

Abstract
The E571K mutation of CRM1 is highly prevalent in some cancers, but its mechanism of tumorigenesis is unclear. Glu571 of CRM1 is located in its nuclear export signal (NES)-binding groove, suggesting that binding of select NESs may be altered. We generated HEK 293 cells with either monoallelic CRM1WT/E571K or biallelic CRM1E571K/E571K using CRISPR/Cas9. We also combined analysis of binding affinities and structures of 27 diverse NESs for wild-type and E571K CRM1 with structure-based bioinformatics. While most NESs bind the two CRM1 similarly, NESs from Mek1, eIF4E-transporter, and RPS2 showed >10-fold affinity differences. These NESs have multiple charged side chains binding close to CRM1 position 571, but this feature alone was not sufficient to predict different binding to CRM1(E571K). Consistent with eIF4E-transporter NES binding weaker to CRM1(E571K), eIF4E-transporter was mislocalized in tumor cells carrying CRM1(E571K). This serves as proof of concept that understanding how CRM1(E571K) affects NES binding provides a platform for identifying cargoes that are mislocalized in cancer upon CRM1 mutation. Finally, we showed that large affinity changes seen with some NES peptides (of Mek1 and RPS2) do not always translate to the full-length cargoes, suggesting limitations with current NES prediction methods. Therefore, comprehensive studies like ours are imperative to identify CRM1 cargoes with real pathogenic potential.
INTRODUCTION
Proper subcellular distribution of macromolecules is critical in maintaining cellular homeostasis, and karyopherin-mediated transport of macromolecules between the nucleus and the cytoplasm determines nuclear-cytoplasmic distribution of many macromolecules in the cell (Gorlich and Kutay, 1999; Chook and Blobel, 2001; Weis, 2003; Cook et al., 2007). The exportin CRM1 (chromosomal region maintenance 1; also known as exportin-1 or XPO1) mediates nuclear export of hundreds of diverse-functioning proteins including many tumor suppressors, cell cycle regulators, and many mRNAs (Xu et al., 2012a, b). Whole exome sequencing across a variety of cancer types have revealed that residue Glu571 of CRM1 is prevalently mutated in lymphoid malignancies such as primary mediastinal B-cell lymphoma (PMBCL), diffuse large B-cell lymphoma (DLBCL) and chronic lymphocytic leukemia, most often to a lysine (Jardin et al., 2016). The E571K mutation of CRM1 was shown to stimulate tumorigenesis in B-cell lymphoma mouse models (Taylor et al., 2019). Several proteins, such as DCD, LRMP, S100A8, TRAF2, and ANKRD62, were found to be mislocalized in these cells (Taylor et al., 2019). While these proteins are not known to interact with CRM1, and their mislocalizations may be secondary effects of the E571K mutation of CRM1, it is clear that there is a gap in knowledge in how CRM1(E571K) interacts with cargoes.
CRM1 binds to nuclear export signals (NESs) in its protein cargoes (Dong et al., 2009; Monecke et al., 2009). NESs are usually 8–15 amino acids long and contain four or five hydrophobic (φ) residues that are characteristically spaced into diverse patterns; hence the organization into 11 NES classes (class 1a–d, 2, 3, 4, and 1a–d reverse) (Kosugi et al., 2008; Fung et al., 2015). In the nucleus, CRM1 binds the NES-containing cargo and RanGTP cooperatively to form the cargo•CRM1•RanGTP complex, which translocates through the nuclear pore complex (NPC) via interactions between CRM1 and phenylalanine–glycine or FG repeats that span the central channel of the NPC. When the cargo•CRM1•RanGTP complex reaches the cytoplasm, RanGAP1 catalyzes hydrolysis of the Ran-bound GTP to GDP. RanGDP no longer binds CRM1, and the loss of Ran binding reduces CRM1-cargo affinity, leading to release of cargo into the cytoplasm (Gorlich and Kutay, 1999; Weis, 2003; Port et al., 2015).
CRM1 is composed of 21 HEAT repeats that are arranged into a ring-shaped solenoid. The NES of a cargo binds to a hydrophobic groove on the convex side of the CRM1 ring formed by HEAT repeats 11 and 12 (Figure 1A) (Dong et al., 2009; Monecke et al., 2009). Structures of CRM1 bound to 13 different NES peptides show that the conformation of the NES-binding groove remains unchanged when the structures are bound to NESs with diverse sequences. The NES-binding groove, which contains five hydrophobic pockets (P0–P4), is wide at pockets P0–P2 and is narrow beyond pocket P3 (Figure 1B). The wide portion of the groove often binds the NES α-helix while the narrow part of the groove is constrained to bind β-strands or extended portions of the NES (Fung et al., 2015, 2017). NESs adopt many different conformations to bind into the CRM1 groove; some NESs bind in one direction (Figure 1, B and C) while others bind in the opposite direction (Figure 1D) (Fung et al., 2015, 2017; Fu et al., 2018). Glu571 of CRM1 is located in the NES-binding groove near the β-strands of many bound NESs (Figure 1, B–D) (Fung et al., 2015, 2017). The proximity of this mutation site to the side chains of the bound NESs, and the switch from the negative charge of the Glu571 side chain to the positive charge of a lysine, suggest that some NESs are likely to bind CRM1(E571K) and WT CRM1 differently.
FIGURE 1:
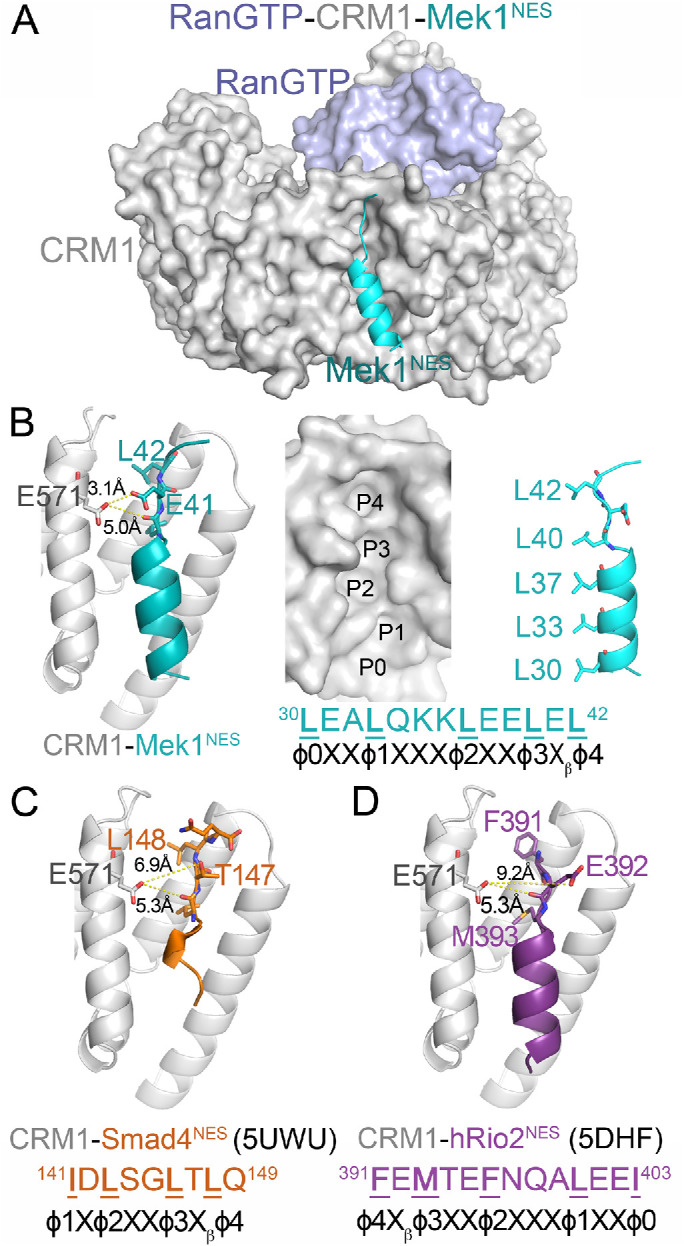
Glu571 of CRM1 is located near the β-strand of bound NESs. (A) Structure of CRM1 (gray) bound to RanGTP (light blue) and the NES of Mek1 (cyan). (B) Left panel: details of the Mek1NES-bound CRM1 groove, showing the Glu571 side chain of CRM1 (gray) and the side chains of the NES β-strand (cyan). Middle panel: surface representation of the NES-binding groove, with the NES peptide removed and rotated along the vertical axis to show the hydrophobic φ0-4 side chains (right panel). (C, D) Details of the Smad4NES-bound (C; brown; 5UWU) and hRio2NES-bound (D; purple; 5DHF) CRM1 grooves, showing the Glu571 side chains of CRM1 and the side chains of the NES β-strands.
To obtain this basic knowledge, we set out to determine how CRM1(E571K) binds diverse cargoes. First, we report cellular models with endogenous heterozygous CRM1WT/E571K and homozygous CRM1E571K/E571K, respectively, which show decreased growth and cell cycle disruption. Second, we measured the affinities of both WT and E571K CRM1 binding to 17 NES peptides with diverse sequences and found two NES peptides from the cargoes Mek1 and eIF4E-transporter (4E-T) that bind the CRM1 proteins very differently. Both NESs have multiple charged side chains in their β-strands, and we used this information in a proteome-wide, structure-based bioinformatics method and identified 184 new NES-like sequences with charged β-strands. We measured binding affinities for 10 of the 184 peptides for WT and E571K CRM1 and found only one peptide, the putative NES of RPS2, that binds very differently to the mutant CRM1. The finding that many NESs with charged β-strands bind similarly to WT and mutant CRM1 suggests that the characteristic of charged NES β-strand is not sufficient to predict CRM1(E571K)-impacted NESs. In addition to charged β-strands, hydrophobic side chains in the β-strand also affect binding to CRM1(E571K). Of the three NESs (Mek1, RPS2, and 4E-T) that bind WT and mutant CRM1 differently, the localization pattern of full-length 4E-T in cells is consistent with the results that its NES binds weaker to CRM1E571K. 4E-T is mostly cytoplasmic in cells carrying WT CRM1 but is evenly distributed across the nucleus and the cytoplasm in cells carrying CRM1(E571K), including in primary tumor cells from chronic lymphocytic leukemia (CLL) patients carrying the E571K mutation. Finally, we showed that large affinity changes seen with NES peptides (such as the NESs of Mek1 and RPS2) do not always translate to the full-length cargo proteins because of inaccuracies in NES prediction, suggesting that comprehensive studies like the one we are presenting are imperative to identify CRM1 cargoes driving pathogenic changes.
