

Abstract
This review of current literature provides background to the COVID-19 pandemic, as well as an examination of potential pathophysiologic mechanisms behind development of thrombosis and ischemic stroke related to COVID-19. SARS-CoV-2 infection is well-documented to cause severe pneumonia, however, thrombosis and thrombotic complications, such as ischemic stroke, have also been documented in a variety of patient demographics. SARS-CoV-2 infection is known to cause a significant inflammatory response, as well as invasion of vascular endothelial cells, resulting in endothelial dysfunction. These factors, coupled with imbalance of ACE2 and RAS axis interactions, have been shown to create a prothrombotic environment, favoring thromboembolic events. Ischemic stroke is a severe complication of COVID-19 and may be a presenting symptom in some patients.
Keywords: Coronavirus, Stroke, Thrombosis, COVID-19
Non-standard Abbreviations and Acronyms: ACE2, Angiotensin converting enzyme 2; ANGII, Angiotensin II; ANG1–7, Angiotensin-(1–7); SARS-CoV-2, SARS coronavirus 2; COVID-19, Coronavirus disease 2019; R0, Basic reproductive number; RAS, renin-angiotensin system; S-protein, Spike protein
1. Introduction
Coronaviruses are positive-sense single stranded RNA viruses of the coronaviridae family [1]. The genome of coronaviruses contains open reading frames for 16 non-structural proteins as well as for spike (S), envelope, membrane, and nucleocapsid structural proteins [1]. Four genera exist within the coronaviridae family based on phylogeny: alpha, beta, gamma, and deltacoronaviruses [1]. Only members of the alpha and betacoronavirus genera are known to infect humans and can cause clinical presentations ranging from the common cold to severe acute respiratory syndrome (SARS) [1]. Viruses of particular note from the betacoronavirus genus are the SARS-coronavirus (SARS-CoV) and Middle East respiratory syndrome coronavirus (MERS-CoV) [1]. SARS-CoV is the etiological agent of the 2002 to 2003 SARS epidemic beginning with zoonotic origin in Southern China and leading to over 8000 confirmed cases with an estimated 9–11% fatality rate [1,2]. MERS-CoV is currently endemic to the Arabian Peninsula and has proven to be a dangerous virus of zoonotic origin with an estimated 36% fatality rate [1].
December 2019 marked the discovery of a new coronavirus in Wuhan, China after an outbreak of severe pneumonia of unknown origin [3]. Isolation and sequencing of this virus from human airway epithelial cells allowed the characterization of the betacoronavirus named SARS coronavirus 2 (SARS-CoV-2) that is the etiologic agent of coronavirus disease 2019 (COVID-19) [3,4]. Further characterization of SARS-CoV-2 has demonstrated close genomic similarity to several types of bat coronavirus, indicating bats as a likely reservoir for this virus of zoonotic origin [4]. SARS-CoV-2 shares approximately 79% sequence identity with SARS-CoV, though SARS-CoV-2 has demonstrated a higher rate of transmission than SARS-CoV [4]. Basic reproductive number (R0) is used to represent the transmissibility of a disease and is defined the average number of new cases caused by a single infective person in an unexposed population [5]. The R0 of SARS-CoV-2 and SARS-CoV are estimated at 2.9 and 1.85 for each virus, respectively [5,6]. This is likely due to the presence of asymptomatic or mildly symptomatic transmission of SARS-CoV-2, and its current prevalence in the human population supports the infective potential of this novel coronavirus [5,6]. Since its December 2019 emergence, SARS-CoV-2 has been declared a global pandemic by the World Health Organization, and the Centers for Disease Control and Prevention reports over 3 million infections resulting in over 130,000 deaths in the United States alone as of 10 July 2020 [7,8].
1.1. Angiotensin Converting Enzyme 2 (ACE2) as the functional receptor of SARS-CoV-2
Viral entry into cells by coronaviruses is mediated through the interaction between a cell-surface receptor protein and the viral S-protein [9]. Cellular tropism of coronaviruses is dependent on the S-protein-receptor interaction [9] and understanding the tropism of SARS-CoV-2 is the beginning to elucidating the myriad effects this virus may have on human physiology.
