

Abstract
The Wnt/β-catenin signaling pathway is central to metazoan development and routinely dysregulated in cancer. Wnt/β-catenin signaling initiates transcriptional reprogramming upon stabilization of the transcription factor β-catenin, which is otherwise posttranslationally processed by a destruction complex and degraded by the proteasome. Since various Wnt signaling components are enriched at centrosomes, we examined the functional contribution of centrosomes to Wnt signaling, β-catenin regulation, and posttranslational modifications. In HEK293 cells depleted of centrosomes we find that β-catenin synthesis and degradation rates are unaffected but that the normal accumulation of β-catenin in response to Wnt signaling is attenuated. This is due to accumulation of a novel high-molecular-weight form of phosphorylated β-catenin that is constitutively degraded in the absence of Wnt. Wnt signaling operates by inhibiting the destruction complex and thereby reducing destruction complex–phosphorylated β-catenin, but high-molecular-weight β-catenin is unexpectedly increased by Wnt signaling. Therefore these studies have identified a pool of β-catenin effectively shielded from regulation by Wnt. We present a model whereby centrosomes prevent inappropriate β-catenin modifications that antagonize normal stabilization by Wnt signals.
INTRODUCTION
The Wnt/β-catenin signaling pathway is a conserved strategy involved in cell proliferation, polarity, and fate specification (Loh et al., 2016). In the absence of signaling, β-catenin is posttranslationally processed and marked for degradation by a destruction complex, chiefly composed of two kinases—GSK3 andCK1—and two scaffolds—APC and axin. β-Catenin processing by the destruction complex begins with phosphorylation of Ser45 by CK1, a priming event for subsequent phosphorylation on Ser33/Ser37/Thr41 by GSK3 (MacDonald et al., 2009; Nusse and Clevers, 2017). The E3 ubiquitin ligase β-Trcp then binds the triply phosphorylated β-catenin, resulting in polyubiquitination and degradation by the proteasome (Liu et al., 1999). The destruction complex is inactivated when Wnt ligand binds to the coreceptors Frizzled and LRP, mediated by the scaffold protein Disheveled (Dvl), which recruits the complex to the intracellular domains of the coreceptor (MacDonald et al., 2009). Reduced β-catenin degradation upon Wnt signaling allows newly synthesized β-catenin to enter the nucleus, bind to the DNA binding protein TCF, and activate transcription of Wnt-responsive target genes (Hernandez et al., 2012; Gerlach et al., 2014). Defects in Wnt signaling and β-catenin regulation promote tumorigenesis and are associated with several cancers (Nusse and Clevers, 2017).
Centrosomes are non–membrane-bound organelles that organize microtubule asters and coordinate the bipolar mitotic spindle, but a growing number of observations suggest additional roles in regulating signaling proteins (Nigg and Stearns, 2011; Arquint et al., 2014; Vora and Phillips, 2016). Centrosomes are composed of two centrioles, cylindrical appendages that duplicate once per cell cycle, and pericentriolar material, a proteinaceaous matrix of centrosomal factors that nucleate microtubules to organize the bipolar spindle. After centriole duplication, the two centrosomes remain attached until they are split and driven to opposite poles of the cell by kinesin motors in G2/M (Nigg and Stearns, 2011). β-Catenin has been shown to participate in at least two distinct centrosomal functions: γ-tubulin recruitment to centrosomes and centrosome splitting (Mbom et al., 2013). In mouse neuronal progenitors, β-catenin loss results in decreased γ-tubulin at centrosomes and thus defects in microtubule organization (Chilov et al., 2011). In addition, cells without β-catenin form a monopolar spindle due to compromised splitting, while β-catenin overexpression results in premature centrosome splitting and the formation of extracentrosomal structures that harbor various centrosomal factors (Kaplan et al., 2004; Bahmanyar et al., 2010). β-Catenin–dependent centrosome splitting requires phosphorylation by Nek2 at a variety of sites including Ser33/Ser37/Thr41, sites also known to be phosphorylated by GSK-3. Unlike GSK3, however, β-catenin phosphorylation by Nek2 is CK1-independent and is not involved in destruction complex–mediated degradation. Nek2 becomes activated at G2/M, where it phosphorylates and stabilizes centrosomal β-catenin to promote centrosome splitting (Mbom et al., 2014).
In addition to the above β-catenin centrosomal localization pattern seen in mammals, observations of β-catenin localization in Caenorhabditis elegans and Platynereis dumerilii demonstrate that its association with the centrosome is conserved (Huang et al., 2007; Phillips et al., 2007; Schneider and Bowerman, 2007; Baldwin and Phillips, 2014; Baldwin et al., 2016; Vora and Phillips, 2015; Lam and Phillips, 2017). In C. elegans, centrosomal localization of the β-catenin homologue SYS-1 promotes dynamic centrosome–associated proteasomal degradation during asymmetric cell division, limiting its retention by daughter cells prior to cell fate specification. Unlike mammalian β-catenin, SYS-1 is dispensable for centrosome separation and mitotic division and thus behaves as a substrate rather than an effector of the centrosome (Vora and Phillips, 2015). A β-catenin regulation mechanism dependent on centrosomal localization is also suggested in mammals by experiments in embryonic stem cells, which showed asymmetric localization of β-catenin as well as many other proteasome substrates to one of two centrosomes after division (Fuentealba et al., 2008). It follows from these studies that although β-catenin’s role as an effector of the centrosome varies between species, its regulation via accumulation at centrosomes may be conserved.
Cell sensitivity to Wnt signals is under complex regulation and has been linked to many aspects of basic cell physiology, including the cell cycle status (Niehrs and Acebron, 2012; Costa et al., 2019; El-Sahli et al., 2019). Cyclin Y-CDK primes LRP6 via phosphorylation of PPPSP motifs to boost Wnt signaling competence during G2/M, which is also when basal cellular β-catenin concentration and Wnt target gene expression is highest (Olmeda et al., 2003; Davidson et al., 2009). The converse is also true: Wnt signaling itself controls the cell cycle status since induction of its target genes c-myc and cyclin D1 promotes G1 progression (Niehrs and Acebron, 2012). That the Wnt signaling pathway is so poorly insulated from other cell processes combined with the fact that Wnt pathway proteins have additional roles in centrosome and spindle organization suggests a link between Wnt signaling and centrosome function.
Indeed, various studies have suggested cross-talk between centrosomal components and Wnt signaling as well as regulation of Wnt components via centrosomal localization. It was recently shown that Dvl localizes to centrosomes and participates in Nek2-mediated release of centrosome linker components to promote splitting during G2/M (Cervenka et al., 2016). Dvl is then released from the centrosome upon phosphorylation by Nek2. Interestingly, depletion of Nek2 results in attenuation of the Wnt response, presumably by increasing centrosomal localization of Dvl and reducing the cytoplasmic pool of Dvl available for signaling (Cervenka et al., 2016). Modulation of Wnt signaling by centrosomal localization of pathway components was also reported by studies on the centrosomal protein Diversin, a known antagonist of β-catenin stability. Diversin shows punctate localization to centrosomes and dynamically relocalizes to cytoplasmic granules upon Wnt signaling, and Diversin alleles unable to localize to the centrosome are unable to antagonize Wnt signaling. Although the explicit mechanism is unclear, Diversin depends on centrosomal localization to appropriately promote β-catenin degradation (Itoh et al., 2009). Together, these studies highlight an intriguing role of mammalian centrosomes in modulating Wnt response, a possibility that warrants further investigation by virtue of the fact that both Wnt signaling and centrosome copy number are often dysregulated in tumors (Gonczy, 2015; Nusse and Clevers, 2017). However, the explicit causal relationship between centrosome function and Wnt signaling behavior is not known.
Here, we determine the net effects of centrosome loss on β-catenin regulation and Wnt signaling in human cell lines by depleting cells of centrosomes entirely. We demonstrate that centrosomes prevent inappropriate β-catenin modifications that result in Wnt-insensitive degradation, allowing cells to efficiently halt β-catenin degradation after Wnt signaling and rapidly increase its concentration. Finally, we discuss possible mechanisms underlying centrosome-enforced β-catenin posttranslational processing and its relevance to disease states.
