

Abstract
The human Ska complex (Ska) localizing to both spindle microtubules and kinetochores is essential for proper chromosome segregation during mitosis. Although several mechanisms have been proposed to explain how Ska is recruited to kinetochores, it is still not fully understood. By analyzing Ska3 phosphorylation, we identified six critical Cdk1 sites, including the previously identified Thr358 and Thr360. Mutations of these sites to phospho-deficient alanine (6A) in cells completely abolished Ska3 localization to kinetochores and Ska functions in chromosome segregation. In vitro, Cdk1 phosphorylation on Ska enhanced WT, not phospho-deficient 6A, binding to Ndc80C. Strikingly, the phosphomimetic Ska 6D complex formed a stable macro-complex with Ndc80C, but Ska WT failed to do so. These results suggest that multisite Cdk1 phosphorylation-enabled Ska–Ndc80 binding is decisive for Ska localization to kinetochores and its functions. Moreover, we found that Ska decrease at kinetochores triggered by the microtubule-depolymerizing drug nocodazole is independent of Aurora B but can be overridden by Ska3 overexpression, suggestive of a role of spindle microtubules in promoting Ska kinetochore recruitment. Thus, based on the current and previous results, we propose that multisite Cdk1 phosphorylation is critical for the formation of Ska–Ndc80 macro-complexes that are essential for chromosome segregation.
INTRODUCTION
Proper kinetochore–microtubule interactions are essential for faithful chromosome segregation and preventing chromosome instability. At early mitosis, the kinetochore is initially captured by spindle microtubules though the KMN (Knl1, the Mis12, and Ndc80 complexes) network, but the initial kinetochore–microtubule interactions on their own are not sufficient to sustain the subsequent chromosome segregation (Tanaka et al., 2005; Varma and Salmon, 2012; Cheerambathur and Desai, 2014). Other critical factors are needed to finalize the KMN-mediated interactions with microtubules. Among them is the Ska complex (Ska), which comprises three subunits, Ska1, 2, and 3 (Guimaraes and Deluca, 2009). Depletion of any subunit in this complex significantly compromised the kinetochore–microtubule interactions and triggered massive mitotic arrest (Hanisch et al., 2006; Daum et al., 2009; Gaitanos et al., 2009; Raaijmakers et al., 2009; Theis et al., 2009; Welburn et al., 2009; Ohta et al., 2010), suggestive of an essential role of Ska in promoting kinetochore–microtubule interactions. The subsequent studies further demonstrated that Ska promotes kinetochore–microtubule interactions by bringing PP1 to the kinetochore–microtubule binding interface (Sivakumar et al., 2016), or by directly binding to spindle microtubules (Welburn et al., 2009; Jeyaprakash et al., 2012; Schmidt et al., 2012; Boeszoermenyi et al., 2014; Abad et al., 2014, 2016; Thomas et al., 2016; Monda et al., 2017). Furthermore, the Ska was shown to interact with microtubule tips in a load-bearing manner (Helgeson et al., 2018) and also track with dynamic microtubule ends (Schmidt et al., 2012; Monda et al., 2017). These interesting observations suggested that the Ska collaborates with the outer kinetochore proteins to perform its functions and also highlighted the importance of the physical interactions between Ska and the outer kinetochore proteins in establishing and/or maintaining proper kinetochore–microtubule interactions. Thus, understanding how Ska interacts with kinetochores would be key to deciphering the molecular mechanisms whereby Ska promotes kinetochore–microtubule interactions.
Extensive studies have shown that Ska localization to kinetochores depends on the outer kinetochore protein Ndc80 (Gaitanos et al., 2009; Raaijmakers et al., 2009; Zhang et al., 2012), suggesting a possible physical interaction between Ska and Ndc80C. Our recent findings validated a direct binding between Ndc80C and Ska3 (Zhang et al., 2017). We further demonstrated that the Ska–Ndc80 interaction depends on Cdk1 phosphorylation in the C-terminal region of Ska3, and the Ndc80 loop together with its flanking regions is responsible for Ska binding. Several other studies also showed that the Ndc80 N-terminal tail and the calponin-homology domain are involved in recruiting Ska to kinetochores (Cheerambathur et al., 2017; Janczyk et al., 2017; Helgeson et al., 2018), suggesting that multiple pathways might coexist to install Ska to kinetochores. However, the key determinant that docks the Ska onto kinetochores still remains elusive. We have previously identified the two important sites Thr358 and Thr360 in Ska3 whose phosphorylation by Cdk1 promoted the Ska–Ndc80 interaction in vitro, but mutations of them to alanine only resulted in moderate defects in chromosome segregation, provoking a question whether Cdk1 phosphorylation-dependent Ska–Ndc80 interactions are the major determinant for Ska functions or other factors exist to promote Ska functions.
By analyzing Cdk1 phosphorylation on Ska3, we identified six critical sites in determining the Ska–Ndc80 interaction, including the previously identified Thr358 and Thr360. We demonstrated that these six sites are decisive for Ska recruitment to kinetochores and its functions. Strikingly, in vitro reconstitution experiments indicate that Cdk1 phosphorylation on these six sites is sufficient for the formation of a stable Ska–Ndc80C macro-complex. Furthermore, our results also suggest spindle microtubules may promote Ska recruitment to kinetochores. Thus, based on the results from the current and previous studies, we propose that with the help of spindle microtubules, Cdk1 phosphorylation installs Ska to Ndc80C.
RESULTS
The Ska3 C-terminal region is heavily phosphorylated at multiple conserved Cdk1 sites
We have recently shown that Cdk1 phosphorylation on Ska3 promotes Ska binding to Ndc80C (Zhang et al., 2017). Surprisingly, the Ska3 mutant (Thr358A Thr360A) that exhibited reduced Ndc80C-binding activity still partially sustained chromosome segregation albeit at a reduced level in comparison to WT, making it unclear whether Cdk1 phosphorylation-dependent Ska–Ndc80 interactions are the major determinant for Ska functions or other factors also exist to promote Ska functions. As in the Ska3 C-terminal region there are a total of 14 potential Cdk1 sites and many of them, including the previously identified two sites Thr358 and Thr360, have been found to be phosphorylated (Zhang et al., 2017); we reasoned that in addition to these two reported sites, other phosphorylation sites might also contribute to the Ska–Ndc80 interaction and Ska functions. We first analyzed the conservation of these Cdk1 sites across species, including human (hs), mouse (mm), chicken (gg), and Xenopus laevis (xl) (Supplemental Figure S1A). Six sites, Thr203, Trh217, Ser283, Thr291, Thr358, and Thr360, were found to distribute within three conserved regions. Among them, Thr203, Ser283, Thr291, and Thr358 are well conserved across the tested species; and the other two sites, Thr217 and Thr360, are less conserved (Figure 1A and Supplemental Figure S1A). Second, we performed mass-spectrometric analyses on immunoprecipitated Myc-Ska3 isolated from nocodazole-arrested HeLa Tet-On cells. Among the above six sites, five were identified being phosphorylated in mitotic cells (Figure 1B). Thr203, although it was not retrieved from our mass-spectrometric analyses, was also included for further analysis because of its high conservation. In addition, all six sites were able to be phosphorylated by Cdk1 in vitro (Supplemental Figure S1B). Thus, these six sites, Thr203, Thr217, Ser283, Thr291, Thr358, and Thr360, were subject to the subsequent analysis in this study. We therefore constructed various mutants containing different sets of mutations on these sites (Figure 1C). As Ska3 phosphorylation significantly reduced its mobility in SDS–PAGE (Zhang et al., 2017), we then examined how mutations of these sites affected its migration behavior. Nocodazole-arrested HeLa Tet-On cells were transfected with GFP-Ska3 WT, 2A, 4A1/2/3, 6A, and 6D, and cell lysates were analyzed with SDS–PAGE followed by Western blots. Consistent with the previous findings, the slower-migration species of Ska3 WT appeared on SDS–PAGE (Figure 1D) (Zhang et al., 2017). Double mutations to nonphosphorylatable alanine (2A) accelerated the gel mobility of the slower species, and quadruple mutations to alanine (4A1/2) that include Thr358 and Thr360 further accelerated the gel mobility. Strikingly, mutations of all the six sites to alanine (6A) almost completely abolished the slower-migration species; conversely, mutations of them to phosphomimetic aspartic acid (6D) largely restored them, suggesting that these six sites are the major phosphorylation sites controlling gel mobility behavior of Ska3. Surprisingly, another quadruple-mutation mutant (4A3) that excludes Thr358 and Thr360 exhibited similar gel mobility to WT, suggesting that these two sites play a more important role in controlling gel mobility behavior of Ska3. Taken together, Ska3 is phosphorylated at multiple sites during mitosis.
