

Abstract
Alzheimer's disease (AD) is tightly linked to oxidative stress since amyloid beta-peptide (Aβ) aggregates generate free radicals. Moreover, the aggregation of Aβ is increased by oxidative stress, and the neurotoxicity induced by the oligomers and fibrils is in part mediated by free radicals. Interestingly, it has been reported that oxidative stress can also induce BACE1 transcription and expression. BACE1 is the key enzyme in the cleavage of the amyloid precursor protein to produce Aβ, and the expression of this enzyme has been previously shown to be enhanced in the brains of Alzheimer's patients. Here, we have found that BACE1 expression is increased in the hippocampi from AD patients at both the early (Braak stage II) and late (Braak stage VI) stages of the disease as studied by immunohistochemistry and western blot. To address the role of Aβ and oxidative stress in the regulation of BACE1 expression, we have analyzed the effect of subtoxic concentrations of Aβ oligomers (0.25 μM) and H2O2 (10 mM) on a human neuroblastoma cell line. Firstly, our results show that Aβ oligomers and H2O2 induce an increase of BACE1 mRNA as we studied by qPCR. Regarding BACE1 translation, it is dependent on the phosphorylation of the eukaryotic initiation factor 2α (eIF2α), since BACE1 mRNA bears a 5′UTR that avoids its translation under basal conditions. BACE1 5′UTR contains four upstream initiating codons (uAUGs), and its translation is activated when eIF2α is phosphorylated. Consistently, we have obtained that Aβ oligomers and H2O2 increase the levels of BACE1 and p-eIF2α assayed by western blot and confocal microscopy. Our results suggest that Aβ oligomers increase BACE1 translation by phosphorylating eIF2α in a process that involves oxidative stress and conforms a pathophysiological loop, where the Aβ once aggregated favors its own production continuously by the increase in BACE1 expression as observed in AD patients.
1. Introduction
Alzheimer's disease (AD) is a neurodegenerative process that occurs in the elderly being the most prevalent type of human dementia. AD symptoms consist in dramatic memory deficits and an irreversible cognitive decline. They start with neuronal death in the hippocampus and the brain structure for learning and memory, and later, the neuronal loss progresses to other cortical areas. The histopathological characteristics of AD patients are extracellular senile plaques and intracellular neurofibrillary tangles. The senile plaques are mainly composed of amyloid β-protein (Aβ) and the neurofibrillary tangles of hyperphosphorylated tau protein [1, 2].
Aβ is a peptide having from 36 to 43 amino acids [3]; however, Aβ1-40 is the most abundant. Aβ is produced by the consecutive enzymatic action of the beta- and gamma-secretase activities on an integral type I transmembrane glycoprotein termed amyloid precursor protein (APP) [4]. Aβ1-42 production increases with aging [5], in patients of familiar AD bearing some mutations in the presenilins (PS1/PS2) [6], which are the catalytic core of the gamma-secretase complex, or due to the nitrotyrosination of the gamma-secretase complex [7]. The pathophysiological relevance of this shift in the Aβ species production is due to the high aggregant properties of the Aβ1-42 [8, 9], which makes it more neurotoxic.
The oligomeric forms are the responsible for the most neurotoxic effects associated to the Aβ [10–12]. Aβ aggregates produce H2O2 [13] inducing oxidative stress and starting an intracellular cascade of reactive oxygen species, which will compromise neuronal viability [14, 15]. Moreover, oxidative stress has been reported to increase the transcription and expression of the enzyme Beta-site Amyloid precursor protein Cleaving Enzyme 1 (BACE1) [16, 17]. BACE1 is the key enzyme that carries out the beta-secretase activity on APP to initiate the Aβ production pathway.
BACE1 expression is physiologically repressed at the transcriptional and translational level. There are different pathways, mostly related to different types of stress, that induce the nuclear translocation of transcription factors, such as cJun/cFos, to bind to BACE1 gene promoter [18, 19]. However, the tightest control of BACE1 is at the translational level since BACE1 mRNA bears a particular 5′UTR that contains four upstream initiation codons (uAUGs) and possesses a rich GC content. These sequence motifs confer a particular secondary structure to that region of the mRNA that impairs ribosomes to reach the main AUG in order to start BACE1 translation [20–23]. In basal conditions, ribosomes remain in one of the uAUG, especially the second one. BACE1 mRNA translation is only activated when the eukaryotic initiation factor 2α (eIF2α) is phosphorylated at serine 51 [19, 24–26]. The physiological relevance of the eIF2α phosphorylation is to block the translation of the most of the proteins under stressful conditions and to induce only the translation of a group of special proteins that bear 3 or more uAUGs in the 5′UTR [27]. These stressful conditions are virus infection, reticular stress, nutrient deprivation, or oxidative stress. There are four eIF2α kinases: heme-regulated eukaryotic initiation (HRI), general control nonderepressible 2 kinase (GCN2), double-stranded RNA-activated protein kinase (PKR), and double-stranded RNA-activated protein kinase-like (PERK), which phosphorylate eIF2α at serine 51 blocking translation initiation [25–30].
