

Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder that is multifactorial in nature. Yet, despite being the most common form of dementia in the elderly, AD’s primary cause remains unknown. As such, there is currently little to offer AD patients as the vast majority of recently tested therapies have either failed in well-controlled clinical trials or inadequately treat AD. Recently, emerging preclinical and clinical evidence has associated the brain renin angiotensin system (RAS) to AD pathology. Accordingly, various components of the brain RAS were shown to be altered in AD patients and mouse models, including the angiotensin II type 1 (AT1R), angiotensin IV receptor (AT4R), and Mas receptors. Collectively, the changes observed within the RAS have been proposed to contribute to many of the neuropathological hallmarks of AD, including the neuronal, cognitive, and vascular dysfunctions. Accumulating evidence has additionally identified antihypertensive medications targeting the RAS, particularly angiotensin receptor blockers (ARBs) and angiotensin-converting enzyme inhibitors (ACEIs), to delay AD onset and progression. In this review, we will discuss the emergence of the RAS’s involvement in AD and highlight putative mechanisms of action underlying ARB’s beneficial effects that may explain their ability to modify the risk of developing AD or AD progression. The RAS may provide novel molecular targets for recovering memory pathways, cerebrovascular function, and other pathological landmarks of AD.
Keywords: Renin angiotensin system, Memory pathways, Cerebrovascular dysfunction, Angiotensin receptor blocker, AT2R, AT4R
Introduction
Alzheimer’s disease (AD) is a debilitating neurodegenerative disease afflicting memory, language, personality, cognition, and executive functioning, whereby patients’ gradual decline becomes severe enough to interfere with their daily activities. With more than 40 million individuals affected worldwide by dementia, and AD being the most common irreversible form of dementia, it has become a growing problem in the health sector due to both the disease’s devastating societal impact and estimated annual cost (Chang et al. 2015). Intriguingly, AD is thought to begin years before symptoms of dementia are present, providing an opportunity to delay and reduce AD prevalence. There remains no cure for AD; thus, identifying risk factors, discovering biological mechanisms, and developing intervention strategies to prevent, slow down, or stop the disease progression are essential research fields.
Dr. Alois Alzheimer first identified the classical behavioral and neuropathological features of AD in 1906 following post-mortem investigations of patient Auguste Deter, who had developed an unusual progressive dementia at age 51 (Goedert and Spillantini 2006). Utilizing Bielschowsky silver staining, Alzheimer observed tangles of fibrils (known today as neurofibrillary tangles (NFTs)) within the cytoplasm of degenerating neurons, widespread presence of tiny silver-stained deposits (senile plaques) scattered throughout the cortex (Goedert and Spillantini 2006), and identified cerebrovascular pathology that he deemed atherosclerosis of the brain (Maurer et al. 1997). The hallmark of this debilitating disease remains the gradual decline in cognitive function and progressive memory loss, disease components associated with AD’s synaptic dysfunction, synaptic loss, and neuronal death (Mucke et al. 2000). Areas of the brain predominately affected by AD are regions required for cognitive function and include the hippocampus, entorhinal and frontal cortices, and subcortical structures. To this day, AD is characterized by NFTs resulting from aggregation of hyperphosphorylated tau, increases in amyloid-β (Aβ) peptides and Aβ plaque formation, glial cell activation, oxidative stress, as well as metabolic and cerebrovascular dysregulation (Gorelick et al. 2011; Serrano-Pozo et al. 2011; Sweeney et al. 2018; Girouard and Iadecola 2006).
AD as a vascular disorder
The cerebrovascular component of AD has often been disregarded due to the distinction made between vascular dementia and AD pathology, a distinction that is no longer tenable (Iadecola 2013; Kisler et al. 2017). Indeed, vascular dysregulation was identified as the earliest biomarker in AD in a multimodality brain imaging study, with vascular dysregulation occurring prior to Aβ deposition, altered glucose metabolism, and cognitive dysfunctions in sporadic AD compared with controls (Iturria-Medina et al. 2016). Despite vascular pathology in AD being first identified in the early 1900s by Alois Alzheimer, research overlooked the cerebrovascular component of AD when the Aβ peptide was suggested to be the initiator of AD due to its presence in the parenchyma in the form of Aβ plaques and in the vasculature as cerebral amyloid angiopathy (CAA) (Iadecola 2013; Kisler et al. 2017; Iadecola 2004). Moreover, vascular alterations affecting both extra- and intracerebral vessels have been reported throughout the vascular architecture in AD and are thought to contribute to cerebrovascular dysfunction (Sweeney et al. 2018). Pericytes, endothelial cells (ECs), and smooth muscle cells (SMCs) undergo degenerative changes throughout the pathogenesis of AD, with vascular changes including EC damage, perivascular leakage, destruction of the cell membrane and cytoplasm, condensed nucleus and swollen mitochondria, basement membrane thickening, vacuolization, and vessel lumen distortions (Kalaria 1996). As such, the impact of cerebrovascular lesions, as well as structural and functional alterations of cerebral blood vessels observed in AD patients (Jellinger 2007), has re-instilled the importance of gaining a better understanding of cerebrovascular pathogenic mechanisms in AD.
The majority of demented patients display mixed AD and vascular pathology with several cerebrovascular alterations (Iadecola 2013; Jellinger 2007). Accumulating evidence supports the idea of a strong vascular contribution to sporadic AD. Studies have demonstrated reduced resting cerebral blood flow (CBF), lessened vasoreactivity and perfusion to activated brain regions (neurovascular coupling dysregulation) in AD patients (Girouard and Iadecola 2006; Iadecola 2013; Kisler et al. 2017; Iturria-Medina et al. 2016), as well as cerebrovascular lesions in 40–50% of clinically diagnosed AD patients (Schneider et al. 2007; Sonnen et al. 2007). Since perfusion deficits are present from an early preclinical phase and persist into the late stages of AD, a progressive hypoperfusion may impede brain structure, function, and result in cognitive deficits in advanced stages of the disease. Chronic brain hypoperfusion is thought to increase the energy demand in neurons, thus, initiating cellular and molecular changes that result in AD pathology (Iadecola 2004; Langbaum et al. 2009; Farkas and Luiten 2001).
The aforementioned alterations in AD cerebrovasculature are further detectable in individuals with mild cognitive impairment (MCI, the prodromal stage of AD) and those expressing the E4 allele of apolipoprotein E, the most consistent susceptibility gene linked with an increased risk of AD (Kisler et al. 2017; Poirier et al. 1993; Grothe et al. 2016). These observations lead to the vascular hypothesis of AD wherein vascular risk factors, altered homeostasis between CBF nutrient and oxygen delivery, and neuronal and glial metabolic demands, and are thought to play a pivotal role in the manifestation and progression of AD (Iadecola 2013; Kisler et al. 2017; Iadecola 2004). Chronic cerebral hypoperfusion was identified as a key mechanism contributing to AD pathology due to its suggested role in decreasing Aβ peptide clearance, inducing oxidative stress and EC dysfunction, and leading to a shortage of essential brain nutrients and contributing to neurotoxic aggregates thereby creating a threat to the brain homeostasis (Langbaum et al. 2009). This molecular cascade is further thought to play a key role in AD synaptic failure, neuronal dysregulation, and neurodegeneration (Sweeney et al. 2018; Iadecola 2004; Farkas and Luiten 2001).
In comparison with the genetic form of AD known as familial AD (FAD), more than 95% of AD cases are sporadic and have a later age of onset. While age is the greatest risk factor, AD is not a normal feature of aging and age alone is not sufficient to cause the disease. Indeed, cardiovascular diseases including hypertension, hypercholesterolemia, stroke, diabetes, and bad lifestyle habits are main risk factors that increase the probability of developing sporadic AD (Duron and Hanon 2008; de la Torre 2010), the latter being enhanced with each additional risk factor (Luchsinger et al. 2005). A large proportion of patients diagnosed with AD possess concurrent cerebrovascular diseases that are thought to promote the conversion of MCI to AD (Li et al. 2011) and worsen the cognitive impact of AD (Esiri et al. 1999). Epidemiological studies further suggest that an improved societal cardiovascular health may contribute to reduced AD incidence (Pase et al. 2017; Claassen 2015), a finding supported by the association between treatment of vascular risk factors with a reduced incidence of AD and a slower cognitive decline in AD patients (Girouard and Iadecola 2006; Iadecola 2013; Villapol and Saavedra 2015). Knowing the association between cardiovascular risk factors and AD, disease-modifying therapies addressing both neuronal and vascular dysfunctions in AD uphold a significant potential for its prevention and treatment.