SARS-CoV-2 shares 73% to 76% amino acid sequence identity in the receptor binding domain of its S-protein with SARS-CoV [4,10], and the amino acid sequence directly interacting with the cell receptor is highly conserved between the viruses [4,10]. SARS-CoV has been previously determined to use the human transmembrane protein angiotensin converting enzyme 2 (ACE2) as its receptor for viral entry, and SARS-CoV-2 had been speculated to use ACE2 for viral entry as well [9,10]. Several studies have confirmed that ACE2 is the functional receptor for SARS-CoV-2 [[11], [12], [13]].
1.2. ACE2: its location and role in the renin-angiotensin system (RAS)
Human ACE2 is a transmembrane zinc metalloprotease that acts as a carboxypeptidase in the metabolic degradation of angiotensin I and angiotensin II (ANGII) [14]. ACE2 mRNA is expressed in most tissues of the body, with highest expression in the GI tract, kidney, testes, heart, and lungs [15]. ACE2 protein is found expressed on the surface of lung alveolar epithelial cells, enterocytes of the small intestine, arterial smooth muscle cells, and both arterial and venous endothelial cells, including intracranial vessels [16]. While soluble ACE2 exists after cleavage of ACE2 from the apical cell surface, it plays little to no physiologic role [16].
ACE2 primarily catalyzes the conversion of ANGII into angiotensin-(1–7) (ANG1–7), a metabolite that opposes the actions of ANGII and the RAS axis through activation of the Mas receptor [17]. Generation of ANG1–7 can take a more circuitous route through ACE2 catalyzation of angiotensin I into angiotensin-(1–9) followed by ACE catalyzation of angiotensin-(1–9) into ANG1–7 [17]. Increased circulating levels of ANG1–7 have been demonstrated to lower blood pressure, improve endothelial function, and attenuate the effects of ANGII in spontaneously hypertensive rats [17]. Additionally, ANG1–7 administered to spontaneously hypertensive rats treated with a nitric oxide synthase inhibitor attenuated the inhibitor's effects on MAP, as well as demonstrated cardioprotective effects in the setting of global cardiac ischemia [18]. ACE2 and ANG1–7 play an essential physiologic role in vasodilation and regulation of endothelial function in opposition to the effects of ANGII. Fig. 1 summarizes the production of ANGII and ANG1–7, as well as their effects. Imbalance of ACE and ACE2 products has the potential to cause significant dysfunction and has been implicated as playing a role in the pathogenesis of SARS [19]. Downregulation of ACE2 expression has been demonstrated after SARS-CoV pulmonary and myocardial infection [19,20] and is linked to the acute pulmonary injury seen in SARS [19].
Fig. 1.
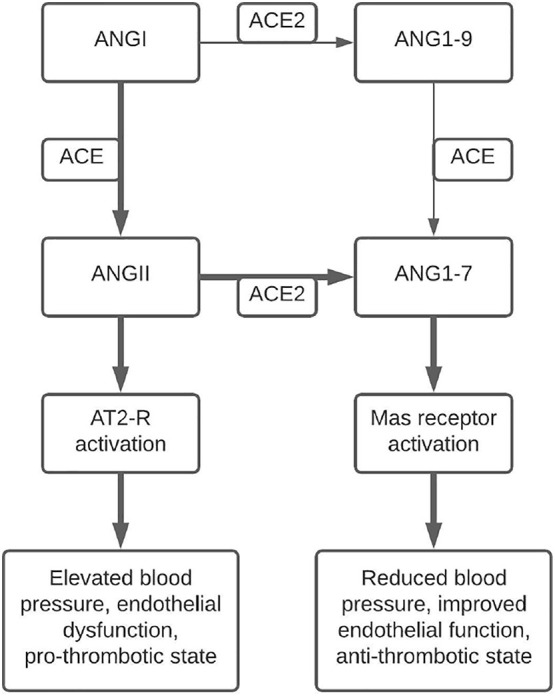
An illustration of the pathway leading from ANGI to ANGII and ANG1–7, as well as their receptor-mediated effects. Bold arrows indicate the major pathway toward metabolite accumulation and their effects. ANGII production leads to increased blood pressure and endothelial dysfunction, while ANG1–7 production leads to decreased blood pressure and improved endothelial function. Abbreviation: ACE – angiotensin converting enzyme, ACE2 – angiotensin converting enzyme 2, ANGII – angiotensin II, ANG1–7 – angiotensin-(1–7), AT2-R – angiotensin II receptor.