FIGURE 1:

The Ska3 C-terminal region is highly phosphorylated by Cdk1 during mitosis. (A) Analysis on the conservation of Cdk1 sites in Ska3. Six Cdk1 sites (*) in human Ska3 are distributed in three conserved domains identified from a sequence alignment across four species, human (hs), mouse (mm), chicken (gg), and Xenopus laevis (xl) (Supplemental Figure S1). Among the six sites, four are highly conserved across the tested species. The other two are conserved at least among human and mouse. In the conserved regions, identical amino acids are marked in red and similar amino acids in orange. (B) Phosphorylated Cdk1 sites identified by mass-spectrometric analyses on immunoprecipitated Myc-Ska3 isolated from nocodazole-arrested HeLa Tet-On cells. The sites first identified are marked in orange and the newly identified ones in the second time are marked in red. A total of 11 Cdk1 sites were identified being phosphorylated. (C) Summary of Ska3 mutants applied in this study. (D) Lysates of nocodazole-arrested HeLa Tet-On cells expressing GFP-Ska3 WT, 2A, 4A1/2/3, 6A, or 6D were resolved with SDS–PAGE and blotted with the indicated antibody.
Multisite Cdk1 phosphorylation determines Ska3 localization to kinetochores
We then sought to determine how mutations on the six sites of Ska3 affected its localization to kinetochores. We first examined Ska3 localization in unperturbed mitotic cells using fixed-cell immunostaining. Thymidine-arrested HeLa Tet-On cells expressing GFP-Ska3 WT, 2A, 6A, and 6D were released to fresh medium, and mitotic cells were collected for immunostaining. As shown in Figure 2, A and B, the fluorescence signals of GFP-Ska3 WT were detected at kinetochores in prometaphase and metaphase cells. The signals of GFP-Ska3 2A were greatly reduced, which is consistent with our previous results (Zhang et al., 2017). Strikingly, GFP-Ska3 6A with all six sites mutated to alanine completely lost its kinetochore signals, suggesting that in addition to Thr358 and Thr360, the other four sites, Thr203, Trh217, Ser283, and Thr291, also contribute to the kinetochore localization of Ska3. Conversely, the signals of the phosphomimetic GFP-Ska3 6D were easily detected at kinetochores, albeit they were about twofold weaker than the ones of WT, suggesting that the phosphomimetic mutant only partially mimics WT in cells. All the above observations support the notion that Cdk1 phosphorylation predominantly determines Ska localization to kinetochores. The observed localization defects for these mutants are unlikely due to distinct protein expression levels as they were all comparable to WT (Figure 1D). Similar localization patterns for these mutants were also observed in MG132-arrested metaphase cells (Supplemental Figure S2, A and B). To rule out the possible artifacts caused by formaldehyde fixation in immunostaining (Teves et al., 2016), we performed live-cell imaging. Consistently, GFP-Ska3 WT was robustly localized to kinetochores and the three mutants, 2A, 6A and 6D, all exhibited reduced kinetochore localization (Figure 2, E–H). Interestingly, the tested mutants were all normally localized to spindle microtubules (Figure 2, E–H). Thus, Cdk1 phosphorylation of Ska3 is the decisive factor for Ska localization to kinetochores, but dispensable for its localization on microtubules.
FIGURE 2:

Cdk1 phosphorylation in Ska3 is essential for Ska localization at kinetochores. (A) Thymidine-arrested HeLa Tet-On cells expressing GFP-Ska3 WT, 2A, 4A1/2, 6A, or 6D were released into fresh medium. Mitotic cells were collected for immunostaining using the indicated antibodies. Representative images were shown here. Scale bars, 5 μm. (B) Quantification of in GFP-Ska3 intensity on kinetochores in A. Detailed description about quantification was recorded in Materials and Methods section. At least 60 kinetochores (6 kinetochores per cell) were analyzed for each condition. Average and SD were shown in lines. ****P < 0.0001; n.s. denotes no significance. (C) Cells expressing GFP-Ska3 WT, 4A3, or 6D were treated similarly to the ones in A. Scale bars, 5 μm. (D) Quantification of GFP-Ska3 intensity on kinetochores in C. At least 60 kinetochores (6 kinetochores per cell) were analyzed for each condition. Average and SD were shown in lines; n.s. denotes no significance. (E, G) Live-cell imaging of HeLa Tet-On cells expressing GFP-Ska3 WT, 2A (E), 6A (E), 4A3 (G), or 6D (G) treated with MG132 for 1 h. Scale bars, 5 μm. (F, H) Quantification of GFP-Ska3 intensity on kinetochores (left panel) and microtubules (right panel) in E and G were normalized to that of cytoplasm. Detailed description about quantification was recorded in Materials and Methods section. For both F and H, at least 10 cells (10 kinetochores and 10 microtubules per cell) were analyzed for each condition. Average and SD were shown in lines. *P < 0.05; ****P < 0.0001; n.s. denotes no significance.
Differential contributions of Ska3 phosphorylation sites to its kinetochore localization
Mutations of the six phosphorylation sites to alanine abolished Ska3 localization to kinetochores, whereas mutations of Thr358 and Thr360 to alanine only reduced it, suggesting that the other four sites also contribute to Ska3 localization to kinetochores. We then tested this hypothesis by examining the kinetochore localization of the Ska3 4A3 mutant that excludes the mutations of Thr358 and Thr360. Unexpectedly, GFP-Ska3 4A3 was still localized to kinetochores as robustly as WT in unperturbed mitotic HeLa Tet-On cells (Figure 2, C and D). This observation was further confirmed by the results from live-cell imaging (Figure 2, G and H). Thus, the six phosphorylation sites contribute differentially to Ska3 localization to kinetochores, and Thr358 and Thr360 play a dominant role in Ska kinetochore localization (Zhang et al., 2017). To further confirm this, we examined the effects of other phospho-defective quadruple mutations (4A1/2) that include Thr358 and Thr360 on the kinetochore localization of Ska3. As expected, the signals of GFP-Ska3 4A1 and 4A2 were also significantly reduced compared with WT, but still stronger than the ones of GFP-Ska3 6A (Figure 2, A and B). To rule out the possibility that the remaining signals of 2A and 4A mutants on kinetochores are due to their dimerization with endogenous Ska, we reexamined their kinetochore localization in cells depleted of endogenous Ska3. Their localization patterns were similar to the ones in cells without depletion of endogenous Ska3 (Supplemental Figure S2, C–E).
We then sought to examine the behavior of their corresponding phosphomimetic quadruple mutants (4D1/2) in Ska3 kinetochore recruitment. Again, the localizations of GFP-Ska3 4D1 and 4D2 were mitigated but could still be detected at the comparable levels to 2D (Supplemental Figure S3, A–C), suggesting that these phosphomimetic Ska3 mutants, including 6D, can only partially mimic WT in cells. Based on these observations, we conclude that Ska3 localization to kinetochores is predominantly determined by Thr358 Thr360-centered multisite phosphorylation. However, it remains unknown how the additional four mutations have a significant effect on Ska3 localization to kinetochores on the base of mutations at Thr358 and Thr360.
Spindle microtubules may directly promote Ska3 recruitment to kinetochores
It was previously shown that nocodazole treatment significantly reduced Ska localization to kinetochores (Chan et al., 2012). We repeated the experiment in HeLa Tet-On and observed a similar phenomenon. Treatment of the Aurora B inhibitor ZM447439 did not restore the kinetochore localization of Ska3 at all while it completely abrogated the fluorescence signals of histone H3pS10 (Figure 3, A–C). Similar results were observed in nontransformed RPE-1 cells (Supplemental Figure S4, A and B). These results suggest that spindle microtubules rather than Aurora B play a role in Ska recruitment to kinetochores. Similarly, the Dam1 complex, the functional Ska analog in budding yeast, was previously shown to be loaded to kinetochores by spindle microtubules (Li et al., 2002). We hypothesized that spindle microtubules may facilitate Ska loading to kinetochores by enriching Ska to the proximity of kinetochores. If this was the case, Ska overexpression could override the nocodazole-induced Ska decrease at kinetochores. To test this, we compared the kinetochore localization of GFP-Ska3 in between the absence and the presence of nocodazole. Western blots confirmed the levels of GFP-Ska3 were more than 10-fold the ones of endogenous Ska3 (Supplemental Figure S4C). Strikingly, GFP-Ska3 intensity on kinetochores was barely altered by the addition of nocodazole (Figure 3, D and E), suggesting that increasing Ska protein concentration is able to bypass the requirement of spindle microtubules for Ska recruitment to kinetochores. Thus, spindle microtubules per se may be important for Ska localization to kinetochores in cells.