2. Materials and Methods
2.1. BACE1 Expression Study by Immunohistochemistry in Human Hippocampi
Human hippocampal samples were supplied by the Neurological Tissue Bank of the Biobank-Hospital Clínic-IDIBAPS, Barcelona, Spain. The procedure was carried out according to the rules of the Helsinki Declaration and to the Ethics Committee of the Institut Municipal d'Investigacions Mèdiques-Universitat Pompeu Fabra (EC-IMIM-UPF). Hippocampal samples were obtained from 3 nondemented controls (1 man and 2 women; mean: 69 years old), 2 AD patients at Braak stage II (2 men; 83 and 76 years old), and 4 AD patients at Braak stage VI (2 men and 2 women; mean: 70 years old). Sections (5 μm) were treated with 4% H2O2 and incubated o.n. at 4°C with 1 : 100 rabbit anti-BACE1 Antibody (Ab; Invitrogen). The secondary Ab was 1 : 500 donkey anti-rabbit peroxidase-conjugated Ab, which was incubated for 1 h at room temperature (RT). A Peroxidase Substrate Kit DAB (Vector) was used. Slides were stained with hematoxylin and fixed. The images were taken by a Leica DMR microscope.
2.2. BACE1 Expression Study by Western Blot in Human Hippocampi
Human brain tissue sections, obtained as indicated in the subsection 2.1., were lysed with 50 μL RIPA buffer: 150 mM sodium chloride, 1% Triton X-100 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 50 mM Tris-HCl, 1 mM dithiothreitol, 1 mM sodium orthovanadate, and protease inhibitor cocktail (Roche), pH 8. Membranes were blocked for 1 h at RT with Tween 20-Tris buffer solution (TTBS; 100 mM Tris-HCl, 150 mM NaCl, pH 7.5) plus 5% skimmed milk. Then, membranes were incubated overnight (o.n.) at 4°C with 1 : 4,000 mouse anti-actin Ab (Sigma) or 1 : 1,000 rabbit anti-BACE1 Ab. 1 : 2,000 secondary Abs were horseradish peroxidase-conjugated donkey anti-mouse and anti-rabbit (GE Healthcare), which were incubated for 1 h at RT. Bands were visualized with Super Signal (Pierce) and analyzed with the Quantity One system in a BioRad Universal Hood II.
2.3. Cell Line
Human neuroblastoma cells (SH-SY5Y cells) were grown with Ham's F12 GlutaMax (F12 medium; Gibco) supplemented with 15% fetal bovine serum (FBS; Gibco) and 1% penicillin/streptomycin (Gibco). Cells were incubated at 37°C in a humidified atmosphere of 5% CO2.
2.4. Aβ Oligomer Formation
1 mg of lyophilized human Aβ1-42 (Anaspec) went to solution with 250 μL of ultrapure MilliQ water. Therefore, we adjusted pH to ≥10.5 with NaOH (1 M) to keep away from the Aβ isoelectric point. The solution was sonicated for 1 min in 250 μL of phosphate buffer (20 mM; pH 7.4). To favor the proper oligomer formation, the Aβ aliquots were dissolved in F12 medium at 0.4 mg/mL and incubated for 24 at 4°C.
2.5. Cell Viability Studies
SH-SY5Y cells were seeded in a 96-well plate at a density of 2.5 × 104 cells/well. After 12 h, the growth medium was removed. Cells were treated with increasing concentrations of Aβ1-42 oligomers or H2O2 (Sigma) in F12 medium. Treatments were carried out along 24 h at 37°C. Then, cell survival was assayed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction method. It consists of the addition of 10% (regarding cell medium volume) of MTT stock solution at 5 mg/mL. Cells were incubated with the MTT for 2 h. Medium was discarded and 100 μL of DMSO were placed per well. MTT absorbance was measured in an absorbance plate reader (BioRad) at A540 nm and A650 nm (as reference). Control cells treated with phosphate buffer saline (PBS) were the 100%. Bridge field images were obtained with a Leica DM IL microscope.
2.6. Apoptosis Studies
Cells were seeded on coverslips in 24-well plates at a density of 3 × 104 cells/well. After 12 h, the growth medium was removed and cells were treated for 24 h with subtoxic concentrations of Aβ1-42 oligomers (0.25 μM) or H2O2 (10 μM) and the toxic ones for Aβ1-42 oligomers (15 μM) and H2O2 (100 μM) in F12 medium. Cells were fixed with 4% paraformaldehyde (PFA) and permeabilized with 0.1% Triton X-100 at RT. Coverslips were incubated o.n. at 4°C with 1 : 100 rabbit anti-cleaved Caspase-3 Ab (Cell Signalling). Cells were washed thrice and incubated with 1 : 2,000 Alexa Fluor 555 goat anti-rabbit Ab (Life Technologies) and Hoechst 1 : 10,000 (Thermo Scientific) for 1 h at RT. Coverslips were mounted with Fluoromount (Southern Biotech). Digital images were taken with a Leica TCS SP confocal microscope and analyzed with the Leica confocal software.
