

Standfirst
Over the last decade, we have witnessed the emergence of ever more machine learning applications in all aspects of the chemical sciences. Here, we highlight specific achievements of machine learning models in the field of computational chemistry by considering selected studies of electronic structure, interatomic potentials, and chemical compound space in chronological order.
Subject terms: Method development, Materials science, Atomistic models
Accurate solutions of the Schrödinger equation for the electrons in molecules and materials would vastly enhance our capability for chemical discovery, but computational cost makes this prohibitive. Since Dirac first exhorted us to find suitable approximations to bypass this cost1, much progress has been made, but much remains out of reach for the foreseeable future. The central promise of machine learning (ML) is that, by exploiting statistical learning of the properties of a few cases, we might leap-frog over the worst bottlenecks in this process.
As visible from the publication record in the field (Fig. 1), over the decade since Nature Communications first appeared, machine learning has gained increasing traction in the hard sciences2, and has found many applications in atomistic simulation sciences3. Here, we focus on the progress achieved in the last decade on three interrelated topics (i) electronic structure theory, broadly defined, (ii) universal force field models, as used for vibrational analysis or molecular dynamics applications, and (iii) first principles-based approaches enabling the exploration of chemical compound space.
Fig. 1. Publications each year from a web of science search with topics of machine learning and either chemistry or materials, July 20, 2020.
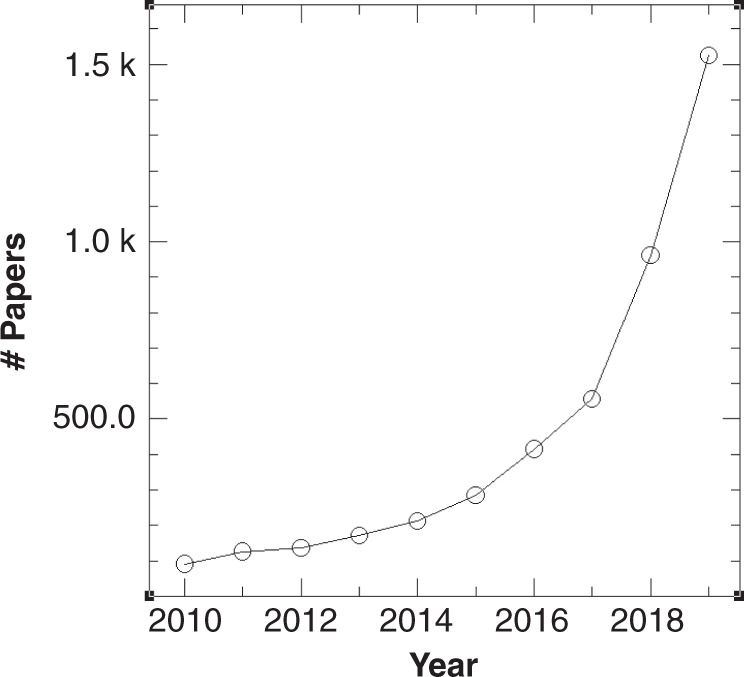
The average number of citations per article is 12. This updates Fig. 1 of ref. 30.
Basic challenges
The central challenge of Schrödinger space is to use supervised learning from examples to find patterns that either accelerate or improve upon the existing human algorithms behind these technologies. In density functional theory (KS-DFT), this most often means improved approximate functionals; in quantum Monte Carlo (QMC), this is faster ways to find variational wavefunctions; in ab initio quantum chemistry such as coupled cluster considering single, double, and perturbative triple excitations (CCSD(T)), this is learned predictions of wavefunction amplitudes instead of recalculation for every system.
In the condensed phase, molecular dynamics simulations yield a vast amount of useful thermodynamic and kinetic properties. Classical force fields cost little to run, but are often accurate only around the equilibrium. The only first-principles alternative is Kohn–Sham density functional theory (DFT), but its computational cost vastly reduces what is practical. A central challenge of configuration space is therefore to produce energies and forces from a classical potential of accuracies comparable to DFT (at least) via training on DFT-calculated samples, possibly for just one element, but with hundreds to thousands of atoms in unique bonding (and bond-breaking) arrangements.
Finally, the challenge of chemical compound space is to explore all useful combinations of distinct atoms. The number of stable combinations is often astronomical. The central aim is to train on quantum-chemical examples, and create a ML algorithm that can, given a configuration of atoms, generate the atomization energy without running, e.g., a DFT calculation, in order to scan the vast unknown of unsynthesized molecules for desirable functionalities.
These challenges are hierarchical. Progress in creating better density functionals clearly impacts finding accurate forces for molecular dynamics and accurate searching of chemical compound space. Finding a way to learn molecular energies with fewer examples is useful for chemical compound space, but forces would also be needed to run molecular dynamics, and self-consistent densities to run orbital-free DFT. The challenges are also overlapping: improved density functionals may be irrelevant if ML force fields can be trained on CCSD(T) energies and forces.
