 [22, 33]. We did not subject the prodrugs to AR binding studies, based on our earlier publication of Besada et al. [25] and other studies of SAR [4], in which acylation of both 2′- and 3′-hydroxyl groups greatly reduces or precludes affinity at ARs. This is also consistent with the steric crowding in the now well-characterized ribose binding site in the A2AAR (which is structurally analogous in other AR subtypes), as the 2′- and 3′-hydroxyl groups are required for H-bond formation with polar groups of the receptor [4]. The cLogP values for the prodrugs compared with the active drugs were − 6.11 (−0.187 as neutral acid) and − 4.02 (1.90 as neutral acid) for 5 and 6, respectively, compared with − 1.42 and 1.54 for 2 and 3, respectively. However, experimental LogP data were not determined for this class of compounds. The enhancement of aqueous solubility with this derivative was particularly pronounced. The maximum solubility of MRS7476 in water was 2.5 mg/ml (3.1 mM), which is > 1000-fold greater than the aqueous solubility of MRS5698 [19]. The maximum solubility of MRS7422 in water was 3.0 mg/ml (3.6 mM), potential off-target activities at 46 receptors were measured by the Psychoactive Drug Screening Program, and only μM affinity was determined for several sites (Table S1, Supporting Information) [34].
The two prodrugs were first tested for the ability to be cleaved by porcine liver esterases. Under the incubation conditions used (37 °C, pH 7.3, 72 h), both were converted to the corresponding parent drug as determined by HPLC, but MRS7422 (Fig. 2B) was more rapidly cleaved than MRS7476 (Figure S3, Supporting information). Thus, we expect that these prodrugs will lead to the generation of the active drug molecule in vivo.
Activity in a chronic pain model
To determine the in vivo efficacy of the novel prodrugs we tested them first in an acute-administration setting using the chronic constriction injury (CCI) mouse model [26], where a single dose of an A3AR agonists can reverse established mechano-allodynia at day 7 post-injury (time of maximal pain), and its efficacy and duration are measurable in real time. The two prodrugs were administered by oral gavage (1 or 3 μmol/kg) in mice in the CCI model, as reported in our previous structure activity relationship (SAR) studies [19, 21]. This allowed the real-time in vivo efficacy of the A3AR agonists at the ipsilateral (nerve injured) paw to be followed. The parent drug Cl-IB-MECA was compared directly to its prodrug MRS7422, and the activity of the prodrug reached a higher efficacy at the same dose (Fig. 3A). However, the effect of MRS7422 at 1 μmol/kg was relatively short lasting. In contrast, the effect of MRS7476 at 3 μmol/kg was still significant at 4-h post-administration (Fig. 3B), which is similar to the time course of pain protection by the parent drug MRS5698 [19]. There was no effect of either nucleoside derivative at the contralateral paw.
Fig. 3.
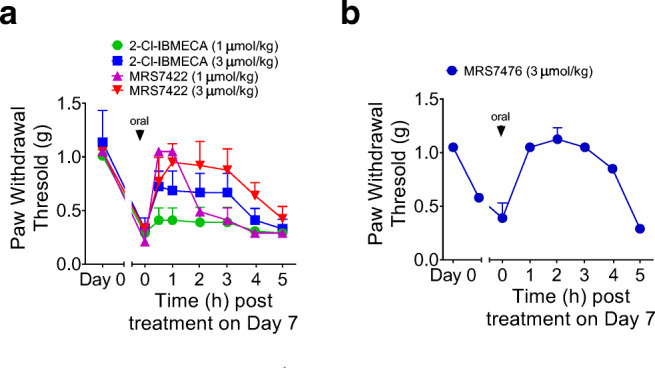
Anti-hyperalgesia effects of orally administered prodrugs in the mouse CCI model. A Effects of Cl-IB-MECA at 1 μmol/kg (n = 5) compared with MRS7422 at 1 μmol/kg (n = 2) and 3 μmol/kg (n = 3). B Effects of MRS7476 at 3 μmol/kg (n = 2)
Given its apparent benefit in the CCI model, it was decided to apply MRS7476 as the prodrug analogue to be used when measuring the response to chronic administration in a model of NASH. However, prior to testing this A3AR agonist prodrug in a mouse therapeutic experiment in a NASH model, we tested and found Adora3 to be expressed in the mouse liver. Specifically, we measured the expression of the receptor in liver tissue and cells. All four ARs were expressed in human liver tissue from NAFLD patients (n = 19; Fig. 4A). When comparing hepatic ADORA3 expression between patients with NAFLD and controls with normal liver (n = 10), we found a 1.9-fold decreased expression in the NAFLD patients (Fig. 4B; p = 0.02). To determine which cells in the liver predominantly express the receptor, we compared its expression between primary mouse hepatocytes and stellate cells, the main drivers of hepatic fibrosis. The expression level of Adora3 was 27.6-fold greater in the stellate cells (p = 0.008; Fig. 4C).