RESULTS
Cells without centrosomes accumulate a distinct high-molecular-weight species of β-catenin that is phosphorylated at Ser33/Ser37/Thr41
To study the influence of centrosomes on β-catenin regulation and Wnt signaling, we depleted HEK293 cells of centrosomes by treating with 200 mM centrinone A (CentA, hereon) for a period of 5–7 d. HEK293 cells bear no known mutations in key Wnt pathway components and are routinely used to study Wnt signaling (Hernandez et al., 2012; Li et al., 2012; Kim et al., 2013). CentA is a selective and reversible inhibitor of the kinase Plk4, whose activity is necessary for centrosome duplication. Chronic treatment with CentA results in a progressive loss of centrosomes throughout cell division cycles and eventually gives rise to a population of cells without centrosomes (Wong et al., 2015). Centrosomal puncta labeled with γ-tubulin were lost after CentA treatment but not in control cells treated with dimethyl sulfoxide (DMSO) using the same regimen. (Figure 1A, top panel). Control cells also showed punctate localization patterns of phosphorylated β-catenin (pβ-catenin) to γ-tubulin–labeled centrosomes, consistent with previous reports of pβ-catenin association with the centrosome (Figure 1B, bottom panel) (Chilov et al., 2011). HEK293 cells treated with CentA did not arrest and were able to proliferate in the absence of centrosomes. Continued cell proliferation during CentA treatment and the presence of mitotic cells lacking centrosomes suggested that HEK293 cells lack a functional centrosome duplication checkpoint. Since HEK293 cells have intact Wnt signaling along with the ability to proliferate in the absence of centrosomes, we reasoned that this cell line could be used as a model to study the effects of centrosome depletion on Wnt/β-catenin signaling and β-catenin regulation.
FIGURE 1:

Cells without centrosomes accumulate a distinct higher-molecular-weight species of β-catenin. (A) HEK293 cells after chronic treatment with CentA lacking γ-tubulin–positive centrosomes (arrowheads) readily observable in cells treated with DMSO (bottom panel). (B) HEK293 cells coiimmunostained for γ-tubulin and pβ-catenin confirming colocalization at centrosomes in control DMSO-treated cells (highlighted by 1.48 μm dashed box) but not CentA cells. An enlarged image of the merged centrosomes appears in the bottom right corner of the DMSO window. (C) HEK293 cells were treated chronically with DMSO or CentA and blotted for β-catenin, pβ-cateninS33/37/41, pβ-cateninS45, active-β-catenin, and tubulin as a loading control. The higher-molecular-weight band (*) corresponds to a migration distance between 95 and 130 kDa (arrowheads). (D) Lysates from HEK293 cells treated with either DMSO, transient CentA (4 h), or chronic CentA blotted for pβ-catenin demonstrate that the high-molecular-weight species accumulates only after chronic treatment associated with centrosome loss. (E) HeLa, HEK293, and NCIH28Δβ-catenin cell lysates were blotted for pβ-catenin and β-catenin showing the absence of the high–molecular–weight species in cells with a homozygous deletion of β-catenin (top panel). Quantification shows up-regulation of higher-molecular-weight forms of both species in HeLa and HEK293 cells but not in NCIH28Δβ-catenin (bottom panel). (F) Accumulation of higher-molecular-weight pβ-catenin species after CentA treatment of HCT116p53–/– cells in whole cell lysates and cytosolic extracts (top panel). Quantification of pβ-cateninHiMW in cytosolic extracts shows a roughly 40-fold increase in CentA cells (p < 0.01, Student’s t test) (bottom panel). (G) Immunoprecipitation of FLAG::β-catenin from HEK293 cells. Higher-molecular-weight species is readily stained by antibodies against FLAG and pβ-catenin (S33/37, T41) but not pβ-catenin (S45). Arrowheads mark 95 kDa except where otherwise mentioned.
Phosphorylation of β-catenin by GSK3 at Ser33, Ser37, and Thr41 is required for β-catenin degradation, and modulation of these posttranslational marks is key to Wnt signaling. Therefore, to examine any potential effects of centrosome depletion on β-catenin regulation, we visualized pβ-cateninSer33/37, Thr41 by Western blot analysis in control versus CentA-treated cells. Lysates from centrosome-depleted cells showed similar levels of total pβ-catenin compared with control but accumulated a higher-molecular-weight band (pβ-cateninHiMW, hereon) of an approximately 10–20 kDa shift. (Figure 1C, CentA lane). Consistent with the fact that pβ-catenin is a low-abundance molecule in cells, pβ-cateninHiMW was generally present in low, variable abundance and was consistently up-regulated by centrosome depletion. As previously shown, most pβ-catenin in control cells exhibited a wild-type migration pattern corresponding to 100 kDa, the approximate molecular weight of β-catenin (pβ-cateninLowMW). pβ-Catenin bands migrating like pβ-cateninHiMW have been observed in previous studies, but their identity and relevance to Wnt signaling have not been investigated (Hernandez et al., 2012; Kim et al., 2013). We were also able to detect this species with antibodies against total β-catenin (under high-exposure settings) but not with antibodies against pβ-cateninS45 (Figure 1C, asterisk). The observation that β-cateninHiMW is detected with antibodies to pβ-cateninS33/37, T41 but not S45 suggests CK1 may not be involved in generating this particular β-catenin species and is addressed below. To test whether pβ-cateninHiMW accumulation results from direct inhibition of Plk4 by CentA rather than the centrosome loss after prolonged treatment, we visualized pβ-catenin in cells treated with CentA acutely (4 h) and chronically (5 d). Only cells treated chronically with CentA up-regulated pβ-cateninHiMW relative to control cells (Figure 1D), demonstrating that its accumulation is not likely due to direct effects of Plk4 inhibition. β-CateninHiMW accumulation was also detected in HeLa cells and HCT116 p53–/– cells but was not detectable in NCI-H28 cells, a malignant mesothelioma line with a homozygous β-catenin deletion (Figure 1, E and F). CentA-induced accumulation of β-cateninHiMW was especially apparent in HCT116p53–/– cytosolic extracts, where β-catenin levels are elevated (Ilyas et al., 1997) (Figure 1F). Furthermore, β-cateninHiMW was detected in low abundance after immunoprecipitation of FLAG::β-catenin in 293 cells (Figure 1G). These experiments confirm that the high-molecular-weight band we observed after centrosome loss is specific to β-catenin. Together, these data demonstrate the accumulation of a higher-molecular-weight β-catenin species in cells without centrosomes.
β-Catenin degradation is not impaired upon centrosome loss
Since pβ-catenin is ordinarily subject to polyubiquitination and degradation, we reasoned that accumulation of pβ-cateninHiMW in centrosome-depleted cells may result from defective degradation. We tested the hypothesis that pβ-catenin is degraded at centrosomes by inhibiting the centrosome-associated proteasome. Centrosome-associated proteasomal degradation is readily disrupted by expression of a dominant negative allele of the proteasomal subunit S5a fused to the centrosomal targeting domain PACT (Puram et al., 2013). We transfected HeLa cells with the proteasome-inhibiting dominant negative allele GFP::S5aC with and without fusion to PACT and visualized total polyubiquitinated proteins by immunofluorescence. As expected, expression of GFP::S5aC led to a dramatic increase in global polyubiquitinated proteins in successfully transfected cells (Supplemental Figure S1A) and expression of S5aC::PACT led to strong punctate polyubiquitinated protein accumulation specifically at S5aC::PACT-marked centrosomes (Supplemental Figure S1B, middle panel), indicative of impaired processing of polyubiquitinated substrates. Importantly, we observed strong centrosomal accumulation of polyubiquitinated proteins in cells where expression of S5aC::PACT was present at centrosomes yet virtually undetectable in the cytoplasm and nucleus, confirming potent inhibition of the centrosome-associated proteasome by this fusion protein (Supplemental Figure S1C). Unlike total polyubiquitinated proteins, pβ-catenin showed no apparent increase at centrosomes in S5aC::PACT-expressing cells relative to untransfected cells (Supplemental Figure S1D). This result suggests that centrosome-associated degradation does not make a major contribution to regulation of β-catenin levels. To confirm this possibility, we measured the degradation rate of cytosolic β-catenin by treating cells with Wnt-conditioned media for 12 h and recovering them in fresh media for various times. Since the majority of β-catenin associates with adherens junctions where it functions independently of Wnt signaling, the Wnt-regulated pool of β-catenin is routinely measured in cytosolic extracts here and in other studies (Li et al., 2012). This assay revealed that the degradation kinetics of cytosolic β-catenin in centrosome-depleted cells was indistinguishable from that in control cells (SupplementalFigure S2, A andB). Together, these data suggest that accumulation of pβ-cateninHiMW in centrosome-depleted cells is not due to attenuated β-catenin degradation.