1.3. Hypercoagulability in SARS-CoV-2 infection
COVID-19 symptomology is diverse, including shortness of breath, cough, and fatigue with many cases progressing into pneumonia requiring oxygen therapy [21,22]. COVID-19 has also been demonstrated to have detrimental effects on extra-pulmonary systems, including the heart and systemic vasculature [[20], [21], [22], [23], [24]]. SARS-CoV-2 infection has been linked to increased risk for venous thromboembolism and arterial thrombosis [23]. Several markers of coagulopathy, including thrombocytopenia, elevated D-dimer and fibrin degradation products, elevated PT and PTT, and fibrinolysis shutdown, were associated with increased rates of thromboembolic events and mortality in COVID-19 [23,[25], [26], [27]]. Anticoagulation given to COVID-19 patients with coagulopathy showed improved mortality whereas patients with non-COVID-19 disease with similar coagulopathy measurements did not have improved survival [27]. This serves to further implicate coagulation defects as a cause of death in COVID-19, though the underlying mechanism has yet to be confirmed.
Given the similar clinical presentation of COVID-19 and SARS, as well as use of the same receptor for viral entry, patient data collected during the SARS epidemic provides a framework to begin studying SARS-CoV-2 mechanisms of pathogenesis. Endothelial dysfunction has been considered to likely contribute to the coagulation abnormalities seen in COVID-19. Post-mortem studies of SARS patients have demonstrated systemic polyangiitis, vasculitis, and infiltration of inflammatory cells with accompanying thrombi in the microcirculation of several organ systems, including the lungs and kidneys [28,29]. An autopsy series of 3 COVID-19 patients indicated systemic viral infection of the endothelium with inflammatory cells associated with virally infected cells [30]. The vascular endothelium has come to be seen as an integral participant in the regulation of vascular homeostasis. Endothelial dysfunction is strongly associated with a pro-coagulant state [31]. The inherent tropism of SARS-CoV-2 for ACE2-expressing tissues, such as the vascular endothelium, raises the possibility that systemic viral infection of the vascular endothelium is a strong contributing factor to COVID-19-related thromboembolism.
Inflammatory and immune effects further compound endothelial dysfunction in the creation of a pro-coagulant state in COVID-19. Immune cell infiltration of virally-infected tissue is evident in both SARS-CoV and SARS-CoV-2 infection [20,[28], [29], [30]]. Immunity and coagulation systems are deeply intertwined, and activation of the immune system during infection will invariably result in a lower threshold for the formation of thrombi [32]. Clinical features of SARS-CoV-2 include increased production of several inflammatory cytokines that are able to predispose thrombus formation, and infiltration of inflammatory cells into the vascular endothelium may lead to endothelial and platelet activation, further increasing the risk of thromboembolic events [22,32]. Antibody responses may also play a role in COVID-19-related coagulation. A series of patients with multiple cerebral infarctions with markers of coagulopathy demonstrated production of antiphospholipid antibodies after SARS-CoV-2 infection [33]. Another COVID-19 patient developed immune thrombocytopenic purpura after heparin treatment was begun [34]. Heparin treatment was ceased after thrombocytopenia developed, though antibody testing for antiplatelet factor 4 and antiplatelet antibodies was negative [34]. These events suggest SARS-CoV-2 infection as the precipitating event for the thrombocytopenia in this case, though causative studies are necessary.
The complement system has also been implicated in the pathophysiology of SARS-CoV-2 infection. A series of patients with severe COVID-19 were determined to have significant deposits of terminal complement proteins and signs of systemic complement activation were present [35]. Complement was also co-localized with SARS-CoV-2 S-protein in these patients, indicating complement targeting of virally-infected endothelium [35]. Previous studies of complement activation in SARS-CoV infection indicated endothelial dysfunction as the source of complement activation, and murine C3 protein knockout models demonstrated less severe infection with SARS-CoV [36]. Activation of the complement system has the potential to increase the risk of thrombus formation, both through C3a stimulation of platelets and insertion of terminal complement components into membranes [37].