2.7. Transcriptional Studies in Cells
Cells were seeded on 60 mm Petri dishes at a density of 6 × 105 cells/dish. After 12 h, the growth medium was removed and cells were treated for 24 h with subtoxic concentrations of 0.25 μM Aβ1-42 oligomers or 10 μM H2O2 in F12 medium. The mRNA was extracted by using the NucleoSpin RNA extraction kit (Macherey Nagel) and quantified with NanoDrop ND-1000 (Thermo Fisher Scientific). BACE1 and HPRT cDNAs were obtained with the SuperScript III Reverse Transcriptase (Invitrogen). Finally, a quantitative PCR was performed by using the fluorophore Sybr Green (Thermo Fisher Scientific). Samples were quantified with QuantStudio 12K Flex Real-Time PCR System (Thermo Fisher Scientific).
2.8. Translational Studies by Western Blot in Cells
Cells were seeded on 6-well plates at a density of 3 × 105 cells/well. After 12 h, the growth medium was removed and cells were treated for 24 h with subtoxic concentrations of 0.25 μM Aβ1-42 oligomers or 10 μM H2O2 in F12 medium. To study phosphorylated proteins, the cells were lysed on ice with lysis solution as indicated in subsection 2.2. Extracts were homogenised using vortex for 30 min at 4°C; afterwards, samples were centrifuged at 10,000 for 5 min to obtain the supernatant. A Bio-Rad kit was used to calculate protein concentrations. Aliquots of 20 μL (for phosphorylated proteins) or 80 μg (for the other proteins) were loaded into a 10% SDS-PAGE gels. Afterwards, proteins were transferred onto 0.2 μm pore nitrocellulose membranes. Membranes were blocked for 1 h at RT with TTBS plus 5% bovine serum albumin (BSA) for phosphorylated proteins or 5% skimmed milk for the other proteins. Then, membranes were incubated o.n. at 4°C with 1 : 1,000 rabbit anti-BACE1 Ab (Invitrogen), 1 : 500 rabbit anti-p-eIF2α (Ser51) Ab (Invitrogen), 1 : 500 mouse anti-eIF2α Ab (Abcam), and 1 : 5,000 mouse anti-tubulin Ab (Sigma). 1 : 2,000 secondary Abs were horseradish peroxidase-conjugated donkey anti-mouse and anti-rabbit (GE Healthcare) for 1 h at RT. Bands were visualized with Super Signal (Pierce) and analyzed with the Quantity One system in a BioRad Universal Hood II.
2.9. Translational Studies by Confocal Microscopy in Cells
Cells were seeded on coverslips in 24-well plates at a density of 3 × 104 cells/well. After 12 h, the growth medium was removed, and cells were treated for 24 h with subtoxic concentrations of 0.25 μM Aβ1-42 oligomers or 10 μM H2O2 in F12 medium plus 15% FBS. Then, cells were fixed with 4% paraformaldehyde (PFA). Cells were permeabilized with 0.1% Triton X-100. Coverslips were incubated o.n. at 4°C with 1 : 100 mouse anti-eIF2α Ab, rabbit anti-p-eIF2α Ab, or rabbit anti-BACE1 Ab. After primary Abs, cells were incubated with 1 : 2,000 Alexa Fluor 555 goat anti-rabbit Ab or 1 : 2,000 Alexa Fluor 647 goat anti-mouse Ab (Life Technologies) for 1 h at RT. Coverslips were mounted with Fluoromount. Digital images were taken with a Leica TCS SP confocal microscope and analyzed with Leica confocal software. Immunofluorescence was quantified by ImageJ program.
2.10. Statistical Analysis
Data are expressed as mean ± SEM of n experiments as indicated in the corresponding figures. Statistical analyses were performed by one-way ANOVA using Tukey's posttest or Student's t-test using the GraphPad software.
3. Results
3.1. BACE1 Expression Is Increased in the Hippocampi from AD Patients
There are previous works that reported increased expression of BACE1 in AD patients [31–33] relating this finding with the enhanced Aβ production that will induce the onset and progression of the disease. Consistently, Aβ1-42 oligomers have been reported to be present in the hippocampi from AD patients since the early stages of the disease [34]. Here, we have studied the expression of BACE1 in the hippocampi from AD patients at two stages of the disease: Braak II (one of the initial stages showing the early symptoms of AD) and Braak VI (the final stage of the disease, when the Aβ is widespread in the brain and dementia is severe). We found in both stages an increased expression of BACE1 (Figure 1(a)), linking the early enhancement of BACE1 expression with Aβ production. Furthermore, this increment in BACE1 expression was also demonstrated through western blot (Figure 1(b)) when the mature glycosylated form (70 kDa) was analysed. AD patient samples showed an increased expression of BACE1 compared with nondemented controls (p < 0.05). The maintained BACE1 expression even in the late stage of the disease suggest that oxidative stress, which has been reported to be significantly increased in AD [14], would be inducing BACE1 expression directly or by the indirect effect of Aβ1-42 oligomers, which generate free radicals, as we address in the present work.
Figure 1.