Cells without centrosomes show normal β-catenin synthesis rate but attenuated accumulation in response to Wnt signaling
Next, we sought to determine whether centrosome loss influenced the response to Wnt signaling by treating normal and centrosome-depleted cells with Wnt and measuring cytosolic β-catenin at various time points. Centrosome-depleted cells showed a dramatically attenuated rate of β-catenin increase throughout the time course and accumulated less β-catenin than control cells (Figure 2, A and B). To test whether this phenomenon was due to decreased β-catenin production, we eliminated β-catenin degradation by proteasome inhibition with MG132, so that β-catenin levels are determined only by synthesis. In this assay, cells without centrosomes showed a pattern of β-catenin accumulation similar to wild type, suggesting that the effects on β-catenin accumulation after Wnt are not due to impaired synthesis (Figure 2, C and D). We also visualized pβ-catenin formation after MG132 treatment, which showed a similar pattern (Figure 2C). In CentA cells, pβ-cateninHiMW increased throughout the time course, suggesting its negative regulation by the proteasome (Figure 2C, asterisk). To test the effects of centrosome loss on Wnt-dependent transcription, we transfected control and CentA-treated cells with the Wnt-responsive reporter TOPFLASH, in which β-catenin–dependent TCF binding sites drive expression of firefly luciferase in response to pathway activation (Veeman et al., 2003). After 5 h of stimulation by Wnt-conditioned media, CentA cells showed a significant reduction (p = 0.0083, Student’s t test) in TOPFLASH reporter activity compared with control cells. The control reporter FOPFLASH, in which TCF binding sites are mutated, showed no significant difference between control and CentA cells (Figure 2E). In CentA cells, reporter activity was dampened ∼1.4-fold, similar to the observed ∼1.3-fold decrease in β-catenin accumulation (Figure 2, B and E). Together, these data indicate that centrosomes are required for proper accumulation of β-catenin in response to Wnt and thus proper Wnt-responsive transcriptional output.
FIGURE 2:

β-Catenin accumulation after Wnt but not synthesis rate is attenuated in cells without centrosomes. (A) Cytosolic extracts from cells treated with Wnt-conditioned media for various time points were blotted for β-catenin, showing rapid increase followed by plateau and attenuated increase in DMSO and CentA cells, respectively. (B) Quantification of Wnt-dependent β-catenin increase from A. (C) Cytosolic extracts from cells treated with MG132 for various time points were blotted for β-catenin, showing increase in β-catenin and characteristic laddering associated with polyubiquitination. (D) Quantification of MG132-induced β-catenin (95–130 kDa) increase from C. (E) DMSO and CentA-treated cells were treated with Wnt for 5 h and assayed for TOPFLASH reporter activity. CentA cells show significantly decreased reporter activity compared with control, consistent with decreased β-catenin accumulation observed in C. All error bars represent SEM. **Student’s t test.
pβ-Catenin HiMW immediately accumulates after Wnt signaling and is not derived from pβ-catenin LowMW
We have demonstrated key abnormalities in β-catenin regulation and Wnt signaling in cells lacking centrosomes. Centrosome-depleted cells: 1) accumulate a distinct higher-molecular-weight β-catenin species that is dynamically cleared by the proteasome and 2) have an attenuated β-catenin accumulation rate after Wnt signaling. We reasoned that both phenomena could be explained by a single model if cells without centrosomes funnel β-catenin into an inappropriate, Wnt-insensitive degradation pathway that yields a higher-molecular-weight β-catenin species and maintains a basal degradation rate of newly synthesized β-catenin even after Wnt signaling.
After Wnt signaling, the pβ-catenin concentration in cells rapidly decreases since GSK3-phosphorylation of β-catenin is inhibited and residual pβ-catenin is rapidly degraded (Hernandez et al., 2012). If these two species are intermediate forms of β-catenin within a single, Wnt-regulated degradation pathway such that pβ-cateninHiMW is derived from pβ-cateninlowMW, we should expect that pβ-cateninHiMW decreases along with pβ-cateninLowMW after Wnt signaling (Figure 3A, Model 1). On the other hand, if centrosome-depleted cells harbored an alternative degradation pathway that competes with the Wnt-regulated degradation for β-catenin capture, we should expect that inhibition of β-catenin flux through the Wnt-regulated pathway would allow increased incorporation into such an alternative degradation pathway. Thus, upon Wnt signaling, pβ-cateninHiMW should increase as pβ-cateninLowMW decreases (Figure 3B, Model 2).
FIGURE 3:

pβ-cateninHiMW immediately accumulates after Wnt signaling and is not derived from pβ-cateninLowMW. (A) Schematic of the two models for pβ-cateninHiMW synthesis. Model 1 posits that pβ-cateninHiMW (red) is directly derived from pβ-cateninLowMW (blue) in the same posttranslational modification cascade (i.e., destruction complex). After Wnt, cells without centrosomes should show a simultaneous decrease in both species (top right). Model 2 posits that pβ-cateninHiMW and pβ-cateninLowMW are both derived from newly synthesized β-catenin (gray) via two distinct degradation cascades that compete for its capture, and pβ-cateninLowMW is processed by the destruction complex. Reducing β-catenin flux into the destruction complex via Wnt signaling should lead to an immediate decrease in pβ-cateninLowMW that is accompanied by a simultaneous increase in pβ-cateninHiMW since its degradation cascade no longer competes with the Wnt-disengaged cascade for capture of newly synthesized β-catenin (bottom right). (B) Lysates from DMSO and CentA cells treated with Wnt for various time points blotted for pβ-catenin and β-catenin. (C) Quantification of pβ-cateninLow in DMSO-treated cells and CentA-treated cells from the blot. (D) Quantification of pβ-cateninHiMW and pβ-cateninLowMW only in CentA-treated cells, showing a pattern consistent with Model 2 (top panel, bottom right).
We tested Model 1 by treating cells with Wnt-conditioned media and measuring relative abundances of pβ-cateninLowMW and pβ-cateninHiMW at various time points. Cells with and without centrosomes all showed the characteristic immediate decrease in pβ-cateninLowMW levels after Wnt signaling, confirming decreased incorporation into the canonical degradation pathway. Interestingly, cells without centrosomes showed a trend of increasing pβ-cateninHiMW as pβ-cateninLowMW levels decreased (Figure 3, B–D). This result demonstrates that pβ-cateninHiMW is not directly derived from pβ-cateninLowMW and—as is predicted by Model 2—suggests a distinct posttranslational modification cascade that competes for capture of newly synthesized β-catenin in cells without centrosomes.
Low-molecular-weight pβ-catenin is reduced after Wnt addition but later recovers to its original concentration as β-catenin concentration rises, thus enforcing a new β-catenin steady-state level after Wnt signaling (Hernandez et al., 2012; Kim et al., 2013). Our measurements are consistent with this phenomenon as pβ-cateninLowMW recovers to its original concentration 4 h after Wnt stimulation in both control and CentA-treated cells. However, a simple yet highly predictive kinetic model by Hernandez et al. (2012) demonstrates that any Wnt-insensitive degradation of β-catenin should prevent pβ-cateninLowMW from fully recovering during Wnt signaling, presenting a potential discrepancy between our data and this model. We address this discrepancy in the Discussion.