Fig. 4.
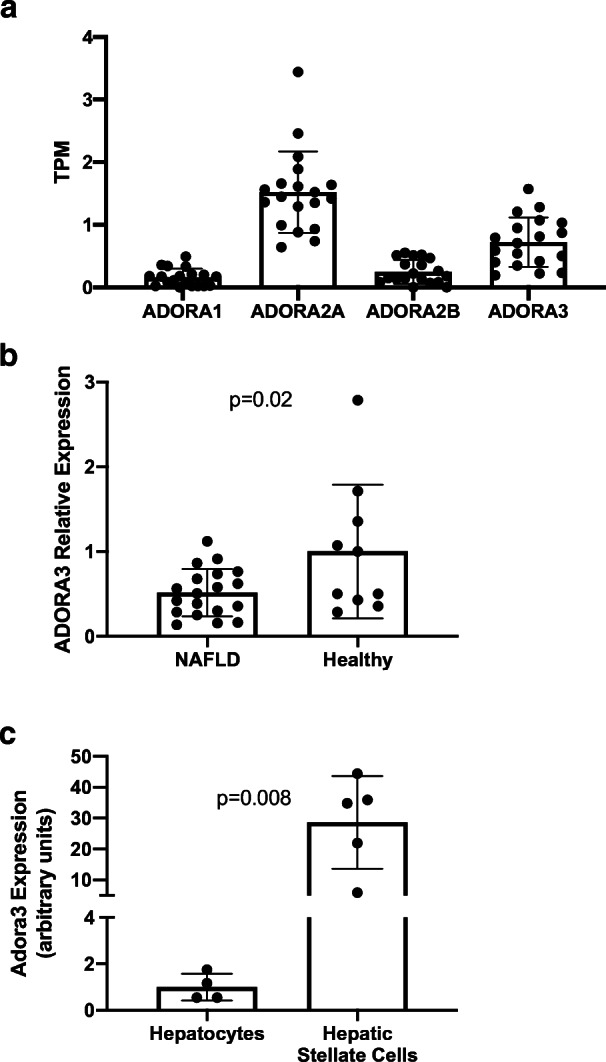
A mRNA expression of all four ARs in liver tissue from human NAFLD patients by RNA sequencing. B Comparative expression of ADORA3 in liver tissue from NAFLD patients and controls with normal liver. C Expression of ADORA3 by qPCR in primary mouse hepatocytes and stellate cells. NAFLD, non-alcoholic fatty liver disease. TPM transcripts per million
Activity in a model of NASH
The mouse STAM NASH model [37–39] was used to test the efficacy of MRS7476. NASH was established in male C57BL/6 mice by a single subcutaneous injection of 200 μg streptozotocin 2 days after birth and feeding with a high fat diet ad libitum after 4 weeks of age (original study plan in Figure S4, Supporting Information). Three groups of mice were examined (treatment beginning at 6 weeks of age, for 3 weeks): vehicle control, MRS7476, and telmisartan (angiotensin II receptor antagonist, 10 mg/kg, p.o., once daily, as a positive control) [40]. The impact of chronic drug treatment on liver histology (Fig. 5A) and other markers were examined (Tables S4–S6 and protocol details in the Supporting Information). MRS7476 (5 mg/kg, p.o., twice daily) had no effect on hepatic triglycerides (Fig. 6C) or histological steatosis scores (Fig. 7A). Similarly, no effect was seen on histological inflammation (Fig. 7B) scores or serum ALT (Fig. 6A), but markedly improved hepatocyte ballooning (Fig. 7C), the hallmark of NASH-associated hepatocyte injury, was observed. The composite NAFLD activity score (NAS) was significantly improved by MRS7476 (Fig. 5B). MRS7476 did not affect liver fibrosis.
Fig. 5.