RESULTS
CRM1(E571K) affects HEK 293 cell growth and causes mitotic defects
We used CRISPR/Cas9-mediated, homology-directed recombination to knock in CRM1(E571K) (GAA > AAA) to the endogenous locus of HEK 293 cells. Isogenic clones were created containing either monoallelic (CRM1WT/E571K) or biallelic (CRM1E571K/E571K) knock in of the E571K mutation (Figure 2A). The status of CRM1, WT or E571K, was validated by genomic extraction, PCR, and sequencing the region around the Glu571 codon.
FIGURE 2:
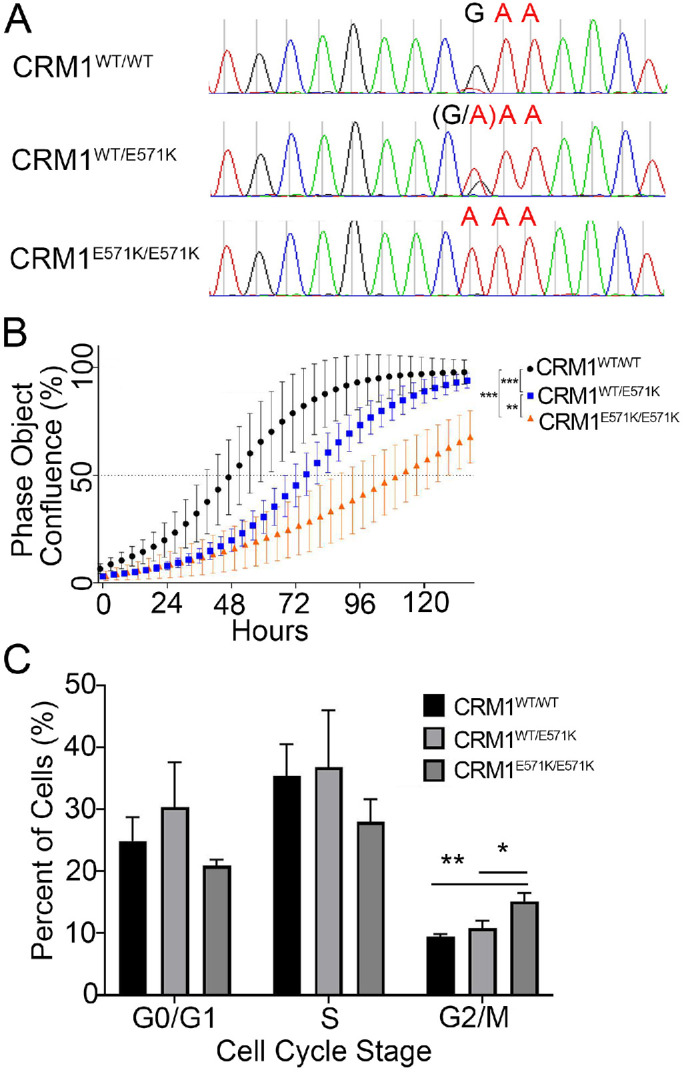
Characterization of HEK 293 CRM1 mutant cell lines. (A) Sequences of HEK 293 genomic DNA confirm successful knock-in of CRM1(E571K) into heterozygous and homozygous isogenic clones. (B) Proliferation curves of HEK 293 cells with CRM1WT/WT, CRM1WT/E571K, and CRM1E571K/E571K. (C) FACS analysis of fixed, asynchronous HEK 293 cells (CRM1WT/WT, CRM1WT/E571K, and CRM1E571K/E571K; n = 1000 cells per each of three replicates) stained with propidium iodide to analyze the population in different cell cycle stages. Statistical significance determined using the Holm–Sidak method, with alpha = 0.05 without a consistent SD.
To compare the proliferation rate of cells containing CRM1(E571K), cell density/confluence was measured as a function of time using light microscopy. Both CRM1WT/E571K and CRM1E571K/E571K cells show significant reduction in growth rates compared with CRM1WT/WT cells (p > 0.001), with CRM1E571K/E571K cells growing slower than CRM1WT/E571K cells (Figure 2B; statistical significance determined using the Holm–Sidak method, with alpha = 0.05). We also examined how progression of cell cycle stages was affected by CRM1(E571K). Cell cycle analysis via fluorescence activated cell sorting (FACS) showed a higher percentage of CRM1E571K/E571K cells in the G2/M stage than CRM1WT or CRM1WT/E571K cells (p = 0.0023) (Figure 2C). There are only slightly fewer CRM1E571K/E571K cells in G0/G1 and S than observed with CRM1WT/WT cells (p = 0.1534 and p = 0.1119, respectively).
A few NES peptides bind WT and E571K CRM1 with different affinities
Effects of the E571K mutation of CRM1 on cell growth and mitotic stability suggest that the single-amino-acid change likely alters CRM1 activity. Location of Glu571 at the NES-binding groove of CRM1 further suggests that the mutation may affect NES binding and hence nuclear-cytoplasmic localization of select NESs/cargoes (Figure 1). The Glu571 side chain is closest to the side chain of the NES residue Xβ, which is located between positions φ3 and φ4 (Figure 1, B–D). The φ3-Xβ-φ4 residues form β-strands in many CRM1-bound NESs. The φ3 and φ4 side chains point into the hydrophobic groove of CRM1 while the Xβ side chain points out toward solvent. The Xβ side chain in some NESs, such as the Mek1NES, contacts the CRM1 Glu571 side chain (Figure 1B). However, NES β-strands have diverse sequences and are positioned differently in the CRM1 groove, and the Xβ side chains of some NESs, such as in Smad4NES and hRio2NES, are too far away to interact with CRM1 Glu571 (Figure 1, C and D). Furthermore, there is no obvious amino acid preference for position Xβ, making it difficult to predict which NES would bind to CRM1(E571K) differently (Xu et al., 2012a).
We selected 17 previously characterized NESs to compare interactions with WT versus E571K CRM1 (Table 1; Supplemental Figures 1 and 2). These peptides were chosen to be diverse in sequence, especially in their β-strands, and cover all NES classes. We measured dissociation constants (Kds) of CRM1-NES interactions using a previously described fluorescent PKINES differential photobleaching assay (Fung et al., 2015, 2017). Kd measurements for each NES were performed in triplicate, and direct PKINES titration was used as quality control for the different preparations of CRM1 protein (Table 1; Supplemental Figures 1 and 2). Experiments where the positive control (CRM1 binding to FITC-PKINES) Kds varied more than twofold from the average Kd were not considered. Kds for several NES peptides binding to WT CRM1 were reported in previous publications (labeled with asterisks in Table 1) (Fung et al., 2015, 2017). Here, we repeated the affinity measurements of WT CRM1 with three of the same NESs (PKINES, Mek1NES, Smad4NES) and found the Kds measured in this study similar to those in the previous reports.
TABLE 1:
Binding affinities (Kd) of a diverse set of NESs for WT and E571K CRM1.
| Binding affinity (Kd) | ||||
|---|---|---|---|---|
| NES | Sequence | CRM1WT | CRM1(E571K) | Kd,E571K/Kd,WT |
| Mek1NES | TNLEALQKKLEELELDEQ | 70 nM[40–130 nM]a | 5 nM[3–8 nM] | 0.071 |
| P73NES | NFEILMKLKESLELMELVP | 2.0 μM[1.7–2.4 μM]a | 1.5 μM[1.0–2.2 μM] | 0.75 |
| hRio2NES | RSFEMTEFNQALEEIKG | 2.4 μM[1.7–4.7 μM]a | 1.9 μM[1.4–2.5 μM] | 0.79 |
| X11L2NES | SSLQELVQQFEALPGDLV | 1.5 μM[1.1–2.0 μM]a | 1.3 μM[0.9–1.8 μM] | 0.87 |
| mDia2NES | SVPEVEALLARLRAL | 1.7 μM[1.0–2.7 μM]a | 1.5 μM[1.0–2.2 μM] | 0.88 |
| IκβαNES | EQMVKELQEIRLEPQ | 22 μM[17–28 μM] | 21 μM[17–27 μM] | 0.95 |
| Snupn1NES | MEELSQALASSFSVSQDLNS | 12 μM[10–15 μM]a | 21 μM[15–31 μM] | 1.8 |
| HIV RevNES | LQLPPLERLTLDC | 1.8 μM[0.9–1.4 μM]a | 4.1 μM[3.6–4.7 μM] | 2.3 |
| Smad4NES | YERVVSPGIDLSGLTLQ | 4.6 μM[3.6–5.9 μM]a | 13 μM[10–17 μM] | 2.8 |
| FMRP1bNES | LKEVDQLRALERLQID | 3 μM[2.3–3.9 μM]a | 9 μM[5–18 μM] | 3.0 |
| Dusp6NES | PLPVLGLGGLRISS | 4.5 μM[3.7–5.1 μM] | 14 μM[10–18 μM] | 3.1 |
| Cpeb4NES | RTFDMHSLESSIDIMR | 590 nM[400–840 nM]a | 1900 nM[1.5–2.3 μM] | 3.2 |
| IκβεNES | SLVLLPFDDLKISG | 3.6 μM[3.3–4.0 μM] | 12 μM[10–14 μM] | 3.3 |
| PaxillinNES | RELDELMASLSDFKFMA | 670 nM[400–1000 nM]a | 3.1 µM[2.5–3.9 μM] | 4.6 |
| PKINES | NSNELALKLAGLDINK | 35 nM[31–39 nM] | 200 nM[170–30 nM] | 6.3 |
| PP2A B56αNES | REELWKKLEELKLKK | 4.4 μM[3.5–5.4 μM] | 38 μM[28–55 μM] | 8.6 |
| eIF4E-transporter (4E-TNES) | SVEEVEAGLKGLKVDQQVK | 1.4 μM[1.2–1.7 μM] | 14 μM[11–18 μM] | 10 |
68.3% confidence intervals are reported in brackets.
aKds measured previously (Fung et al., 2015, 2017; Fu et al., 2018).