Cells without centrosomes accumulate distinct profiles of posttranslationally modified β-catenin species
GSK3-phosphorylated β-catenin is polyubiquitinated before degradation by the proteasome, and polyubiquitinated β-catenin can be visualized after proteasomal inhibition, which gives rise to a laddered migration pattern approaching 130 kDa (Hernandez et al., 2012) (Figure 2C). Our model invoking an alternative β-catenin degradation cascade predicts that cells without centrosomes should accumulate an abnormal profile of β-catenin species upon proteasome inhibition. Indeed, centrosome-depleted cells showed a markedly different laddering pattern involving preferential accumulation of β-cateninHiMW, which continued to increase with proteasome inhibition time. We observed the same pattern when visualizing pβ-cateninSer33/37/Thr41 (Figure 4A). Interestingly, in anti–pβ-cateninS45 Western blots of centrosome-depleted cells, there was no β-cateninHiMW band (Figure 4, A and B). Furthermore, total pβ-cateninS45 levels in centrosome-depleted cells were consistently weaker relative to control cells throughout the MG132 treatment, suggesting reduced CK1-dependent phosphorylation in cells without centrosomes. Together, these data further support the idea that cells without centrosomes up regulate an alternate, Wnt-independent β-catenin degradation cascade, which likely does not involve phosphorylation by CK1.
FIGURE 4:
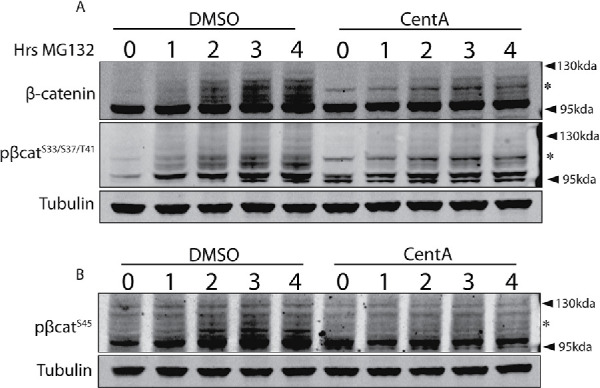
Cells without centrosomes accumulate distinct profiles of β-catenin species. Lysates from DMSO and CentA cells treated with MG132 for various time points were blotted for β-catenin and β-cateninS33/37/41 (A) or β-cateninS45 (B). For β-catenin and β-cateninS33/37/41, a ladder of various molecular weights accumulates with MG132, but is mostly restricted to β-cateninHiMW(*) in CentA cells. β-CateninS45 also accumulates in a ladder after MG132 but its accumulation is attenuated in CentA cells and is not enriched with a distinct high-molecular-weight species like the other forms of β-catenin.
GSK3 and Nek2 are dispensable for β-catenin HiMW phosphorylation
What is the posttranslational modification status of the accumulated β-cateninHiMW species in cells without centrosomes? β-CateninHiMW is readily visualized with antibodies against pβ-cateninSer33/S37/Thr41 but not pβ-cateninSer45, suggesting its phosphorylation by either GSK3 or NEK2, but not CK1. We first confirmed that β-cateninHiMW is, indeed, phosphorylated by analyzing pβ-cateninSer33/S37/Thr41 in lysates harvested in the absence of phosphatase inhibitors, which showed decreased staining of both the higher- and lower-molecular-weight bands compared with lysates containing phosphatase inhibitors (Supplemental Figure S3A). Furthermore, pβ-cateninSer33/S37/Thr41 higher-molecular-weight species increased in abundance after treatment of cells with 50 nM okadaic acid, which inhibits protein phosphatase 2A (Supplemental Figure S3B). To test whether NEK2 is required for β-cateninHiMW phosphorylation, we treated cells with INH1, a small molecule known to down-regulate NEK2 protein level after overnight treatment (Cervenka et al., 2016). Treatment of cells with 37.5 μM INH1 to a dramatic reduction in Nek2 levels but did not reduce the reactivity of anti–pβ-cateninSer33/S37/Thr41 with β-cateninHiMW, indicating that Nek2 is not required for its phosphorylation (Supplemental Figure S3C). We then tested whether β-cateninHiMW is phosphorylated by GSK3 by treating cells with 6-bromoindirubin-3′-oxime (BIO), a reversible inhibitor of GSK3 activity (Meijer et al., 2003). Treatment of cells with 10 μM BIO for 4 h led to robust GSK3 inhibition—indicated by an increase in total β-catenin accompanied by loss of lower-molecular-weight pβ-cateninSer33/S37/Thr41—yet failed to decrease pβ-cateninHiMW abundance (Supplemental Figure S3D). Rather, GSK3 inhibition by BIO led to an increase in pβ-cateninHiMW levels, consistent with increased β-catenin flux into an alternate degradation cascade in the absence of processing by the destruction complex as we observed with Wnt treatment (Supplemental Figure S3D and Figure 3, A and B, Model 2). Thus, β-cateninHiMW is phosphorylated at Ser33/37/Thr41 in a Nek2- and GSK3-independent manner.
Mass spectrometry reveals distinct posttranslational modifications on β-catenin HiMW
To identify posttranslational modifications on β-cateninHiMW, we resolved control and CentA pβ-catenin immunoprecipitates by SDS–PAGE and performed liquid chromatography–mass spectrometry (LC-MS/MS) on a region of the gel harboring a band corresponding to β-cateninHiMW. Both samples gave a distinct band in the region corresponding to β-cateninHiMW (∼110–120 kDa) after silver staining (Figure 5A, left panel). However, our LC-MS/MS analysis detected β-catenin peptides in our CentA samples but not in our control samples, consistent with up-regulation of β-cateninHiMW after centrosome loss. We used LC-MS/MS data from FLAG::β-catenin immunoprecipitates prepared in parallel to serve as a reference for unmodified β-catenin to compare with any CentA-specific posttranslational modifications (PTMs) (Figure 5A, right panel). In CentA samples, LC-MS/MS identified 14 unique peptides that mapped exclusively to the β-catenin protein sequence, obtaining a 16% amino acid coverage (Figure 5, B and C). Further analysis of these peptides revealed 10 PTMs with perfect ambiguity scores (1000), eight of which were not present on immunoprecipitated FLAG::β-catenin, despite a greater number of identified peptides and nearly full coverage of the overexpressed FLAG::β-catenin. We detected a range of PTMs in the pβ-cateninHiMW sequences with instances of amino acid substitutions (e.g., Met → Xle, Arg→Lys) and residue modifications (Carbamidomethylation) (Figure 5B). Though the amino acid substitutions could be simple translational misincorporations or artifacts from sample processing, one modification of particular interest was a predicted ubiquitination tag at K394, a residue previously known to be ubiquitinated by the ubiquitin conjugating enzyme Rad6B (Gerard et al., 2012) (Figure 5Bj). Combined with the fact that pβ-cateninHiMW displays an approximately 10–20 kDa shift (Figure 1C) in apparent molecular weight, this finding suggests that the accumulation of pβ-cateninHiMW in centrosome-depleted cells could be due to β-catenin monoubiquitination, which would theoretically add 8.5 kDa to its mass. These data identify distinct PTMs present on pβ-cateninHiMW and offer a potential explanation for its apparent molecular weight shift.
FIGURE 5:

Mass spectrometry identifies distinct PTMs on β-cateninHiMW. (A) Silver-stained SDS–PAGE gel resolving anti–pβ-cateninS33/37, S45 immunoprecipitates in CentA and DMSO-treated cells as well as anti-FLAG immunoprecipitate after FLAG::β-catenin expression (used as a reference “unmodified species” for analysis of posttranslational modifications). Dashed boxes correspond to approximate locations of gel slices abscised for LC-MS/MS analysis. (B) List of modified residues identified by LC-MS/MS showing residue identity and number as well as modification. Letters in gray represent modifications that were either detected in the FLAG::β-catenin control band or have ambiguity scores less than 20. “Frequency” corresponds to #occurences/#peptides spanning residue. (C) Sequence coverage histogram for pβ-cateninHiMW with β-catenin amino acids on X-axis (represented to scale with domain schematic; R1-R12 correspond to β-catenin armadillo repeats). (D) Sequence coverage histogram for FLAG::β-catenin (control band). Letters from list in B are mapped onto the protein schematic at approximate positions in C and D.