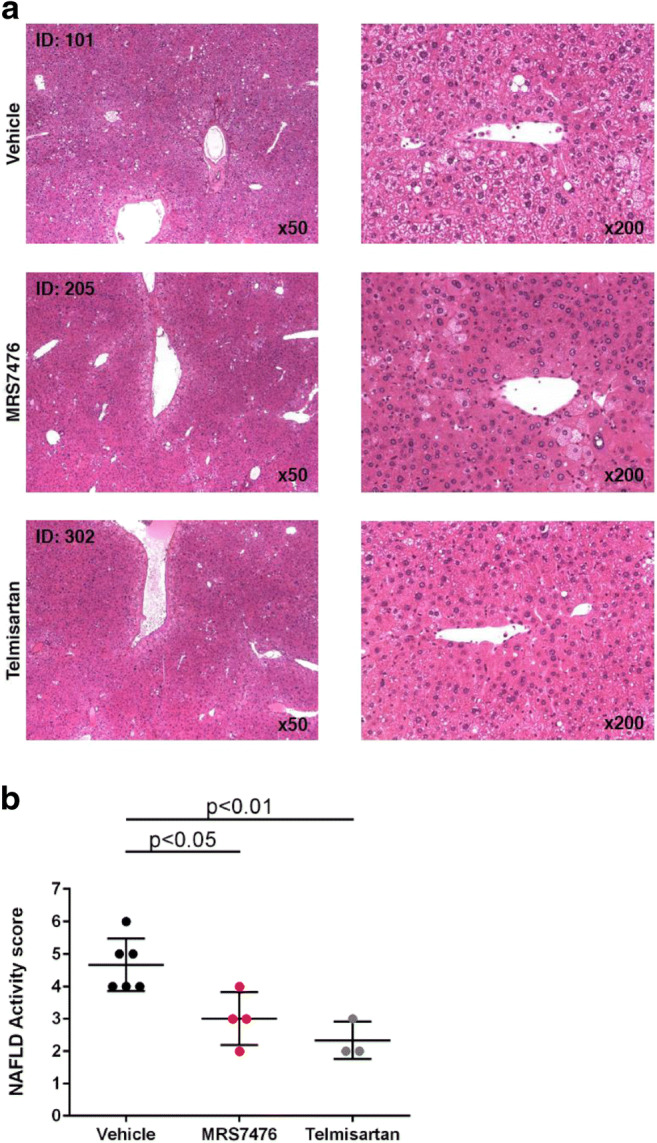
Histological effects in the liver in STAM mice following administration of MRS7476 (5 mg/kg, p.o., twice daily, for 3 weeks) or telmisartan included as a positive control. Data corrected using the Bonferroni multiple comparison test
Fig. 6.

Biochemical measures of damage in the liver of STAM mice following administration of MRS7476 (5 mg/kg, p.o., twice daily, for 3 weeks) or telmisartan included as a positive control. Statistics used the Bonferroni multiple comparison test
Fig. 7.

Scoring of liver damage in STAM mice. The A3AR agonist prodrug MRS7476 was compared with vehicle alone or telmisartan included as a positive control
There was no impact of treatment on hepatic TGFβ and ACTA2 expression [35, 36]. Plasma IL-10 levels were increased in MRS7476-treated animals (Fig. 6B; p = 0.01). Two of 6 animals treated with MRS7476 (and none treated with vehicle) developed significant weight loss starting at day 10 and 14 of treatment and were found dead on day 17 and 18, respectively (Figure S5, Supporting Information). Notably, these two mice had markedly lower baseline body weights compared with the other animals and to the control group. The cause for excess mortality is not clear, and the dead animals were not included in analyses. The weight drop on the last day in the other animals was likely due to the blood volume required for the OGTT test (~1% of body weight), rather than an A3 agonist-related effect, as the safety of chronically administered A3 agonists has been demonstrated [7–9, 11]..
Enhanced hepatic inflammation and steatosis in A3Δ/Δ mice fed HFD
Obesity is associated with enhanced risk of developing nonalcoholic fatty liver disease (NAFLD). Development of liver steatosis and secondary chronic liver inflammation are key factors contributing to metabolic disorders. To examine the effect of the A3AR on the development of liver steatosis and inflammation associated with obesity, we subjected control and A3AR knockout mouse lines to HFD for the period of 32 weeks. mRNA levels of inflammatory markers in liver tissue of A3Δ/Δ mice and WT mice maintained on HFD were examined by RT-PCR. Monocyte chemoattractant protein 1 (Mcp1) mRNA levels were increased in the liver of A3Δ/Δ mice (Fig. 8). Accordingly, the gene expression levels of macrophage marker, cluster of differentiation 68 (Cd68), was increased in the liver of A3Δ/Δ mice. Whole body knockout of A3AR also resulted in significant increase in mRNA levels of macrophage inflammatory protein-1 alpha (Mip1a) in liver tissue (Fig. 8). Gene expression of pro-inflammatory cytokines such as interleukin 6 (Il6) and tumor necrosis factor alpha (Tnfa) was increased in the liver of A3Δ/Δ mice (Fig. 8). No differences were observed in mRNA levels of genes coding for interferon-gamma (Ifng), interleukin 1-alpha (Il1a), interleukin 1-beta (Il1b), and macrophage inflammatory protein-1 (Mip1b) (Fig. 8). Our data suggest enhanced expression of inflammation markers in livers of mice lacking functional A3AR. Further, liver tissues from HFD A3Δ/Δ mice and WT mice were sectioned and HE stained. Thus, the lack of A3AR may enhance the severity of liver steatosis and secondary hepatic inflammation associated with obesogenic diet.