Of the 17 NESs studied, six bind WT and mutant CRM1 with similar affinities (Kd,E571K/Kd,WT ∼0.5–2.0 or < twofold differences in affinities), eight others show only marginal (2–8.6-fold) differences in affinities, and two NESs show large (>10-fold) differences in affinities between WT and mutant CRM1 (Table 1; Supplemental Figures 1 and 2). Most of the NES peptides bind CRM1(E571K) with lower affinity than WT CRM1. The two NESs that bind WT and mutant CRM1 very differently are the NESs of the eIF4E transporter (4E-T; also known as EIF4ENIF1) and the Mek1 kinase. The 4E-TNES binds 10-fold weaker to CRM1(E571K) than to WT CRM1 (Supplemental Figure 1E). In contrast, the Mek1NES shows a striking and unusual 14-fold-stronger binding to CRM1(E571K) than to WT CRM1 (Supplemental Figure 2A).
Structural analysis to explain NESs that bind CRM1(E571K) very differently
We solved 10 new crystal structures of NES-bound CRM1 to understand how CRM1(E571K) interacts with NESs. First, we solved the structures of unliganded WT CRM1 and CRM1(E571K), which are almost identical (root-mean-square deviation [r.m.s.d] 0.102 for 1219 Cα atoms; Supplemental Tables 1 and 2; Supplemental Figure 3). Both NES-binding grooves are closed, with Glu571 in WT CRM1 and the mutated lysine side chain of CRM1(E571K) extending out toward solvent without making any contacts that might affect the stability of the closed groove (Supplemental Figure 3).
Next, we solved the structures of CRM1(E571K) bound to three different NES peptides (from Mek1, PKI, and mDia2; Supplemental Table 2) and compared them to the structures of WT CRM1 bound to the same NESs (Mek1 and PKI in Supplemental Table 1, mDia2 previously solved; Fung et al., 2017). We also solved the structure of WT CRM1 bound to 4E-TNES (Supplemental Table 1). We compared these new structures to 13 previously reported structures of WT CRM1 bound to NESs to explain CRM1(E571K) binding affinity differences (Fung et al., 2015, 2017). We will first discuss the Mek1NES- and 4E-TNES-CRM1 structures since both NES peptides showed large affinity differences for E571K versus WT CRM1. This will be followed by structural analysis of NESs with modest or no changes in affinities.
Mek1NES binds CRM1(E571K) 14-fold tighter than WT CRM1. The structure of Mek1NES bound to WT CRM1 shows the NES Xβ (Glu41) side chain 3.1 Å from CRM1 Glu571 (Figure 3A). The same Glu41 (Xβ) side chain of Mek1NES makes electrostatic interactions with the lysine in position 571 of CRM1(E571K) (Figure 3B). This electrostatic lysine–glutamate interaction CRM1(E571K) likely contributes to the 14-fold tighter binding to the CRM1 mutant. We mutated Glu41 of Mek1NES to alanine, to test its importance in contributing to affinity differences between CRM1(E571K) and WT CRM1. The Mek1NES(E41A) peptide binds 14-fold tighter to WT CRM1 than the WT Mek1NES (Kd,Mek1NES(E41A) = 5 nM vs. Kd,Mek1NES = 70 nM; Supplemental Figure 4A), possibly due to removal of electrostatic repulsion between the NES and CRM1 Glu571. Mek1NES(E41A) binds similarly to WT and E571K CRM1 (Kd,WT CRM1 = 5 nM vs. Kd,CRM1(E571K) = 6 nM; Supplemental Figure 4A), supporting the idea that the NES Xβ position is important in determining whether the NES will bind differently to CRM1(E571K) (Figure 3C).
FIGURE 3:

NESs with charged β-strands bind either stronger or weaker to CRM1 E571K. (A) Details of the NES-binding grooves of NES-bound CRM1 from crystal structures of WT (left) and E571K (right) CRM1 bound to the Mek1NES. CRM1 is in gray, and the NES is in cyan. (B) Details of the NES-binding grooves of eIF4E-TNES-bound WT CRM1. CRM1 is in gray, and the NES is in green. Electron density from composite omit maps (2Fo-Fc contoured in blue mesh to 1σ) are shown for key residues in A andB. (C) Binding affinities of interactions between wild type and mutated Mek1NES, eIF4E-TNES, and PaxillinNES with WT and E571K CRM1.
If a glutamate at the Xβ position confers tighter binding to CRM1(E571K), then a lysine there should cause the NES to bind weaker to the CRM1 mutant. Indeed, all NESs in Table 1 with a Xβ lysine, such as the 4E-TNES, PP2A B56αNES, and PaxillinNES, show 4–10-fold weaker binding to CRM1(E571K) than to WT CRM1 (Supplemental Figures 1, D and E, and 2I). In the crystal structure of WT CRM1 bound to the 4E-TNES, which binds the CRM1(E571K) 10-fold weaker, the Xβ side chain (Lys446) of 4E-TNES points toward solvent and has poor electron density (average side chain B-factor ∼1252), but is nonetheless near CRM1 Glu571 (Figure 3B; Supplemental Table 1). Mutation of the 4E-T Lys446 attenuated the affinity differences between WT and mutant CRM1 (Kd,E571K/Kd,WT is 5.8 for 4E-TNES[K446A] vs. 10 for 4E-TNES; Supplemental Figure 4B), thus providing additional support that the Xβ position is important in determining the impact of the E571K mutation in CRM1 (Figure 3C).
Structural analysis of NES with modest or no affinity differences for WT versus E571K CRM1
We organize structural analysis of NESs with modest or no affinity differences for CRM1(E571K) into three groups: 1) NESs with charged β-strand side chains, 2) NESs with polar β-strand side chains, and 3) one NES with no β-strand. Several NES peptides, including the PKINES, have negatively charged β-strand side chains but show little difference in affinity for E571K versus WT CRM1. When bound to WT CRM1, the Asp46 (Xβ) side chain of PKINES makes intra-NES interactions with the backbone amides of Asn48 and Lys49 and is thus positioned far from CRM1 Glu571 (Supplemental Figure 5A; Supplemental Table 1). When bound to CRM1(E571K), the PKINES Asp46 repositions toward the CRM1 lysine at position 571 (although there is no electron density for the side chain) and no longer participates in intra-NES interactions. The modest approximately sixfold binding affinity decrease for E571K versus WT CRM1 may be explained by a potentially modest gain of binding energy from the new long-range Asp46 (NES) –Lys571 (CRM1) interaction that is offset by the loss of intra-NES interactions (Supplemental Figure 5B; Supplemental Table 2).
PP2A B56αNES, PaxNES, Dusp6NES, IκβεNES, IκβαNES, P73NES, CPEB4NES, and hRio2NES all have charged residues in their β-strands but show only modest affinity differences when binding CRM1(E571K). The first six NES peptides have positively charged Xβ side chains but bind <10-fold weaker to CRM1(E571K) (Kd,E571K/Kd,WT = 8.6, 4.6, 3.1, 3.3, 0.95, and 0.75; Supplemental Figures 1, A–D, and 2D) and none is suitable for structural analysis because of either solubility issues or weak affinities for WT CRM1. We therefore studied PaxillinNES, which also has an Xβ lysine (Lys277) and binds WT CRM1 well (Kd 670 nM). Lys277 of PaxillinNES makes electrostatic interactions with Glu571 of WT CRM1 (Supplemental Figure 5C). Like 4E-TNES, the affinity difference for E571K versus WT CRM1 is attenuated when Lys277 of PaxillinNES is mutated to alanine (Supplemental Figure 4C and Figure 3C; Kd,E571K/Kd,WT is 2.1 for PaxNES[Xβ,K277A] vs. 4.6 for PaxNES), suggesting that the proximity the Xβ lysine to the lysine at position 571 of CRM1(E571K) likely destabilizes binding to CRM1(E571K). CPEB4NES and hRio2NES are reverse–Class 1a NESs, and their peptide chain orientations in the CRM1 groove position the Xβ side chains (Asp382 in CPEB4NES and Glu392 in hRio2NES) far from Glu571 of WT CRM1 (Supplemental Figure 5, D and E). Arg379 of CPEB4NES, which is two positions away from φ4, contacts Glu571 of WT CRM1 instead, suggesting that Arg379 may also be close to the lysine in position 571 of CRM1(E571K), and thus slightly destabilizes the CRM1-NES(E571K) interactions (Supplemental Figure 5D; Kd,E571K/Kd,WT = 3.2) (Fung et al., 2015). There is no interaction between any hRio2NES residues with Glu571 of WT CRM1 (Supplemental Figure 5E), explaining why the peptide does not bind CRM1(E571K) differently (Table 1; Kd,E571K/Kd,WT = 0.79).