BACE1 expression increases in the hippocampi from AD patients. (a) The expression of BACE1 was studied by immunohistochemical analysis in hippocampal sections from nondemented controls and AD patients at the initial stages of the disease (Braak stage II) and late stages of the disease (Braak stage VI). Here, we show the representative images obtained in two different individuals from each group. The expression of BACE-1 (brown) was predominant along the axons from AD patients as indicated by black arrows. Bars represent 200 μm. (b) BACE1 expression was also studied by western blot from hippocampal samples of nondemented controls and AD patients. Data are expressed as A.U. and represent mean ± SEM of 4 samples per each group. ∗p < 0.05vs. nondemented controls by Student's t-test. BACE1 bands correspond to the mature glycosylated enzyme (70 kDa) and the immature enzyme (~60 kDa).
3.2. Aβ1-42 Oligomers and H2O2 Decrease Cell Viability at High Concentrations
Human neuroblastoma cells were treated with increased concentration of oligomeric Aβ1-42 for 24 h (Figure 2(a)) in order to study their neurotoxic abilities and to find the range of concentrations that do not produce a significant cytotoxicity to carry out the experiments on BACE1 regulation. Aβ1-42 oligomers were significantly neurotoxic at 5 μM (p < 0.005), 10 μM (p < 0.001), and 15 μM (p < 0.001). The production of H2O2 by the Aβ1-42 oligomers is a continuous process, while the Aβ1-42 is present, along 24 h in our experiments. Therefore, we have also studied the effect of the treatment with H2O2 on cell viability (Figure 2(d)). We have obtained that H2O2 is cytotoxic at 100 μM (p < 0.001).
Figure 2.

Aβ1-42 oligomers and H2O2 decrease cell viability. Human neuroblastoma cells were treated with increasing concentrations of (a) Aβ1-42 oligomers or (d) H2O2 for 24 h. Cell survival was measured by MTT reduction. Data are mean ± SEM of 4 independent experiments performed in triplicate. ∗∗p < 0.005, ∗∗∗p < 0.001vs. control by one-way ANOVA using Tukey's posttest. Images of cells with the MTT reduced to blue formazan were taken after 24 h of treatment with (b) Aβ1-42 oligomers or (e) H2O2. Bars represent 20 μm. The proapoptotic state of the cells was studied by the identification of cleavaged caspase-3 (green; marked with white arrows) after 24 h of treatment with (c) Aβ1-42 oligomers or (f) H2O2. Nuclei were counterstained with Hoechst (pink). Bars represent 30 μm.
The cell cytotoxicity was also studied by images obtained after the reduction of the MTT to blue formazan by bright field microscopy (Figures 2(b) and 2(e)) and with an Ab against active caspase-3 (Figures 2(c) and 2(f)). In both studies, 15 μM Aβ1-42 oligomers and 100 μM H2O2 showed a low reduction of MTT and high caspase activation as expected.
In order to mimic the pathophysiological effects of a continuous and insidious damage on neurons along the life, we decided to carry out the experiments on BACE1 regulation with the subtoxic concentration of 0.25 μM Aβ1-42 oligomers and 10 μM H2O2. These concentrations were producing neither cytotoxicity when assayed by MTT reduction (Figures 2(a), 2(d), 2(b), and 2(e)) nor significant caspase activation (Figures 2(c) and 2(f)).
3.3. Aβ1-42 Oligomers and H2O2 Increase BACE1 Transcription and Translation
Human neuroblastoma cells were treated with 0.25 μM Aβ1-42 oligomers for 24 h, and BACE1 mRNA showed a significant transcriptional increase (Figure 3(a); p < 0.05). The treatment with 10 μM H2O2 for 24 h produce the same effect on BACE1 mRNA transcription (Figure 3(b); p < 0.05). These results suggest that both Aβ1-42 oligomers and H2O2 share common mechanisms at the transcriptional level.
Figure 3.
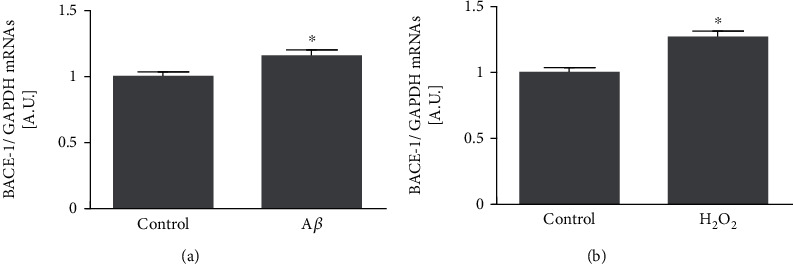
Aβ1-42 oligomers and H2O2 increase BACE1 transcription. Human neuroblastoma cells were treated with subtoxic concentrations of Aβ1-42 oligomers (0.25 μM) or H2O2 (10 μM) for 24 h. BACE1 mRNAs were quantified by qPCR after (a) Aβ1-42 oligomers or (b) H2O2 treatments. Data are expressed as arbitrary units (A.U.) and represent mean ± SEM of 3 independent experiments performed by triplicate. ∗p < 0.05vs. control cells by Student's t-test.