Targeting cardiovascular diseases in AD therapy
AD and cardiovascular diseases share important cardiometabolic and lifestyle risk factors that occur in middle aged and elderly individuals. Findings from observational studies have associated several vascular risk factors with increased risk of late-life cognitive deficits and AD (de la Torre 2010; O'Brien and Markus 2014). Indeed, treatments of cardiovascular risk factors have been associated with reduced incidence of AD and cognitive decline in AD patients (Iadecola 2013; Villapol and Saavedra 2015). Since many cardiovascular diseases have well-established therapeutic interventions, they may provide insight on pathways to prevent or delay AD in aged individuals (de la Torre 2010; Khachaturian et al. 2006; Chiu et al. 2014). While statins and antidiabetic drugs have been considered to have benefits for AD patients, the best evidence seems to be related to antihypertensive medication. Hypertension (HT), defined by an elevated arterial blood pressure, is the leading risk factor for dementia and AD. HT causes thickening of the blood vessel wall, reduced vessel elasticity, narrowing of vessel lumen, EC dysfunction, and alters blood-brain-barrier (BBB) integrity (Skoog 2000). There seems to be a synergistic relationship between HT and AD pathological hallmarks, namely Aβ and NFTs, suggesting an ability for HT to accelerate AD pathophysiology (Cifuentes et al. 2015). Longitudinal and neuropathological studies have drawn a correlation between individuals with mid-life HT and AD (Khachaturian et al. 2006; Barnes and Yaffe 2011; Skoog and Gustafson 2006), with growing evidence suggesting that mid-hypertensive patients have a greater conversion rate to AD than healthy older adults (Barnes and Yaffe 2011; Skoog and Gustafson 2006). Midlife HT has also been associated with an increase of white matter lesions, smaller brain volumes, as well as a key risk factor for cognitive decline (Lane et al. 2019). The FINMONICA study associated higher systolic blood pressure in midlife to a significantly greater risk of developing MCI, followed by AD conversion. Correspondingly, the longitudinal Cache County Study of memory and aging identified elderly patients with mild-HT (140–159 mmHg systolic, 90–99 mmHg diastolic) as having a significantly higher conversion rate to AD (8.9%) than healthy elderly controls (1–2%) (Khachaturian et al. 2006). Likewise, in a systematic review and meta-analysis exploring the relationship between midlife HT and AD, midlife systolic HT (> 140 and > 160 mmHg) was associated with an increased risk of AD by 18 and 25%, respectively (Lennon et al. 2019). Knowing that HT is modifiable, its treatment represents an opportunity for the prevention of AD.
The renin angiotensin system (RAS) appears to be a key component in AD due to its role in blood pressure regulation and HT, as well as its involvement in numerous neuropathological hallmarks of AD, including AD’s progressive cognitive decline. For these reasons, the present review will discuss the potential for targeting components of the RAS in relation to memory, cerebrovascular and neuropathological hallmarks of AD, particularly addressing the beneficial effects of ARB treatment. In this respect, we seek to describe how the brain RAS may provide novel molecular targets for recovering memory pathways and cerebrovascular function in AD patients.
The renin angiotensin system
The RAS is well known for its involvement in regulation of water and electrolyte balance, systemic vascular resistance, and blood pressure control (Gebre et al. 2018). There is a complete independent RAS within the CNS (Jackson et al. 2018), as the BBB restricts peripheral RAS components from accessing most brain regions. The brain RAS is a complex biochemical pathway possessing all necessary precursors and enzymes required for the synthesis of its main biologically active peptides: angiotensin II (AngII), angiotensin (1-7), and angiotensin IV (AngIV) (Fig. 1) (Wright and Harding 2011). In the CNS, AngII has been found in astrocytes, microglia, and neurons and has a high binding affinity for two receptors, AngII type 1 (AT1R) and type 2 (AT2R) receptors, both mediating different effects (Fig. 1), described in the subsequent sections (Wright and Harding 2011). AngII is converted to either angiotensin (1-7) by ACE2, where it binds to MAS receptors, or angiotensin III, followed by AngIV by cleavage via aminopeptidases. AngIV is an endogenous hexapeptide, located in neurons, and is the last known active metabolite in the RAS. AngIV binds with low affinity to AT1Rs and AT2Rs (Bosnyak et al. 2011) and is highly selective to the AngIV receptor (AT4R), also known as insulin-regulated aminopeptidase (IRAP) (Fig. 1) (Albiston et al. 2001).
Fig. 1.

Schematic representation of the biological actions and the sites of action of pharmacological agents targeting the RAS. Losartan and one of its active metabolite EXP3174 are potent antagonists acting on AT1Rs; thereby, blocking the action of angiotensin II (AngII), while its second active metabolite EXP3179 mediates pleiotropic effects. Losartan treatment has been shown to rescue AD dysfunctions in AD mouse models. This restorative benefit may be mediated by AngII activation of AT2Rs or through its conversion into other active peptides, including angiotensin IV (AngIV) that binds to the AngIV receptor (AT4R, also known as insulin-regulated aminopeptidase (IRAP)) and angiotensin (1-7) that binds to Mas receptors. Pharmacological intervention of the RAS has improved our understanding of the mechanisms of action mediating losartan’s beneficial effects in AD
Angiotensin II type 1 receptors
AT1Rs are G protein-coupled receptors located on neurons, astrocytes, oligodendrocytes, and microglia within the cortex, hippocampus, hypothalamus, basal ganglia, and on cerebral blood vessels (Jackson et al. 2018). AT1R is a major activator of the NADPH-oxidase complex, leading to pro-inflammatory and pro-oxidative effects (Fig. 1) (Labandeira-Garcia et al. 2017). AT1R knockout mice exhibit smaller infarct size following MCA occlusion, benefits thought to be due to the receptors’ role in activating pro-inflammatory pathways and mediating cerebrovascular pathological growth (Nishimura et al. 2000). Activation of AT1Rs has also been associated with superoxide formation (Prusty et al. 2017), vasoconstriction, CBF regulation (Wright and Harding 2011; Stromberg et al. 1993), and cognitive impairment (Fig. 1) (Ahmed et al. 2018). AT1R blockers, known as ARBs, have attenuated cognitive impairment in a variety of diseases, including HT, AD, and stroke, identifying them as key players in cognitive impairment.
Angiotensin II type 2 receptors
AT2Rs are also G protein-coupled receptors, sharing 34% sequence homology with AT1Rs, and located on neurons, astrocytes, and microglia within the cortex, hippocampus, locus coeruleus, basal ganglia, and blood vessels (Jackson et al. 2018). AT2R activation is thought to counteract AT1R-mediated actions. AT2Rs play a role in mediating vasodilatation (Matavelli and Siragy 2015), with AT2R activation increasing production of cyclic guanosine monophosphate (cGMP), nitric oxide (NO), and bradykinin (Fig. 1) (Widdop et al. 2003; Siragy and Carey 1996). AT2R-mediated vasodilatory effects have been linked to cross-talk between AT1Rs and AT2Rs, whereby NO/cGMP activation has been shown to decrease AT1R-mediated vasoconstriction (Savoia et al. 2006), a mechanism associated with AT2R’s ability to regulate CBF (Fig. 1) (Matavelli and Siragy 2015). Activation of AT2Rs by the selective AT2R agonist compound 21 (C21) increased CBF in mice injected with Aβ1–40 (Jing et al. 2012) and demonstrated anti-inflammatory and antioxidant properties (Fig. 1) (Sampson et al. 2016), likely by reducing inflammation and reactive oxygen species (ROS) production (Goel et al. 2018). AT2Rs have also been suggested to play an important role in mediating cognitive function since C21 administration prevented cognitive deficits post-stroke (Ahmed et al. 2019), in aged hypertensive mice (Ahmed et al. 2018) and in mice injected with Aβ1–40 (Jing et al. 2012).