Fig. 8.

Enhanced inflammation and steatosis in the liver of HFD A3Δ/Δ mice. A Real-time PCR analysis of genes coding for hepatic inflammatory markers. 18s RNA was taken as a normalization control. All data are given as mean + SEM with *p < 0.05 using two-tailed, unpaired Student’s t test (n = 3–5, each group). B HE Stain of liver sections from WT and A3Δ/Δ mice kept on HFD. Microscopic analysis using × 20 magnification
Discussion
The objective was to demonstrate that we could reversibly mask, in a highly water-soluble form, known A3AR agonists using a common chemical approach that potentially applies to both riboside agonists and (N)-methanocarba derivatives. One of the prodrugs was tested by oral administration in two in vivo mouse models to demonstrate its action in two known activities of A3AR agonists, i.e., anti-hyperaglesia in the chronic pain state and protection in NASH. We showed that the otherwise poorly soluble parent drug, MRS5698, was formed by the action of esterases, such as are plentiful in vivo. In vitro, MRS7422 was more readily hydrolyzed enzymatically by liver esterase in vitro compared with the rigid bicyclic methanocarba derivative MRS7476. We did not determine the rate of cleavage by intestinal or gastric epithelial esterases, but these tissues are likely sites for hydrolysis and absorption, by analogy to a published study of such esterases acting on chlorogenic acid esters present in coffee [41]. Also, the rate of hydrolysis within mouse tissues leading to the real time reversal of chronic neuropathic pain within 1 h cannot be compared quantitatively to the slower kinetics in the model in vitro study of PLE hydrolysis of MRS7476.
NASH is a progressive disease often associated with obesity that can lead to cirrhosis and liver cancer, and its incidence is rapidly increasing [42]. Thus, the cost to society will be enormous, and novel, effective pharmacological treatments are needed. Drugs in clinical trials were recently reviewed [43]. NASH patients typically display histological liver pathology, such as cytoskeletal damage (hepatocyte ballooning), steatosis, and inflammation. Fibrosis is often associated with the later stages of the condition. The chemically induced STAM model has become a standard in the field of drug development for NASH treatment [44, 45]. In a study of the effects of the parent drug Cl-IB-MECA in the STAM model, NASH was decreased with reduction of both inflammatory and steatotic parameters [15]. In addition, use of the same agonist in the carbon tetrachloride model, improved liver inflammation and fibrosis and normalized alanine aminotransferase (ALT), to clearly demonstrate an anti-NASH effect of a selective A3AR agonist [15]. One of the A3AR agonist prodrugs synthesized here, MRS7476, was active in vivo in improving NASH. We have shown here that hepatic stellate cells, the main effectors of hepatic fibrosis, have much higher expression levels of mRNA encoding the A3AR than hepatocytes. We did not establish if these expression levels are altered in the NASH mouse model or in A3AR agonist-treated mice.
In the STAM model of NASH, treatment with MRS7476 showed a significant improvement in the NAFLD activity score (NAS), driven mainly by a remarkable reduction in hepatocyte ballooning. In addition, plasma IL-10 level was increased in the MRS7476 group compared with the vehicle group. This result suggests that MRS7476 has anti-NASH potential in this model. We have not determined whether the prodrug derivatization of MRS5698 led to its lack of hepatoprotective effects on liver enzymes and reduced fibrosis in this NASH model, as was seen with another A3AR agonist, Cl-IB-MECA [15]. Further characterization including in vivo pharmacodynamic analysis of prodrug MRS7476 would be appropriate.