Attending to the effects on BACE1 translation, we have obtained that 0.25 μM Aβ1-42 oligomers increased BACE1 expression after 24 h as it was analyzed by western blot (Figure 4; p < 0.05 by western blot and p < 0.001 by immunofluorescence). Similar results were obtained when cells were treated with 10 μM H2O2 for 24 h (Figure 5; p < 0.05 by western blot and p < 0.001 by immunofluorescence). Interestingly, the phosphorylation of eIF2α was increased with both 0.25 μM Aβ1-42 oligomers and 10 μM H2O2 (Figures 4, 5; p < 0.05 by western blot) supporting that BACE1 expression is due to an increase in its translation. These results also correlated with the increased fluorescence showed by p-eIF2α after the treatment with 0.25 μM Aβ1-42 oligomers and 10 μM H2O2 (Figures 4(d) and 5(d)).
Figure 4.

Subtoxic Aβ1-42 oligomers increase BACE1 translation. Neuroblastoma cells were treated with subtoxic concentrations of Aβ1-42 oligomers (0.25 μM) for 24 h. BACE1, p-eIF2α, and eIF2α expressions were studied by (a) western blot and (d) immunofluorescence. The bands corresponding to (b) BACE1/tubulin and (c) p-eIF2α/eIF2α were quantified and expressed as A.U. Data are expressed as arbitrary units (A.U.) and represent mean ± SEM of 4-7 independent experiments. ∗p < 0.05vs. control cells by Student's t-test. BACE1 expression by immunofluorescence analysis was quantified (e) and expressed as fluorescence intensity (F.I.). Data are mean ± SEM of 3 independent experiments. ∗∗∗p < 0.001vs. control cells by Student's t-test. Bars represent 5 μm.
Figure 5.

Subtoxic H2O2 increases BACE1 translation. Human neuroblastoma cells were challenged with subtoxic concentrations of H2O2 (10 μM) for 24 h. BACE1, p-eIF2α, and eIF2α expressions were studied by (a) western blot and (d) immunofluorescence. The bands corresponding to (b) BACE1/tubulin and (c) p-eIF2α/eIF2α were quantified and expressed as arbitrary units (A.U.). Data are mean ± SEM of 3-9 independent experiments. ∗p < 0.05vs. control cells by Student's t-test. BACE1 expression by immunofluorescence analysis was quantified (e) and expressed as fluorescence intensity (F.I.). Data are mean ± SEM of 3 independent experiments. ∗∗∗p < 0.001vs. control cells by Student t-test. Bars represent 5 μm.
4. Discussion
There are many evidences that relate oxidative stress with the ethiopathogenesis of AD, a devastating neurodegenerative disease whose major risk factor is aging. This tight relationship starts with the aggregation of Aβ, which increases by oxidative stress [35]. Once the fibrils are formed, they also produce H2O2 [13] and hydroxyl radical [36, 37]. In fact, the neurotoxicity of Aβ aggregates has been reported to be mediated by oxidative stress [14, 15, 38, 39]. Moreover, oxidative stress has been demonstrated to increase BACE1 transcription and translation [16, 17]. We have demonstrated in this work that BACE1 expression in increased even in the early stages of the disease. Since oxidative stress is a concomitant process with aging and Aβ aggregation is considered the key factor of AD, according to the amyloid cascade hypothesis, we have studied the effect of Aβ oligomers and oxidative stress in BACE1 expression and the mechanisms that control its pathophysiological expression.
BACE1 is an enzyme that accomplishes some physiological functions as dendritic spine growth in the hippocampus where contributes to memory formation [26]. In fact, BACE1 knockout mice show synaptic plasticity deficits and cognitive impairment [40, 41]. However, the dysregulation of BACE1 has harmful effects. In our study, we have found that long treatments with low nanomolar concentration of Aβ1-42 oligomers, mimicking the environment of neurons in AD patients, increase BACE1 transcription and expression in vitro. The relevance of this finding is supported by the increased expression of BACE1 found in AD hippocampi. This would suggest the existence of a loop of amyloid production that will activate BACE1 to release more amyloid contributing to accelerate the dysregulation of BACE1 and the characteristic amyloidosis of AD. This effect should be based in the capability of Aβ1-42 oligomers to produce oxidative stress since similar results has been obtained when we used hydrogen peroxide.
BACE1 expression is controlled physiologically by the phosphorylation of eIF2α carried out by the enzyme HRI [26]. HRI is activated throughout the nitric oxide released after NMDA receptors activation in glutamatergic neurons. However, aging produces an increase in oxidative stress in brain [42, 43] and would result in the loss of BACE1 physiological control. It is due to the fact that HRI, PKR, and PERK, three of the four kinases that phosphorylates eIF2α, can be activated by oxidative stress [44–48]. In our study, we have found that both oxidative stress and Aβ1-42 oligomers yield to the phosphorylation of the eIF2α in vitro. The dysregulation of the different eIF2α kinases by oxidative stress would explain these results.
Summarizing, our results suggest that oxidative stress induced by Aβ oligomers increase BACE1 transcription and translation. Our data support that the mechanism involved in the increased BACE1 expression is the dysregulation of the phosphorylation of the eIF2α that would generate the amyloid burden in AD.