Angiotensin IV receptors
AngIV is distributed throughout the brain, predominately in structures important to cognitive function, including the hippocampus, neocortex, basal ganglia, amygdala, and blood vessels (Thomas and Mendelsohn 2003). AngIV has been shown to enhance cognition in rodents (Braszko et al. 1988; Benoist et al. 2011; Paris et al. 2013) and restore scopolamine-induced spatial working memory deficits (Fig. 1) (McCoy et al. 2013). Correspondingly, divalinal, an AT4R antagonist, produced deficits in spatial memory acquisition in rodents (Wilson et al. 2009). The AngIV peptide has also been related to CBF regulation (Kramar et al. 1998; Naveri et al. 1994) and vasodilatory responses (Fig. 1) (Kramar et al. 1998). Intriguingly, chronic blockade of AT1Rs has been suggested to favor conversion of AngII to AngIV (Braszko et al. 2006), allowing AT4Rs to exert positive effects on memory. The mechanism of AngIV remains under debate, but it is known to bind to insulin-regulated aminopeptidase (IRAP) (Albiston et al. 2003), while not all researchers accept IRAP as the AT4R (Benoist et al. 2014).
MAS receptors
A bifurcation to the classic RAS incorporates the production of Ang(1-7) by ACE2 whereby Ang(1-7) mediates its action by binding to the G protein coupled receptor MAS (Santos et al. 2003). Neuroprotective effects mediated by enhancing the ACE2/Ang(1-7) axis has been shown in various animal models of hypertensive encephalopathy, ischemia and hemorrhagic stroke, chronic hypoperfusion, and diabetes (Jiang et al. 2013; Chen et al. 2014; Xie et al. 2014; Chen et al. 2017), spurring interest for neuroprotective mechanisms of MAS (Fig. 1). Infusions of Ang(1-7) were found to induce cerebral ischemic tolerance by promoting brain angiogenesis through an endothelial nitric oxide synthase (eNOS)-dependent manner (Jiang et al. 2014). Interestingly, enhancing systemic Ang(1-7) attenuated the development of HT, reduced the risk of ischemic stroke and the size of cerebral infarct, and ameliorated neurological outcome (Feng et al. 2010; Mecca et al. 2011), benefits attributed to MAS antioxidant and anti-inflammatory effects (Fig. 1).
The renin angiotensin system and AD
Clinical studies
Hypertensive patients treated with ARBs over a 3-year period exhibited superior memory performance compared with hypertensive patients treated with other antihypertensive medications, suggesting that this subset of antihypertensive medication has neuroprotective pleiotropic beneficial effects (Ho and Alzheimer’s Disease Neuroimaging Initiative 2017). Correspondingly, antihypertensive medications specifically targeting the RAS, such as ACEIs and ARBs, were not only shown to preserve cognitive function but have been linked to a significant reduction of AD progression, MCI onset, and AD onset (Chiu et al. 2014; Li et al. 2010). This association was further supported by a recent meta-analysis that found that ARB use was significantly associated with a reduced risk of AD incidence (Oscanoa et al. 2020). ARBs have also been suggested to purportedly have a greater reduction in the incidence and progression of AD compared with ACEIs thereby indicating that utilizing ARBs may have more clinical benefits and be more efficacious for AD intervention (Li et al. 2010). When patients taking ACEIs were switched to ARBs, they demonstrated greater benefits, supporting the therapeutic efficacy of ARBs (Li et al. 2010). Despite these observations, few clinical trials have been conducted to explore the ability of ARBs to reduce AD onset and progression, with ongoing trials assessing telmisartan (NCT02085265; NCT02471833), losartan (NCT02913664), and candesartan (NCT01984164; NCT02646982).
Preclinical studies
Preclinical studies in AD mouse models generally support the use of ARBs (Fig. 2) (Takeda et al. 2009; Liu et al. 2014; Wang et al. 2007; Mogi et al. 2008; Tsukuda et al. 2009; Ongali et al. 2014; Ferrington et al. 2012; Danielyan et al. 2010). Specifically, ARB treatment in AD mouse models prevented and rescued cerebrovascular (Fig. 3a), neuropathological, and cognitive deficits (Fig. 3b), with varied abilities to alter Aβ pathology, actions independent of their intended blood pressure-lowering effects (Fig. 3c) (Takeda et al. 2009; Wang et al. 2007; Mogi et al. 2008; Ongali et al. 2014; Ferrington et al. 2012). ARBs were demonstrated to improve spatial learning and memory, as well as working memory (Takeda et al. 2009; Wang et al. 2007; Mogi et al. 2008; Ongali et al. 2014; Trigiani et al. 2018; Royea et al. 2020a), benefits associated with improved long-term potentiation (LTP), increased neurogenesis, and elevated peroxisome proliferator-activated receptor gamma (PPAR-γ) activity (Takeda et al. 2009; Trigiani et al. 2018). ARBs also restored CBF (Takeda et al. 2009; Tsukuda et al. 2009; Ongali et al. 2014), improved autoregulation (Takeda et al. 2009), rescued EC- and SMC-mediated vasodilatory function, and increased NO bioavailability within the vessel wall (Ongali et al. 2014; Trigiani et al. 2018), together with potent anti-inflammatory and antioxidant properties (Ongali et al. 2014; Trigiani et al. 2018; Torika et al. 2017).
Fig. 2.

Preclinical ARB effects on cerebrovascular, cognitive, and pathological deficits in AD mouse models. Schematic representation of preclinical ARB interventions, including telmisartan, losartan, olmesartan, valsartan, and candesartan, and their respective effects on cerebrovascular, cognitive, and pathological deficits on a variety of AD mouse models. CBF, cerebral blood flow; EC, endothelial cell; SMC, smooth muscle cell; NO, nitric oxide; LTP, long-term potentiation; ROS, reactive oxygen species; PPAR-γ, peroxisome proliferator-activated receptor gamma; Aβ, amyloid-β; TNFα, tumor necrosis factor-α; SOD2, superoxide dismutase 2
. Fig. 3.
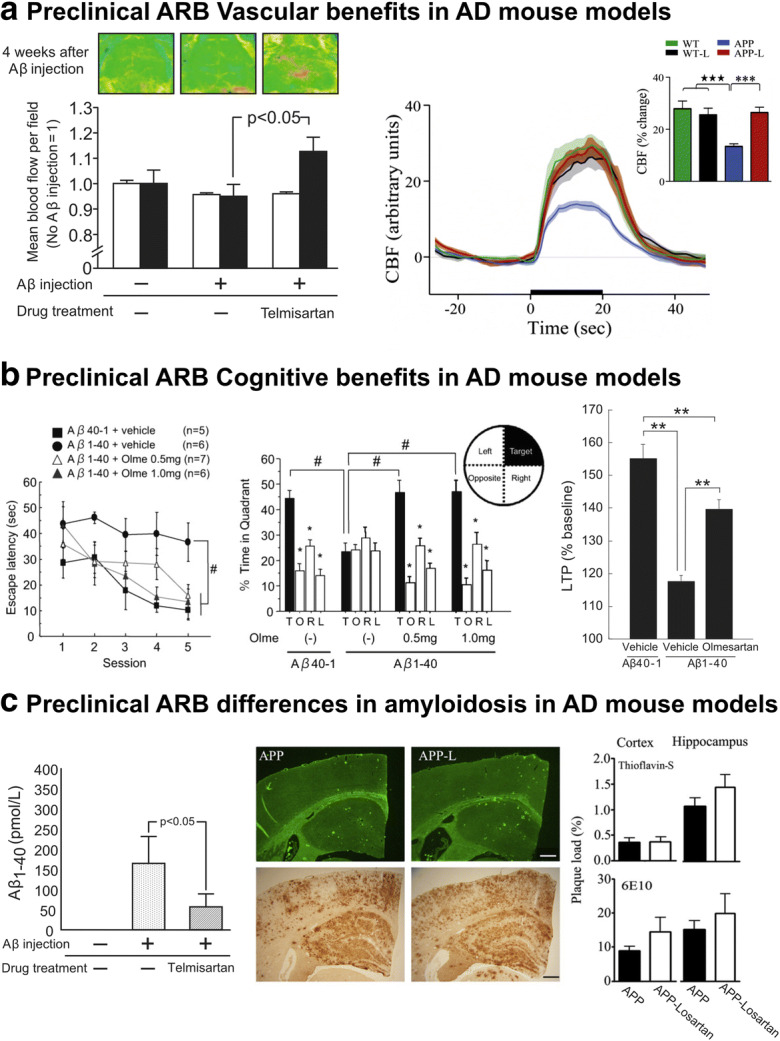
Preclinical ARB effects on cerebrovascular, cognitive and pathological deficits in AD mouse models. The Aβ-induced cerebrovascular deficits associated with oxidative stress and endothelial dysfunction are reminiscent observed in cardiovascular diseases, including AD. Telmisartan (right, Tsukuda et al. 2009) and losartan (left, Ongali et al. 2014) restored neurovascular coupling in Aβ-injected and APP J20 mice, respectively (a). The ability of ARBs to restore cognitive function, as demonstrated by olmesartan’s spatial learning and memory rescue, as well as its associated increase in long-term potentiation in Aβ-injected mice (Takeda et al. 2009) is illustrated in (b). Despite the consistency observed in preclinical cerebrovascular and cognitive benefits, preclinical ARB interventions have exhibited discrepancies on brain Aβ levels and plaque load with various AD mouse models (Tsukuda et al. 2009; Ongali et al. 2014), respectively, ranging from significant reductions to no effect (c). #p < 0.05; **p < 0.01; ***p < 0.001. Reproduced with permission
To complicate the matter, ARBs may differ in their therapeutic value as demonstrated by differences in each ARB’s ability to restore cognitive and cerebrovascular function in AD mouse models (Takeda et al. 2009; Mogi et al. 2008; Ongali et al. 2014; Danielyan et al. 2010; Trigiani et al. 2018). This could possibly explain why some human cohorts demonstrated no significant difference in the age of AD onset while taking ARBs (Anderson et al. 2011). ARBs being pharmacologically heterogenous, their actions may not only be due to AT1R blockade but also to additional mechanisms that may be selective for specific compounds (Villapol and Saavedra 2015). Whereas the complete pharmacological profile and therapeutic efficacy of each ARB has yet to be compared in controlled clinical studies; preclinical evidence indicates that repurposing ARBs for the treatment of AD possesses major translational value.