Supplemental Figure 5, F–J, shows structures of WT CRM1 bound to five NESs that bind similarly to WT and E571K CRM1 and also have polar Xβ side chains. All the Xβ side chains are too far from Glu571 for interactions, likely explaining why NES binding is not affected by the E571K mutation. Finally, the structures of WT CRM1(PDB 5UWP) and CRM1(E571K) bound to the entirely helical mDia2NES are almost identical (Supplemental Figure 5L). The NES helix occupies only the wide part of the CRM1 groove (P0–P3), leaving the portion of the CRM1 groove that normally binds the NES β-strand unoccupied (Supplemental Figure 5K), explaining why the NES binds WT and E571K CRM1 similarly (Table 1; Kd,E571K/Kd,WT = 0.88).
In summary, the NESs that we have studied can be separated into three groups: 1) those with charged or 2) those with polar residues in their β-strand, or 3) those with no β-strand. Some NESs with charged β-strand residues can bind E571K and WT with quite different affinities because of differences in electrostatic interactions between CRM1 Glu or Lys 571 and the NES. Other NESs have modest to no changes because their charged residues are too far away. NESs with polar β-strands are also too far to contact CRM1 residue 571 and therefore are modestly or not affected by the CRM1 mutation. Class 3 NESs, which have no β-strand, do not contact residue 571 of CRM1 and are thus also not affected by the mutation.
Identification of novel NESs with charged β-strands
We used a recently developed structure-based NES prediction tool to search for new NESs that may be substantially affected by CRM1(E571K) (Lee et al., 2019, 2020). The Rosetta-based predictor models putative NES sequences into multiple CRM1-NES structure templates, allows for structural rearrangement, and then calculates the predicted binding energy of the docked peptide, which is used to score the likelihood of binding CRM1 (Figure 4A). It was previously shown that predicted CRM1-NES binding energies for different NESs correlate well with measured Kds (Lee et al., 2019). We used this method to search the human reference proteome for sequences that match the patterns of Class 1a NESs that carry charged side chains in their β-strands (sequence pattern: Φ1-X-X-X-Φ2-X-X-Φ3-[E,K]-Φ4-[D,E,K]) because we have shown above that such NESs are most likely to bind WT and E571K CRM1 differently. The search produced a list of 184 novel potential NESs with charged β-strands (Supplemental Table 3). Gene ontology analysis suggests that many of these sequences are found in proteins involved in mitotic spindle checkpoint (false discovery rate [FDR] = 4.00e-02), regulation of mitotic metaphase/anaphase transition (FDR = 4.71e-02), cell differentiation (FDR = 1.31e-04), cell death (FDR = 2.51e-09) and chromosome segregation (FDR = 1.56e-03) (Supplemental Table 3).
FIGURE 4:

Structure-based NES prediction: proteome search for NESs with charged β-strands. (A) Schematic of the structure-based NES prediction method used to identify putative NES sequences that contain a charged NES motif (Φ3 X1 Φ4 X2 X3, where X1 = E/K and X2 = D/E/K) in their β-strands. (B) Binding affinities of CRM1 WT and E571K binding to 10 different NESs with positive or negative charges in their β-strands that were identified by the method in A.
We selected 10 of these new putative NESs, five each with negatively charged and positively charged β-strands, to measure Kds for WT and E571K CRM1 using a higher throughput fluorescence polarization assay (Supplemental Figures 6 and 7). To rule out variations of Kd values across different assays (fluorescence polarization vs. photobleaching assays), we measured the Kds of PKINES and Dusp6NES and found their binding affinities to be the same as those measured by the MST photobleaching assay (Table 1). As with the MST photobleaching experiments, we control for consistency across different preparations of proteins by measuring the Kd of CRM1 for the FITC-PKINES in all fluorescence polarization experiments. We also used the same preparations of proteins to measure the Kds for both WT and CRM1(E571K).
Only one newly identified putative NES, from RPS2 (40S ribosomal protein S2), showed a large affinity difference for E571K versus WT CRM1. RPS2NES binds CRM1(E571K) 16.0-fold weaker than WT CRM1 (Figure 4B). The other nine NESs showed modest to no affinity differences. NESs that bind CRM1(E571K) with modest differences follow the trends described above: NESs with negatively charged β-strands bind CRM1(E571K) slightly tighter, and those with positively charged β-strands bind slightly weaker (Table 1 and Figure 4B). In summary, of the 21 NES peptides with charged β-strands that we tested for WT versus E571K CRM1 binding (Table 1 and Figure 4B), only three showed greater than 10-fold affinity differences for CRM1(E571K), suggesting that although charges in the NES β-strands are important, this feature alone is not sufficient to predict that an NES will be affected by the E571K mutation in CRM1.
Other NES features that affect binding to E571K versus WT CRM1
We analyzed several CRM1-NES structures to understand how other residues in the NES β-strand may influence interactions with Glu571 of CRM1, in order to explain why charges on the NES β-strands are not sufficient to predict differences in binding CRM1(E571K). The Mek1NES, which binds CRM1(E571K) 14-fold tighter than WT CRM1, has a β-strand with a 40φ3Eφ4DE44 sequence pattern (Figure 1B). The NES Asp43 side chain is 8.1 Å away from the lysine in position 571 of CRM1(E571K) and may participate in long-range electrostatic interactions and contribute to increased affinity for the mutant CRM1. We mutated 43DE44 of Mek1NES to Asn-Gln (Mek1NESNQ) to test the importance of these C-terminal residues. Mek1NESNQ binds weaker to CRM1(E571K) than Mek1NES, suggesting that residues beyond the φ4 position can contribute to interactions with WT and E571K CRM1 (Figure 5, A–C; Supplemental Figure 4D; Supplemental Table 1).
FIGURE 5:

Additional NES β-strand residues contribute interactions with WT and E571K CRM1. (A) Details of the NES-binding grooves of Mek1NES (green, left) and (B) Mek1NQNES (pink) bound to WT CRM1 (gray). (C) Binding affinities of interactions between wild type and mutated Mek1NES with WT and E571K CRM1. (D, E) Details of the NES-binding grooves of PKINES (cyan, D) and PKINESDE (pink, E) bound to WT CRM1 (gray). (F) Binding affinities of interactions between wild type PKINES and PKINESDE with WT and E571K CRM1. Electron density from composite omit maps (2Fo-Fc contoured in blue mesh to 1σ) are shown for β-strands in A, B, D, and E.
C-terminal NES residues are diverse in sequence and their contributions to interactions with WT and E571K CRM1 are difficult to predict. For example, the PKINES has an acidic residue in its β-strand (45φ3Dφ4NK49) but binds weaker to CRM1(E571K). Intra-NES interactions between Asp46 and backbone amides of Asn48 and Lys49 may be destabilized by the lysine in position 571 of CRM1(E571K) (Table 1 and Figure 5D). We mutated 48NK49 of PKINES to DE (PKINESDE) to mimic the acidic C-terminus of Mek1NES. The structure of PKINESDE bound to WT CRM1 shows that Asp46 no longer makes intra-NES interactions and instead points toward Glu571 of CRM1 (Figure 5E). While PKINES binds CRM1(E571K) sixfold weaker, PKINESDE binds threefold tighter to CRM1(E571K) (Figure 5, D–F; Supplemental Figure 4E; Supplemental Table 1), again suggesting that side chains beyond the φ4 position can contribute to interactions with WT and E571K CRM1
NES-like sequences from BAZ1A (φ3Eφ4DE) and Nup62 (φ3Kφ4DQ) have the same charged residues in their β-strands as Mek1NES (φ3Eφ4DE) and 4E-TNES (φ3Kφ4DQ), respectively, but they do not bind CRM1(E571K) as differently as Mek1NES and 4E-TNES (Supplemental Figure 8A and Table 1). Both BAZ1ANES and Nup62NES have hydrophobic side chains (MEIDE and VKLDQ, respectively) in their β-strands different from Mek1 and 4E-T (LELDE and LKVDQ, respectively) which result in different CRM1-bound conformations of NES β-strands (Supplemental Figure 8B) (Fung et al., 2015). Bulky aromatic side chains like phenylalanine increase the distance of between the NES backbone and the floor of the CRM1 groove. Different hydrophobic side chains should affect the position of the NES β-strand relative to CRM1 Glu571.
In summary, multiple nonhydrophobic residues in the NES β-strand, especially side chains beyond the φ4 position, can contribute to interactions with WT and E571K CRM1. Hydrophobic side chains in the φ3 and φ4 positions also affect NES structure and therefore interactions with WT and E571K CRM1.
Analysis of Mek1, RPS2, and 4E-transporter as cargoes of WT and E571K CRM1
Of all the NESs and putative NESs that we have studied, only Mek1NES, RPS2NES, and 4E-TNES bind WT and E571K CRM1 with substantially different binding affinities. We therefore analyzed the three full-length proteins as cargoes of E571K versus WT CRM1. First, we considered the locations of the NESs within the full-length proteins.
The structure of full-length Mek1 (PDB 3W8Q) shows that its NES α-helix (30LEALQKKLEE39) is disordered and thus should be accessible to CRM1. However, the NES β-strand and especially the φ4 Leu42 side chain appear buried in the kinase domain (Figure 6A). This observation suggests that CRM1 can likely bind the NES helix of Mek1 but not its β-strand.
FIGURE 6:

Accessibility of the NESs of Mek1 and RPS2 in the full-length cargoes. (A) Left panel: residues in the predicted NES of Mek1 (cyan) are shown in the structure of the full-length protein (gray; PDB 3W8Q). Middle panel: the CRM1-bound Mek1NES peptide (cyan). Right panel: binding affinities of the Mek1NES peptide and the full-length Mek1 protein for WT and E571K CRM1. (B) Residues in the predicted NES of RPS2 (cyan) shown in the structure of full-length RPS2 in the 40S ribosome (green; PDB 4V88). (C) Localization of endogenous RPS2 was detected by immunostaining with anti-RPS2 and a fluorescently labeled secondary antibody and is predominantly cytoplasmic in HEK 293 cells in the presence and absence of LMB. (D) The cytoplasmic/nuclear ratio of endogenous RPS2 in HEK 293 CRM1WT/WT, CRM1WT/E571K, and CRM1E571K/E571K cells.