Acknowledgments
This research was funded by the Spanish Ministry of Economy and Business through the grant Plan Estatal SAF2017-83372-R and SAF2014-52228-R (FEDER funds/UE) to FJM and RV, Chilean Government through Fondecyt 11611065 and AFB170005 to AA and REDES 180084 to AA and FJM, and MDM-2014-0370 through the María de Maeztu Programme for Units of Excellence in R&D to Departament de Ciències Experimentals i de la Salut. Silvia Menéndez is supported by the Health Deparment of the Generalitat de Catalunya, Spain (PERIS SLT006/17/00040). This work was also supported by the Advanced Microscopy Facility UC.
Abbreviations
- Ab:
Antibody
- Aβ:
Amyloid-β peptide
- AD:
Alzheimer's disease
- APP:
Amyloid precursor protein
- A.U:
Arbitrary units
- BACE1:
β-site APP Cleaving Enzyme 1
- BSA:
Bovine serum albumin
- EIF2α:
Eukaryotic initiation factor 2-α
- FBS:
Fetal bovine serum
- GCN2:
General control nonderepressible-2 kinase
- HRI:
Heme-regulated eukaryotic initiation factor eIF2 α kinase
- MTT:
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PBS:
Phosphate buffer saline
- o.n:
Overnight
- PERK:
PKR-like endoplasmic reticulum-related kinase
- PKR:
Double-stranded RNA-activated protein kinase
- PS:
Presenilin
- RT:
Room temperature
- SDS:
Sodium dodecyl sulphate
- TTBS:
Tween 20-Tris buffer solution
- 5′UTR:
5′untranslated region
Data Availability
All the data used to support the findings of this study are available from the corresponding author upon request.
Conflicts of Interest
The authors declare that there is no conflict of interest regarding the publication of this paper.
Authors' Contributions
Pol Picón-Pagès and Daniela A. Gutiérrez have contributed equally to the present work.
References
- 1.Glenner G. G., Wong C. W. Alzheimer’s disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochemical and Biophysical Research Communications. 1984;120(3):885–890. doi: 10.1016/S0006-291X(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 2.Masters C. L., Simms G., Weinman N. A., Multhaup G., McDonald B. L., Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proceedings of the National Academy of Sciences of the United States of America. 1985;82(12):4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu X. Gamma-secretase catalyzes sequential cleavages of the AbetaPP transmembrane domain. Journal of Alzheimer's Disease. 2009;16(2):211–224. doi: 10.3233/JAD-2009-0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toro D., Coma M., Uribesalgo I., Guix F., Munoz F. The amyloid β-Protein precursor and Alzheimers disease. Therapeutic approaches. Current Medicinal Chemistry-Central Nervous System Agents. 2005;5(4):271–283. doi: 10.2174/156801505774913053. [DOI] [Google Scholar]
- 5.Funato H., Yoshimura M., Kusui K., et al. Quantitation of amyloid beta-protein (A beta) in the cortex during aging and in Alzheimer’s disease. The American Journal of Pathology. 1998;152(6):1633–1640. [PMC free article] [PubMed] [Google Scholar]
- 6.Scheuner D., Eckman C., Jensen M., et al. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature Medicine. 1996;2(8):864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 7.Guix F. X., Wahle T., Vennekens K., et al. Modification of γ-secretase by nitrosative stress links neuronal ageing to sporadic Alzheimer’s disease. EMBO Molecular Medicine. 2012;4(7):660–673. doi: 10.1002/emmm.201200243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jarrett J. T., Berger E. P., Lansbury P. T. The carboxy terminus of the beta amyloid protein is critical for the seeding of amyloid formation: implications for the pathogenesis of Alzheimer’s disease. Biochemistry. 2002;32(18):4693–4697. doi: 10.1021/bi00069a001. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki N., Cheung T. T., Cai X. D., et al. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264(5163):1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 10.Klein W. L., Krafft G. A., Finch C. E. Targeting small Abeta oligomers: the solution to an Alzheimer’s disease conundrum? Trends in Neurosciences. 2001;24(4):219–224. doi: 10.1016/S0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- 11.Dinamarca M. C., Ríos J. A., Inestrosa N. C. Postsynaptic receptors for Amyloid-β oligomers as mediators of neuronal damage in Alzheimer’s disease. Frontiers in Physiology. 2012;3:p. 464. doi: 10.3389/fphys.2012.00464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Valls-Comamala V., Guivernau B., Bonet J., et al. The antigen-binding fragment of human gamma immunoglobulin prevents amyloid β-peptide folding into β-sheet to form oligomers. Oncotarget. 2017;8(25):41154–41165. doi: 10.18632/oncotarget.17074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang X., Atwood C. S., Hartshorn M. A., et al. The Aβ peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38(24):7609–7616. doi: 10.1021/bi990438f. [DOI] [PubMed] [Google Scholar]