DISCUSSION
Our experiments suggest that centrosomes prevent inappropriate β-catenin modifications that antagonize the normal stabilization of β-catenin by Wnt signals. We show that cells without centrosomes undergo attenuated response to Wnt and also accumulate a distinct, higher-molecular-weight species of pβ-catenin (Figures 1C and 2, A–E). This species is turned over by the proteasome and is apparently distinct from Wnt-regulated pβ-catenin as well as polyubiquitinated β-catenin derived by the destruction complex (Figures 3B and 4). We propose that centrosomes inhibit a Wnt-insensitive β-catenin degradation cascade that generates a higher-molecular-weight phosphorylated β-catenin species as an intermediate.
Wnt-insensitive degradation fully accounts for the observed formation and dynamics of a higher-molecular-weight β-catenin species as well as reduced β-catenin accumulation after Wnt signaling but is inconsistent with two observations from our data:
As previously mentioned, Wnt-insensitive β-catenin degradation should theoretically prevent pβ-cateninLowMW levels from fully recovering during late Wnt signaling, yet we observe robust recovery in CentA-treated cells 4 h after Wnt addition (Figure 3, B and C).
The addition of a second β-catenin modification/degradation cascade should theoretically increase its overall degradation rate, simply due to its consumption by two degradation cascades rather than one. Yet CentA-treated cells exhibit β-catenin degradation kinetics similar to those of control (Figure S2, A and B).
We investigate these discrepancies by formalizing our postulate into a kinetic model (simplified from Hernandez et al., 2012) for the behavior of β-catenin species in cells with and without centrosomes (schematized in Figure 6A). The model for control cells is composed of a simple linear synthesis→modification→degradation cascade and recapitulates the Wnt-induced concentration dynamics of various β-catenin species observed here and in other studies (Figure 6B, left and right panels) (Hernandez et al., 2012; Kim et al., 2013). To model CentA-treated cells, we included a second modification branch to simulate β-cateninHiMW formation and degradation, whose catalytic rate constants are denoted by k2 and kDEG2, respectively (Figure 6A, top right). Given that this alternative modification branch is Wnt-independent, we expect k2 and kDEG2 to be equal when [β-cateninHiMW] is at steady state, which we assumed in our model under both +/– Wnt conditions (Supplemental Figure S4A). We varied the strength of Wnt-insensitive degradation by simultaneously changing k2 and kDEG2 relative to k1 (hereon referred to as the Wnt-insensitive degradation rate) and show that β-cateninLowMW recovery approaches 100% as Wnt-insensitive degradation approaches zero (Supplemental FigureS4B). Interestingly, we find a stable parameter range of Wnt-insensitive degradation rates that simultaneously resolve the two discrepancies mentioned above and also recapitulate our four key observations in CentA cells: 1) Moderate β-cateninLowMW recovery after Wnt addition, 2) β-cateninHiMW increase after Wnt addition, 3) β-catenin accumulation after Wnt addition, and 4) intact total β-catenin degradation rate compared with control. Simulations within this parameter range suggest that all of our observations of the unique β-catenin dynamics exhibited by CentA cells are parsimoniously accounted for under a simple model assuming a low rate of Wnt-insensitive degradation during Wnt signaling (Supplemental Figure S4). An example corresponding to a β-cateninHiMW degradation rate 1/20th of that of β-cateninLowMW is shown in Figure 6, C and D. Importantly, these kinetics predict that a low level of Wnt-insensitive β-catenin degradation exerts minimal effects on the overall β-catenin degradation rate (Supplemental Figure S2 and Figure 6D) yet dramatically reduces β-catenin accumulation after Wnt addition, as we measured in cells lacking centrosomes (Figure 2, A and B). This model is also consistent with our observation that pβ-cateninHiMW is in low abundance relative to pβ-cateninLowMW (Figure 1C). Thus, even a mild Wnt-insensitive degradation/modification cascade that produces a higher-molecular-weight intermediary in low abundance results in attenuated total β-catenin increase after Wnt signaling. In a broader sense, the apparent sensitivity of β-catenin dynamics to Wnt-insensitive degradation suggests that Wnt-insensitive degradation can very powerfully tune the Wnt response, indicating that it may serve as a key modulator and a potential interface for cross-talk between the Wnt pathway and any signaling events with links to global β-catenin metabolism.
FIGURE 6:

Wnt-insensitive β-catenin degradation model recapitulates the behaviors of β-catenin species in centrosome-depleted cells. (A) Model for β-catenin degradation in control (left panel) and CentA (right) cells. β-Catenin is synthesized (with rate S), modified (k1), unmodified (k-1), and degraded (kDEG). A second modification (k2) and degradation (kDEG2) reaction is added to simulate Wnt-insensitive degradation in CentA cells. Model assumptions at steady state are listed below each panel. (B, C) Comparison of β-catenin species behavior after Wnt addition in control (B) and CentA (C) cells between kinetic simulations (left panels) and experimental data (right panel). (D) Comparison between kinetic simulations (left) and experimental data (right) of β-catenin degradation rates. Simulations in CentA cells correspond to a k2/k1 ratio of 1/20. Note that experimental data from the Wnt washout assay only approximates β-catenin degradation rate since residual destruction complex inhibition is expected after Wnt washout. B–D were taken from Figures 2B (top) and 3B (middle) and Supplemental Figure S2B (bottom) and replotted with the same scale as simulations.
Many examples of Wnt-insensitive β-catenin degradation have been revealed over the past decade and have been proposed as possible drug targets in the treatment of malignant cells with overactive Wnt signaling (Nusse and Clevers, 2017). Recently, the E3 ligase Huwe1/Arf-BP1 (Mule) has been shown to trigger β-catenin degradation in murine intestinal organoids after prolonged Wnt hyperactivation due to APC loss-of-function mutations, thus limiting β-catenin levels during aberrant Wnt signaling (Dominguez-Brauer et al., 2017). Siah1, an E3 ligase up-regulated by p53 activation after genotoxic stress, was shown to bind APC to recruit ubiquitination machinery to β-catenin independent of GSK3 activity (Liu et al., 2001). Centrosome loss may likewise lead to genotoxic stress, as cells without centrosomes exhibit chromosome segregation defects. However, Siah-1–mediated β-catenin degradation is unlikely to be the mechanism underlying the effects on Wnt signaling in CentA cells because β-cateninHiMW accumulated in HCT116 cells with a homozygous p53 deletion (Figure 1F).
Our LC-MS/MS data show pβ-cateninHiMW ubiquitination at K394 in cells without centrosomes. Given the apparent molecular weight of the hiMW species, we propose that it is monoubiquitinated (Figure 5, A and C). The effect of this species in promoting Wnt-insensitive degradation (Figures 2, A and B, and 3B) distinguishes it from the Rad6B polyubiquitinated K394 (Gerard et al., 2012), which is thought to have a stabilizing effect on the protein (Gerard et al., 2012). A recent study supports the possibility that monoubiquitination can trigger proteasomal degradation (Braten et al., 2016). As polyubiquitin chains have higher affinity for the proteasome than single ubiquitin moieties, monoubiquitination is consistent with our model’s prediction that Wnt-insensitive degradation is slow relative to Wnt-sensitive degradation involving polyubiquitination by β-Trcp (Supplemental Figure S4, A and B) (Thrower et al., 2000; Lu et al., 2015). Weak Wnt-insensitive degradation is also indicated by the lack of β-catenin reduction upon centrosome depletion in HCT116p53–/– cells, which have chronic Wnt overactivation due to a heterozygous β-catenin deletion of Ser45, the CK1 phosphorylation site (Figure 1F) (Ilyas et al., 1997).
More recently, β-catenin was shown to be subjected to citrullination, an amino acid modification in which arginine residues are deiminated to form citrulline, promoting protein unfolding and degradation. Further, inhibition of peptidyl arginine deiminase 2 (PAD2) results in increased citrullination and degradation, an effect shown to decrease β-catenin levels after constitutive Wnt signaling (Qu et al., 2018). Although citrullination is not indicated by our mass spectrometry data per se, amino acid modifications we detected on β-cateninHiMW (such as Asn carbidomethylation and threonine thiophosphorylation) might also promote destabilization and degradation (Figure 5B). Further studies are needed to determine the role of each of the CentA–up-regulated PTMs on β-cateninHiMW abundance and Wnt-dependent β-catenin stabilization.