Consistent with this idea, full-length Mek1 binds CRM1 with a Kd of 4.5 µM (Supplemental Figure 9) or 64-fold weaker than the affinity of Mek1NES for CRM1 (Kd 70 nM; Table 1). Furthermore, full-length Mek1 binds both WT and E571K CRM1 proteins with the same affinity, which is also consistent with the prediction that the NES β-strand (41EL42) does not interact with CRM1. Nuclear-cytoplasmic localization of Mek1 is the same in CRM1WT/WT and CRM1E571K/E571K HEK 293 cells (p = 0.5916; Supplemental Figure 10). Even though the predicted Mek1NES binds CRM1(E571K) 14-fold tighter, in reality, the NES in full-length Mek1 is shorter than predicted and does not include the β-strand that interacts differently with E571K versus WT CRM1.
We examined the putative RPS2NES in a structure of the Saccharomyces cerevisiae 40S ribosome that contains RPS2 (PDB 4V88; human and S. cerevisiae RPS2 have high sequence conservation; Figure 6B) (Ben-Shem et al., 2011) and found that RPS2NES is buried within the folded RPS2 protein. Disorder prediction using the RPS2 sequence found the putative RPS2NES to be in the boundary between the ordered and disordered regions; hence it is considered to be accessible to CRM1. However, RPS2 is folded when assembled into the 40S ribosome, and the putative NES sequence is not accessible to CRM1 in that context. Furthermore, the subcellular localization of endogenous RPS2 is not affected by CRM1 inhibition by leptomycin B (LMB), suggesting that RPS2 is not a CRM1 cargo (Figure 6C). Consistent with this knowledge, localization of RPS2 is the same in HEK 293 cells with CRM1WT/WT CRM1WT/E571K or CRM1E571K/E571K (Figure 6D).
The 4E-T protein is predicted to be mostly intrinsically disordered (Xu et al., 2012b), and no structural information for this protein is available at this time. 4E-T is also not amenable to overexpression and purification; the little recombinant protein obtained is not soluble, making the protein unsuitable for biochemical studies with CRM1. We have not been able to test the binding of full-length 4E-T to WT or E571K CRM1. However, since 4E-T is a well-established CRM1 cargo, with LMB-sensitive nuclear-cytoplasmic localization in mammalian cells and mutations in the NES that disrupt cytoplasmic localization (Dostie et al., 2000), we proceeded to examine its localization in cells with CRM1(E571K).
The 4E-T protein is mislocalized to the nucleus in cells with CRM1(E571K)
The NES of 4E-T binds CRM1(E571K) 10-fold weaker than WT CRM1. We examined the localization of endogeneous 4E-T protein in HEK 293 cells expressing either CRM1WT/WT, CRM1WT/E571K or CRM1E571K/E571K. We used YFP2-SV40NLS-mDia2NES as a negative control since the mDia2NES, which does not have a β-strand, binds with similar affinities to both E571K and WT CRM1 and has similar cytoplasmic-nuclear (C/N) distribution in HEK 293 cells expressing either WT or E571K CRM1 (Supplemental Figure 12). Previous studies have shown that 4E-T is important in the formation of processing bodies (P-bodies) and is localized to P-bodies in the cytoplasm (Ferraiuolo et al., 2005). In line with previous reports, 4E-T shows a preferentially cytoplasmic signal (C/N = 1.7) in HEK 293 WT cells and localizes to cytoplasmic puncta. However, endogenous 4E-T is more nuclear in cells expressing CRM1(E571K) than in those expressing WT CRM1 (Figure 7, A and B). In HEK 293 CRM1WT/E571K cells, 4E-T has a C/N = 1.1, which is significantly more nuclear than in WT cells (C/N = 1.7; p < 0.0001). 4E-T is also more nuclear in HEK 293 CRM1E571K/E571K cells (C/N = 1.4) than in WT cells (p < 0.0001).
FIGURE 7:

4E-transporter is mislocalized to the nucleus in HEK 293 and CLL patient cells with CRM1(E571K). (A) Cells were fixed and immunostained using an anti–4E-T antibody. Localization of endogenous 4E-T is mostly cytoplasmic in HEK 293 CRM1WT/WT cells but is more evenly distributed across the nucleus and the cytoplasm in HEK 293 CRM1WT/E571K and CRM1E571K/E571K cells. (B) Quantification of cytoplasmic/nuclear signal of endogenous 4E-T in HEK 293 cells. (C) Immunostaining using an anti–4E-T antibody shows the localization of endogenous 4E-T is mostly cytoplasmic in CLL patient samples with WT CRM1 but is more distributed across the nucleus and cytoplasm in patient cells with the CRM1(E571K) mutation. (D) Quantification of cytoplasmic/nuclear signal of endogenous 4E-T in CLL patient cells.
Previous studies have reported that inhibiting CRM1 with LMB shifted significant amounts of 4E-T from the cytoplasm into the nucleus, but did not change the number of P-bodies in the cells (Ferraiuolo et al., 2005). We counted the P-bodies in HEK 293 CRM1WT/WT, CRM1WT/E571K, and CRM1E571K/E571K cells by setting a high pixel intensity threshold of 60,000 for the images using the ReyniEntropy tool. WT cells have an average of three puncta per cell, which is statistically the same as averages of 3.2 and 3.5 puncta in HEK 293 CRM1WT/E571K or HEK 293 CRM1E571K/E571K cells, respectively (Supplemental Figure 11; p = 0.7114 and p = 0.4668).
The CRM1(E571K) mutation has been found in a variety of cancer types, with enrichment in advanced hematologic diseases. We examined the subcellular localization of 4E-T in tumor cells from CLL patients with and without a heterozygous E571K CRM1 mutation (Supplemental Figure 5). Patient samples across a range of cytogenetic profiles were selected (Supplemental Table 4). Patient samples WT-5 and K-5 are the only ones with normal cytogenetics. Patient cells were thawed and immunostained for endogenous 4E-T as was done for HEK 293 cells. Confocal microscopy imaging of immunostained CLL cells showed significantly lower C/N ratios of 4E-T (more nuclear) in patient cells with CRM1(E571K) compared with patient cells with WT CRM1: average C/N values for 4E-T in WT-1, WT-2, WT-3, and WT-4 cells range from 1.2 to 2.6 compared with average C/N values for K-1, K-2, K-3, and K-4 cells that range from 0.3 to 0.9 (p < 0.0001). Patient cells with normal cytogenetics, WT-5 and K-5, show an average C/N of 8.6 in WT-5 versus 3.0 in K-5 cells (p = 0.0017; Figure 7, C and D).
DISCUSSION
In our study to understand how the E571K mutation of CRM1 affects cargo binding, we found that only a small subset of highly charged NESs bind significantly differently to the cancer mutant versus WT CRM1. Most NESs bind similarly to WT and E571K CRM1. Because other factors in the NESs control structural positioning of the highly charged NES β-strands and therefore interactions with position 571 (glutamate or lysine) of CRM1, the simple presence of charged residues in NES β-strands cannot predict binding differences to CRM1(E571K). We also showed that large affinity changes seen with NES peptides do not always translate to the full-length cargo proteins because of inaccuracies in NES prediction. For example, the predicted NESs Mek1 and RPS2 are partially and completely inaccessible, respectively, in the full-length proteins. One CRM1 cargo, the 4E-T protein, which has an NES that bound much weaker to CRM1(E571K) and is very likely accessible to CRM1 in the full-length protein, shows substantial mislocalization to the nucleus in cells that carry CRM1(E571K).
Of the 27 NES or putative NES peptides that we studied, only three showed a significant difference in affinities for WT versus E571K CRM1. Although it is striking that so few NESs are affected, this result is not entirely surprising as it was previously shown through protein mass spectrometry of cytoplasmic and nuclear fractions that around 50 proteins are enriched or depleted in the cytoplasm or nucleus in Nalm-6 cells engineered to carry CRM1(E571K) (Taylor et al., 2019). If CRM1(E571K) significantly affects the nuclear export of many cargoes, one would expect many more proteins to have altered localization in that study. Since relatively few CRM1 cargoes are affected by the E571K mutation, it is difficult but imperative to determine which of the hundreds of CRM1 cargoes are driving the pathogenic changes. We showed here that the 4E-T protein is one such good candidate.
The search for CRM1 cargoes or NESs that are affected by the E571K mutation is difficult because charges in the NES alone is not a sufficient handle or criteria to predict whether an NES will bind differently to WT versus E571K CRM1. Even NESs with the same charged β-strand residues, such as Mek1NES and the putative BAZ1ANES, which share the same charged residues, and 4E-TNES and the putative Nup62NES, which share the same charged residues have very different effects for CRM1(E571K). The putative NESs BAZ1ANES and Nup62NES bind much weaker to CRM1, and it is possible that their β-strand binds in the CRM1 groove weaker and more transiently than those of high-affinity NESs like the Mek1NES and 4E-TNES. Transient interactions of weaker NESs would cause them to be near E571 side chains less often, therefore decreasing the effects of CRM1(E571K). It is possible that NESs that are affected by CRM1(E571K) tend to bind WT CRM1 in the medium to strong binding affinity regime. Regardless, a strongly predictive β-strand consensus sequence could not be derived and we do not have a straightforward way to predict NES that are affected by the CRM1 mutation. New tools are needed to accurately predict NESs that bind significantly differently to CRM1(E571K). One possibility is to adapt our Rosetta-based NES structural prediction tool to model how WT CRM1 and CRM1(E571K) bind NESs, which could then predict which NESs are stabilized or destabilized by the cancer mutant.