Losartan, a commonly prescribed ARB that readily crosses the BBB (Culman et al. 1999), has been associated with reduced incidence of AD in humans (Li et al. 2010). Pharmacologically, losartan is metabolized to form two active derivatives: EXP3174, which has a higher affinity for AT1R than losartan (Michel et al. 2013), and EXP3179, which has no AT1R blocking activity but inhibits endothelial cyclooxygenase (COX)-2 and increases PPAR-γ activity (Fig. 1) (Rossi 2009). Losartan has also been recognized for its memory-enhancing properties, ability to normalize CBF and vasodilatory function, as well as possessing anti-inflammatory and antioxidant properties in various mouse models of AD, benefits unrelated to its blood pressure-reducing effects (Ongali et al. 2014; Danielyan et al. 2010; Royea et al. 2017, 2020a). Knowing that AD mouse models based on familial AD mutations of the amyloid precursor protein (APP) are not hypertensive, they provide a suitable model to explore pleiotropic effects of ARBs that are independent of blood pressure-lowering effects, and for deciphering the underlying mechanism(s) depicting how ARB treatment functions in AD pathology.
Overall, the convergence of clinical and preclinical epidemiological and molecular data and the availability of drugs to effectively inactivate ACE or antagonize RAS receptors provides valuable information on chronic ARB use that is thought to increase endogenous AngII that can either (i) increase its binding to AT2R or (ii) be converted to AngIV, leading to increased AT4R activation. It is thus most pertinent to understand the underlying mechanism(s) of action mediating ARB losartan’s reported multifaceted beneficial effects to better prevent or treat AD, hence opening opportunity to investigate whether AT2Rs and AT4Rs mediate losartan’s beneficial effects in well-characterized AD mouse models, like the APP J20 mice that overexpress a mutated form of the human amyloid precursor protein (APPSwe/Ind) (Mucke et al. 2000).
Losartan’s cognitive and cerebrovascular benefits in APP J20 mice: AT2Rs and AT4Rs as possible contributors
Losartan intervention in adult, aged, and elderly APP J20 mice completely restored the spatial learning and memory dysfunction in adult and aged mice, while improving memory for elderly mice (Ongali et al. 2014; Royea et al. 2017, 2020a), as also observed with losartan or telmisartan in Aβ1–40-injected mice (Mogi et al. 2008; Tsukuda et al. 2009). Furthermore, losartan rescued the cerebrovascular deficits (Ongali et al. 2014; Royea et al. 2017) that occur early in APP J20 mice and attributed mainly to Aβ-induced oxidative stress with increased ROS production by NADPH oxidase (Hamel et al. 2008; Park et al. 2005). Specifically, losartan rescued neurovascular coupling, EC- and SMC-mediated dilatory responses in cerebral arteries, as well as baseline NO bioavailability in the vessel wall of APP J20 mice, a benefit equally effective regardless of age and treatment duration (Ongali et al. 2014). Similar neurovascular coupling benefits were found with other sartans in transgenic APP23 or Aβ1–40-injected mice (Takeda et al. 2009; Tsukuda et al. 2009). In order to assess whether AT2Rs or AT4Rs were involved in mediating losartan’s cognitive and cerebrovascular benefits, APP J20 mice were treated with losartan until cognitive benefits appeared and then were concomitantly administered the AT2R antagonist PD123319 or the AT4R antagonist divalinal. Robust evidence implicated AT4Rs (Fig. 4b) and, to a smaller extent, AT2Rs (Fig. 4a) in mediating spatial learning and memory beneficial effects of losartan in APP J20 mice (Royea et al. 2017, 2020a, b). In relation to cerebrovascular function, AT2R antagonism did not oppose losartan’s EC- and SMC-mediated dilatory rescue but did block losartan’s benefits on baseline NO bioavailability and neurovascular coupling (Royea et al. 2020a). AT4R blockade, in contrast, countered all losartan’s benefits on neurovascular coupling, EC- and SMC-dependent vasodilatory function, and NO bioavailability within the vessel wall (Royea et al. 2017). The ARB’s positive effects were independent of their intended blood pressure-lowering effects and provided a rationale for examining whether administration of a selective AT2R or AT4R agonist, C21 or AngIV, could mimic the benefits observed with losartan treatment.
Fig. 4.
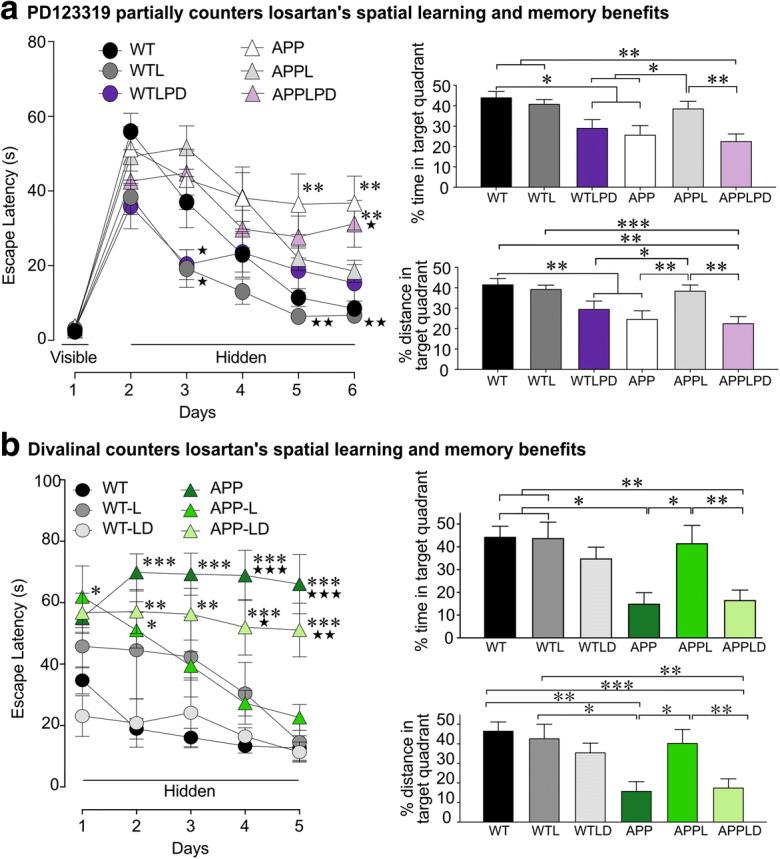
Both AT2R and AT4R antagonists countered losartan’s cognitive benefits in APP J20 mice. Losartan (L) treatment improved spatial learning and memory retention in APP J20 mice, as shown in both Morris water mazes following 5 (a) and 4 months (b) of losartan treatment. AT2R J20 antagonism with PD123319 (PD) partially countered, while AT4R antagonism with divalinal (d) prevented spatial learning and memory retention benefits in L-treated APP J20 mice, as shown by a significantly longer escape latency during the hidden platform test and reduced time and distance swam within the target quadrant during the probe trial in LPD- (a) and LD-treated (b) APP J20 mice compared with L-treated APP mice. *p < 0.05; **p < 0.01; ***p < 0.001 for comparisons with WT or *p < 0.05; **p < 0.01; ***p < 0.001 comparisons with APP-L treated mice. From Royea et al. (2017, 2020a), reproduced with permission
Testing the efficacy of novel therapeutic targets for AD
Since AT2R and, more convincingly, AT4R blockade in APP J20 mice identified these receptors as potential targets for AD intervention following chronic ARB treatment (Royea et al. 2017, 2020b), we investigated the efficacy of ATR2 and AT4R respective selective agonists, C21 (Royea et al. 2020a) and AngIV (Royea et al. 2020b). Short (1 month, 1.6 nmol day−1) intracerebroventricular (icv) or 7-month-long oral C21 (10 mg kg−1 day−1) C21 intervention failed to restore cognitive (Fig. 5a) and vasodilatory deficits in APP J20 mice (Fig. 6) (Royea et al. 2020a), indicating that, at the selected doses, C21 was incapable of mediating the same benefits as losartan treatment. The lack of benefits following C21 intervention may be due to the absence of AT1R antagonism. Indeed, our findings suggest that the benefits obtained following losartan treatment may only be mitigated through AT2Rs when AT1Rs are antagonized as supported by our findings since AT2R antagonism countered losartan’s beneficial effects, whereas AT2R agonism exerted limited benefits regardless of treatment administration or duration (Royea et al. 2020a). However, previous studies with intraperitoneal C21 (1, 3, and 10 μg kg−1 day−1) or oral C21 (0.12 mg kg−1 day−1) implicated AT2Rs in preserving cognitive function in Aβ1–40-injected mice (Jing et al. 2012) and in rats post-stroke (Ahmed et al. 2019). These findings indicated that solely targeting AT2Rs is insufficient to restore cognitive deficits in APP J20 mice, memory impairments being the main hallmark of AD (Fig. 5a) (Royea et al. 2020a). Further studies should investigate whether combined losartan and C21 therapeutic intervention could exhibit a greater therapeutic outcome than losartan alone.