How does β-cateninHiMW become phosphorylated at Ser33/S37/Thr41? Our data show that GSK3 and Nek2 are dispensable for β-cateninHiMW phosphorylation (Supplemental Figure S4). β-Catenin is known to be phosphorylated by protein kinase C (PKC) at these sites to promote its degradation in a manner independent of Wnt but dependent on the E3 ligase β-Trcp (Gwak et al., 2006). Although our data suggest that β-cateninHiMW accumulates at the expense of β-Trcp–polyubiquitinated β-catenin, it is possible that PKC somehow participates in Wnt-insensitive degradation in centrosome-lacking cells (Figure 4A). PKC involvement is also supported by the fact that its proper function depends on localization to centrosomes (Passalacqua et al., 1999; Chen et al., 2004).
How might centrosome loss trigger Wnt-insenstive β-catenin degradation? We imagine that a protein responsible for promoting β-cateninHiMW formation and degradation is inhibited or sequestered upon localization to the centrosomes but is active upon centrosome loss. Previous studies have shown centrosomal localization of many β-catenin regulators (Itoh et al., 2009; Mbom et al., 2014; Vora and Phillips, 2015; Cervenka et al., 2016). Any bifunctional protein that promotes core centrosome functions but also β-catenin degradation will become available to degrade β-catenin when it is no longer sequestered by the centrosome. If this is true, the presence of supernumerary centrosomes in colorectal cancers may synergize with Wnt hyperactivation to further enhance β-catenin stability. CentA treatment of different hyperactive Wnt colorectal tumors with supernumerary centrosomes might test this possibility. Of course, the opposite scenario in which centrosomes are completely lacking may also be considered. Diseases in which centrosome organization is disrupted such as Down syndrome, primordial dwarfism, and primary recessive microcephaly—all of which share severe defects in cortical development—may have attenuated Wnt signaling as a contributing pathophysiological factor (Bettencourt-Dias et al., 2011).
Another possibility is that cell cycle checkpoints disrupted upon centrosome loss affect β-catenin regulation. The discovery of cell cycle checkpoints has revealed basic cellular processes such as chromosome segregation and cell cycle progression halt in the case of stress-related insults to the cell (Barnum and O’Connell, 2014). A p53-dependent checkpoint is known to induce G1 arrest in cells with abnormal centrosome number or composition (Mikule et al., 2007). The accumulation of β-cateninHiMW in HCT116p53–/– cells indicates that p53-downstream signaling events such as the centrosome-integrity checkpoint are not likely involved (Figure 1F). Nevertheless, it remains possible that similar mechanisms triggered by centrosome loss may function to modulate output from Wnt and other signaling pathways. Indeed, data by the Niehrs group indicate an intimate link between Wnt and cell cycle regulation (Niehrs and Acebron, 2012; Huang et al., 2015). It is thus possible that engagement of cell cycle checkpoints upon centrosome loss increases the activity of Wnt signaling components and other β-catenin regulators and contributes to attenuated Wnt output in CentA-treated cells. Furthermore, since centrosomal regulation of β-catenin would be predicted to peak at mitosis when β-catenin and pericentriolar material accumulation reach their maximum, these mechanisms could contribute to the reported increased sensitivity of mitotic cells to Wnt ligand (Davidson et al., 2009; Mbom et al., 2014).
Future studies might address the explicit effects of centrosome loss on the ability of cells to execute a broad range of signaling events as other effectors may be dysregulated in response to centrosome abnormalities. Since the GSK3 phosphorylation site that is phosphorylated in β-cateninHiMW is also included in other phosphorylation target motifs, we searched the centrosomal proteome sequences bearing tandem [S/T]XXX[S/T] sites (Amanchy et al., 2011). Bioinformatic analysis showed that more than 20% (40/191) of proteins within the known centrosomal proteome are predicted to contain this motif (Supplemental Figure S5) (Nogales-Cadenas et al., 2009; Beurel et al., 2015). Phosphotargets identified by our analysis are known to participate in a variety of biological functions including cell cycle regulation, stress response, and protein degradation (Supplemental Figure S5). Thus, there may exist additional centrosomal proteins whose posttranslational processing may be disrupted by centrosome abnormalities, and future studies might address this possibility. A more thorough understanding of the systemic cellular effects of centrosome abnormalities would greatly inform therapeutic strategies targeting malignancies with compromised centrosome function and reveal how the subcellular arrangement of signaling components modulates their signaling function.
MATERIALS AND METHODS
Cell culture and treatment with Wnt-conditioned media
HEK293 and HeLa cells were cultured in DMEM, supplemented with 10% fetal bovine serum, and 100 U/ml penicillin–streptomycin and were incubated at 37°C with 5% CO2. Wnt-conditioned media was obtained from L-cells stably expressing Wnt3a. Cells were initially cultured at 10% confluency, and media was harvested at 4 and 8 d of growth prior to filter-sterilization. Wnt-conditioned media was combined with DMEM and used at a final concentration of 50%. HCT116p53–/– cells (kindly provided by Bert Vogelstein) were cultured with McCoy’s 5a medium with 10% fetal bovine serum. TOPFLASH assays were performed by transfecting cells via lipotransfection (Lipofectamine 2000; Thermo Fisher) with either Super 8X TOPFLASH plasmid or Super 8x FOPFLASH control plasmid along with pRL for general expression of renilla luciferase as a transfection control. Passive lysates were collected and analyzed for firefly and renilla luciferase activity using the Promega Dual luciferase reporter assay system according to the manufacturer’s instructions.
Immunofluorescence
Cells were plated on 12 mm circular coverslips precoated with poly-d-lysine 24 h before fixation with 100% ice-cold methanol for 10 min at –20°C. Cells were washed 2X in phosphate-buffered saline (PBS), permeabilized with 0.25% Triton X-100 in PBS for 10 min, and blocked for 1 h with blocking buffer composed of 10% goat serum in PBS. Cells were stained overnight at 4°C with primary antibodies diluted in blocking buffer at the appropriate dilutions (mouse anti–γ-tubulin [ab11317], 1:500, Abcam; rabbit anti–GSK3-phosphorylated β-catenin [9561], 1:500, Cell Signaling Technologies) followed by 3X wash in PBS and 1 h incubation with appropriate secondary antibodies diluted in blocking buffer (oat anti-rabbit Alexa Fluor 594 [A-11008], 1:500, Thermo Fisher Scientific; and goat anti-mouse Alexa Fluor 488 [ab150105], 1:500, Abcam). After 3X wash with PBS, cells were counterstained with 4′,6-diamidino-2-phenylindole (50 μg/ml), mounted in Fluorogel with DABCO (Electron Microscopy Sciences), sealed with nail polish, and analyzed with a Zeiss D1 epifluorescence microscope with a 63X oil-immersion objective lens (NA = 1.4). Fluorescent images were processed and using ImageJ (National Institutes of Health).
Western blotting analysis and immunoprecipitation
Cells were scraped in lysis buffer containing 25 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 5% glycerol, 1X Halt phosphatase, and protease inhibitor (Thermo Fisher Scientific) and 5 mM N-ethylmalemide. Protein concentrations of lysates were determined by Red 660 assay (BG Biosciences). For immunoprecipitation, 1 mg of lysate was precleared with agarose beads, incubated with antibodies overnight at 4°C (2 μg mouse anti–β-catenin [clone 14], BD Transduction Labs; or mouse anti-FLAG M2 [Sigma Aldrich]), and immunoprecipitated with protein A/G agarose beads for 1 h at 4°C.