We saw that NES peptides, such as the Mek1NES and the putative RPS2NES, do not always behave the same in the full-length folded proteins. This is disconcerting since our structure-based NES prediction method used protein domain databases to examine whether a putative NES sequence is in a folded domain or is outside of a domain and therefore more likely to be accessible to bind CRM1. However, domain identification is not always accurate at identifying whether a sequence is accessible; NESs (such as Mek1NES and the putative RPS2NES) can be located in the boundary region between a domain and disordered region. We must therefore incorporate structural information in the protein database (PDB) to examine the location of an NES and its potential accessibility, to improve NES prediction. However, structures from the PDB do not cover all possible conformational states of proteins and therefore do not provide information on NES accessibility when it is bound to other proteins or modified posttranslationally.
Mek1 (and Mek2 by similarity) is an interesting study of NES accessibility and NES prediction. The Mek1NES is located at the N-terminal end of the kinase domain and is mostly but not entirely accessible to CRM1. The MK2 kinase also has an NES at the end of its kinase domain, and some cellular contexts can induce regulated disorder of the NES and make it available for CRM1-mediated export (Engel et al., 1998). The influenza NS1 protein also has an NES at the boundary of its effector domain that can be revealed under certain cellular conditions (Li et al., 1998). It was previously reported that Mek1 is regulated differently in suspension versus adherent cells (Slack-Davis et al., 2003) and that Mek2 undergoes regulation different from that of Mek1 (Xu et al., 1997). It is possible that Mek1 or Mek2 NESs can become fully revealed under certain signaling conditions and can then bind much stronger to CRM1(E571K), much more like the Mek1NES peptide. Therefore, it is possible that MAP kinases might actually be affected under specific signaling conditions in CRM1(E571K) cells, but they are more difficult to study than cargoes like 4E-T that have clearly accessible NESs.
The 4E-TNES binds much weaker to E571K than WT CRM1, and the 4E-T protein is mislocalized to the nucleus in CRM1(E571K) cells compared with the strong punctate cytoplasmic localization in cells with WT CRM1. However, it is unclear whether mislocalization of 4E-T in CRM1(E571K) cells perturbs its cellular function and contributes to pathogenesis caused by the CRM1 mutation. 4E-T plays crucial roles in the formation of P-bodies in cells and inhibits translation of a variety of mRNAs (Ferraiuolo et al., 2005). Recent work that characterized mRNAs in P-bodies found that many mRNAs for cell type–specific factors, centrosome and kinetochore proteins, and for PI3K, preferentially associate with P-bodies (Hubstenberger et al., 2017). If defects in 4E-T nuclear export drive CRM1(E571K) phenotypes, then mutations in 4E-T to weaken its NES in cells carrying WT CRM1 should result in the same 4E-T protein mislocalization that we see with WT 4E-T in CRM1(E571K) cells. This type of functional cellular experiments must be done on cargoes affected by CRM1(E571K), in future studies, to understand its direct contribution to the cancer phenotypes.
Since NESs can bind substantially tighter or weaker to CRM1(E571K), we speculate that the mutation could drive oncogenesis by changing (increasing and/or decreasing) the nuclear versus cytoplasmic localization of a select small subset of cargoes. We will need to know the functional consequences of nuclear export defect of a particular cargo in order to understand how the defect leads to oncogenesis. For example, 4E-T is known to store silenced AU-rich mRNAs in the cytoplasm and repress translation (Rasch et al., 2020), and decreased 4E-T levels in the cytoplasm may relieve translational inhibition of some mRNAs that are especially important for B-cell physiology. Conversely, 4E-T may have nuclear functions (not defined at this time) that are enhanced upon an increase of 4E-T levels in the nucleus. Engineering CRM1(E571K) into several B-cell lines to compare functional changes across cell types may shed light on why the mutation is found at a very high frequency in B-cell malignancies compared with solid tumors.
Although we focused on identifying NESs with large binding affinity changes for E571K versus WT CRM1, it is possible that smaller affinity changes (<10-fold) could also alter functions in cells. Previous studies showed that NFκβ signaling is amplified in cells with CRM1(E571K) (Taylor et al., 2019). We found that an inhibitor of NFκβ signaling, IKBαNES, does not bind differently to CRM1(E571K), suggesting that other factors are driving the increased amplitude of NFκβ signaling in B-cells carrying CRM1(E571K). Another NFκβ inhibitor, Iκβε, binds modestly weaker to the cancer mutant (Kd,E571K/Kd,WT = 3.3) (Table 1). Although these changes are modest, they may be sufficient to contribute to the increased NFκβ signaling. Further studies are necessary in the future to explore whether impaired nuclear export of Iκβε drives amplified NFκβ signaling in cells with CRM1(E571K).
We have focused only on CRM1 function in nuclear export, but CRM1 also has important roles in mitosis after when the nuclear envelope is dismantled (Arnaoutov et al., 2005). Severe mitotic defects found in our homozygous HEK 293 CRM1E571K/E571K cells raise the question of whether the roles of CRM1 in recruiting proteins to kinetochores during the metaphase-to-anaphase transition are affected by the E571K mutation. Given the highly timed processes in mitosis, even small perturbations in CRM1 binding may lead to chromosome segregation defects. As noted in the results above, the highly charged putative NESs that were predicted by our structure-based prediction methods are enriched with proteins involved in mitotic metaphase/anaphase transition and chromosome segregation (Supplemental Table 3). The severe mitotic defects in our homozygous HEK 293 CRM1E571K/E571K cells may explain why only the heterozygous E571K mutation has been seen in patients, as a homozygous mutational burden may have lethal consequences for cell growth and effectively remove that clone from the tumor population (Taylor et al., 2019). Further studies are needed to provide a rationale for the CRM1(E571K) mutation decreasing proliferative potential in HEK 293 cells since it contrasts what is observed in Nalm-6 cells (Taylor et al., 2019). Since genomic instability is a well-known hallmark of cancer pathogenesis, it is intriguing to speculate that the up-regulation of DNA repair pathways seen in Nalm-6 cells carrying the CRM1(E571K) mutation may be a consequence of subtle DNA damage in heterozygous cells from CRM1 dysregulation in mitosis (Taylor et al., 2019). We look forward to future studies to investigate direct functional consequences of the cancer mutant and to address whether mitotic defects observed in CRM1(E571K) cells are due to CRM1 function in interphase, mitosis, or both.
MATERIALS AND METHODS
Generation of HEK 293 CRM1(E571K) cell lines
The pSpCas9(BB)-2A-GFP plasmid was received from Addgene. The short guide RNA (sgRNA) (AAGCTGTTCGAATTCATGCA) was cloned into the vector using Bbs1 and sequenced. HEK 293 early-passage cells were electroporated using the LonzaT electroporation kit with manufacturer protocols with 2 μg of plasmid and 3 μM ssODN (TTTTTGAGAGCTCACTGGAAATTTCTGAAGACTGTAGTTAACAAGCTGTTCAAATTCATGCATGGTAAATCTCTTTCTTTACTATATTTTGCTTTTATTTTTATTGAAGAAAATA). Cells recovered for 24 h in a single six-well plate and were trypzined, collected, and sorted by FACS (130 μm nozzle) for GFP signal. Single cells were collected in 96-well plates containing DMEM 40% fetal bovine serum (FBS) with penicillin and streptomycin. Viable clones were expanded into six-well plates, and genomic DNA was extracted by salting out. Cells were washed with phosphate-buffered saline (PBS) and then incubated with 200 μl of 10 mM Tris, pH 7.5, 10 mM EDTA, 10 mM NaCl, 0.5% SDS, and 1 mg/ml Proteinase K to lyse the cells at 65°C overnight. Cold ethanol with 75 mM NaCl (400 μl) was added, followed by incubation at room temperature (RT) for 1 h to precipitate genomic DNA. The genomic DNA was removed with a pipette tip and washed with 500 μl of 70% ethanol three times by centrifugation (12,000 rpm, 5 min, RT). The DNA was air dried at 37°C for 15 min and resuspended using 100 μl TE buffer (10 mM Tris-HCl, pH 8.0, with 1 mM EDTA) with 0.1 mg/ml RNase A. A 500–base pair amplicon containing the E571 codon was generated by PCR and sequenced.
Measurements of cell viability
Low-passage (<10) HEK 293 cells were seeded in a 24-well culture plate (Cellstar; #662160) at densities of 500 or 1000 cells/well in DMEM (Life Technologies; #11995-065) supplemented with 1% FBS (VWR #97068-085) and penicillin/streptomycin/l-glutamine (pen/strep/l-glut) (10,000 U/ml/10 mg/ml/200 mM) (Life Technologies; #15140-122, #25030-081) for 16 h in an incubation chamber at 37°C with 5% CO2. One percent DMEM was aspirated and replaced with 10% DMEM with pen/strep/L-glut and cultured for 5 d (136 h). Phase object confluence (percent) was monitored in 4 h intervals using the IncuCyte ZOOM Live-Cell Imaging System (Essen Biosciences).
FACS cell cycle analysis
Low-passage HEK 293 cells (∼2,000,000) were collected into 1.5 ml sterile centrifuge tubes, centrifuged (100 × g, 5 min at room temperature), and washed with 1 ml PBS. The cells were resuspended in 1 ml of prechilled 70% ethanol/PBS and fixed overnight at –20°C. Cells were centrifuged and washed with 1 ml of PBS to remove ethanol and resuspended in 500 μl nuclear staining buffer (1x PBS, 0.1% Triton X-100, 20 μg/ml propidium iodide, and 2 µg RNase A) at RT for 90 min. One thousand single cells were gated and analyzed for DNA content via FACS from three separate preparations. Data were processed using cell cycle analysis in FlowJo.