Fig. 5.
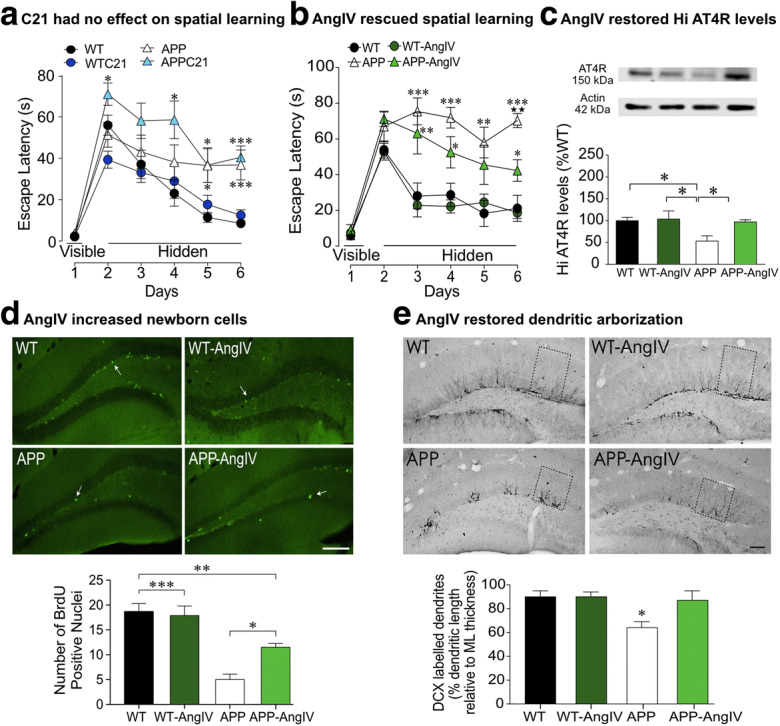
AT2R agonism had no effect on cognitive deficits, whereas AT4R agonism rescued spatial learning, elevated AT4R expression, and stimulated newborn cell formation and dendritic arborization in APP J20 mice. AT2R agonist Compound 21 (C21) had no effect on spatial learning (a), as shown by a similar escape latency to APP J20 controls in the Morris water maze (from Royea et al. 2020a). AT4R agonist, AngIV improved spatial learning in APP J20 mice (b), as shown by a significantly reduced escape latency compared with APP J20 controls in the Morris water maze. Cognitive benefits following AngIV treatment were attributed to the ability of AngIV to normalize hippocampal AT4R expression in APP J20 mice (c), stimulate neurogenesis as shown by an increase in the number of BrdU-positive nuclei (d) and significant increase in the dendritic length projecting in the molecular layer (e) in AngIV-treated APP J20 mice compared with APP J20 controls (from Royea et al. 2020b). *p < 0.05; **p < 0.01; ***p < 0.001 for comparisons with WT, **p < 0.01 for comparison with AngIV-treated APP mice. Reproduced with permission
Fig. 6.
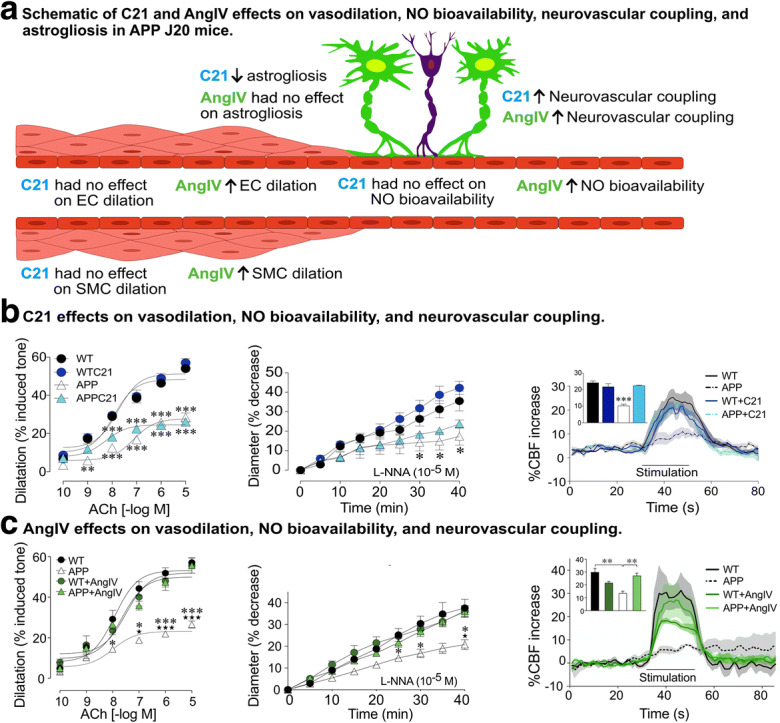
AT2R and AT4R effects on cerebrovascular vasodilation, NO bioavailability, and neurovascular coupling in APP J20 mice. a A schematic representation of the effects of both AT2R (compound 21, C21) and AT4R (AngIV) agonism on elements regulating cerebral blood flow (CBF). Endothelial-mediated vasodilatory dysfunction was not altered following AT2R agonism, as shown by similar ex vitro-isolated posterior cerebral artery acetylcholine (ACh)-mediated dilatory responses in untreated APP J20 mice (b). The influence of NO on the vessel basal tone was assessed by the administration of l-nitro-arginine (l-NNA), a compound that inhibits endothelial NO synthesis, a response reduced in APP J20 mice. C21 normalized the reduced NO bioavailability within the vessel wall in APP J20 mice (b). Intriguingly, the reduced whisker-evoked CBF response in APP J20 mice was normalized to WT levels following C21 administration (b) (from Royea et al. 2020a). AngIV treatment rescued endothelial-mediated vasodilatory dysfunction and reduced the production of baseline NO in APP J20 mice to WT levels (c). Correspondingly, AngIV treatment restored whisker-evoked CBF increases in APP J20 mice (c) (from Royea et al. 2020b). *p < 0.05; **p < 0.01; ***p < 0.001 for comparisons with WT. Reproduced with permission
Contrary with C21 interventions, AngIV’s ability to fully restore cognitive (Fig. 5b) and cerebrovascular pathology in APP J20 mice strongly supports targeting AT4Rs as a promising therapeutic target to combat AD (Royea et al. 2020b). At the selected dose (1.3 nmol day−1), AngIV was capable of producing cognitive, neurogenic, vascular, and antioxidant benefits (Royea et al. 2020b). A major caveat for AngIV treatment, however, remains the inability of AngIV analogs to readily cross the BBB, identifying an important future research endeavor for AD intervention. Despite this caveat, developing new AngIV analogs as a promising AD treatment has been considered (Wright et al. 2015) and may be more potent than losartan due to its capability of manifesting similar pleiotropic benefits within a shorter treatment duration, 1 vs. 4 months, respectively (Royea et al. 2017, 2020b). Intriguingly, AngIV treatment normalized AT1R expression in APP mice, confirming a mechanism that may play a fundamental role in mediating AD recovery following ARB use.