Imbalance of the interactions between ACE2 and the RAS axis may also contribute to the thromboembolic events seen in SARS-CoV-2 infection. ANG1–7, the major product of ACE2, and ANGII have competing effects on blood pressure and endothelial activation: where ANGII serves to increase blood pressure and activate the endothelium, ANG1–7 reverses these actions through the Mas receptor (Fig. 1) [17,38]. ANG1–7 has also been shown to decrease thrombus formation through the production of nitric oxide and prostacyclin by both platelets and endothelial cells [[39], [40], [41]]. This is contrasted by the actions of ANGII, which has been shown to accelerate thrombus formation through induction of tissue factor production and generation of free radicals that scavenge free nitric oxide [42,43]. Downregulation of ACE2 by SARS-CoV-2 infection [20] may result in an imbalance of these systems, leading to predisposition to thromboembolic events.
1.4. Cerebrovascular events in COVID-19
Cerebrovascular events can be a significant consequence of uncontrolled thrombotic states, and represent a global burden to both quality of life and national economics [44,45]. Ischemic stroke due to occlusion of large arteries has been a documented complication of SARS-CoV infection in patients with minimal to no risk factors [46]. SARS-CoV-2 infection seems to also increase risk of developing ischemic stroke, among other neurological consequences. 78 of 214 patients in a retrospective case series of hospitalized patients in Wuhan, China demonstrated nervous system dysfunction (CNS, peripheral nervous system, and/or skeletal muscle dysfunction) [47]. Of these 78 patients, 6 developed ischemic strokes; 5 of these patients had been categorized as severe COVID-19 and 1 had been categorized as non-severe [47]. Unexplained encephalopathic features in 13 of 58 patients were seen in another case series of COVID-19 patients [48]. Ischemic stroke was diagnosed in 3 of these 13 patients (2 small acute and 1 sub-acute strokes) [48]. A 40-year-old woman without significant medical history diagnosed with severe COVID-19 pneumonia was noted to develop significant left middle cerebral artery territory stroke after ICU hospitalization [49].
Concerningly, severe respiratory disease is not necessarily requisite to the development of cerebral ischemia. In a retrospective case review at NYU Langone Health, 17 patients undergoing neurologic imaging (CT, MRI, angiography) for reasons unrelated to COVID-19 tested positive for SARS-CoV-2 infection; 4 of these 17 patients had been presenting for symptoms of stroke [50]. Further, stroke has been a presenting or complicating factor in mild COVID-19 disease of young patients. Separate case series involving a total of 11 COVID-19 patients all under 55-years-old with minimal respiratory involvement developed large vessel ischemic stroke [51,52]. A 33-year-old male presenting to the ED for occipital headache, nausea/vomiting, and balance disorder was discovered to have thrombosis of his left vertebral artery with concomitant SARS-CoV-2 infection [53]. A 52-year-old male with history of hypertension was discharged from the ED after being given antibiotics for a presumed respiratory infection before returning days later for stroke symptoms caused by occlusion of the left internal carotid artery [54].
2. Conclusions
While hypertension is known to produce a prothrombotic state [55], lack of many typical stroke risk factors seems to indicate a need for surveillance of stroke in COVID-19 patients, as well as the consideration of SARS-CoV-2 infection in patients presenting with cerebrovascular events. Elevated D-dimer, as well as other markers of coagulopathy, has been noted as a potential prognostic factor for COVID-19 [23,[25], [26], [27]], and these series of case reports provide anecdotal evidence further supporting the use of D-dimer [47,49,[51], [52], [53], [54]]. Ischemic stroke is an uncommon, though severe, complication of SARS-CoV-2 infection, and Fig. 2 details the theoretical progression and contributing factors leading from viral infection to stroke. Thrombus formation is a risk that cannot be completely ruled out in COVID-19 patients given the prothrombotic milieu precipitated by this viral syndrome. Whether as a result of embolization of distant thrombus or thrombus formation in situ, ischemic stroke in the setting of COVID-19 is a significant sequalae that warrants further research on its pathophysiologic origins, as well as clinical consideration in patients of diverse demographic origins and risk factor profiles.
Fig. 2.

A theoretical pathway beginning from SARS-CoV-2 to the activation of the coagulation cascade through several mechanisms, including immune activation, direct endothelial infection, and downregulation of ACE2. Hypercoagulable states then lead to ischemic stroke through both embolus formation and thrombosis formation in-situ. Abbreviations: ANGII – angiotensin II, ANG1–7 – angiotensin-(1–7).