For Western blot analysis, NuPage LDS buffer (Thermo Fisher Scientific) was added to samples at a final concentration of 1X and lysates were incubated at 70°C for 10 min and run on 4–12% NuPage Bis-Tris Gels (Thermo Fisher Scientific) followed by overnight transfer at 4°C onto nitrocellulose membranes. Blots were washed in PBS, blocked in Licor TBS-based blocking buffer (ab166952; Abcam), and incubated with primary antibodies diluted in blocking buffer at appropriate concentrations (mouse anti–β-catenin [12F7], 1:5000, Abcam; rabbit anti–GSK3-phosphorylated β-catenin [#9561], 1:1000, Cell Signaling Technologies; rabbit anti–CK1-phosphorylated β-catenin [#9564], 1:1000, Cell Signaling Technologies; mouse anti-tubulin [12G10], 1 μg/ml, Developmental Studies Hybridoma Bank, University of Iowa). Blots were washed 4X in PBS with 0.1% Tween, incubated with secondary antibodies for 1 h at room temperature (goat anti-mouse DyLight 800, 1:10,000, Rockland; goat anti-rabbit Alexa Fluor 680, 1:10,000, Thermo Fisher Scientific), washed 4X in PBS with 0.1% Tween, and scanned using the LI-COR Odyssey imaging system. Western blots were quantified using LI-COR Image Studio Lite software. A rectangular box was drawn around each band to be quantified, and marginal fluorescence slightly outside each box (background) was subtracted from fluorescence inside each box (signal + background). For each blot, tubulin intensity was used to quantify relative protein abundance per lane and each band subject to quantification was normalized accordingly.
Cytosolic extracts were obtained by scraping cells in hypotonic lysis buffer containing 10 mM Tris, pH 7.5, 10 mM KCl, 2 mM EDTA, 5 mM N-ethylmalemide, and 1x Halt proteasome and phosphatase cocktail. Cells suspensions were passed 10 times through a 26-gauge needle, and nuclei and membrane were pelleted by centrifugation at 13,000 × g for 30 min.
Liquid chromatography and mass s
Samples from control and CentA-treated cells were subject to immunoprecipitation using anti-FLAG M2 (Sigma Aldrich) or rabbit anti–GSK3-phosphorylated β-catenin (#9561; Cell Signaling Technologies) as described above. Samples were resolved using SDS–PAGE, and bands corresponding to regions of interest were excised for trypsin digest following LC-MS/MS analysis. LC-MS/MS data were analyzed using Scaffold and Scaffold PTM (Proteome Software).
Kinetic modeling
Ordinary differential equations based on Hernandez et al. (2012) were used to model the dynamics of β-catenin species over time. The following equations were used to model β-catenin dynamics in the “DMSO” and “CentA” scenarios (terms schematized in Figure 6):
DMSO:
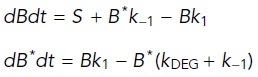 |
where B = β-catenin, B* = β-cateninLowMw (destruction complex–modified); k1 and k -1 = forward and reverse formation rates, respectively; and kDEG = degradation rate.
CentA:
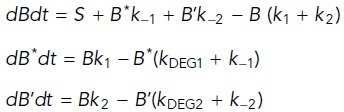 |
where B* = β-cateninLowMW (destruction complex–modified), B’ = β-cateninHiMw; k1 and k –1 = forward and reverse formation rates for β-cateninLowMw, respectively; k2 and k –2 = forward and reverse formation rates for β-cateninHiMw, respectively; and kDEG1 and kDEG2 = degradation rates for β-cateninLowMW and β-cateninHiMw, respectively.
Supplementary Material
Acknowledgments
We thank Fenghuang Zhan for providing the INH1 Nek2 inhibitor and Bert Vogelstein for the HCT116 p53–/– cell line as well as Christopher Stipp and Vena Prahlad for technical assistance with cell culture. We also thank the University of Iowa Proteomics Core for assistance with liquid chromatography/mass spectrometry experiments. Funding was provided by a National Institutes of Health grant (5 R01 GM114007) awarded to B.T.P.
Abbreviations used:
- APC
adenomatous polyposis coli
- GFP
green fluorescent protein
- LRP6
low-density lipoprotein receptor-related protein 6
- Nek2
Never in Mitosis (NIMA) Related Kinase 2
- TCF
T cell factor.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-02-0139) on June 17, 2020.
REFERENCES
- Amanchy R, Kandasamy K, Mathivanan S, Periaswamy B, Reddy R, Yoon WH, Joore J, Beer MA, Cope L, Pandey A. (2011). Identification of novel phosphorylation motifs through an integrative computational and experimental analysis of the human phosphoproteome. J Proteomics Bioinform , 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arquint C, Gabryjonczyk AM, Nigg EA. (2014). Centrosomes as signalling centres. Philos Trans R Soc Lond B Biol Sci , 20130464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahmanyar S, Guiney EL, Hatch EM, Nelson WJ, Barth AI. (2010). Formation of extra centrosomal structures is dependent on beta-catenin. J Cell Sci , 3125–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AT, Clemons AM, Phillips BT. (2016). Unique and redundant beta-catenin regulatory roles of two Dishevelled paralogs during C. elegans asymmetric cell division. J Cell Sci , 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baldwin AT, Phillips BT. (2014). The tumor suppressor APC differentially regulates multiple beta-catenins through the function of axin and CKIalpha during C. elegans asymmetric stem cell divisions. J Cell Sci , 2771–2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnum KJ, O’Connell MJ. (2014). Cell cycle regulation by checkpoints. Methods Mol Biol , 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt-Dias M, Hildebrandt F, Pellman D, Woods G, Godinho SA. (2011). Centrosomes and cilia in human disease. Trends Genet , 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Grieco SF, Jope RS. (2015). Glycogen synthase kinase-3 (GSK3): regulation, actions, and diseases. Pharmacol Ther , 114–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braten O, Livneh I, Ziv T, Admon A, Kehat I, Caspi LH, Gonen H, Bercovich B, Godzik A, Jahandideh S, et al. (2016). Numerous proteins with unique characteristics are degraded by the 26S proteasome following monoubiquitination. Proc Natl Acad Sci USA , E4639–E4647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cervenka I, Valnohova J, Bernatik O, Harnos J, Radsetoulal M, Sedova K, Hanakova K, Potesil D, Sedlackova M, Salasova A, et al. (2016). Dishevelled is a NEK2 kinase substrate controlling dynamics of centrosomal linker proteins. Proc Natl Acad Sci USA , 9304–9309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Purohit A, Halilovic E, Doxsey SJ, Newton AC. (2004). Centrosomal anchoring of protein kinase C betaII by pericentrin controls microtubule organization, spindle function, and cytokinesis. J Biol Chem , 4829–4839. [DOI] [PubMed] [Google Scholar]
- Chilov D, Sinjushina N, Rita H, Taketo MM, Makela TP, Partanen J. (2011). Phosphorylated beta-catenin localizes to centrosomes of neuronal progenitors and is required for cell polarity and neurogenesis in developing midbrain. Dev Biol , 259–268. [DOI] [PubMed] [Google Scholar]
- Costa R, Peruzzo R, Bachmann M, Monta GD, Vicario M, Santinon G, Mattarei A, Moro E, Quintana-Cabrera R, Scorrano L, et al. (2019). Impaired mitochondrial ATP production downregulates Wnt signaling via ER stress induction. Cell Rep , 1949–1960.e1946. [DOI] [PubMed] [Google Scholar]
- Davidson G, Shen J, Huang YL, Su Y, Karaulanov E, Bartscherer K, Hassler C, Stannek P, Boutros M, Niehrs C. (2009). Cell cycle control of wnt receptor activation. Dev Cell , 788–799. [DOI] [PubMed] [Google Scholar]