FIGURE 3:

Spindle microtubules facilitate Ska recruitment to kinetochores. (A) Aurora B kinase activity is dispensable for Ska3 localization at kinetochores. Thymidine-arrested HeLa Tet-On cells were released into fresh medium. At 7 h after release, nocodazole with a final concentration of 5 μM was added. Aurora B inhibitor ZM447439 (ZM) was then added 2 h later. Mitotic cells with 1-h ZM treatment were collected for staining with DAPI and the indicated antibodies. (B) Quantifications of Ska3 intensity (left) and H3pS10 intensity (right) on kinetochores in A. Detailed description about quantification was recorded in Materials and Methods section. At least 60 kinetochores (6 kinetochores per cell) were analyzed for each condition. Average and SD were shown in lines. ****P < 0.0001; ***P < 0.001; n.s. denotes no significance. (C) Cell lysates in A were separated with SDS–PAGE and then blotted with the indicated antibodies. (D) Ska3 overexpression overrides the effects of nocodazole on its kinetochore localization. HeLa Tet-On cells transfected with plasmids containing GFP-Ska3 were treated with DMSO or nocodazole. Mitotic cells were collected for staining with DAPI and the indicated antibodies. (E) Quantification of GFP-Ska3 intensity on kinetochores in D. Only prometaphase cells were selected for analysis. Detailed description about quantification was recorded in Materials and Methods section. At least 60 kinetochores (6 kinetochores per cell) were analyzed for each condition. Average and SD were shown in lines. ****P < 0.0001; n.s. denotes no significance. (F) HeLa Tet-On cells were treated under the same conditions described in Figure 2A, except that nocodazole was added at 7 h after thymidine release. Scale bars, 5 μm. (G) Quantification of GFP-Ska3 intensity on kinetochores in F. Detailed description about quantification was recorded in Materials and Methods section. At least 60 kinetochores (6 kinetochores per cell) were analyzed for each condition. Average and SD were shown in lines. ****P < 0.0001; n.s. denotes no significance.
We then examined how spindle microtubules affected the kinetochore localization of these Ska3 mutants. GFP-Ska3 WT, 2A, 4A1/2/3, 6A, and 6D were expressed in nocodazole-treated HeLa Tet-On cells and kinetochore localization was examined. In the presence of nocodazole, GFP-Ska3 WT was detected at kinetochores. Surprisingly, all the tested mutants except for 4A-3 completely lost the kinetochore signals (Figure 3, F and G), seemingly in contrast to the results from cells with intact microtubules (Figure 2). Considering that all the mutants but 4A-3 had more or less defects in kinetochore localization in the unperturbed mitosis (Figure 2, A and B), the above observations suggest that spindle microtubules may help stabilize the weak Ska–Ndc80 interactions in these mutants. Similar results were also obtained in cells depleted of endogenous Ska3, excluding the interference of endogenous Ska3 in GFP-Ska3 recruitment to kinetochores (Supplemental Figure S4, D and E). Interestingly, with nocodazole treatment, the kinetochore signals of GFP-Ska3 4D1 and 4D2 were almost abolished while the ones of 2D were still detected at reduced levels (Supplemental Figure S4F), further supporting the above notion that these phosphomimetic Ska3 mutants can only partially mimic WT. Taken together, our results here suggest that although Cdk1-mediated multisite phosphorylation plays a decisive role in Ska localization to kinetochores, spindle microtubules may regulate this process. In addition, the contrasting behavior of 4A3 further confirms that our previously identified Thr358 and Thr360 are major sites contributing to Ska localization to kinetochores (Zhang et al., 2017).
Ska functions in chromosome segregation are predominantly determined by multisite Cdk1 phosphorylation
As the Ska is required for chromosome segregation, we next determined how Cdk1 phosphorylation on Ska3 affected chromosome segregation. GFP-Ska3 WT, 6A, and 6D were expressed in Ska3-depleted HeLa Tet-On cells stably expressing mCherry-H2B, and then chromosome dynamics was monitored by time-lapse microscopy. As shown in Figure 4, A and B, mock cells spent an average of 48 min from nuclear envelope breakdown (NEB) to anaphase onset. Depletion of Ska3 gave rise to two major groups of cellsࣧ one with prolonged metaphase followed by anaphase onset and the other with prolonged metaphase followed by cohesion fatigue and cell death. We quantified mitotic duration (from NEB to anaphase onset or from NEB throughout cohesion fatigue to cell death) that these two groups of cells spent and found an average of 307 min. Expression of GFP-Ska3 WT largely rescued the defects caused by Ska3 depletion and significantly reduced the mitotic duration with an average of 55 min, which exhibited no significant difference from mock cells. Strikingly, expression of GFP-Ska3 6A almost failed to rescue the delay caused by Ska3 depletion with an average mitotic duration of 234 min. As Ska3 6A completely lost its kinetochore localization (Figure 2), the above results strongly support the notion that Ska localization to kinetochore is crucial for its functions in chromosome segregation. These results also reveal a decisive role of Cdk1 phosphorylation in activating Ska functions. In addition, we also found that while phosphomimetic GFP-Ska3 6D (150 min) cells exhibited a significantly shorter mitotic duration than cells expressing 6A (234 min), it did so poorly in comparison to WT (55 min) (Figurs 4, A and B), suggesting that Ska3 6D is not a good phosphomimetic in cells. The underlying reason will be discussed in the Discussion.
FIGURE 4:

Cdk1 phosphorylation in Ska3 is essential for chromosome segregation. (A, C) HeLa Tet-On cells stably expressing H2B-mCherry were cotransfected with siSka3 and vectors (V) or plasmids containing GFP-Ska3 WT, 2A (C), 4A2 (C), 4A3 (C), 6A (A), or 6D (A). Time-lapse microscopic analysis was performed. Scale bars, 5 μm. (B, D) Quantification of the duration from NEB to anaphase onset in A and C. At least 100 mitotic cells in A and 80 mitotic cells in C were analyzed for each condition. Average and SD were shown in lines. *P < 0.05, **P < 0.01; ****P < 0.0001; n.s. denotes no significance. Scale bars, 5 μm. (E) Cdk1 phosphorylation in Ska3 is required for stability of kinetochore–microtubule attachments. siSka3-treated HeLa Tet-On cells transfected with vectors or plasmids containing GFP-Ska3 WT, 2A, 4A2, or 6A were treated with MG132 for 1 h after 9 h release from thymidine, incubated on ice for 5 min, and then subject to staining with DAPI and the indicated antibodies. Representative images are shown here. (F) Quantification of microtubule intensity in E normalized to that of DNA. At least 15 mitotic cells (10 microtubules per cell) were analyzed for each condition. Average and SD are shown in lines. **P < 0.01; ****P < 0.0001; n.s. denotes no significance.
We also examined chromosome segregation in siSka3-treated cells expressing GFP-Ska3, 2A, 4A2, and 4A3 (Figure 4, C and D). Consistent with our previous findings (Zhang et al., 2017), expression of GFP-Ska3 2A showed a mild delay with an average mitotic duration of 82 min. Cells expressing GFP-Ska3 4A that includes the mutations of T358A and T360A were also delayed in chromosome segregation with an average mitotic duration of 154 min. Strikingly, expression of GFP-Ska3 4A3 that excludes the mutations of Thr358A and Thr360A did not delay chromosome segregation with an average mitotic duration of 70 min, compared with GFP-Ska3 WT (64 min). Again, these results support that Thr358 and Thr360 are the major sites contributing to Ska functions. Of note, cells with endogenous Ska3 depletion or expressing GFP-Ska3 6A only exhibited a 5-min delay from NEB to metaphase compared with mock cells (Supplemental Figure S5), suggesting that the significant delay in our experiments is mainly derived from metaphase-to-anaphase transition (Figures 4, A–D) and thus the Ska may be dispensable for chromosome congression to metaphase plates from initial attachments, which is consistent with the previous findings (Daum et al., 2009; Auckland et al., 2017).