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
No support was used in the creation of this review.
Declaration of Competing Interest
None.
Acknowledgements
None.
References
- 1.Su S., Wong G., Shi W., et al. Epidemiology, genetic recombination, and pathogenesis of coronaviruses. Trends Microbiol. 2016;24(6):490–502. doi: 10.1016/j.tim.2016.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chan-Yeung M., Xu R. SARS: epidemiology. Respirology. 2003;8(Suppl. 1):S9–S14. doi: 10.1046/j.1440-1843.2003.00518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhu N., Zhang D., Wang W., et al. A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med. 2020;382(8):727–733. doi: 10.1056/NEJMoa2001017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lu R., Zhao X., Li J., et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet. 2020;395(10224):565–574. doi: 10.1016/S0140-6736(20)30251-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y., Gayle A.A., Wilder-Smith A., Rocklöv J. The reproductive number of COVID-19 is higher compared to SARS coronavirus. J Travel Med. 2020;27(2) doi: 10.1093/jtm/taaa021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu T., Hu J., Kang M., et al. Transmission dynamics of 2019 novel coronavirus (2019-NCoV) Soc Sci Res Netw. 2020 doi: 10.2139/ssrn.3526307. [DOI] [Google Scholar]
- 7.CDC . Centers for Disease Control and Prevention; 2020. Coronavirus Disease 2019 (COVID-19)https://www.cdc.gov/coronavirus/2019-ncov/cases-updates/us-cases-deaths.html Published June 17, 2020. Accessed July 10, 2020. [Google Scholar]
- 8.WHO Director-General's opening remarks at the media briefing on COVID-19 - 11 March 2020. 2020. https://www.who.int/dg/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020
- 9.Fehr A.R., Perlman S. Coronaviruses: an overview of their replication and pathogenesis. Coronaviruses. 2015;1282:1–23. doi: 10.1007/978-1-4939-2438-7_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wan Y., Shang J., Graham R., Baric R.S., Li F. Receptor recognition by the novel coronavirus from Wuhan: an analysis based on decade-long structural studies of SARS coronavirus. J Virol. 2020;94(7) doi: 10.1128/JVI.00127-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou P., Yang X.-L., Wang X.-G., et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270–273. doi: 10.1038/s41586-020-2012-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walls A.C., Park Y.-J., Tortorici M.A., Wall A., McGuire A.T., Veesler D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell. 2020;181(2):281–292.e6. doi: 10.1016/j.cell.2020.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffmann M., Kleine-Weber H., Schroeder S., et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181(2):271–280.e8. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tipnis S.R., Hooper N.M., Hyde R., Karran E., Christie G., Turner A.J. A human homolog of angiotensin-converting enzyme cloning and Functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275(43):33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 15.Harmer D., Gilbert M., Borman R., Clark K.L. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett. 2002;532(1–2):107–110. doi: 10.1016/S0014-5793(02)03640-2. [DOI] [PubMed] [Google Scholar]
- 16.Hamming I., Timens W., Bulthuis M., Lely A., Navis G., van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631–637. doi: 10.1002/path.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brit Rentzsch, Mihail Todiras, Radu Iliescu, et al. Transgenic angiotensin-converting enzyme 2 overexpression in vessels of SHRSP rats reduces blood pressure and improves endothelial function. Hypertension. 2008;52(5):967–973. doi: 10.1161/HYPERTENSIONAHA.108.114322. [DOI] [PubMed] [Google Scholar]
- 18.Benter I.F., Yousif M.H.M., Anim J.T., Cojocel C., Diz D.I. Angiotensin-(1–7) prevents development of severe hypertension and end-organ damage in spontaneously hypertensive rats treated with l-NAME. Am J Physiol Heart Circ Physiol. 2006;290(2):H684–H691. doi: 10.1152/ajpheart.00632.2005. [DOI] [PubMed] [Google Scholar]