Conclusions
We have introduced 2′,3′-di-succinylation of potent and selective A3AR agonists as a reversible prodrug strategy. This approach greatly increased water solubility in an otherwise difficult to dissolve compound and allowed us to observe salutary activity of masked A3AR agonists, i.e., prodrugs for unmasking in vivo, in disease models already associated with beneficial A3AR action. We have shown that both ester bonds on each molecule are cleaved by liver esterase in vitro, and by deduction in vivo. Of the two compounds prepared, the riboside MRS7422 was more readily hydrolyzed enzymatically in vitro compared with the rigid bicyclic methanocarba derivative MRS7476. We then subjected this prodrug approach to models of two disease conditions, in which the drug was administered orally to simulate this treatment modality for chronic human diseases. However, the longer duration of protection by MRS7476 compared with MRS7422 in a real-time in vivo assay of mechano-allodynia prompted us to examine the methanocarba derivative MRS7476 in a test of protection against NASH [46], and a benefit was observed. The unusually high selectivity of the parent drug MRS5698 offers confidence that A3AR is responsible, along with evidence of receptor expression in liver stellate cells and a gene expression pattern consistent with increased injury in A3AR knockout mouse liver. Selective A3AR agonists are already in clinical trials for inflammatory diseases (IB-MECA) and liver diseases (Cl-IB-MECA), including NASH, and are proposed for the future treatment of chronic pain. Thus, we have expanded the toolbox of useful nucleoside derivatives for studying these and other actions of A3AR agonists in vivo.
Electronic supplementary material
(PDF 2.35 MB)
Abbreviations
- ACTA2
Smooth muscle α actin
- ALT
Alanine transaminase
- AR
Adenosine receptor
- AUC
Area under the curve
- CCI
Chronic constriction injury
- Cl-IB-MECA
2-Chloro-N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide
- GPCR
G protein-coupled receptor
- HCC
Hepatocellular carcinoma
- IB-MECA
N6-(3-iodobenzyl)-adenosine-5′-N-methyluronamide
- IL-10
Interleukin 10
- MRS5698
(1S,2R,3S,4R,5S)-4-(6-((3-chlorobenzyl)amino)-2-((3,4-difluorophenyl)ethynyl)-9H-purin-9-yl)-2,3-dihydroxy-N-methylbicyclo[3.1.0]hexane-1-carboxamide
- NAFLD
Non-alcoholic fatty liver disease
- NAS
NAFLD activity score
- NASH
Non-alcoholic steatohepatitis
- NLRP3
Nucleotide-binding oligomerization domain-like receptor protein 3 (inflammasome)
- OGTT
Oral glucose tolerance test
- SAR
Structure activity relationship
- TGFβ
Transforming growth factor β
Funding information
This work has been supported by funds from SLU (Salvemini Start-Up Funds) and NIDDK Intramural Research Program (Jacobson, ZIADK31117; Rotman, ZIADK75013).
Compliance with ethical standards
Conflict of interest
All other authors claim no conflicts of interest.
Ethical approval
All procedures were conducted in accordance with the International Association for the Study of Pain, the National Institutes of Health guidelines on laboratory animal welfare guidelines, and the approval of Saint Louis University and NIDDK Institutional Animal Care and Use Committees.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
R. Rama Suresh, Email: rama.ravi@nih.gov.
Shanu Jain, Email: shanu.jain@nih.gov.
Zhoumou Chen, Email: zhoumou.chen@health.slu.edu.
Dilip K. Tosh, Email: dilipkumar.tosh@nih.gov
Yanling Ma, Email: mayl2007@126.com.
Maren C. Podszun, Email: maren.podszun@gmail.com
Yaron Rotman, Email: rotmany@niddk.nih.gov.
Daniela Salvemini, Email: daniela.salvemini@health.slu.edu.
Kenneth A. Jacobson, Email: kennethj@niddk.nih.gov
References
- 1.Cekic C, Linden J. Purinergic regulation of the immune system. Nat Rev Immunol. 2016;16:177–192. doi: 10.1038/nri.2016.4. [DOI] [PubMed] [Google Scholar]
- 2.Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nature Rev Drug Disc. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varani K, Vincenzi F, Merighi S, Gessi S, Borea PA (2017) Biochemical and pharmacological role of A1 adenosine receptors and their modulation as novel therapeutic strategy. Adv Exp Med Biol - Protein Reviews pp. 193–232, DOI 10.1007/5584_2017_61 [DOI] [PubMed]