- 14.Miranda S., Opazo C., Larrondo L. F., et al. The role of oxidative stress in the toxicity induced by amyloid beta-peptide in Alzheimer’s disease. Progress in Neurobiology. 2000;62(6):633–648. doi: 10.1016/S0301-0082(00)00015-0. [DOI] [PubMed] [Google Scholar]
- 15.Picón-Pagès P., Garcia-Buendia J., Muñoz F. J. Functions and dysfunctions of nitric oxide in brain. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2019;1865(8):1949–1967. doi: 10.1016/j.bbadis.2018.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Tamagno E., Bardini P., Obbili A., et al. Oxidative stress increases expression and activity of BACE in NT2 neurons. Neurobiology of Disease. 2002;10(3):279–288. doi: 10.1006/nbdi.2002.0515. [DOI] [PubMed] [Google Scholar]
- 17.Coma M., Guix F. X., Ill-Raga G., et al. Oxidative stress triggers the amyloidogenic pathway in human vascular smooth muscle cells. Neurobiology of Aging. 2008;29(7):969–980. doi: 10.1016/j.neurobiolaging.2007.01.009. [DOI] [PubMed] [Google Scholar]
- 18.Sambamurti K., Kinsey R., Maloney B., Ge Y., Lahiri D. K. Gene structure and organization of the human β-secretase (BACE) promoter. The FASEB Journal. 2004;18(9):1034–1036. doi: 10.1096/fj.03-1378fje. [DOI] [PubMed] [Google Scholar]
- 19.Christensen M. A., Zhou W., Qing H., Lehman A., Philipsen S., Song W. Transcriptional regulation of BACE1, the beta-amyloid precursor protein beta-secretase, by Sp1. Molecular and Cellular Biology. 2004;24(2):865–874. doi: 10.1128/MCB.24.2.865-874.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tonelli D. D. P., Mihailovich M., Di Cesare A., Codazzi F., Grohovaz F., Zacchetti D. Translational regulation of BACE-1 expression in neuronal and non-neuronal cells. Nucleic Acids Research. 2004;32(5):1808–1817. doi: 10.1093/NAR/GKH348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lammich S., Schöbel S., Zimmer A.-K., Lichtenthaler S. F., Haass C. Expression of the Alzheimer protease BACE1 is suppressed via its 5’-untranslated region. EMBO Reports. 2004;5(6):620–625. doi: 10.1038/sj.embor.7400166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou W., Song W. Leaky scanning and reinitiation regulate BACE1 gene expression. Molecular and Cellular Biology. 2006;26(9):3353–3364. doi: 10.1128/MCB.26.9.3353-3364.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mihailovich M., Thermann R., Grohovaz F., Hentze M. W., Zacchetti D. Complex translational regulation of BACE1 involves upstream AUGs and stimulatory elements within the 5’ untranslated region. Nucleic Acids Research. 2007;35(9):2975–2985. doi: 10.1093/nar/gkm191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Connor T., Sadleir K. R., Maus E., et al. Phosphorylation of the translation initiation factor eIF2α increases BACE1 levels and promotes amyloidogenesis. Neuron. 2008;60(6):988–1009. doi: 10.1016/j.neuron.2008.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.ILL-Raga G., Köhler C., Radiske A., et al. Consolidation of object recognition memory requires HRI kinase-dependent phosphorylation of eIF2α in the hippocampus. Hippocampus. 2013;23(6):431–436. doi: 10.1002/hipo.22113. [DOI] [PubMed] [Google Scholar]
- 26.ILL-Raga G., Tajes M., Busquets-García A., et al. Physiological control of nitric oxide in neuronal BACE1 translation by Heme-regulated eIF2α kinase HRI induces synaptogenesis. Antioxidants & Redox Signaling. 2015;22(15):1295–1307. doi: 10.1089/ars.2014.6080. [DOI] [PubMed] [Google Scholar]
- 27.de Haro C., Méndez R., Santoyo J. The eIF‐2α kinases and the control of protein synthesis1. The FASEB Journal. 1996;10(12):1378–1387. doi: 10.1096/fasebj.10.12.8903508. [DOI] [PubMed] [Google Scholar]
- 28.ILL-Raga G., Palomer E., Wozniak M. A., et al. Activation of PKR causes amyloid ß-peptide accumulation via de-repression of BACE1 expression. PLoS One. 2011;6(6):p. e21456. doi: 10.1371/journal.pone.0021456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Palomer E., ILL-Raga G., Tajes M., et al. PKR and PP1C polymorphisms in Alzheimer’s disease risk. Neuroscience and Medicine. 2011;2(3):226–231. doi: 10.4236/nm.2011.23031. [DOI] [Google Scholar]