Progress with machine learning
Schrödinger space
Within DFT, the focus is usually on the ever-elusive exchange-correlation (XC) energy4, which is needed as a functional of the spin densities. An ‘easier’ target is orbital-free (OF) DFT, which tries to find the kinetic energy of Kohn–Sham electrons, to bypass the need to solve the Kohn-Sham equations. A primary question is: can machines find better density functional approximations than those created by people? Two distinct approaches are to improve the accuracy of existing human-designed approximations or to create entirely new machine-learned approximations that overcome qualitative failures of our present approximations. Often tests are first performed on model systems, and later applied to more realistic first-principles Hamiltonians.
In orbital-free DFT, Snyder et al.5 used Kernel-Ridge-Regression (KRR) on a one-dimensional model of a molecule an machine-learned functional for OF DFT that breaks bonds correctly, which has been successively built upon6. Brockherde et al.7 showed how KRR could be applied by finding densities directly from potentials (the Hohenberg-Kohn map) avoiding functional derivatives. The problem of XC is harder. Nagai et al.8 showed that accurate densities of just three small molecules are sufficient to create machine-learned approximations that are comparable to those created by people. In ab initio quantum chemistry, Welborn et al.9 have shown how to use features from Hartree-Fock calculations to accurately predict CCSD energies, while an intriguing alternative is to map to spin problems and use a restricted Boltzmann machine10. In the last year, two new applications for finding wavefunctions within QMC have appeared11,12.
While many avenues are being explored, there is as yet no clearly improved, general-purpose ML-designed density functional, ML-powered QMC, or ML approach to ab initio quantum chemistry available to the general user. But for such a complex problem, progress is measured in decades, and we are reasonably confident that such codes could appear over the next five years.
Configuration space
Machine learning models for exploring configurational spaces yield rapid force predictions for extended molecular dynamics simulations. While surrogate models of interatomic potentials using neural networks were firmly established before 201013, Csanyi, Bartok and co-workers used KRR in their seminal ’Gaussian-Approximated Potential’ (GAP) method, relying on Gaussian kernel functions and an atom index invariant bispectrum representation14. In 2013, the first flavor of the smooth overlap of atomic positions (SOAP) representation for KRR based potentials was published15. First stepping stones towards universal force-field, trained ‘on-the-fly’ or throughout the chemical space of molecules displaced along their normal modes, were established in ref. 16,17. KRR based force-field models with CCSD(T) accuracy were introduced in 201718, and based on Behler’s atom-centered symmetry function representations in neural network-based potentials tremendous progress was made19 enabling Smith et al. to train an Accurate Neural network engIne (ANI) on millions of configurations of tens of thousands of organic molecules distorted along aforementioned normal mode displacements20. Impactful applications include KRR potentials used to model challenging processes in ferromagnetic iron21, or Weinan E, Car and co-workers using the Summit supercomputer to simulate 100 million atoms of water with ab initio accuracy using convolutional neural networks22.
Chemical compound space
The idea of using machine learning to mine ab initio materials data bases dates back to 2010 in seminal work by Hautier et al.23. Starting with the Coulomb-matrix24, the development of a selection of ever improved machine learning models (due to improved representations and/or regressor architectures) is exemplified25 on atomization energies of the Quantum Mechanics results for organic molecules with up to 9 heavy atoms (QM9) data set26, as shown in Fig. 2 “QM9-IPAM-challenge”. Such single-point energy calculations typically dominate the cost of quantum chemistry compute campaigns, and therefore a vital minimal target for surrogate models.
Fig. 2. Learning curves of atomization energies of organic molecules, showing out-of-sample prediction error (mean absolute error) decays with increasing number of training molecules drawn at random from QM9 dataset26.
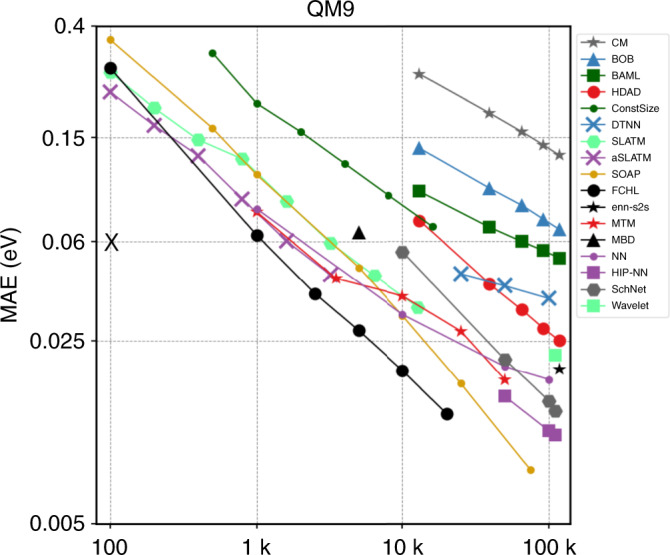
Models shown differ by representation and architecture. The black X denotes the "QM9 challenge'' of achieving 1 kcal/mol accuracy on the QM9 dataset using only 100 molecules for training3. Adapted from ref. 25, Springer Nature Limited.