- 19.Kuba K., Imai Y., Rao S., et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat Med. 2005;11(8):875–879. doi: 10.1038/nm1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oudit G.Y., Kassiri Z., Jiang C., et al. SARS-coronavirus modulation of myocardial ACE2 expression and inflammation in patients with SARS. Eur J Clin Invest. 2009;39(7):618–625. doi: 10.1111/j.1365-2362.2009.02153.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guan W., Ni Z., Hu Y., et al. Clinical characteristics of coronavirus disease 2019 in China. N Engl J Med. 2020;382(18):1708–1720. doi: 10.1056/NEJMoa2002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang C., Wang Y., Li X., et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497–506. doi: 10.1016/S0140-6736(20)30183-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Klok F.A., Kruip M.J.H.A., van der Meer N.J.M., et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145–147. doi: 10.1016/j.thromres.2020.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marone E.M., Rinaldi L.F. Upsurge of deep venous thrombosis in patients affected by COVID-19: preliminary data and possible explanations. J Vasc Surg Venous Lymphat Disord. 2020;8(4):694–695. doi: 10.1016/j.jvsv.2020.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang N., Li D., Wang X., Sun Z. Abnormal coagulation parameters are associated with poor prognosis in patients with novel coronavirus pneumonia. J Thromb Haemost. 2020;18(4):844–847. doi: 10.1111/jth.14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhou F., Yu T., Du R., et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: a retrospective cohort study. Lancet. 2020;395(10229):1054–1062. doi: 10.1016/S0140-6736(20)30566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wright F.L., Vogler TO, Moore E.E., et al. Fibrinolysis shutdown correlation with thromboembolic events in severe COVID-19 infection. J Am Coll Surg. 2020 doi: 10.1016/j.jamcollsurg.2020.05.007. Published online May 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiang-hua Y., Le-min W., Ai-bin L., et al. Severe acute respiratory syndrome and venous thromboembolism in multiple organs. Am J Respir Crit Care Med. 2010;182(3):436–437. doi: 10.1164/ajrccm.182.3.436. [DOI] [PubMed] [Google Scholar]
- 29.Ding Y., Wang H., Shen H., et al. The clinical pathology of severe acute respiratory syndrome (SARS): a report from China. J Pathol. 2003;200(3):282–289. doi: 10.1002/path.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Varga Z., Flammer A.J., Steiger P., et al. Endothelial cell infection and endotheliitis in COVID-19. Lancet. 2020;395(10234):1417–1418. doi: 10.1016/S0140-6736(20)30937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonetti P.O., Lerman L.O., Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23(2):168–175. doi: 10.1161/01.atv.0000051384.43104.fc. [DOI] [PubMed] [Google Scholar]
- 32.Esmon C.T. Interactions between the innate immune and blood coagulation systems. Trends Immunol. 2004;25(10):536–542. doi: 10.1016/j.it.2004.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y., Xiao M., Zhang S., et al. Coagulopathy and Antiphospholipid antibodies in patients with Covid-19. N Engl J Med. April 8, 2020 doi: 10.1056/NEJMc2007575. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zulfiqar A.-A., Lorenzo-Villalba N., Hassler P., Andrès E. Immune thrombocytopenic Purpura in a patient with Covid-19. N Engl J Med. April 15, 2020 doi: 10.1056/NEJMc2010472. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Magro C., Mulvey J.J., Berlin D., et al. Complement associated microvascular injury and thrombosis in the pathogenesis of severe COVID-19 infection: a report of five cases. Transl Res. 2020;220:1–13. doi: 10.1016/j.trsl.2020.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Campbell C.M., Kahwash R. Will complement inhibition be the new target in treating COVID-19–related systemic thrombosis? Circulation. 2020;141(22):1739–1741. doi: 10.1161/CIRCULATIONAHA.120.047419. [DOI] [PubMed] [Google Scholar]
- 37.Ruf W. Links between complement activation and thrombosis. Blood. 2019;134(Supplement_1):SCI–SCI-40. doi: 10.1182/blood-2019-121113. [DOI] [Google Scholar]