- 4.Jacobson KA, Merighi S, Varani K, Borea PA, Baraldi S, Tabrizi MA, Romagnoli R, Baraldi PG, Ciancetta A, Tosh DK, Gao ZG, Gessi S. A3 adenosine receptors as modulators of inflammation: from medicinal chemistry to therapy. Med Res Rev. 2017;38:1031–1072. doi: 10.1002/med.21456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Preti D, Baraldi PG, Moorman AR, Borea PA, Varani K. History and perspectives of A2A adenosine receptor antagonists as potential therapeutic agents. Med Res Rev. 2015;35:790–848. doi: 10.1002/med.21344. [DOI] [PubMed] [Google Scholar]
- 6.Gao ZG, Balasubramanian R, Kiselev E, Wei Q, Jacobson KA. Probing biased/partial agonism at the G protein-coupled A2B adenosine receptor. Biochem Pharmacol. 2014;90:297–306. doi: 10.1016/j.bcp.2014.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cohen S, Barer F, Itzhak I, Silverman MH, Fishman P. Inhibition of IL-17 and IL-23 in human keratinocytes by the A3 adenosine receptor agonist piclidenoson. J Immunol Res. 2018;2018:2310970. doi: 10.1155/2018/2310970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoilov RM, Licheva RN, Mihaylova MK, Reitblat T, Dimitrov EA, Shimbova KM, Bhatia G, Pispati A, Gurman-Balbir A, Bagaria BR, Oparanov BA, Fishman S, Harpaz Z, Farbstein M, Cohen S, Bristol D, Silverman MH, Fishman P. Therapeutic effect of oral CF101 in patients with rheumatoid arthritis: a randomized, double-blind, placebo-controlled phase II study. Immunome Res. 2014;11:1. doi: 10.4172/1745-7580.1000087. [DOI] [Google Scholar]
- 9.Stemmer M, Benjaminov F, Medalia G, Ciuraru NB, Silverman MH, Bar-Yehuda S, Fishman S, Harpaz Z, Farbstein M, Cohen S, Patoka R, Singer B, Kerns WD, Fishman P. CF102 for the treatment of hepatocellular carcinoma: a phase I/II, open-label, dose-escalation study. Oncologist. 2013;18:25–26. doi: 10.1634/theoncologist.2012-0211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohana G, Cohen S, Rath-Wolfson L, Fishman P. A3 adenosine receptor agonist, CF102, protects against hepatic ischemia/reperfusion injury following partial hepatectomy. Mol Med Rep. 2016;14(5):4335–4341. doi: 10.3892/mmr.2016.5746. [DOI] [PubMed] [Google Scholar]
- 11.David M, Gospodinov DK, Gheorghe N, Mateev GS, Rusinova MV, Hristakieva E, Solovastru LG, Patel RV, Giurcaneanu C, Hitova MC, Purcaru AI, Horia B, Tsingov II, Yankova RK, Kadurina MI, Ramon M, Rotaru M, Simionescu O, Benea V, Demerdjieva ZV, Cosgarea MR, Morariu HS, Michael Z, Cristodor P, Nica C, Silverman MH, Bristol DR, Harpaz Z, Farbstein M, Cohen S, Fishman P. Treatment of plaque-type psoriasis with oral CF101: data from a phase II/III multicenter, randomized, controlled trial. J Drugs Dermatol. 2016;15(8):931–938. [PubMed] [Google Scholar]
- 12.Fishman P, Cohen S. The A3 adenosine receptor (A3AR): therapeutic target and predictive biological marker in rheumatoid arthritis. Clin Rheumatol. 2016;35:2359–2362. doi: 10.1007/s10067-016-3202-4. [DOI] [PubMed] [Google Scholar]
- 13.Ochaion A, Bar-YehudaS CS, Barer F, Patoka R, Amital H, Reitblat T, Reitblat A, Ophir J, Konfino I, Chowers Y, Ben-Horin S, Fishman P. The anti-inflammatory target A3 adenosine receptor is over-expressed in rheumatoid arthritis, psoriasis and Crohn’s disease. Cell Immunol. 2009;258:115–122. doi: 10.1016/j.cellimm.2009.03.020. [DOI] [PubMed] [Google Scholar]
- 14.Varani K, Padovan M, Vincenzi F, Vincenzi F, Targa M, Trotta F, Govoni M, Borea PA. A2A and A3 adenosine receptor expression in rheumatoid arthritis: upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res Ther. 2011;13:R197. doi: 10.1186/ar3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fishman P, Cohen S, Itzhak I, Amer J, Salhab A, Barer F, Safadi R. The A3 adenosine receptor agonist, namodenoson, ameliorates non-alcoholic steatohepatitis in mice. Int J Mol Med. 2019;44(6):2256–2264. doi: 10.3892/ijmm.2019.4364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fishman P, Cohen SA (2017) A3 adenosine receptor ligand for use in treating ectopic fat accumulation. WO2017090036 (A1)