Cognitive function, RAS agonism, and AD
When looking for potential mechanisms of action that could depict how the centrally acting ARB losartan exerts blood pressure-independent neuroprotective effects; both AT2Rs and AT4Rs were identified as mediating losartan’s cognitive benefits based on their respective ability to counteract losartan’s spatial learning and memory benefits when co-administered in APP J20 mice (Fig. 4a, b) ( Royea et al. 2017, 2020a). Yet, the complete failure of AT2R agonist C21 to rescue spatial learning and memory in APP J20 mice (Fig. 5b) (Royea et al. 2020a) as opposed to the high efficacy of AT4R agonist AngIV (Royea et al. 2020b), strongly identified AngIV intervention as a most exciting research avenue. Such conclusion further supports prior research identifying AngIV as facilitating memory performance in various paradigms (Braszko et al. 1988; Paris et al. 2013). The cognitive benefits of AngIV further provide urgency for the development of BBB penetrant new analogs (Wright et al. 2015).
Since the hippocampus is rich in AT4Rs, and APP J20 mice have reduced AT4R expression (Ongali et al. 2014; Royea et al. 2020b), the ability of both losartan and AngIV to restore AT4R expression supports a commonality among these two therapies (Fig. 5c) (Royea et al. 2017, 2020b). Enhanced cognitive function by AT4R activation has been imputed to benefits on synaptic transmission, LTP (Kramar et al. 2001), and hippocampal spinogenesis and synaptogenesis (McCoy et al. 2013). One possible mechanism of action for AngIV’s beneficial effects relates to neurogenesis. Indeed, dysfunctional neurogenesis may exacerbate neuronal vulnerability and contribute to memory impairment during AD (Chevallier et al. 2005; Verret et al. 2007). The majority of AD transgenic animal models expressing a mutated form of human APP or presenilin (PS1 or PS2) have diminished neurogenesis in the dentate gyrus (DG) or both the DG and subventricular zone (SVZ) (Chevallier et al. 2005; Verret et al. 2007). Likewise, post-mortem examinations of AD patients have revealed reductions in the number of neurons within the hippocampal formation compared with healthy elderly individuals (Simic et al. 1997), suggesting that impaired neuronal proliferation or its deregulation is a likely consequence of AD and may represent a causative factor for the observed cognitive alterations associated with AD; thereby, offering a target site for therapeutic intervention. In this regard, underlying cognitive benefits may be the ability of AngIV to increase newborn cell formation (Fig. 5d) and proliferation in APP J20 mice, and normalize dendritic extension from remaining maturing (doublecortin (DCX)-positive) neurons within the DG (Fig. 5e) (Royea et al. 2020b). This would favor a role for AngIV-mediated neuroprotection and neurogenesis as contributing towards cognitive recovery. Prior studies that showed the ability of AngIV to enhance LTP in the CA1 region of the rat hippocampus in vitro (Kramar et al. 2001) and DG in vivo (Wayner et al. 2001) further support a role for neurogenesis in AngIV-mediated cognitive recovery.
Another possible mechanism explaining AngIV’s cognitive recovery could be its ability to potentiate cholinergic transmission. Specifically, AT4Rs and cholinergic neurons have been closely linked within brain regions involved in cognitive processing, including the hippocampus and neocortex, and AT4R activation has resulted in acetylcholine (ACh) release in hippocampal slices (Lee et al. 2001). Despite APP J20 mice not possessing a cholinergic deficit at 6 months of age (Aucoin et al. 2005), the common mechanistic links between the cholinergic system and AT4Rs, and the fact that the majority of current AD therapies target the cholinergic system, identify promise for AT4R intervention in AD patients. Another pathway associating AT4Rs with AD is the phosphatidylinositol 3-kinase (PI3K)/Akt pathway, whereby AngIV has been shown to increase Akt phosphorylation in a rat model of ischemia, a pathway that could potentially mediate AT4R’s neuroprotective effects in AD mouse models. It cannot be excluded, however, that Ang(1-7) could be a target for cognitive recovery in AD due to its neuroprotective effects in animal models of hypertensive encephalopathy, stroke, and chronic cerebral hypoperfusion (Jiang et al. 2013; Chen et al. 2014; Xie et al. 2014), and evidence of reduced Ang(1-7) levels in animal models of AD during disease progression (Jiang et al. 2016).
Brain vasculature in RAS agonism and AD
The importance of a functional brain perfusion can be highlighted from the fact that the brain is responsible for 20–25% of the body’s oxygen and glucose consumption, despite representing only approximately 2% of the body’s total mass (Iadecola 2013). Due to the high-energy demand and lack of fuel reserves within the brain, CBF interruption results in loss of brain function within seconds and neuronal damage within minutes (Moskowitz et al. 2010). The brain requires a functional cerebrovasculature to meet its metabolic demands for proper functionality; hence, alterations within the cerebrovasculature can alter brain integrity and have detrimental consequences (Kisler et al. 2017), as observed in AD.
Intriguingly, AT2R-dependent NO production was previously observed following AT1R blockade with losartan, suggesting an inhibitory effect of AT1Rs on AT2Rs. Since AT1Rs are upregulated in APP mice, it is possible that AT2R-dependent NO production benefits were not recovered following C21 intervention as AT1Rs were still functional (Fig. 6a, b). AT2Rs were not identified as mediating losartan’s benefits on vasodilatory recovery in pial arteries (Royea et al. 2020a). Yet, in vitro AT2R activation was previously shown to induce vasodilation in preconstricted renal arterioles and mesenteric arteries vessels via a NO/cGMP signaling pathway and bradykinin release (Carey 2017), or vasoconstriction in large renal arteries (Hayashi et al. 1993). Since AT2R antagonism did not counter losartan’s EC- or SMC-mediated vasodilatory benefits and, similarly, AT2R agonist C21 had no benefits on vasomotor function (Fig. 6a, b), AT2Rs were not identified as key players in cerebral vasodilatory dysfunction in APP mice, despite their capacity to dilate brain arterioles and capillaries (Henrion et al. 2001). It is thus possible that heterogeneity in AT2Rs in vessels from different sizes and locations act differently as supported by our findings in APP J20 mice of PD123319 countering and C21 interventions recapitulating losartan’s neurovascular coupling recovery (Royea et al. 2020a), a response initiated in the microvascular bed. Future studies should elucidate the roles of AT2Rs in brain arteries, arterioles, and capillaries to determine whether such heterogeneity exists.
AT4Rs were identified as a key mediator of losartan’s cerebrovascular benefits, restoring EC- and SMC-mediated vasodilatory function and NO bioavailability in APP J20 mice (Fig. 6a, c). These findings were supported by divalinal’s ability to counter losartan-mediated benefits of cerebrovascular reactivity and neurovascular coupling and AngIV’s positive effects on both EC function and neurovascular coupling (Fig. 6a, c). These findings coincide with several studies demonstrating AngIV’s ability to dilate, via NO-dependent mechanisms, rabbit pial arteries (Haberl et al. 1991) and internal carotid arteries (Kramar et al. 1998). Likewise, AngIV induced EC-dependent dilation and increased endothelial NO synthesis in porcine pulmonary cells (Patel et al. 1998). Moreover, chronic AngIV treatment also normalized diabetes-induced EC dysfunction, vascular hypertrophy, and deficits in NO bioavailability (Nasser et al. 2014) and reverse endothelial dysfunction in ApoE-deficient mice fed a high-fat diet (Vinh et al. 2008). These observations, together with findings in APP J20 mice, identify AT4Rs as a key therapeutic target for EC dysfunction and highlight the need for developing new brain penetrant agonists to improve AD-related cerebrovascular dysfunction.