Measurements of CRM1-NES binding affinities
Protein reagents for affinity measurements were obtained as previously reported in Fung et al. (2015). NES peptides (sequences in Supplemental Table 5) were cloned into pMAL-TEV vectors, expressed in Escherichia coli (DE3) cells with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 3 h at 37°C, collected, and lysed in 40 mM HEPES (pH 7.5), 2 mM magnesium acetate, 200 mM NaCl, 10 mM dithiothreitol (DTT), and protease inhibitors for purification. Maltose binding protein (MBP)-NES fusion proteins were purified from lysates first by affinity chromatography using amylose resin (New England Biolabs), followed by ion exchange chromatography HiTrap Q (GE Healthcare Life Sciences), and finally gel filtration chromatography using Superdex 75 10/300 GL (GE Healthcare Life Sciences). The pGEX-TEV vector was used to express full-length human CRM1 and CRM1(E571K). CRM1(E571K) was cloned by quick-change mutagenesis of the full-length human CRM1 plasmid. The CRM1 proteins were expressed in E. coli (DE3) cells with 0.5 mM IPTG at 25°C for 12 h. CRM1 was purified from lysates in GF buffer (20 mM HEPES, pH 7.5, 100 mM NaCl, 5 mM MgOAc, 2 mM DTT, and protease inhibitors) by Glutathione Sepharose 4B beads (GE Healthcare Life Sciences, PA), cleaved with TEV protease, and purified by HiLoad 16/600 Superdex 200 pg (GE Healthcare Life Sciences) chromatography. pET-21d GSP1 (1-179, Q71L) was expressed the same way as MBP-NESs but lysed in a different buffer (50 mM HEPES [pH 8.0], 2 mM magnesium acetate, 200 mM NaCl, 10% [vol/vol] glycerol, 5 mM imidazole [pH 7.8], 2 mM DTT) and purified using Ni-NTA Agarose (Qiagen, Hilden, Germany), loaded with GTP as previously described (Fung et al., 2015), and then further purified by anion exchange and size exclusion chromatography into TB buffer (20 mM HEPES, pH 7.5, 110 mM KOAc, 2 mM MgOAc, 10% glycerol, and 2 mM DTT).
Differential photobleaching experiments, used to generate data in Table 1, were all performed as described in Fung et al. (2015) using 20 nM FITC-PKINES peptide (Genscript) in TB buffer. A triplicate 16-fold 1:1 dilution scheme was used in both direct and competition titrations. For direct assays, CRM1 was titrated from 10 µM. For competition assays, MBP-NESs were titrated from 50 µM with a constant 150 nM CRM1 WT. For competition assays, 300 nM CRM1(E571K) was used instead of 150 nM WT CRM1 for better curve generation given weaker probe binding. The data were fitted and analyzed in PALMIST (Scheuermann et al., 2016). For quality control, we determined the affinity for every preparation of CRM1 binding to the FITC-PKINES probe. We did not use any data obtained with a particular preparation of CRM1 if affinity for the probe varied twofold from the average affinity. MBP alone was used as a negative control.
For higher-throughput measurements, fluorescence polarization was measured using the same competition assay scheme and parameters as the photobleaching experiments. These assays were performed in a total volume of 15 µl in Corning 384-well low volume microplates (black, flat bottom), in triplicate. A CLARIOstar Plus plate reader was used to measure FITC-PKI polarization with the following parameters: 50 flashes per well, excitation 482-16 nm, emission 530-40 nm, gain A: 1500, gain B: 1507, focal height 5.1 mm, and 0.1 s settling time. PALMIST was adapted to process and analyze these data sets as previously defined in Fung et al. (2015).The binding affinities were similar for PKINES and Dusp6NES in both fluorescence polarization and photobleaching assays.
Crystallization of CRM1-Ran-RanBP1-NES complexes
Humanized yeast CRM1 (ScCRM1 residues 1–1058, ∆377–413, 537-DLTVK-541 to GLCEQ, V441D, ±E582K) was cloned and purified as for binding affinity measurements. Yrb1p (yeast RanBP1 residues 62–201) was expressed in a pGEX-TEV construct, expressed in E. coli (DE3) cells with 0.5 mM IPTG for 12 h at 25°C, and purified from lysates using the same purification scheme as for CRM1. pET-15b-human Ran was expressed and purified as for GSP1 above except that GMPPNP was used instead of GTP (Fung et al., 2015). CRM1-NES-Ran-RanBP1 complexes were assembled in a 20:1:3:3 M ratio and purified by gel filtration using Superdex 75 10/300 GL (GE Healthcare Life Sciences). The complex was concentrated to 10 mg/ml and crystallized at 20°C in 17% (wt/vol) PEG3350, 100 mM Bis-Tris (pH 6.4), 200 mM ammonium nitrate, and 10 mM spermine HCl. Crystals were cryoprotected with the same crystallization condition supplemented with up to 23% PEG3350 and 12% glycerol and flash frozen in liquid nitrogen. Data collection, processing, and solving of the structures were performed in the same manner as previously described (Fung et al., 2015). In brief, x-ray diffraction data were collected at the APS 19ID beamline at the Argonne National Laboratory and processed using HKL-3000 (Minor et al., 2006). Structures were determined by molecular replacement using the unliganded CRM1-Ran-Ranbp1 structure (PDB 4HB2) as search model in PHENIX with reiterative modeling in Coot (Chen et al., 2010; Emsley et al., 2010).
Immunostaining: reagents and antibodies
Antibodies were obtained for Mek1 (Abcam; E342), 4E-T (Bethyl; ICH-00127, Abcam; ab168098), PP2A B56alpha (Santa Cruz; 271151), RPS2 (Abcam; ab155961), mouse immunoglobulin G H&L (Abcam; ab6785). Secondary antibodies used include anti-rabbit or goat anti-mouse (Abcam; ab150113) secondary antibody conjugated with Alexa488.
Patient cell samples
Blood was obtained from CLL patients following written informed consent under a protocol approved by the Institutional Review Board (IRB) of The Ohio State University (OSU; Columbus, OH) in accordance with the Declaration of Helsinki. All patients examined had CLL as defined by the 2008 IWCLL criteria (Hallek et al., 2008), and cells were isolated and cultured as previously described (Lapalombella et al., 2012).
Immunostaining and confocal microscopy to visualize CRM1 cargoes
HEK 293 cells were counted by hemocytography, and 700,000 cells were seeded on black, glass-bottom plates coated with poly-l-lysine (Cellvis). Twenty-four hours later, the cells were fixed with 4% paraformaldehyde for 20 min at RT, washed with PBS, permeabilized for 10 min with 0.3% Triton X-100 in PBS, blocked with 0.1% Triton X-100, 5% bovine serum albumin (BSA) (Sigma), and PBS for 1–2 h, and stained with primary antibodies at manufacturer recommended dilutions for 16 h at 4°C. Cells were washed twice with 500 µl 0.1% Triton X-100 in PBS and incubated with 1:1000 secondary antibody conjugated with Alexa488 for 1 h at room temperature in the dark. Cells were washed twice with 500 µl 0.1% Triton X-100 in PBS and then incubated with 3 µg/ml Hoescht 33342 (Thermo Fisher) for 1 h in the dark. For the negative control, the pEYFP2-SV40NLS-mDia2NES was transiently transfected with lipofectamine 2000 as instructed by the manufacturer and previously described (Fu et al., 2018).
Human CLL cells were thawed from cryovial preservation and plated on glass microscope slides precoated with 0.01% poly-l-lysine (Sigma; P8920) and allowed to settle for 4 h. Cells were fixed with 4% paraformaldehyde for 20 min at RT and washed twice with PBS, permeabilized, and blocked (PBS with 0.1% Triton X-100 and 1% BSA) for 20 min, and stained with primary 4E-T antibody at manufacturer recommended dilutions for 1 h in the dark at RT. Slides were washed three times in PBS and stained with fluorescein isothiocyanate (FITC)-conjugated anti-mouse antibody for 1 h in the dark. Slides were washed three times with PBS and stained with 4’,6-diamidino-2-phenylindole (DAPI) infused mounting medium (Vector Laboratories; #H-1200) and stored at 4°C. Slides were stained in a closed environment to avoid evaporation.
HEK 293 cell images were collected with a Nikon-4 confocal microscope with 40× oil magnification, and Zen image analysis was used to collect 10 z-stacks of 1 µm. Human CLL cell images were collected with an Olympus FV 3000 Spectral Confocal Microscope with 60× oil magnification, and Zen image analysis was used to collect 10 z-stacks of ∼1 µm. Cytoplasmic-to-nuclear ratios were quantified as previously described (Fu et al., 2018). Two z-stacks in the center of the nucleus were projected by mean fluorescence intensity. A nuclear boundary was established in ImageJ using the DNA stain images. A ring-shaped area of the cytoplasm was sampled just outside of the nuclear region.
Purification of full-length Mek1
Pet(29) codon optimized stag-FL hMek1-6XHis was a gift from the Goldsmith lab (UT Southwestern) and transformed in BL21 DE3 gold E. coli cells. Cells were grown in 6 l of Luria broth (LB) media with ampicillin to an OD = 0.8, the temperature was lowered to 25°C, and cells were induced with 0.5 mM IPTG for 12 h. Cells were collected by centrifugation and resuspended in 50 mM HEPES (pH 8.0), 2 mM magnesium acetate, 200 mM NaCl, 10% (vol/vol) glycerol, 5 mM imidazole (pH 7.8), 2 mM DTT and frozen at –80°C until purification. Cells were homogenized, and the clarified lysate was applied to a 5 ml Ni-NTA Agarose (Qiagen, Hilden, Germany) column for affinity purification followed by further purification by size exclusion chromatography using a S200-Increase 10/30 column in TB buffer.
Accession codes
Coordinates for structures and crystallographic data have been deposited at the PDB and are found in Supplemental Tables 1 and 2.
Statistical analysis
Statistical significance for cell cycle and viability analysis was determined using the Holm–Sidak method, with alpha = 0.05. Each row was analyzed individually, without assuming a consistent SD. t tests were utilized to compare cytoplasmic/nuclear ratio for protein localization from three separate experiments.