Stability of kinetochore–microtubule attachments is determined by multisite Cdk1 phosphorylation on Ska3
Ska depletion has been shown to significantly decrease the stability of kinetochore–microtubule attachments (Gaitanos et al., 2009; Raaijmakers et al., 2009; Zhang et al., 2017). As Ska localizes on both microtubules and kinetochores, we wanted to know which pool of Ska could be more important for the stability of kinetochore–microtubule attachments. We previously demonstrated that the Ska3 2A (Thr358A Thr360A) mutant that was localized less to kinetochores and normally to spindle microtubules exhibited a similar microtubule cold sensitivity as WT (Zhang et al., 2017). The result seemingly suggested that the microtubule pool of Ska might play an important role in promoting the stability of kinetochore–microtubule attachments (Zhang et al., 2018). However, the Ska3 2A mutant did not complete lose its localization from kinetochores, compromising the above notion. To clarify it, we examined the microtubule cold sensitivity for cells expressing the Ska3 6A mutant that completely lost its kinetochore localization. As a comparison, the 2A and 4A2 mutants that retained partial kinetochore localization were also included. Consistently, Ska3 depletion significantly increased the microtubule cold sensitivity (Figure 4, E and F). Expression of GFP-Ska3 WT and 2A largely rescued the sensitivity and no significant difference between WT and 2A was observed, which is consistent with our previous findings. Expression of GFP-Ska3 4A2 also rescued the cold sensitivity but at a reduced level compared with WT. Strikingly, expression of GFP-Ska3 6A did not rescue the cold sensitivity at all. Considering that the 6A mutant completely lost its kinetochore localization but retained normally on microtubules, the above observations strongly indicate that the Cdk1 phosphorylation-dependent kinetochore pool of Ska is the major determinant for the kinetochore–microtubule stability. The full rescue of microtubule cold sensitivity by Ska3 2A might suggest that there is a threshold in the Ska levels on kinetochores, above which kinetochore–microtubule attachments become much more stable. Alternatively, the implemented assay here may not be sensitive enough to detect the moderate defects caused by Ska3 2A.
Multisite Cdk1 phosphorylation on Ska3 promotes Ska binding to the Ndc80-Nuf2 complex in vitro
Using recombinant proteins of the Ska3 C-terminal region (141-412), we have found that Cdk1 phosphorylation is able to promote Ska3 binding to Ndc80C in vitro (Zhang et al., 2017); however, it was not clear whether this would be the case in the context of the full-length Ska. To test this, we reconstructed and purified the full-length Ska and the GST-Nuf2-Ndc80 complex and performed in vitro Cdk1 phosphorylation followed by GST–pull-down assays. As shown in Figure 5A, an interaction was detected between the Ska and the GST-Nuf2-Ndc80 even without Cdk1 treatment. This basal interaction does not require Cdk1 phosphorylation, which was also demonstrated in an in vitro cross-linking experiment (Helgeson et al., 2018), and its nature is not quite understood. Cdk1 treatment significantly enhanced the interaction between Ska WT and GST-Nuf2-Ndc80. Treatment of RO3306, a potent Cdk1 inhibitor, completely abolished the increased binding between these two complexes, suggesting that the increased binding is Cdk1 phosphorylation-dependent. As a comparison, we also examined the interaction between the Ska 6A and the GST-Nuf2-Ndc80 complexes under the same conditions. Again, the basal interaction was detected as well between these two complexes (Figure 5A). Neither Cdk1 treatment nor Cdk1 together with RO3306 treatment altered the interaction between Ska 6A and GST-Nfu2-Ndc80. Western blots using our homemade phospho-specific antibody against Thr358 and Thr360 showed that Cdk1 kinase was effective in our experiments (Zhang et al., 2017). Taken the above results together, we conclude that Cdk1 phosphorylation on the six sites in Ska3 promotes Ska binding to Ndc80C.
FIGURE 5:
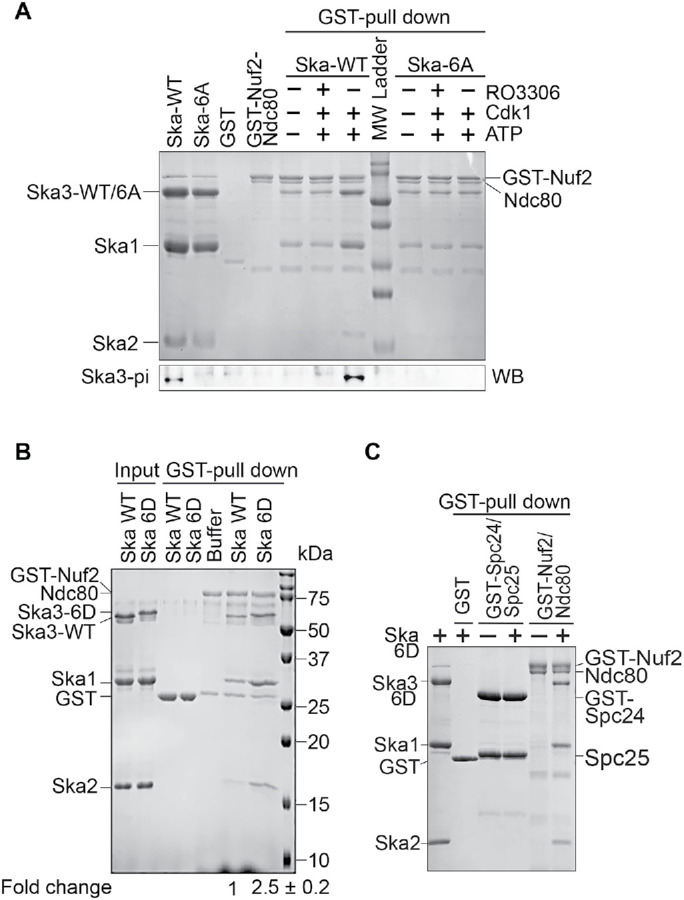
Cdk1 phosphorylation on Ska3 promotes the formation of a sable Ska–Ndc80 macro-complex. (A) Cdk1 phosphorylation promotes Ska binding to Ndc80C. The Ska1-Ska2-Ska3 WT or 6A complex treated with Cdk1/Cyclin B1 with or without RO3306 were mixed with the GST-Nuf2-Ndc80 complex and GST beads. The bead-bound proteins were resolved with SDS–PAGE and then stained with Coomassie Blue or blotted with the indicated antibody. (B) The Ska 6D complex binds to Ndc80C stronger than the WT complex. The Ska1-Ska2-Ska3 WT or 6D complex was mixed with the GST-Nuf2-Ndc80 complex and GST beads. The bead-bound proteins were resolved with SDS–PAGE and then stained with Coomassie Blue. The binding activity in fold change was derived from the amount of pelleted Ska2 by GST-Nuf2-Ndc80 sequentially normalized to the ones of input Ska2 and pelleted GST-Nuf2-Ndc80. The average and SD were calculated from two independent experiments and are shown in the bottom panel. (C) Spc24 and Spc25 are dispensable for Ska binding. The Ska1-Ska2-Ska3 6D complex was mixed with the GST-Nuf2-Ndc80 or GST-Spc24-Spc25 complex and GST beads. The bead-bound proteins were resolved with SDS–PAGE and then stained with Coomassie Blue.
As Ska3 6D partially mimicked the behavior of Ska3 WT in kinetochore localization (Figure 2), we wanted to know if the Ska containing this phosphomimetic Ska3 mutant (Ska 6D) could as well exhibit enhanced binding to Ndc80C. To test this, we examined the interaction between the Ska 6D and the GST-Nuf2-Ndc80 complexes using GST–pull-down assays. Consistent with our previous results, the similar basal interaction was again detected between the Ska WT complex and the GST-Nuf2-Ndc80 (Figure 5B). Interestingly, an enhanced interaction (about 2.5-fold increase) between the Ska 6D and the GST-Nuf2-Ndc80 complexes was also observed, further confirming the phospho-dependent interaction between Ska and Ndc80C. As Ndc80C contains the other two subunits Spc24 and Spc25, we then tested if these two subunits could also interact with the Ska 6D complex. We found that the GST-Spc24-Spc25 complex did not bind the Ska 6D complex at all (Figure 5C), suggesting that the subunits Ndc80 and Nuf2 in Ndc80C are mainly responsible for the interaction with Ska.