- 17.Miao CG, Yang YY, He X, Huang C, Huang Y, Zhang L, Lv XW, Jin Y, Li J. Wnt signaling in liver fibrosis: Progress, challenges and potential directions. Biochimie. 2013;95:2326–2335. doi: 10.1016/j.biochi.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 18.Janes K, Symons-Liguori AM, Jacobson KA, Salvemini D. Identification of A3 adenosine receptor agonists as novel non-narcotic analgesics. Br J Pharmacol. 2016;173:1253–1267. doi: 10.1111/bph.13446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tosh DK, Padia J, Salvemini D, Jacobson KA. Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain. Purinergic Signal. 2015;11:371–387. doi: 10.1007/s11302-015-9459-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wahlman C, Doyle T, Little JW, Luongo L, Janes K, Chen Z, Espostio E, Tosh DK, Cuzzocrea S, Jacobson KA, Salvemini D. Chemotherapy-induced pain is promoted by enhanced spinal adenosine kinase levels via astrocyte-dependent mechanisms. Pain. 2018;159:1025–1034. doi: 10.1097/j.pain.0000000000001177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tosh DK, Finley A, Paoletta S, Moss SM, Gao ZG, Gizewski E, Auchampach J, Salvemini D, Jacobson KA. In vivo phenotypic screening for treating chronic neuropathic pain: modification of C2-arylethynyl group of conformationally constrained A3 adenosine receptor agonists. J Med Chem. 2014;57:9901–9914. doi: 10.1021/jm501021n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tosh DK, Deflorian F, Phan K, Gao ZG, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Structure-guided design of A3 adenosine receptor-selective nucleosides: combination of 2-arylethynyl and bicyclo[3.1.0]hexane substitutions. J Med Chem. 2012;55:4847–4860. doi: 10.1021/jm300396n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rautio J, Meanwell N, Di L, Hageman MJ. The expanding role of prodrugs in contemporary drug design and development. Nat Rev Drug Discov. 2018;17:559–587. doi: 10.1038/nrd.2018.46. [DOI] [PubMed] [Google Scholar]
- 24.Sheahan TP, Sims AC, Zhou S, Graham RL, Pruijssers AJ, Agostini ML, Leist SR, Schäfer A, Dinnon KH, III, Stevens LJ, Chappell JD, Lu X, Hughes TM, George AS, Hill CS, Montgomery SA, Brown AJ, Bluemling GR, Natchus MG, Saindane M, Kolykhalov AA, Painter G, Harcourt J, Tamin A, Thornburg NJ, Swanstrom R, Denison MR, Baric RS. An orally bioavailable broad-spectrum antiviral inhibits SARS-CoV-2 in human airway epithelial cell cultures and multiple coronaviruses in mice. Sci Transl Med. 2020;12(541):eabb5883. doi: 10.1126/scitranslmed.abb5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Besada P, Mamedova LK, Palaniappan KK, Gao ZG, Joshi BV, Jeong LS, Civan MM, Jacobson KA. Nucleoside prodrugs of A3 adenosine receptor agonists and antagonists. Collect Czechoslov Chem Commun. 2006;71:912–928. doi: 10.1135/cccc20060912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- 27.Jain S, Tosh DK, Reitman ML, Jacobson KA (2019) Role of A1 and A3 adenosine receptors in whole body glucose metabolism. ADA abstract 280-LB. 10.2337/db19-280-LB
- 28.Dittert LW, Caldwell HC, Adams HJ, Irwin GM, Swintosky JV. Acetaminophen prodrugs I. synthesis, physicochemical properties, and analgesic activity. J Pharm Sci. 1968;57(5):774–780. doi: 10.1002/jps.2600570510. [DOI] [PubMed] [Google Scholar]
- 29.Fu Q, Wang Y, Ma Y, Zhang D, Fallon JK, Yang X, Liu D, He Z, Liu F. Programmed hydrolysis in designing paclitaxel prodrug for nanocarrier assembly. Sci Rep. 2015;5(1):12023. doi: 10.1038/srep12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Biasutto L, Marotta E, Bradaschia A, Fallica M, Mattarei A, Garbisa S, Zoratti M, Paradisi C. Soluble polyphenols: synthesis and bioavailability of 3,4′,5-tri (alpha-D-glucose-3-O-succinyl) resveratrol. Bioorg Med Chem Lett. 2009;19:6721–6724. doi: 10.1016/j.bmcl.2009.09.114. [DOI] [PubMed] [Google Scholar]