- 38.Santos R.A.S., Sampaio W.O., Alzamora A.C., et al. The ACE2/angiotensin-(1–7)/MAS Axis of the renin-angiotensin system: focus on angiotensin-(1–7) Physiol Rev. 2018;98(1):505–553. doi: 10.1152/physrev.00023.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang C., Stavrou E., Schmaier A.A., et al. Angiotensin 1-7 and mas decrease thrombosis in Bdkrb2−/− mice by increasing NO and prostacyclin to reduce platelet spreading and glycoprotein VI activation. Blood. 2013;121(15):3023–3032. doi: 10.1182/blood-2012-09-459156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fraga-Silva R.A., Costa-Fraga F.P., De Sousa F.B., et al. An orally active formulation of angiotensin-(1-7) produces an antithrombotic effect. Clinics (Sao Paulo) 2011;66(5):837–841. doi: 10.1590/S1807-59322011000500021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fraga-Silva R.A., Pinheiro S.V.B., Gonçalves A.C.C., Alenina N., Bader M., Santos R.A.S. The antithrombotic effect of angiotensin-(1–7) involves mas-mediated NO release from platelets. Mol Med. 2008;14(1–2):28–35. doi: 10.2119/2007-00073.Fraga-Silva. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Senchenkova E.Y., Russell J., Esmon C.T., Granger D.N. Roles of coagulation and fibrinolysis in angiotensin II enhanced microvascular thrombosis. Microcirculation. 2014;21(5):401–407. doi: 10.1111/micc.12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brown N.J., Vaughan D.E. Prothrombotic effects of angiotensin. Adv Intern Med. 2000;45:419–429. [PubMed] [Google Scholar]
- 44.Mukherjee D., Patil C.G. Epidemiology and the global burden of stroke. World Neurosurg. 2011;76(6 Suppl):S85–S90. doi: 10.1016/j.wneu.2011.07.023. [DOI] [PubMed] [Google Scholar]
- 45.Di Carlo A. Human and economic burden of stroke. Age Ageing. 2009;38(1):4–5. doi: 10.1093/ageing/afn282. [DOI] [PubMed] [Google Scholar]
- 46.Umapathi T., Kor A.C., Venketasubramanian N., et al. Large artery ischaemic stroke in severe acute respiratory syndrome (SARS) J Neurol. 2004;251(10):1227–1231. doi: 10.1007/s00415-004-0519-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mao L., Jin H., Wang M., et al. Neurologic manifestations of hospitalized patients with coronavirus disease 2019 in Wuhan, China. JAMA Neurol. 2020;77(6):1–9. doi: 10.1001/jamaneurol.2020.1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Helms J., Kremer S., Merdji H., et al. Neurologic features in severe SARS-CoV-2 infection. N Engl J Med. April 15, 2020 doi: 10.1056/NEJMc2008597. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gunasekaran K., Amoah K., Rajasurya V., Buscher M.G. Stroke in a young COVID−19 patient. QJM. May 22, 2020 doi: 10.1093/qjmed/hcaa177. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jain R., Young M., Dogra S., Kennedy H., Nguyen V., Raz E. Surprise diagnosis of COVID-19 following neuroimaging evaluation for unrelated reasons during the pandemic in hot spots. Am J Neuroradiol. May 28, 2020 doi: 10.3174/ajnr.A6608. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ashrafi F., Zali A., Ommi D., et al. COVID-19-related strokes in adults below 55 years of age: a case series. Neurol Sci. June 24, 2020:1–5. doi: 10.1007/s10072-020-04521-3. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Oxley T.J., Mocco J., Majidi S., et al. Large-vessel stroke as a presenting feature of Covid-19 in the young. N Engl J Med. April 28, 2020 doi: 10.1056/NEJMc2009787. Published online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cavallieri F., Marti A., Fasano A., et al. Prothrombotic state induced by COVID-19 infection as trigger for stroke in young patients: a dangerous association. eNeurological Sci. 2020;20 doi: 10.1016/j.ensci.2020.100247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Valdes Valderrama Eduard, Kelley Humbert, Aaron Lord, Jennifer Frontera, Shadi Yaghi. Severe acute respiratory syndrome coronavirus 2 infection and ischemic stroke. Stroke. 2020;51(7):e124–e127. doi: 10.1161/STROKEAHA.120.030153. [DOI] [PubMed] [Google Scholar]
- 55.Nadar S., Lip G.Y.H. The prothrombotic state in hypertension and the effects of antihypertensive treatment. Curr Pharm Des. 2003;9(21):1715–1732. doi: 10.2174/1381612033454559. [DOI] [PubMed] [Google Scholar]