The Ska 6D complex forms a stable macro-complex with the Ndc80-Nuf2 complex in vitro
Although we have shown that Cdk1 phosphorylation significantly enhanced the interaction between the Ska and the Ndc80 complexes, it is tempting to know whether they could form a stable macro-complex in vitro, and if so, could Cdk1 phosphorylation be the decisive factor for the formation of this macro-complex? To overcome the potential heterogeneity of Ska3 phosphorylation by Cdk1, we utilized the phosphomimetic Ska3 6D mutant as a substitute for phosphorylated Ska3. Because Spc24 and Spc25 are dispensable for Ska binding (Figure 5C), only Nuf2-Ndc80 complexes were applied in our in vitro reconstitution experiments. We found that the Nuf2-Ndc80 complex was eluted at a high molecular weight, which was in the middle of 669 and 440 kDa (Supplemental Figure S6A). Both Ska WT and 6D complexes were also eluted at high molecular weights, slightly more than 440 kDa (Supplemental Figure S6, B and C). The mixture of the Ska 6D and Nuf2-Ndc80 complexes was eluted as a major peak appearing at a molecular weight slightly less than 440 kDa (Figure 6A). Strikingly, SDS–PAGE analysis on the major peak demonstrated that the Ska 6D and Nuf2-Ndc80 complexes were perfectly coeluted, revealing the formation of a stable macro-complex. These findings are consistent with a very recent study showing that Cdk1 treatment is able to promote the formation of a stable Ska–Ndc80 macro-complex in vitro (Huis In ‘t Veld et al., 2019). In contrast, the mixture of the Ska WT and Nuf2-Ndc80 complexes was eluted as two major peaks appearing at the molecular weights that were slightly higher and lower than 440 kDa (Figure 6B). SDS–PAGE analysis on these two major peaks demonstrated that the Ska WT and Nuf2-Ndc80 complexes were not coeluted well with each other. All these interesting observations strongly suggest that Cdk1 phosphorylation is the decisive factor for the formation of the stable Ska–Ndc80 macro-complex in vitro. Thus, to our knowledge, it is the first time successfully reconstituting this macro-complex using phosphomimetic mutants. In addition, the Ska 6D complex was not coeluted with the Ndc80 Bosai that lacks the Ndc80 internal loop and the flanking regions (Figure 6C and Supplemental Figure S6D), although it did so with the full-length Nuf2-Ndc80 complex, further confirming the previous findings that the Ndc80 internal loop together with the flanking regions contains main binding sties for the Ska (Zhang et al., 2012, 2017). Interestingly, the Ndc80 loop is well conserved across species and also responsible for recruiting other important factors (Hsu and Toda, 2011; Maure et al., 2011; Varma et al., 2012; Tang et al., 2013;). Notably, a very recent study further demonstrated that the flanking regions of the Ndc80 internal loop rather than the loop itself plays a more important role in Ska binding in vitro (Huis In ‘t Veld et al., 2019).
FIGURE 6:

A stable macro-complex is formed by the Ska 6D and Ndc80-Nuf2 complexes in vitro. (A–C) The Ska 6D, not WT complex, forms a stable macro-complex with the full-length Ndc80C, not Bonsai. Size-exclusion chromatographic analysis on the mixer of the Ska1-Ska2-Ska3 6D and Nuf2-Ndc80 complexes (A), or the one of the Ska1-Ska2-Ska3 WT and Nuf2-Ndc80 complexes (B), or the one of the Ska1-Ska2-Ska3 6D and the Ndc80 Bosai (C). The thicker lines marked the eluted fractions that were analyzed by SDS–PAGE. The arrows indicate the peaks of the analyzed complexes.
DISCUSSION
The Ska localizes to both kinetochores and microtubules. Our previous and current studies have provided strong evidence to support that Ska recruitment to kinetochores is essential for its functions in chromosome segregation (Zhang et al., 2017). Especially, in this study, we isolated a Ska mutant (Ska3 6A) that completely loses kinetochore localization and does not support Ska functions at all, suggesting that loading Ska onto kinetochores is the only way to enable Ska functions in chromosome segregation. As these sites are phosphorylated by Cdk1, our findings also suggest that Cdk1 phosphorylation is decisive for activating Ska functions.
How is the Ska recruited to kinetochores? Our previous and current findings demonstrate that Cdk1 phosphorylation-enabled Ska–Ndc80 binding is required for docking Ska to kinetochores. Recent studies indicated that the Ndc80 tail and/or the calponin-homology domain facilitate Ska recruitment to kinetochores (Cheerambathur et al., 2017; Janczyk et al., 2017). In addition, our current study indicates that spindle microtubules also promote Ska recruitment to kinetochores. Thus, multiple mechanisms may coexist to recruit Ska to kinetochores. As the binding between Cdk1-phosphorylated Ska3 and the Ndc80 loop with its flanking regions is sufficient for forming a stable Ska–Ndc80 macro-complex in vitro, and it is so predominant for Ska localization at kinetochores in cells, we believe that spindle microtubules and the Ndc80 N-terminal tail and/or the calponin-homology domain play regulatory roles in Ska recruitment to kinetochores. We therefore propose a model to explain how the Ska is recruited to kinetochores, in which spindle microtubules facilitate the delivery of Ska to the kinetochore–microtubule interface, and then Ska binds to Ndc80 in a multisite Cdk1 phosphorylation-dependent manner. Based on this model, one predicts that perturbations on Ska–microtubule interactions could affect Ska localization to kinetochores. Actually, mutations on Ska1 and depletion of other microtubule-interacting proteins, which both impair Ska–microtubule interactions, have been shown to moderately decrease Ska at kinetochores (Abad et al., 2016; Thomas et al., 2016).
Why is multisite phosphorylation needed? The Ska starts to accumulate at kinetochores in prophase, peaks at metaphase, and disappears at middle anaphase (Hanisch et al., 2006; Auckland et al., 2017), suggesting that the binding affinity of Ska to kinetochores may increase with mitotic progression. Multisite phosphorylation may provide such regulation. In support of this idea, we found that the state of Cdk1 phosphorylation on Ska3 is positively correlated with the robustness of Ska3 localization to kinetochores. Although mutations on all six Cdk1 sites abolished Ska localization to kinetochores and its functions in chromosome segregation, their contributions seem to be different. Mutations (2A and 4A1/2) that include the two sites Thr358 and Thr360 are always associated with the phenotypes of decreased kinetochore localization and prolonged mitotic progression, whereas mutations (4A3) that exclude Thr358 and Thr360 resulted in no detectable defects in kinetochore localization and normal mitotic progression. Thus, phosphorylation on the two sites, Thr358 and Thr360, likely lays a foundation for Ska localization to kinetochores, and the additional phosphorylation on the other sites could further enhance its kinetochore recruitment. In spite of this, some inconsistencies were still observed between Ska kinetochore localization and functions. Thus, the precise contributions of the four additional phospho-sites to Ska localization and functions are not fully understood. Of note, although the Ska 6D complex bound to Ndc80C stronger than WT, it exhibited partial defects in kinetochore localization and chromosome segregation, suggesting that Ska3 6D is not a perfect mimic to phosphorylated Ska3 in cells. Alternatively, as a recent study showed that dynamic phosphorylation–dephosphorylation cycles on Ska3 are important for proper Ska functions (Maciejowski et al., 2017), it is possible that similar phosphorylation–dephosphorylation cycles are also critical for proper Ska functions in chromosome segregation (Redli et al., 2016; Sivakumar and Gorbsky, 2017). It is worth mentioning here that at early anaphase when Cdk1 activity has significantly declined, the Ska is still robustly retained at kinetochores (Hanisch et al., 2006), suggesting that either the Cdk1 phosphorylation-dependent Ska–Ndc80 binding is preserved or other unknown mechanisms tether Ska onto kinetochores. It would be interesting to test these possibilities in future.