- 31.Wichitnithad W, Nimmannit U, Wacharasindhu S, Rojsitthisak P. Synthesis, characterization and biological evaluation of succinate prodrugs of curcuminoids for colon cancer treatment. Molecules. 2011;16:1888–1900. doi: 10.3390/molecules16021888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vollmann K, Qurishi R, Hockemeyer J, Müller CE. Synthesis and properties of a new water-soluble prodrug of the adenosine A2A receptor antagonist MSX-2. Molecules. 2008;13(2):348–359. doi: 10.3390/molecules13020348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Melman A, Gao ZG, Kumar D, Wan TC, Gizewski E, Auchampach JA, Jacobson KA. Design of (N)-methanocarba adenosine 5′-uronamides as species-independent A3 receptor-selective agonists. Bioorg Med Chem Lett. 2008;18:2813–2819. doi: 10.1016/j.bmcl.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Besnard J, Ruda GF, Setola V, Abecassis K, Rodriguiz RM, Huang XP, Norval S, Sassano MF, Shin AI, Webster LA, Simeons FR, Stojanovski L, Prat A, Seidah NG, Constam DB, Bickerton GR, Read KD, Wetsel WC, Gilbert IH, Roth BL, Hopkins AL. Automated design of ligands to polypharmacological profiles. Nature. 2012;492:215–220. doi: 10.1038/nature11691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gupta P, Sata TN, Yadav AK, Mishra A, Vats N, Hossain MM, Sanal MG, Venugopal SK. TGF-β induces liver fibrosis via miRNA-181a-mediated down regulation of augmenter of liver regeneration in hepatic stellate cells. PLoS One. 2019;14(6):e0214534. doi: 10.1371/journal.pone.0214534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rockey DC, Du Q, Shi Z. Smooth muscle α-actin deficiency leads to decreased liver fibrosis via impaired cytoskeletal signaling in hepatic stellate cells. Am J Pathol. 2019;189(11):2209–2220. doi: 10.1016/j.ajpath.2019.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yokohama K, Fukunishi S, Ii M, Nakamura K, Ohama H, Tsuchimoto Y, Asai A, Tsuda Y, Higuchi K. Rosuvastatin as a potential preventive drug for the development of hepatocellular carcinoma associated with non-alcoholic fatty liver disease in mice. Int J Mol Med. 2016;38:1499–1506. doi: 10.3892/ijmm.2016.2766. [DOI] [PubMed] [Google Scholar]
- 38.Takakura K, Koido S, Fujii M, Hashiguchi T, Shibazaki Y, Yoneyama H, Katagi H, Kajihara M, Misawa T, Homma S, Ohkusa T, Tajiri H. Characterization of non-alcoholic steatohepatitis-derived hepatocellular carcinoma as a human stratification model in mice. Anticancer Res. 2014;34:4849–4855. [PubMed] [Google Scholar]
- 39.Takakura K, Oikawa T, Tomita Y, Mizuno Y, Nakano M, Saeki C, Torisu Y, Saruta M. Mouse models for investigating the underlying mechanisms of nonalcoholic steatohepatitis-derived hepatocellular carcinoma. World J Gastroenterol. 2018;24(18):1989–1994. doi: 10.3748/wjg.v24.i18.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park JG, Mok JS, Han YI, Park TS, Kang KW, Choi CS, Park HD, Park J. Connectivity mapping of angiotensin-PPAR interactions involved in the amelioration of non-alcoholic steatohepatitis by Telmisartan. Sci Rep. 2019;9:4003. doi: 10.1038/s41598-019-40322-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Farrell TL, Dew TP, Poquet L, Hanson P, Williamson G. Absorption and metabolism of chlorogenic acids in cultured gastric epithelial monolayers. Drug Metab Dispos. 2011;39(12):2338–2346. doi: 10.1124/dmd.111.040147. [DOI] [PubMed] [Google Scholar]
- 42.Bedossa P. Pathology of non-alcoholic fatty liver disease. Liver Int. 2017;37:85–89. doi: 10.1111/liv.13301. [DOI] [PubMed] [Google Scholar]
- 43.Romero FA, Jones C, Xu Y, Fenaux M, Halcomb RL. The race to bash NASH: emerging targets and drug development in a complex liver disease. J Med Chem. 2020;63:5031–5073. doi: 10.1021/acs.jmedchem.9b01701. [DOI] [PubMed] [Google Scholar]
- 44.Hansen HH, Feigh M, Veidal SS, Rigbolt KT, Vrang N, Fosgerau K. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today. 2017;22:1707–1718. doi: 10.1016/j.drudis.2017.06.007. [DOI] [PubMed] [Google Scholar]
- 45.Oniciu DC, Hashiguchi T, Shibazaki Y, Bisgaier CL. Gemcabene downregulates inflammatory, lipid-altering and cell-signaling genes in the STAM™ model of NASH. PLoS One. 2018;13(5):e0194568. doi: 10.1371/journal.pone.0194568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanyal AJ, Brunt EM, Kleiner DE, Kowdley KV, Chalasani N, Lavine JE, Ratziu V, McCullough A. Endpoints and clinical trial design for nonalcoholic steatohepatitis. Hepatology. 2011;54:344–353. doi: 10.1002/hep.24376. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PDF 2.35 MB)