- Dominguez-Brauer C, Khatun R, Elia AJ, Thu KL, Ramachandran P, Baniasadi SP, Hao Z, Jones LD, Haight J, Sheng Y, et al. (2017). E3 ubiquitin ligase Mule targets beta-catenin under conditions of hyperactive Wnt signaling. Proc Natl Acad Sci USA , E1148–E1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sahli S, Xie Y, Wang L, Liu S. (2019). Wnt signaling in cancer metabolism and immunity. Cancers (Basel) , 904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba LC, Eivers E, Geissert D, Taelman V, De Robertis EM. (2008). Asymmetric mitosis: unequal segregation of proteins destined for degradation. Proc Natl Acad Sci USA , 7732–7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerard B, Sanders MA, Visscher DW, Tait L, Shekhar MP. (2012). Lysine 394 is a novel Rad6B-induced ubiquitination site on beta-catenin. Biochim Biophys Acta , 1686–1696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach JP, Emmink BL, Nojima H, Kranenburg O, Maurice MM. (2014). Wnt signalling induces accumulation of phosphorylated beta-catenin in two distinct cytosolic complexes. Open Biol , 140120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonczy P. (2015). Centrosomes and cancer: revisiting a long-standing relationship. Nat Rev Cancer , 639–652. [DOI] [PubMed] [Google Scholar]
- Gwak J, Cho M, Gong SJ, Won J, Kim DE, Kim EY, Lee SS, Kim M, Kim TK, Shin JG, et al. (2006). Protein-kinase-C-mediated beta-catenin phosphorylation negatively regulates the Wnt/beta-catenin pathway. J Cell Sci , 4702–4709. [DOI] [PubMed] [Google Scholar]
- Hernandez AR, Klein AM, Kirschner MW. (2012). Kinetic responses of beta-catenin specify the sites of Wnt control. Science , 1337–1340. [DOI] [PubMed] [Google Scholar]
- Huang S, Shetty P, Robertson SM, Lin R. (2007). Binary cell fate specification during C. elegans embryogenesis driven by reiterated reciprocal asymmetry of TCF POP-1 and its coactivator beta-catenin SYS-1. Development , 2685–2695. [DOI] [PubMed] [Google Scholar]
- Huang YL, Anvarian Z, Doderlein G, Acebron SP, Niehrs C. (2015). Maternal Wnt/STOP signaling promotes cell division during early Xenopus embryogenesis. Proc Natl Acad Sci USA , 5732–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilyas M, Tomlinson IP, Rowan A, Pignatelli M, Bodmer WF. (1997). Beta-catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci USA , 10330–10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Jenny A, Mlodzik M, Sokol SY. (2009). Centrosomal localization of Diversin and its relevance to Wnt signaling. J Cell Sci , 3791–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan DD, Meigs TE, Kelly P, Casey PJ. (2004). Identification of a role for beta-catenin in the establishment of a bipolar mitotic spindle. J Biol Chem , 10829–10832. [DOI] [PubMed] [Google Scholar]
- Kim SE, Huang H, Zhao M, Zhang X, Zhang A, Semonov MV, MacDonald BT, Zhang X, Garcia Abreu J, Peng L, et al. (2013). Wnt stabilization of beta-catenin reveals principles for morphogen receptor-scaffold assemblies. Science , 867–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam AK, Phillips BT. (2017). Wnt signaling polarizes C. elegans asymmetric cell divisions during development. Results Probl Cell Differ , 83–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li VS, Ng SS, Boersema PJ, Low TY, Karthaus WR, Gerlach JP, Mohammed S, Heck AJ, Maurice MM, Mahmoudi T, et al. (2012). Wnt signaling through inhibition of beta-catenin degradation in an intact Axin1 complex. Cell , 1245–1256. [DOI] [PubMed] [Google Scholar]
- Liu C, Kato Y, Zhang Z, Do VM, Yankner BA, He X. (1999). beta-Trcp couples beta-catenin phosphorylation-degradation and regulates Xenopus axis formation. Proc Natl Acad Sci USA , 6273–6278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Stevens J, Rote CA, Yost HJ, Hu Y, Neufeld KL, White RL, Matsunami N. (2001). Siah-1 mediates a novel beta-catenin degradation pathway linking p53 to the adenomatous polyposis coli protein. Mol Cell , 927–936. [DOI] [PubMed] [Google Scholar]
- Loh KM, van Amerongen R, Nusse R. (2016). Generating cellular diversity and spatial form: Wnt signaling and the evolution of multicellular animals. Dev Cell , 643–655. [DOI] [PubMed] [Google Scholar]
- Lu Y, Lee BH, King RW, Finley D, Kirschner MW. (2015). Substrate degradation by the proteasome: a single-molecule kinetic analysis. Science , 1250834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. (2009). Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell , 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbom BC, Nelson WJ, Barth A. (2013). Beta-catenin at the centrosome: discrete pools of beta-catenin communicate during mitosis and may co-ordinate centrosome functions and cell cycle progression. Bioessays , 804–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbom BC, Siemers KA, Ostrowski MA, Nelson WJ, Barth AI. (2014). Nek2 phosphorylates and stabilizes beta-catenin at mitotic centrosomes downstream of Plk1. Mol Biol Cell , 977–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, et al. (2003). GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol , 1255–1266. [DOI] [PubMed] [Google Scholar]
- Mikule K, Delaval B, Kaldis P, Jurcyzk A, Hergert P, Doxsey S. (2007). Loss of centrosome integrity induces p38-p53-p21-dependent G1-S arrest. Nat Cell Biol , 160–170. [DOI] [PubMed] [Google Scholar]
- Niehrs C, Acebron SP. (2012). Mitotic and mitogenic Wnt signalling. EMBO J , 2705–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigg EA, Stearns T. (2011). The centrosome cycle: centriole biogenesis, duplication and inherent asymmetries. Nat Cell Biol , 1154–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogales-Cadenas R, Abascal F, Diez-Perez J, Carazo JM, Pascual-Montano A. (2009). CentrosomeDB: a human centrosomal proteins database. Nucleic Acids Res , D175–D180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R, Clevers H. (2017). Wnt/beta-catenin signaling, disease, and emerging therapeutic modalities. Cell , 985–999. [DOI] [PubMed] [Google Scholar]
- Olmeda D, Castel S, Vilaro S, Cano A. (2003). Beta-catenin regulation during the cell cycle: implications in G2/M and apoptosis. Mol Biol Cell , 2844–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passalacqua M, Patrone M, Sparatore B, Pedrazzi M, Melloni E, Pontremoli S. (1999). Protein kinase C-theta is specifically activated in murine erythroleukaemia cells during mitosis. FEBS Lett , 249–253. [DOI] [PubMed] [Google Scholar]
- Phillips BT, Kidd AR, 3rd, King R, Hardin J, Kimble J. (2007). Reciprocal asymmetry of SYS-1/beta-catenin and POP-1/TCF controls asymmetric divisions in Caenorhabditis elegans. Proc Natl Acad Sci USA , 3231–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puram SV, Kim AH, Park HY, Anckar J, Bonni A. (2013). The ubiquitin receptor S5a/Rpn10 links centrosomal proteasomes with dendrite development in the mammalian brain. Cell Rep , 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y, Olsen JR, Yuan X, Cheng PF, Levesque MP, Brokstad KA, Hoffman PS, Oyan AM, Zhang W, Kalland KH, et al. (2018). Small molecule promotes beta-catenin citrullination and inhibits Wnt signaling in cancer. Nat Chem Biol , 94–101. [DOI] [PubMed] [Google Scholar]
- Schneider SQ, Bowerman B. (2007). beta-Catenin asymmetries after all animal/vegetal-oriented cell divisions in Platynereis dumerilii embryos mediate binary cell-fate specification. Dev Cell , 73–86. [DOI] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, Pickart CM. (2000). Recognition of the polyubiquitin proteolytic signal. EMBO J , 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. (2003). Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Curr Biol , 680–685. [DOI] [PubMed] [Google Scholar]
- Vora S, Phillips BT. (2015). Centrosome-associated degradation limits beta-catenin inheritance by daughter cells after asymmetric division. Curr Biol , 1005–1016. [DOI] [PubMed] [Google Scholar]
- Vora SM, Phillips BT. (2016). The benefits of local depletion: the centrosome as a scaffold for ubiquitin-proteasome-mediated degradation. Cell Cycle , 2124–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong YL, Anzola JV, Davis RL, Yoon M, Motamedi A, Kroll A, Seo CP, Hsia JE, Kim SK, Mitchell JW, et al. (2015). Cell biology. Reversible centriole depletion with an inhibitor of Polo-like kinase 4. Science , 1155–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.