Our in vitro reconstitution results showed the phosphomimetic Ska 6D complex is able to form a stable macro-complex with the Ndc80-Nuf2 complex, suggesting that a tight Ska–Ndc80 interaction can be formed in cells albeit its existence might be transient. As the Ndc80 internal loop together with its flanking regions are mainly responsible for Ska binding and the loop confers structural flexibility to Ndc80C (Wang et al., 2008; Varma et al., 2012; Zhang et al., 2012, 2017), such a tight Ska–Ndc80 interaction might be able to alter the conformation of Ndc80C, thus promoting proper kinetochore–microtubule interactions. In this sense, the Ska not only functions as a scaffold to bring PP1 or other factors in proximity to the kinetochore–microtubule interface (Sivakumar et al., 2016) but also might serve as a structural modifier (Zhang et al., 2018). In the future, it is tempting to test if this could be the case; and if so, how does it affect the behavior of Ndc80C on dynamic microtubules (Ye and Maresca, 2013)? In budding yeast, the DASH/Dam1 complex, the functional Ska homolog, contains 10 subunits and shares no structural and sequence similarity to Ska. The Dam1 complex was reconstituted in vitro and its structure was solved using cryo-EM (Jenni and Harrison, 2018). It was also shown that the Ndc80 complex bridges two Dam1 complex rings (Kim et al., 2017). Although integrating the structure of the Ndc80 complex and published interaction data into that of the Dam1 complex yielded an interesting molecular view of kinetochore–microtubule interactions (Jenni and Harrison, 2018), how these complexes indeed interact with each other still remains unclear. In the future, by solving the structure of the Ska (6D)-Ndc80 macro-complex using cryo-EM, we would gain profound insights into how Cdk1 phosphorylation on Ska3 promotes the Ska–Ndc80 interaction at the atomic level.
In summary, our findings unveil the essential role of multisite Cdk1 phosphorylation-enabled Ska–Ndc80 macro-complexes in controlling Ska localization and functions during mitosis and also reveal that coordinated actions of microtubules and kinases load the Ska to kinetochores.
MATERIALS AND METHODS
Mammalian cell culture, siRNAs, and transfection
HeLa Tet-On cells (Clontech) were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum and 10 mM L-glutamine. To arrest cells at G1/S, cells were incubated in medium containing 2 mM thymidine (Sigma) for 16 h. Nocodazole, MG132, and RO3306 were purchased from Sigma-Aldrich.
Plasmid transfection was done using the Effectene reagent (Qiagen) according to the manufacturer’s protocols. For H2B-mCherry stable cells, HeLa Tet-On cells were transfected with pIRES vectors encoding H2B-mCherry and selected with 0.4 μg ml−1 puromycin (Invitrogen). For Myc-Ska3 stable cells, HeLa Tet-On cells were transfected with pTRE2 vectors encoding RNAi-resistant MYC-Ska3 and selected with 350 μg ml−1 hygromycin (Invitrogen). The surviving clones were screened for expression of the desired proteins in the presence of 1 μg ml−1 doxycycline (Invitrogen). Expression of Myc-Ska3 was also induced with 1 μg ml−1 doxycycline in the subsequent experiments.
For RNAi experiments, the siRNA oligonucleotides were purchased from Thermo Scientific. HeLa cells were transfected using Lipofectamine RNAiMax (Invitrogen) and analyzed at 32–40 h after transfection. The sequences of the siRNAs used in this study are Ska3 siRNA, GGAUAUAGUCCACGUGUCA (D-015700-17, MQ-015700-01-0002, Thermo Scientific).
Antibodies, immunoblotting, and immunoprecipitation
Antibodies used in this study are listed as the following: anti-centromere antibody (ACA or CREST-ImmunoVision, HCT-0100), anti-Tubulin (Thermo Scientific, 62204), anti-Actin (Thermo Scientific, MA5-11869) and anti-Ska3 (Bethyl, A304-215A), and anti-GFP (Abcam, ab1218; Aves, GFP-1010). Anti-pSka3 and anti-APC3 antibodies were made in-house as described previously (Zhang et al., 2017).
Antibody dilution for immunoblotting was often 1:1000 unless specified.
Immunoprecipitation was performed as follows. HeLa Tet-On cells stably expressing Myc-Ska3 cells were lysed with lysis buffer (25 mM Tris–HCl at pH 7.5, 50 mM NaCl, 5 mM MgCl2, 0.1% NP-40, 1 mM DTT, 0.5 μM okadaic acid, 5 mM NaF, 0.3 mM Na3VO4, and 100 U ml−1 Turbo-nuclease (Accelagen). After a 1-h incubation on ice and then a 10-min incubation at 37 °C, the lysate was cleared by centrifugation for 15 min at 4 °C at 20,817 × g. The supernatant was incubated with the antibody beads for 2 h at 4 °C. The Myc-antibody coupled beads (Thermo Scientific, 20168) were washed four times with wash buffer (25 mM Tris–HCl at pH 7.5, 50 mM NaCl, 5 mM MgCl2, 0.1% NP-40, 1 mM DTT, 0.5 μM okadaic acid, 5 mM NaF, 0.3 mM Na3VO4). The proteins bound to the beads were dissolved in SDS sample buffer, resolved by SDS–PAGE, and subject to mass-spectrometric analysis.
Immunofluorescence and chromosome spread
For regular immunostaining in Figures 2, A and C, 3, A and D, and 4E, and Supplemental Figures S2A, S3A, and S4, A and F, mitotic cells were collected and then spun with a Shandon Cytospin centrifuge. Cells were immediately fixed with 4% ice-cold paraformaldehyde for 4 min and then extracted with ice-cold phosphate-buffered saline (PBS) containing 0.2% Triton X-100 for 2 min. Cells were washed with PBS containing 0.1% Triton X-100 and then incubated with primary antibodies (1:1000 dilution) overnight at 4 °C. After washing with PBS containing 0.1% Triton X-100, cells were incubated at room temperature for 1 h with the appropriate secondary antibodies conjugated to fluorophores (Molecular Probes, 1:1000 dilution). After incubation, cells were washed again with PBS containing 0.1% Triton X-100, stained with 1 μg ml−1 DAPI, and mounted with Vectashield.
For chromosome spreads and immunostaining in Figures 3F, S2C, S4D, S4E and S4F, mitotic cells were swelled in a prewarmed hypotonic solution containing 50 or 75 mM KCl for 15 min at 37 °C and then spun onto slides with a Shandon Cytospin centrifuge. Cells were first extracted with ice-cold PBS containing 0.2% Triton X-100 for 2 min and then fixed in 4% ice-cold paraformaldehyde for 4 min. Cells were washed with PBS and then incubated with primary antibodies (1:1000 dilution) overnight at 4 °C. Cells were washed with PBS containing 0.1% Triton X-100 and incubated at room temperature for 1 h with the appropriate secondary antibodies conjugated to fluorophores (Molecular Probes, 1:1000 dilution). After incubation, cells were washed again with PBS containing 0.1% Triton X-100, stained with 1 μg ml−1 DAPI, and mounted with Vectashield.
The images were taken with a Nikon confocal microscope with a 60× objective. Image processing was carried out with ImageJ and Adobe Photoshop. Quantification was carried out with ImageJ.
Microtubule cold-sensitivity assay
For microtubule cold-sensitivity assay, mitotic cells were collected and then spun onto with a Shandon Cytospin centrifuge. Cells on slides were treated with ice for 5 min and then cells were immediately fixed with methanol precooled at –20°C for 5 min, followed by extraction with ice-cold PBS containing 0.2% Triton X-100 for 2 min. Cells were washed with PBS containing 0.1% Triton X-100 and then incubated with primary antibodies (1:1000 dilution) overnight at 4 °C. Cells were washed with PBS containing 0.1% Triton X-100 and then incubated at room temperature for 1 h with the appropriate secondary antibodies conjugated to fluorophores (Molecular Probes, 1:1000 dilution). After incubation, cells were washed again with PBS containing 0.1% Triton X-100, stained with 1 μg ml−1 DAPI, and mounted with Vectashield.
The images were taken by a Nikon confocal microscope with a 60× objective. Image processing was carried out with ImageJ and Adobe Photoshop. Quantification was carried out with ImageJ.
Time-lapse microscopy
HeLa-Teton cells stably expressing H2B-mCherry were treated as indicated (Figure 4), and then long-term imaging was performed. Images were collected every 6 min for 12–15 h using a Nikon confocal microscope equipped with an environment chamber that controls temperature and CO2, and 20× objective. Image panels displaying the elapsed time between consecutive frames were assembled using the software designed for Nikon confocal microscope. The time taken for each cell to progress from NEB to anaphase onset (chromatid separation) was calculated in minutes and plotted in GraphPad Prism. The experiments in Figure 4 were repeated at least two times and the results were highly reproducible. Quantification was performed based on the results from a single experiment. Average and SD were calculated using GraphPad Prism.