It is important to emphasize, however, that only restoring cerebrovascular function in APP mice is not sufficient to restore memory. Specifically, adult APP J20 mice treated with pioglitazone, a PPAR-γ agonist, or the statin simvastatin recovered vascular function independent of cognitive benefits (Nicolakakis et al. 2008; Tong et al. 2009). Likewise, the potent ARB candesartan failed to restore cognitive function in APP J20 mice, while possessing the ability to recover EC and SMC function, as well as reducing neuroinflammation (Trigiani et al. 2018). In contrast, losartan exhibits potent vascular, anti-inflammatory, and antioxidant properties and also countered Aβ-mediated cognitive deficits in APP J20 mice (Ongali et al. 2014; Royea et al. 2017). This discrepancy could be attributed to different pharmacological profiles among ARBs, but also highlights the ability of a compound to restore vascular function independent of cognitive recovery. Correspondingly, the inability of candesartan to recover cognitive function coincided with the downregulation of AT4Rs in treated mice (Trigiani et al. 2018), further identifying an important role for AT4Rs in memory recovery. In this regard, therapeutic interventions need to target more than just cardiovascular health but also mediate neuronal benefits related to neuronal function within the hippocampus.
Cognitive benefits irrespective of changes in amyloidosis
The amyloid cascade hypothesis of AD dominated therapeutic research focus for several years. Yet, clinical trials utilizing Aβ aggregation inhibitors, monoclonal anti-amyloid antibodies as targets for Aβ clearance and aggregation, or γ-secretase inhibitors to prevent the formation and aggregation of Aβ peptides all failed to alter cognitive dysfunction and prevent AD progression (De Strooper and Karran 2016). Immunotherapy utilizing synthetic Aβ1–42 held promise following successful elimination of Aβ plaques within APP mice (Schenk et al. 1999); yet, the amyloid peptide vaccine (AN1792) was unable to alter AD progression or cognitive dysfunctions despite successful clearance of Aβ peptides (Holmes et al. 2008). Nevertheless, clinical trials providing evidence for real benefits following Aβ-targeted immunotherapy have been very limited (Wisniewski and Goni 2015). While next generations of amyloid vaccines may show greater promise and be more efficacious, it is possible that clearing Aβ from brain tissue may not be required for cerebrovascular and cognitive recovery despite Aβ’s detrimental effects on the cerebrovasculature and cognitive performance (Li et al. 2014). Particularly, discrepancies on Aβ levels or plaque load have been observed in various APP mouse models treated with ARBs, findings having ranged from no effect (Takeda et al. 2009; Ongali et al. 2014; Ferrington et al. 2012; Royea et al. 2017, 2020a) to significant reductions (Wang et al. 2007; Danielyan et al. 2010). Correspondingly, the failure of C21 to normalize cognitive function coincided with a significant reduction of dense core plaques within the cortex and hippocampus in APP J20 mice (Fig. 7a) (Royea et al. 2020a). Contrarily, AngIV (Fig. 7b) and losartan treatments restored cognitive deficits despite persistent amyloidosis. Based on these results, the lack of relationship between Aβ plaque load and cognitive recovery was sustained. These findings further support that reducing amyloidosis does not correlate with improved cognitive function, as observed in AD patients who received Aβ1–42 immunization (Holmes et al. 2008). In vitro studies have shown a role of ACE in Aβ peptide degradation (Oba et al. 2005). Interestingly, ACEIs promoted Aβ aggregation and cognitive benefits in AD patients (Rygiel 2016), again stressing the lack of correlation between amyloid pathology and cognitive dysfunction. Together our results suggest that despite the ability of some proponents of RAS to reduce Aβ plaque deposition via the degradation of Aβ species, Aβ reductions are not linked to cognitive recovery.
Fig. 7.

AT2R activation decreased dense core Aβ plaque load, while AT4R agonism had no effect on Aβ plaque load. Cortical and hippocampal dense core Aβ plaque load measured by thioflavin S was significantly reduced following AT2R agonism with compound 21 (C21) (a) (from Royea et al. 2020a). AngIV treatment had no effect on cortical or hippocampal dense core Aβ plaque load measured by thioflavin S (from Royea et al. 2020b). *p < 0.05; **p < 0.01. Reproduced with permission
Oxidative stress and neuroinflammation
Hyperactivation of AT1Rs has been shown to induce NADPH oxidase activity that leads to ROS production; thereby, prompting oxidative stress, a pathway activated by Aβ in AD (Park et al. 2005). Knowing that AT1Rs are upregulated in AD patients and APP mice (Jackson et al. 2018), it is probable that AT1R elevation increases NADPH oxidase activity in AD. Interestingly, AT2Rs have been associated with antioxidant signaling through their ability to inhibit NADPH oxidase and ROS generation (Lu et al. 2015) and downregulate p38 and p44/42 MAP kinase phosphorylation (Dandapat et al. 2008). These findings were supported by the ability of AT2R blockade to counteract losartan’s antioxidant benefits since their blockade counteracted SOD2 downregulation in losartan-treated APP J20 mice (Royea et al. 2020a). Yet, administration of the AT2R agonist C21 upregulated brain SOD2 protein levels in treated APP J20 mice compared with controls (Fig. 8a), supporting a potential role for the ability of AT2Rs to increase superoxide production through NADPH oxidase activation (Park et al. 2013). Alternatively, it is possible that this pro-oxidant effect relates to the persistent upregulation of AT1Rs in C21-treated APP J20 mice, whereby, the antioxidant benefits of AT2Rs was negated by the ROS-generating action of AngII through the activation of AT1Rs in brain tissue and vasculature. Future studies should investigate whether concomitant AT1R blockade and AT2R activation could induce greater antioxidant benefits.
Fig. 8.

Effects of AT2R and AT4R agonism on oxidative stress and astrogliosis. AT2R agonism exacerbated the cortical superoxide dismutase 2 (SOD2) upregulation in APP J20 mice (a) (from Royea et al. 2020a), suggesting a pro-oxidant role for AT2R activation. In this respect, the SOD2 upregulation by C21 may represent an attempt to protect against both Aβ- and AT2R-generated free radicals. AT4R agonism reduced cortical SOD2, indicating an antioxidant for AT4R activation (b) (from Royea et al. 2020b). AT2R agonism attenuated astrogliosis in APP J20 mice, as shown by a reduction in the surface area occupied by GFAP immunopositive astrocytes (c). AT4R agonism had no effect on astrogliosis in APP mice (d), a finding consistent with AT4Rs’ pro-inflammatory role. *p < 0.05; **p < 0.01; ***p < 0.001. Reproduced with permission
Contrary with AT2Rs, AT4R blockade failed to prevent the antioxidant benefits of losartan treatment as measured with brain and vascular SOD2 protein levels, whereas AngIV treatment demonstrated potent antioxidant properties in APP J20 mice (Fig. 8b). AngIV was previously associated with increased intracellular calcium shown to enhance NOS in order to modulate superoxide production (Jackson et al. 2018). In this regard, dysfunctional AT4R signaling in APP J20 mice may result in a shift towards AT1R mediated prooxidant signaling since reduced AT4Rs would be unable to possess antioxidant benefits. This could explain why concomitant AT1R and AT4R antagonism failed to re-establish oxidative stress. AngIV activation exerted antioxidant benefits comparable with losartan, likely through inhibition of NADPH activity by reducing NADPH oxidase cytosolic component p67phox as found in AngIV-treated APP J20 mice (Royea et al. 2020b). A complex balance among RAS receptors seems to exist in order to maintain a synergistic oxidative environment within the brain milieu in AD pathology.