Examples of improvements of understanding compound space include the discovery of an elpasolite crystal containing aluminum atoms with negative oxidation state27, polarizability models using tensorial learning28, or predicting solvation and acidity in complex mixtures29.
Summary and outlook
Much has happened over the last decade, touching on nearly all aspects of atomistic simulations. Our selection of areas (electronic structure, interatomic potentials, and chemical space) and studies mentioned does not do justice to the overall impact machine learning has had on nearly all branches of the atomistic sciences. Much of the more important work first appeared in rather technical journals such as the Journal of Chemical Physics or Physical Review Letters and is already heavily cited. More recent advances were published in broader journals such as Science, PNAS or Nature and Nature Communications. Some of the outstanding challenges in the field include (i) improved quantum chemistry methods which can reliably cope with reaction barriers, d- and f-elements, magnetic and excited states, as well as redox properties of systems in any aggregation state, (ii) extensive high-quality data sets covering many properties over wide swaths of structural and compositional degrees of freedom, and (iii) the removal of hidden and unconscious biases. Extrapolating from the past, the future looks bright: Long-standing problems have been and are being tackled successfully, and new capabilities are always appearing. Likely, the community will soon address challenges that previously were simply considered to be prohibitively complex or demanding, such as automatized experimentation or synthesis of new materials and molecules on demand.
Acknowledgements
O.A.v.L. acknowledges funding from the Swiss National Science foundation (407540_167186 NFP 75 Big Data, 200021_175747, NCCR MARVEL) and from the European Research Council (ERC-CoG grant QML). K.B. is supported by NSF CHE 1856165.
Author contributions
Both authors conceived, discussed, and wrote this article.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
O. Anatole von Lilienfeld, Email: anatole.vonlilienfeld@unibas.ch.
Kieron Burke, Email: kieron@uci.edu.
References
- 1.Dirac PAM. Quantum mechanics of many-electron systems. Proc. R. Soc. Lond. Series A, Containing Papers of a Mathematical and Physical Character. 1929;123:714–733. [Google Scholar]
- 2.Schütt, K. et al. Machine Learning Meets Quantum Physics. Lecture Notes in Physics (Springer International Publishing, 2020).
- 3.von Lilienfeld, O. A., Müller, K.-R. & Tkatchenko, A. Exploring chemical compound space with quantum-based machine learning. Nat. Rev. Chem.4, 347–358 (2020). [DOI] [PubMed]
- 4.Kohn W, Sham LJ. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965;140:A1133–A1138. doi: 10.1103/PhysRev.140.A1133. [DOI] [Google Scholar]
- 5.Snyder JC, et al. Orbital-free bond breaking via machine learning. J. Chem. Phys. 2013;139:224104. doi: 10.1063/1.4834075. [DOI] [PubMed] [Google Scholar]
- 6.Yao K, Parkhill J. Kinetic energy of hydrocarbons as a function of electron density and convolutional neural networks. J. Chem. Theory Comput. 2016;12:1139–1147. doi: 10.1021/acs.jctc.5b01011. [DOI] [PubMed] [Google Scholar]
- 7.Brockherde F, et al. Bypassing the Kohn-Sham equations with machine learning. Nat. Commun. 2017;8:872. doi: 10.1038/s41467-017-00839-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nagai R, Akashi R, Sugino O. Completing density functional theory by machine learning hidden messages from molecules. npj Comput. Mater. 2020;6:43. doi: 10.1038/s41524-020-0310-0. [DOI] [Google Scholar]
- 9.Welborn M, Cheng L, Miller TF. Transferability in machine learning for electronic structure via the molecular orbital basis. Journal of Chem. Theory Comput. 2018;14:4772–4779. doi: 10.1021/acs.jctc.8b00636. [DOI] [PubMed] [Google Scholar]
- 10.Choo K, Mezzacapo A, Carleo G. Fermionic neural-network states for ab-initio electronic structure. Nat. Commun. 2020;11:2368. doi: 10.1038/s41467-020-15724-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pfau, D., Spencer, J. S., de G. Matthews, A. G. & Foulkes, W. M. C. Ab-initio solution of the many-electron Schrödinger equation with deep neural networks. Preprint athttp://arXiv.org/abs/1909.02487 (2019).