For live-cell imaging in Figure 2, E and G, GFP-Ska3 WT, 2A, 4A-3, 6A, and 6D were expressed in HeLa Tet-On cells. After being treated with MG132 for 1 h, cells were immediately subjected to imaging using a Nikon confocal microscope. Images were processed in ImageJ and Adobe Photoshop.
Protein purification
The full-length Ska3 was subcloned into pGEX-6p-1(GE Healthcare) vectors with an N-terminal GST tag with a 3C-cleavage site. The full-length Ska1-Ska2 were subcloned into the modified pET vector (Novagen) with or without 6× His tag. All Ska3 mutants were generated with standard two-step methods using PCR and Dpn1 and confirmed by DNA sequencing. For full-length Ska, GST-Ska3 was expressed in Escherichia coli BL21 (DE3) cells cultured in Luria-Bertani medium. Untagged or 6His-tagged Ska1-Ska2 was expressed in E. coli BL21 (DE3) cells cultured in Terrific Broth medium. All cells were induced by 0.2 mM IPTG at 16°C overnight. Two sorts of cells were harvested and mixed for copurified and then disrupted by high-pressure homogenizer (ATX Engineering) in the PBS buffer. The cell lysates were clarified by centrifuged at 35,000 × g for 30 min at 4°C. The supernatant was added GST agarose beads (GE Healthcare) and rotated at 8°C for 1.5 h, and the protein-bound beads were then transferred into gravity columns, washed with PBS extensively, and treated with 3C protease overnight at 4°C to remove the GST tags. After being dialyzed against 25 mM Tris pH 8.0, 50 mM NaCl and 1 mM DTT, the harvested Ska complex was further purified by anion exchange chromatography (HiTrap Q FF, GE Healthcare) and gel filtration chromatography (Superdex 200 10/300 GL, GE Healthcare). The purified Ska was finally concentrated to 15–20 mg/ml in buffer containing 20 mM Tris, pH 8.0, 150 mM NaCl, and 1 mM DTT and stored at –80°C for later use.
GST-Nuf2-Hec1 complexes, GST-Spc24-Spc25 complexes, and Ndc80 Bosai were purified as previously described (Zhang et al., 2017). To remove GST tags, cell lysates of these complexes were first treated with GST agarose beads (GE Healthcare), and then the protein-bound beads were transferred into gravity columns and treated with 3C protease overnight at 4°C. The eluted complexes were subjected to gel filtration chromatography (Superdex 200 10/300 GL, GE Healthcare) for further purification.
In vitro binding and kinases assays
In Figure 5, B and C, 5–10 µg GST fusion protein (GST-Nuf2:Hec1/GST-Spc24:Spc25) in buffer containing 20 mM Tris, pH 8.0, 150 mM NaCl, and 1 mM DTT was bound to previously equilibrated GST beads at 8°C for 1 h, then incubated with Ska or mutants proteins at 8°C for 1.5 h, and washed three times with buffer containing 20 mM Tris, pH 8.0, 300 mM NaCl, 1 mM DTT, and 0.02% TritonX-100. The proteins retained on the beads were resolved with SDS–PAGE and stained with Coomassie Brilliant Blue. GST proteins were also included as control.
In Figure 5A, purified Skaes (WT and 6A) were treated with CDK1-CyclinB1 plus 10 mM ATP with or without 100 µM RO3306 in kinase buffer containing 20 mM Tris, pH 8.0, 50 mM NaCl, 10 mM MgCl2, and 1 mM DTT at room temperature for 1 h. Then, Ska complexes were incubated with GST beads prebound with the GST-Nuf2-Hec1 complex for 1 h at 8°C. After being washed with kinase buffer containing 0.02%Trition, the proteins bound with beads were resolved by SDS–PAGE stained with Coomassie Brilliant Blue.
Quantification and statistical analysis
ImageJ was used to obtain numeric intensities of experimental subjects under investigation. In the experiments shown in Figures 2, A and C, and 3, A, D, and F; and Supplemental Figures S2, A and C, S3A, S4, A and D–F, six kinetochores were randomly selected from each cell. A mask was generated to mark centromeres on the basis of ACA staining in the projected image. After background subtraction, the intensities of GFP-Ska3 and ACA fluorescence signals within the mask were obtained in number. Relative intensity in Figures 2, B and, D, and 3, B, E, and G; and Supplemental Figures S2B, S3B, and S4, B and D–F was derived from the intensity of GFP-Ska3 signals normalized to the one of ACA signals and plotted with the GraphPad Prizm software. All experiments were repeated at least twice or three times.
For quantification of H3pS10 intensity on kinetochores in Figure 3A and Supplemental Figure S4A, a mask was generated to mark the whole DNA mass, After background subtraction, the intensities of H3pS10 and DAPI fluorescence signals within the mask were obtained in number, Relative intensity in Figure 3B and Supplemental Figure S4B was derived from the intensity of H3pS10 signals normalized to the one of DAPI signals and plotted with the GraphPad Prizm software.
For quantification of GFP-Ska3 intensity on kinetochores and microtubules in Figure 2, E and G, masks were generated to mark the cytoplasm, kinetochores, and microtubules. After background subtraction, the intensities of GFP-Ska3 fluorescence signals within the masks were obtained in number. Relative intensity in Figure 2, F and H was derived from the intensity of GFP-Ska3 signals on kinetochores or microtubules normalized to one of the GFP-Ska3 signals in cytoplasm and plotted with the GraphPad Prism software.
For quantification of microtubule intensity in Figure 4E, a mask was generated to mark the nucleus within a cell. After background subtraction, the intensities of microtubules and DAPI fluorescence signals within the mask were obtained in number. Relative intensity in Figure 4F was derived from the intensity of microtubule signals normalized to the one of DAPI signals and plotted with the GraphPad Prism software.
Quantification was usually performed based on the results from a single experiment unless specified. Differences were assessed using ANOVA followed by pairwise comparisons using Tukey’s test. All samples analyzed were included in quantification. Sample size was recorded in figures and their corresponding legends. No specific statistical methods were used to estimate sample size. No methods were used to determine whether the data met assumptions of the statistical approach. Please refer to Supplemental Table S1.
Supplementary Material
Acknowledgments
This work was supported by the Tulane startup funds and Carol Lavin Bernick Faculty Grants awarded to H.L., the Program of HUST Academic Frontier Youth Team (2018QYTD02) awarded to W.T., and the China Postdoctoral Science Foundation (2019M662588) awarded to LQ.H.. We also thank Dr. Iain Cheeseman (MIT) for the plasmids containing Ska1 and Ska2. Identification of Ska3 phosphorylation sites was performed by LSUHSC Proteomics Core facility.
Abbreviations used:
- NEB
nuclear envelop breakdown
- PBS
phosphate-buffered saline
- Ska
Ska complex.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-10-0569) on June 3, 2020.