Supplementary Material
Acknowledgments
We thank Ho Yee Joyce Fung, Szu-Chin Fu, Zhe Chen, Ke Zhang, Diana Tomchick, and Tolga Cagatay for technical training and support. We thank Jan Erzberger for access to the fluorimeter used in fluorescence polarization assays. We thank Elizabeth Goldsmith for her gift of the plasmid to express full-length CRM1. We thank the Structural Biology Laboratory and Macromolecular Biophysics Resource at the University of Texas Southwestern Medical Center) for their assistance with crystallographic and biophysical data collection. Crystallographic results are derived from work performed at Argonne National Laboratory, Structural Biology Center at the Advanced Photon Source. Argonne is operated by UChicago Argonne, LLC, for the U.S. Department of Energy (DOE), Office of Biological and Environmental Research (BER), under contract DE-AC02-06CH11357. We acknowledge the Texas Advanced Computing Center (TACC; www.tacc.utexas.edu) at The University of Texas at Austin for providing HPC resources. We thank the UTSW life cell imaging core, especially Abhijit Bugde, and the UTSW flow cytometry core, especially Mason Perry. We are grateful to the patients who provided blood for the above studies. Images presented in this report were generated using the instruments and services at the Campus Microscopy and Imaging Facility, The Ohio State University. This facility is supported in part by grant P30 CA016058, National Cancer Institute, Bethesda, MD. We would also acknowledge Matthew Summers for assistance with IncuCyte live-cell imaging. Research reported in this publication was supported in part by the College of Medicine at The Ohio State University. The content is solely the responsibility of the authors and does not necessarily represent the official views of the College of Medicine at The Ohio State University. This work is funded by Cancer Prevention Research Institute of Texas (CPRIT) Grants RP180410 (Y.M.C.) and RP170170 (N.V.G. and Y.M.C.), National Institutes of Health Grants (GM127390 to N.V.G. and R01CA192928 to R.L.), Welch Foundation Grants (I-1532 to Y.M.C. and I-1505 to N.V.G.), and the Mabel and Alfred Gilman Chair for Molecular Pharmacology.
Abbreviations used:
- CLL
chronic lymphocytic leukemia
- C/N
cytoplasmic-nuclear
- CRM1
chromosomal region maintenance 1
- DAPI
4’,6-diamidino-2-phenylindole
- DLBCL
diffuse large B-cell lymphoma
- 4E-T
eIF4E-transporter
- FDR
false discovery rate
- FITC
fluorescein isothiocyanate
- FACS
fluorescence activated cell sorting
- IPTG
isopropyl β- d-1-thiogalactopyranoside
- LMB
leptomycin B
- LB
Luria broth
- MBP
maltose binding protein
- NESs
nuclear export signals
- NPC
nuclear pore complex
- PMBCL
primary mediastinal B-cell lymphoma
- r.m.s.d
root-mean-square deviation
- sg
short guide.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-04-0233) on June 10, 2020.
REFERENCES
- Arnaoutov A, Azuma Y, Ribbeck K, Joseph J, Boyarchuk Y, Karpova T, McNally J, Dasso M. (2005). Crm1 is a mitotic effector of Ran-GTP in somatic cells. Nat Cell Biol , 626–632. [DOI] [PubMed] [Google Scholar]
- Ben-Shem A, Garreau de Loubresse N, Melnikov S, Jenner L, Yusupova G, Yusupov M. (2011). The structure of the eukaryotic ribosome at 3.0 A resolution. Science , 1524–1529. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr , 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chook YM, Blobel G. (2001). Karyopherins and nuclear import. Curr Opin Struct Biol , 703–715. [DOI] [PubMed] [Google Scholar]
- Cook A, Bono F, Jinek M, Conti E. (2007). Structural biology of nucleocytoplasmic transport. Annu Rev Biochem , 647–671. [DOI] [PubMed] [Google Scholar]
- Dong X, Biswas A, Suel KE, Jackson LK, Martinez R, Gu H, Chook YM. (2009). Structural basis for leucine-rich nuclear export signal recognition by CRM1. Nature , 1136–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostie J, Ferraiuolo M, Pause A, Adam SA, Sonenberg N. (2000). A novel shuttling protein, 4E-T, mediates the nuclear import of the mRNA 5’ cap-binding protein, eIF4E. EMBO J , 3142–3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. (2010). Features and development of Coot. Acta Crystallogr D Biol Crystallogr , 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel K, Kotlyarov A, Gaestel M. (1998). Leptomycin B-sensitive nuclear export of MAPKAP kinase 2 is regulated by phosphorylation. EMBO J , 3363–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraiuolo MA, Basak S, Dostie J, Murray EL, Schoenberg DR, Sonenberg N. (2005). A role for the eIF4E-binding protein 4E-T in P-body formation and mRNA decay. J Cell Biol , 913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu SC, Fung HYJ, Cagatay T, Baumhardt J, Chook YM. (2018). Correlation of CRM1-NES affinity with nuclear export activity. Mol Biol Cell , 2037–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung HY, Fu SC, Brautigam CA, Chook YM. (2015). Structural determinants of nuclear export signal orientation in binding to exportin CRM1. eLife , e10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung HY, Fu SC, Chook YM. (2017). Nuclear export receptor CRM1 recognizes diverse conformations in nuclear export signals. eLife , e23961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorlich D, Kutay U. (1999). Transport between the cell nucleus and the cytoplasm. Annu Rev Cell Dev Biol , 607–660. [DOI] [PubMed] [Google Scholar]
- Hallek M, Cheson BD, Catovsky D, Caligaris-Cappio F, Dighiero G, Dohner H, Hillmen P, Keating MJ, Montserrat E, Rai KR, et al. (2008). Guidelines for the diagnosis and treatment of chronic lymphocytic leukemia: a report from the International Workshop on Chronic Lymphocytic Leukemia updating the National Cancer Institute-Working Group 1996 guidelines. Blood , 5446–5456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubstenberger A, Courel M, Benard M, Souquere S, Ernoult-Lange M, Chouaib R, Yi Z, Morlot JB, Munier A, Fradet M, et al. (2017). P-body purification reveals the condensation of repressed mRNA regulons. Mol Cell , 144–157.e145. [DOI] [PubMed] [Google Scholar]
- Jardin F, Pujals A, Pelletier L, Bohers E, Camus V, Mareschal S, Dubois S, Sola B, Ochmann M, Lemonnier F, et al. (2016). Recurrent mutations of the exportin 1 gene (XPO1) and their impact on selective inhibitor of nuclear export compounds sensitivity in primary mediastinal B-cell lymphoma. Am J Hematol , 923–930. [DOI] [PubMed] [Google Scholar]
- Kosugi S, Hasebe M, Tomita M, Yanagawa H. (2008). Nuclear export signal consensus sequences defined using a localization-based yeast selection system. Traffic , 2053–2062. [DOI] [PubMed] [Google Scholar]
- Lapalombella R, Sun Q, Williams K, Tangeman L, Jha S, Zhong Y, Goettl V, Mahoney E, Berglund C, Gupta S, et al. (2012). Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood , 4621–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Baumhardt JM, Pei J, Chook YM, Grishin NV. (2020). pCRM1exportome: database of predicted CRM1-dependent nuclear export signal (NES) motifs in cancer-related genes. Bioinformatics , 961–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Pei J, Baumhardt JM, Chook YM, Grishin NV. (2019). Structural prerequisites for CRM1-dependent nuclear export signaling peptides: accessibility, adapting conformation, and the stability at the binding site. Sci Rep , 6627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Yamakita Y, Krug RM. (1998). Regulation of a nuclear export signal by an adjacent inhibitory sequence: the effector domain of the influenza virus NS1 protein. Proc Natl Acad Sci USA , 4864–4869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, Chruszcz M. (2006). HKL-3000: the integration of data reduction and structure solution— from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr , 859–866. [DOI] [PubMed] [Google Scholar]
- Monecke T, Guttler T, Neumann P, Dickmanns A, Gorlich D, Ficner R. (2009). Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science , 1087–1091. [DOI] [PubMed] [Google Scholar]
- Port SA, Monecke T, Dickmanns A, Spillner C, Hofele R, Urlaub H, Ficner R, Kehlenbach RH. (2015). Structural and functional characterization of CRM1-Nup214 interactions reveals multiple FG-binding sites involved in nuclear export. Cell Rep , 690–702. [DOI] [PubMed] [Google Scholar]
- Rasch F, Weber R, Izaurralde E, Igreja C. (2020). 4E-T-bound mRNAs are stored in a silenced and deadenylated form. Genes Dev , 847–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuermann TH, Padrick SB, Gardner KH, Brautigam CA. (2016). On the acquisition and analysis of microscale thermophoresis data. Anal Biochem , 79–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slack-Davis JK, Eblen ST, Zecevic M, Boerner SA, Tarcsafalvi A, Diaz HB, Marshall MS, Weber MJ, Parsons JT, Catling AD. (2003). PAK1 phosphorylation of MEK1 regulates fibronectin-stimulated MAPK activation. J Cell Biol , 281–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor J, Sendino M, Gorelick AN, Pastore A, Chang MT, Penson AV, Gavrila EI, Stewart C, Melnik EM, Herrejon Chavez F, et al. (2019). Altered nuclear export signal recognition as a driver of oncogenesis. Cancer Discov , 1452–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis K. (2003). Regulating access to the genome: nucleocytoplasmic transport throughout the cell cycle. Cell , 441–451. [DOI] [PubMed] [Google Scholar]
- Xu D, Farmer A, Collett G, Grishin NV, Chook YM. (2012a). Sequence and structural analyses of nuclear export signals in the NESdb database. Mol Biol Cell , 3677–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu D, Grishin NV, Chook YM. (2012b). NESdb: a database of NES-containing CRM1 cargoes. Mol Biol Cell , 3673–3676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Khoo S, Dang A, Witt S, Do V, Zhen E, Schaefer EM, Cobb MH. (1997). Differential regulation of mitogen-activated protein/ERK kinase (MEK)1 and MEK2 and activation by a Ras-independent mechanism. Mol Endocrinol , 1618–1625. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.