- 12.Hermann, J., Schätzle, Z. & Noé, F. Deep neural network solution of the electronic Schrödinger equation. Preprint at http://arXiv.org/abs/1909.08423 (2019). [DOI] [PubMed]
- 13.Behler J, Parrinello M. Generalized neural-network representation of high-dimensional potential-energy surfaces. Phys. Rev. Lett. 2007;98:146401. doi: 10.1103/PhysRevLett.98.146401. [DOI] [PubMed] [Google Scholar]
- 14.Bartók AP, Payne MC, Kondor R, Csányi G. Gaussian approximation potentials: The accuracy of quantum mechanics, without the electrons. Phys. Rev. Lett. 2010;104:136403. doi: 10.1103/PhysRevLett.104.136403. [DOI] [PubMed] [Google Scholar]
- 15.Bartók AP, Kondor R, Csányi G. On representing chemical environments. Phys. Rev. B. 2013;87:184115. doi: 10.1103/PhysRevB.87.184115. [DOI] [Google Scholar]
- 16.Li Z, Kermode JR, De Vita A. Molecular dynamics with on-the-fly machine learning of quantum-mechanical forces. Phys. Rev. Lett. 2015;114:096405. doi: 10.1103/PhysRevLett.114.096405. [DOI] [PubMed] [Google Scholar]
- 17.Rupp M, Ramakrishnan R, von Lilienfeld OA. Machine learning for quantum mechanical properties of atoms in molecules. J. Phys. Chem. Lett. 2015;6:3309. doi: 10.1021/acs.jpclett.5b01456. [DOI] [Google Scholar]
- 18.Chmiela S, et al. Machine learning of accurate energy-conserving molecular force fields. Sci. Adv. 2017;3:e1603015. doi: 10.1126/sciadv.1603015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Behler J. First principles neural network potentials for reactive simulations of large molecular and condensed systems. Angewandte Chemie Int. Edn. 2017;56:12828–12840. doi: 10.1002/anie.201703114. [DOI] [PubMed] [Google Scholar]
- 20.Smith JS, Isayev O, Roitberg AE. Ani-1: an extensible neural network potential with dft accuracy at force field computational cost. Chem. Sci. 2017;8:3192–3203. doi: 10.1039/C6SC05720A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dragoni D, Daff TD, Csányi G, Marzari N. Achieving dft accuracy with a machine-learning interatomic potential: thermomechanics and defects in bcc ferromagnetic iron. Phys. Rev. Mater. 2018;2:013808. doi: 10.1103/PhysRevMaterials.2.013808. [DOI] [Google Scholar]
- 22.Jia, W. et al. Pushing the limit of molecular dynamics with ab initio accuracy to 100 million atoms with machine learning. Preprint at http://arXiv.org/abs/2005.00223 (2020).
- 23.Hautier G, Fischer CC, Jain A, Mueller T, Ceder G. Finding nature’s missing ternary oxide compounds using machine learning and density functional theory. Chem. Mater. 2010;22:3762. doi: 10.1021/cm100795d. [DOI] [Google Scholar]
- 24.Rupp M, Tkatchenko A, Müller K-R, von Lilienfeld OA. Fast and accurate modeling of molecular atomization energies with machine learning. Phys. Rev. Lett. 2012;108:058301. doi: 10.1103/PhysRevLett.108.058301. [DOI] [PubMed] [Google Scholar]
- 25.Faber, F. A., Christensen, A. S. & von Lilienfeld, O. A. In Machine Learning Meets Quantum Physics, (eds Schütt, K. T. et al.) 155–169 (Springer, 2020).
- 26.Ramakrishnan R, Dral P, Rupp M, von Lilienfeld OA. Quantum chemistry structures and properties of 134 kilo molecules. Scientific Data. 2014;1:140022. doi: 10.1038/sdata.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Faber FA, Lindmaa A, von Lilienfeld OA, Armiento R. Machine learning energies of 2 million elpasolite (ABC2D6) crystals. Phys. Rev. Lett. 2016;117:135502. doi: 10.1103/PhysRevLett.117.135502. [DOI] [PubMed] [Google Scholar]
- 28.Wilkins DM, et al. Accurate molecular polarizabilities with coupled cluster theory and machine learning. Proc. Natl Acad. Sci. USA. 2019;116:3401–3406. doi: 10.1073/pnas.1816132116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rossi, K. et al. Simulating solvation and acidity in complex mixtures with first-principles accuracy: The case of CH3SO3H and H2O2 in phenol. J. Chem. Theory Comput.16, 5139–5149 (2020). [DOI] [PubMed]
- 30.Rupp M, von Lilienfeld OA, Burke K. Guest editorial: Special topic on data-enabled theoretical chemistry. J. Chem. Phys. 2018;148:241401. doi: 10.1063/1.5043213. [DOI] [PubMed] [Google Scholar]