REFERENCES
- Abad MA, Medina B, Santamaria A, Zou J, Plasberg-Hill C, Madhumalar A, Jayachandran U, Redli PM, Rappsilber J, Nigg EA, et al. (2014). Structural basis for microtubule recognition by the human kinetochore Ska complex. Nat Commun , 2964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abad MA, Zou J, Medina-Pritchard B, Nigg EA, Rappsilber J, Santamaria A, Jeyaprakash AA. (2016). Ska3 ensures timely mitotic progression by interacting directly with microtubules and Ska1 microtubule binding domain. Sci Rep , 34042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auckland P, Clarke NI, Royle SJ, McAinsh AD. (2017). Congressing kinetochores progressively load Ska complexes to prevent force-dependent detachment. J Cell Biol , 1623–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeszoermenyi A, Schmidt JC, Cheeseman IM, Oberer M, Wagner G, Arthanari H. (2014). Resonance assignments of the microtubule-binding domain of the C. elegans spindle and kinetochore-associated protein 1. Biomol NMR Assign , 275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YW, Jeyaprakash AA, Nigg EA, Santamaria A. (2012). Aurora B controls kinetochore-microtubule attachments by inhibiting Ska complex-KMN network interaction. J Cell Biol , 563–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheerambathur DK, Desai A. (2014). Linked in: formation and regulation of microtubule attachments during chromosome segregation. Curr Opin Cell Biol , 113–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheerambathur DK, Prevo B, Hattersley N, Lewellyn L, Corbett KD, Oegema K, Desai A. (2017). Dephosphorylation of the Ndc80 Tail Stabilizes Kinetochore-Microtubule Attachments via the Ska Complex. Dev Cell , 424–437 e424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum JR, Wren JD, Daniel JJ, Sivakumar S, McAvoy JN, Potapova TA, Gorbsky GJ. (2009). Ska3 is required for spindle checkpoint silencing and the maintenance of chromosome cohesion in mitosis. Curr Biol , 1467–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaitanos TN, Santamaria A, Jeyaprakash AA, Wang B, Conti E, Nigg EA. (2009). Stable kinetochore-microtubule interactions depend on the Ska complex and its new component Ska3/C13Orf3. EMBO J , 1442–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes GJ, Deluca JG. (2009). Connecting with Ska, a key complex at the kinetochore-microtubule interface. EMBO J , 1375–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanisch A, Sillje HH, Nigg EA. (2006). Timely anaphase onset requires a novel spindle and kinetochore complex comprising Ska1 and Ska2. EMBO J , 5504–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgeson LA, Zelter A, Riffle M, MacCoss MJ, Asbury CL, Davis TN. (2018). Human Ska complex and Ndc80 complex interact to form a load-bearing assembly that strengthens kinetochore-microtubule attachments. Proc Natl Acad Sci USA , 2740–2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu KS, Toda T. (2011). Ndc80 internal loop interacts with Dis1/TOG to ensure proper kinetochore-spindle attachment in fission yeast. Curr Biol , 214–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huis In ‘t Veld PJ, Volkov VA, Stender ID, Musacchio A, Dogterom M. (2019). Molecular determinants of the Ska-Ndc80 interaction and their influence on microtubule tracking and force-coupling. Elife . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janczyk PL, Skorupka KA, Tooley JG, Matson DR, Kestner CA, West T, Pornillos O, Stukenberg PT. (2017). Mechanism of Ska recruitment by Ndc80 complexes to kinetochores. Dev Cell , 438–449 e434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenni S, Harrison SC. (2018). Structure of the DASH/Dam1 complex shows its role at the yeast kinetochore-microtubule interface. Science , 552–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeyaprakash AA, Santamaria A, Jayachandran U, Chan YW, Benda C, Nigg EA, Conti E. (2012). Structural and functional organization of the Ska complex, a key component of the kinetochore-microtubule interface. Mol Cell , 274–286. [DOI] [PubMed] [Google Scholar]
- Kim JO, Zelter A, Umbreit NT, Bollozos A, Riffle M, Johnson R, MacCoss MJ, Asbury CL, Davis TN. (2017). The Ndc80 complex bridges two Dam1 complex rings. Elife . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Bachant J, Alcasabas AA, Wang Y, Qin J, Elledge SJ. (2002). The mitotic spindle is required for loading of the DASH complex onto the kinetochore. Genes Dev , 183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Drechsler H, Grundner-Culemann K, Ballister ER, Rodriguez-Rodriguez JA, Rodriguez-Bravo V, Jones MJK, Foley E, Lampson MA, Daub H, et al. (2017). Mps1 regulates kinetochore-microtubule attachment stability via the Ska Complex to ensure error-free chromosome segregation. Dev Cell , 143–156 e146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maure JF, Komoto S, Oku Y, Mino A, Pasqualato S, Natsume K, Clayton L, Musacchio A, Tanaka TU. (2011). The Ndc80 loop region facilitates formation of kinetochore attachment to the dynamic microtubule plus end. Curr Biol , 207–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monda JK, Whitney IP, Tarasovetc EV, Wilson-Kubalek E, Milligan RA, Grishchuk EL, Cheeseman IM. (2017). Microtubule tip tracking by the spindle and kinetochore protein Ska1 requires diverse tubulin-interacting surfaces. Curr Biol , 3666–3675 e3666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta S, Bukowski-Wills JC, Wood L, de Lima Alves F, Chen Z, Rappsilber J, Earnshaw WC. (2010). Proteomics of isolated mitotic chromosomes identifies the kinetochore protein Ska3/Rama1. Cold Spring Harb Symp Quant Biol , 433–438. [DOI] [PubMed] [Google Scholar]
- Raaijmakers JA, Tanenbaum ME, Maia AF, Medema RH. (2009). RAMA1 is a novel kinetochore protein involved in kinetochore-microtubule attachment. J Cell Sci , 2436–2445. [DOI] [PubMed] [Google Scholar]
- Redli PM, Gasic I, Meraldi P, Nigg EA, Santamaria A. (2016). The Ska complex promotes Aurora B activity to ensure chromosome biorientation. J Cell Biol , 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt JC, Arthanari H, Boeszoermenyi A, Dashkevich NM, Wilson-Kubalek EM, Monnier N, Markus M, Oberer M, Milligan RA, Bathe M, et al. (2012). The kinetochore-bound Ska1 complex tracks depolymerizing microtubules and binds to curved protofilaments. Dev Cell , 968–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumar S, Gorbsky GJ. (2017). Phosphatase-regulated recruitment of the spindle- and kinetochore-associated (Ska) complex to kinetochores. Biol Open , 1672–1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sivakumar S, Janczyk PL, Qu Q, Brautigam CA, Stukenberg PT, Yu H, Gorbsky GJ. (2016). The human SKA complex drives the metaphase-anaphase cell cycle transition by recruiting protein phosphatase 1 to kinetochores. Elife . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Mukae N, Dewar H, van Breugel M, James EK, Prescott AR, Antony C, Tanaka TU. (2005). Molecular mechanisms of kinetochore capture by spindle microtubules. Nature , 987–994. [DOI] [PubMed] [Google Scholar]
- Tang NH, Takada H, Hsu KS, Toda T. (2013). The internal loop of fission yeast Ndc80 binds Alp7/TACC-Alp14/TOG and ensures proper chromosome attachment. Mol Biol Cell , 1122–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves SS, An L, Hansen AS, Xie L, Darzacq X, Tjian R. (2016). A dynamic mode of mitotic bookmarking by transcription factors. Elife . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theis M, Slabicki M, Junqueira M, Paszkowski-Rogacz M, Sontheimer J, Kittler R, Heninger AK, Glatter T, Kruusmaa K, Poser I, et al. (2009). Comparative profiling identifies C13orf3 as a component of the Ska complex required for mammalian cell division. EMBO J , 1453–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GE, Bandopadhyay K, Sutradhar S, Renjith MR, Singh P, Gireesh KK, Simon S, Badarudeen B, Gupta H, Banerjee M, et al. (2016). EB1 regulates attachment of Ska1 with microtubules by forming extended structures on the microtubule lattice. Nat Commun , 11665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma D, Chandrasekaran S, Sundin LJ, Reidy KT, Wan X, Chasse DA, Nevis KR, DeLuca JG, Salmon ED, Cook JG. (2012). Recruitment of the human Cdt1 replication licensing protein by the loop domain of Hec1 is required for stable kinetochore-microtubule attachment. Nat Cell Biol , 593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma D, Salmon ED. (2012). The KMN protein network–chief conductors of the kinetochore orchestra. J Cell Sci , 5927–5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HW, Long S, Ciferri C, Westermann S, Drubin D, Barnes G, Nogales E. (2008). Architecture and flexibility of the yeast Ndc80 kinetochore complex. J Mol Biol , 894–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welburn JP, Grishchuk EL, Backer CB, Wilson-Kubalek EM, Yates JR, 3rd, Cheeseman IM. (2009). The human kinetochore Ska1 complex facilitates microtubule depolymerization-coupled motility. Dev Cell , 374–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye AA, Maresca TJ. (2013). Cell division: kinetochores SKAdaddle. Curr Biol , R122–R124. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Chen Y, Yang L, Liu H. (2018). Multitasking Ska in chromosome segregation: its distinct pools might specify various functions. Bioessays . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Sivakumar S, Chen Y, Gao H, Yang L, Yuan Z, Yu H, Liu H. (2017). Ska3 phosphorylated by Cdk1 binds Ndc80 and recruits Ska to kinetochores to promote mitotic progression. Curr Biol , 1477–1484.e1474. [DOI] [PubMed] [Google Scholar]
- Zhang G, Kelstrup CD, Hu XW, Kaas Hansen MJ, Singleton MR, Olsen JV, Nilsson J. (2012). The Ndc80 internal loop is required for recruitment of the Ska complex to establish end-on microtubule attachment to kinetochores. J Cell Sci , 3243–3253. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.