- 30.Ramos-Fernández E., Tajes M., ILL-Raga G., et al. Glutamatergic stimulation induces GluN2B translation by the nitric oxide-Heme-Regulated eIF2α kinase in cortical neurons. Oncotarget. 2016;7(37):58876–58892. doi: 10.18632/oncotarget.11417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang L.-B., Lindholm K., Yan R., et al. Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nature Medicine. 2003;9(1):3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- 32.Fukumoto H., Cheung B. S., Hyman B. T., Irizarry M. C. β-secretase protein and activity are increased in the neocortex in Alzheimer disease. Archives of Neurology. 2002;59(9):1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- 33.Holsinger R. M. D., McLean C. A., Beyreuther K., Masters C. L., Evin G. Increased expression of the amyloid precursor ?-secretase in Alzheimer's disease. Annals of Neurology. 2002;51(6):783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- 34.Scheff S. W., Price D. A., Schmitt F. A., Dekosky S. T., Mufson E. J. Synaptic alterations in CA1 in mild Alzheimer disease and mild cognitive impairment. Neurology. 2007;68(18):1501–1508. doi: 10.1212/01.wnl.0000260698.46517.8f. [DOI] [PubMed] [Google Scholar]
- 35.Multhaup G., Ruppert T., Schlicksupp A., et al. Reactive oxygen species and Alzheimer’s disease. Biochemical Pharmacology. 1997;54(5):533–539. doi: 10.1016/S0006-2952(97)00062-2. [DOI] [PubMed] [Google Scholar]
- 36.Tomiyama T., Shoji A., Kataoka K., et al. Inhibition of amyloid protein aggregation and neurotoxicity by rifampicin. Its possible function as a hydroxyl radical scavenger. The Journal of Biological Chemistry. 1996;271(12):6839–6844. doi: 10.1074/jbc.271.12.6839. [DOI] [PubMed] [Google Scholar]
- 37.Tabner B. J., Turnbull S., El-Agnaf O. M. A., Allsop D. Formation of hydrogen peroxide and hydroxyl radicals from A(beta) and alpha-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radical Biology & Medicine. 2002;32(11):1076–1083. doi: 10.1016/S0891-5849(02)00801-8. [DOI] [PubMed] [Google Scholar]
- 38.Quintanilla R. A., Munoz F. J., Metcalfe M. J., et al. Trolox and 17 -estradiol protect against amyloid -peptide neurotoxicity by a mechanism that involves modulation of the Wnt signaling pathway. The Journal of Biological Chemistry. 2005;280(12):11615–11625. doi: 10.1074/jbc.M411936200. [DOI] [PubMed] [Google Scholar]
- 39.Ill-Raga G., Ramos-Fernández E., Guix F. X., et al. Amyloid-β peptide fibrils induce nitro-oxidative stress in neuronal cells. Journal of Alzheimer's Disease. 2010;22(2):641–652. doi: 10.3233/JAD-2010-100474. [DOI] [PubMed] [Google Scholar]
- 40.Laird F. M., Cai H., Savonenko A. V., et al. BACE1, a major determinant of selective vulnerability of the brain to amyloid- amyloidogenesis, is essential for cognitive, emotional, and synaptic functions. The Journal of Neuroscience. 2005;25(50):11693–11709. doi: 10.1523/JNEUROSCI.2766-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang H., Song L., Laird F., Wong P. C., Lee H.-K. BACE1 knock-outs display deficits in activity-dependent potentiation of synaptic transmission at mossy fiber to CA3 synapses in the hippocampus. The Journal of Neuroscience. 2008;28(35):8677–8681. doi: 10.1523/JNEUROSCI.2440-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benzi G., Pastoris O., Marzatico F., Villa R. F. Age-related effect induced by oxidative stress on the cerebral glutathione system. Neurochemical Research. 1989;14(5):473–481. doi: 10.1007/BF00964863. [DOI] [PubMed] [Google Scholar]
- 43.Sohal R. S., Sohal B. H., Brunk U. T. Relationship between antioxidant defenses and longevity in different mammalian species. Mechanisms of Ageing and Development. 1990;53(3):217–227. doi: 10.1016/0047-6374(90)90040-M. [DOI] [PubMed] [Google Scholar]
- 44.Lu L., Han A.-P., Chen J.-J. Translation initiation control by heme-regulated eukaryotic initiation factor 2alpha kinase in erythroid cells under cytoplasmic stresses. Molecular and Cellular Biology. 2001;21(23):7971–7980. doi: 10.1128/MCB.21.23.7971-7980.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gale M., Blakely C. M., Darveau A., Romano P. R., Korth M. J., Katze M. G. P52rIPK regulates the molecular cochaperone P58IPK to mediate control of the RNA-dependent protein kinase in response to cytoplasmic stress. Biochemistry. 2002;41(39):11878–11887. doi: 10.1021/bi020397e. [DOI] [PubMed] [Google Scholar]
- 46.Ho H. K., White C. C., Fernandez C., et al. Nrf2 activation involves an oxidative-stress independent pathway in tetrafluoroethylcysteine-induced cytotoxicity. Toxicological Sciences. 2005;86(2):354–364. doi: 10.1093/toxsci/kfi205. [DOI] [PubMed] [Google Scholar]
- 47.Avivar-Valderas A., Salas E., Bobrovnikova-Marjon E., et al. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment. Molecular and Cellular Biology. 2011;31(17):3616–3629. doi: 10.1128/MCB.05164-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cullinan S. B., Diehl J. A. Coordination of ER and oxidative stress signaling: the PERK/Nrf2 signaling pathway. The International Journal of Biochemistry & Cell Biology. 2006;38(3):317–332. doi: 10.1016/j.biocel.2005.09.018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All the data used to support the findings of this study are available from the corresponding author upon request.