Regardless of the initiator, increased oxidative stress leads to the release of pro-inflammatory cytokines that further exacerbate cell dysfunction and, eventually, leads to cell death (Jackson et al. 2018). RAS modulation has been associated with balancing prooxidant and antioxidant states, inflammation, and modulating synthesis of several molecules involved in inflammatory pathways including cytokines, chemokines, and transcription factors. Most of the pro-inflammatory effects are mediated by AT1Rs through an upregulation of different pathways such as nuclear factor kappa B (NF-κB), PPAR-γ, endothelin-1, redox pathways, and tumor necrosis factor-α (TNFα) (Capettini et al. 2012). Despite the role of AT2Rs in inflammation remaining controversial, AT2R activation has been suggested to play an anti-inflammatory role through its ability to inhibit NF-κB activity (Rompe et al. 2010), reduce TNFα expression (Sampson et al. 2016) and regulate PPAR-γ activity (Li et al. 2016). Losartan’s anti-inflammatory benefits were found to be AT2R-independent since concomitant AT2R antagonism failed to counter these benefits in APP J20 mice. Intriguingly, however, selective AT2R activation with C21 reduced astrogliosis in APP J20 mice (Fig. 8c), independent of persisting microgliosis, suggesting that C21 benefits on astrogliosis may act through a separate mechanism. The ability of AT4R antagonism to counter losartan’s anti-inflammatory effects support prior work that demonstrated an anti-inflammatory role for AT4Rs in rats with chronic cerebral hypoperfusion (Wang et al. 2018) and in a rat model of ischemia (Park et al. 2016). Unlike these studies that infused AngIV centrally for 6 weeks, 1 month of similarly delivered AngIV failed to reduce astrogliosis (Fig. 8d) and microgliosis in APP J20 mice, suggesting that the treatment regimen was ineffective at altering gliosis in APP J20 mice.
Conclusions and implications for AD patients and novel therapeutic targets
The ability to develop successful novel therapies requires a greater understanding of the molecular and cellular pathophysiology of AD. That is why investigating novel targets aimed to advance AD therapy to a level whereby slowing, ending, or reversing the disease progression is a major research endeavor. The studies summarized in this review suggest that identifying mechanisms underlying the beneficial effects of ARBs in AD patients may bear potential for new therapeutic targets. Specifically, translating the benefits summarized here in well-established AD mouse models to AD patients strongly suggests that modulating the RAS may be capable to restore cognitive and cerebrovascular benefits independent of changes in amyloidosis or blood pressure (Fig. 9). The reported studies further recognize the potential for refining RAS pharmacological manipulations in AD by making compounds capable of crossing the BBB. Overall, despite mediating some ARB benefits, we deemed solely targeting AT2Rs as an inefficient pharmacological therapeutic target for AD. Rather, the AngIV/AT4R cascade appears to be a more promising avenue.
Fig. 9.

Schematic representation of AT2R and AT4R agonism on cognitive and cerebrovascular function, amyloidosis, and gliosis in APP J20 mice. AT2R agonism had no effect on spatial learning and memory, vasodilatory function, or nitric oxide (NO) bioavailability. AT2R agonism normalized neurovascular coupling, reduced dense core Aβ plaques, and reduced astrogliosis independent of persisting microgliosis. AT4R agonism rescued spatial learning and memory, vasodilatory function, NO bioavailability, and neurovascular coupling. AT4R agonism benefits were independent of persisting dense core Aβ plaque load, astrogliosis, and microgliosis. Overall, pharmacological manipulations of the RAS identify the AngIV/AT4R cascade as a promising target for AD intervention
Funding information
The work from our laboratory referred in this review was funded by grants from the Canadian Institute of Health Research (MOP 126001), the Heart and Stroke Foundation of Québec, and the Alzheimer Society of Canada. JR received studentships from the Canadian Vascular Network-Hypertension and les Fonds de Recherche du Québec - Santé (FRQS).
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Ahmed HA, Ishrat T, Pillai B, Bunting KM, Patel A, Vazdarjanova A, Waller JL, Arbab AS, Ergul A, Fagan SC. Role of angiotensin system modulation on progression of cognitive impairment and brain MRI changes in aged hypertensive animals - a randomized double-blind pre-clinical study. Behav Brain Res. 2018;346:29–40. doi: 10.1016/j.bbr.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed HA, Ishrat T, Pillai B, Bunting KM, Vazdarjanova A, Waller JL, Ergul A, Fagan SC. Angiotensin receptor (AT2R) agonist C21 prevents cognitive decline after permanent stroke in aged animals-a randomized double- blind pre-clinical study. Behav Brain Res. 2019;359:560–569. doi: 10.1016/j.bbr.2018.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albiston AL, McDowall SG, Matsacos D, Sim P, Clune E, Mustafa T, Lee J, Mendelsohn FAO, Simpson RJ, Connolly LM, Chai SY. Evidence that the angiotensin IV (AT(4)) receptor is the enzyme insulin-regulated aminopeptidase. J Biol Chem. 2001;276(52):48623–48626. doi: 10.1074/jbc.C100512200. [DOI] [PubMed] [Google Scholar]
- Albiston AL, Mustafa T, McDowall SG, Mendelsohn FAO, Lee J, Chai SY. AT4 receptor is insulin-regulated membrane aminopeptidase: potential mechanisms of memory enhancement. Trends Endocrinol Metab. 2003;14(2):72–77. doi: 10.1016/s1043-2760(02)00037-1. [DOI] [PubMed] [Google Scholar]
- Anderson C, Teo K, Gao P, Arima H, Dans A, Unger T, Commerford P, Dyal L, Schumacher H, Pogue J, Paolasso E, Holwerda N, Chazova I, Binbrek A, Young J, Yusuf S, ONTARGET and TRANSCEND Investigators Renin-angiotensin system blockade and cognitive function in patients at high risk of cardiovascular disease: analysis of data from the ONTARGET and TRANSCEND studies. Lancet Neurol. 2011;10(1):43–53. doi: 10.1016/S1474-4422(10)70250-7. [DOI] [PubMed] [Google Scholar]
- Aucoin JS, Jiang P, Aznavour N, Tong XK, Buttini M, Descarries L, Hamel E. Selective cholinergic denervation, independent from oxidative stress, in a mouse model of Alzheimer’s disease. Neuroscience. 2005;132(1):73–86. doi: 10.1016/j.neuroscience.2004.11.047. [DOI] [PubMed] [Google Scholar]
- Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10(9):819–828. doi: 10.1016/S1474-4422(11)70072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoist CC, Wright JW, Zhu M, Appleyard SM, Wayman GA, Harding JW. Facilitation of hippocampal synaptogenesis and spatial memory by C-terminal truncated Nle1-angiotensin IV analogs. J Pharmacol Exp Ther. 2011;339(1):35–44. doi: 10.1124/jpet.111.182220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoist CC, Kawas LH, Zhu M, Tyson KA, Stillmaker L, Appleyard SM, Wright JW, Wayman GA, Harding JW. The procognitive and synaptogenic effects of angiotensin IV-derived peptides are dependent on activation of the hepatocyte growth factor/c-met system. J Pharmacol Exp Ther. 2014;351(2):390–402. doi: 10.1124/jpet.114.218735. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Bosnyak S, Jones ES, Christopoulos A, Aguilar MI, Thomas WG, Widdop RE. Relative affinity of angiotensin peptides and novel ligands at AT1 and AT2 receptors. Clin Sci (Lond) 2011;121(7):297–303. doi: 10.1042/CS20110036. [DOI] [PubMed] [Google Scholar]
- Braszko JJ, Kupryszewski G, Witczuk B, Wiśniewski K. Angiotensin II-(3-8)-hexapeptide affects motor activity, performance of passive avoidance and a conditioned avoidance response in rats. Neuroscience. 1988;27(3):777–783. doi: 10.1016/0306-4522(88)90182-0. [DOI] [PubMed] [Google Scholar]
- Braszko JJ, Walesiuk A, Wielgat P. Cognitive effects attributed to angiotensin II may result from its conversion to angiotensin IV. J Renin-Angiotensin-Aldosterone Syst. 2006;7(3):168–174. doi: 10.3317/jraas.2006.027. [DOI] [PubMed] [Google Scholar]
- Capettini LS, et al. Role of renin-angiotensin system in inflammation, immunity and aging. Curr Pharm Des. 2012;18(7):963–970. doi: 10.2174/138161212799436593. [DOI] [PubMed] [Google Scholar]
- Carey RM. AT2 receptors: potential therapeutic targets for hypertension. Am J Hypertens. 2017;30(4):339–347. doi: 10.1093/ajh/hpw121. [DOI] [PubMed] [Google Scholar]
- Chang F, Patel T, Schulz ME. The “rising tide” of dementia in Canada: what does it mean for pharmacists and the people they care for? Can Pharm J (Ott) 2015;148(4):193–199. doi: 10.1177/1715163515588107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhao Y, Chen S, Wang J, Xiao X, Ma X, Penchikala M, Xia H, Lazartigues E, Zhao B, Chen Y. Neuronal over-expression of ACE2 protects brain from ischemia-induced damage. Neuropharmacology. 2014;79:550–558. doi: 10.1016/j.neuropharm.2014.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JL, Zhang DL, Sun Y, Zhao YX, Zhao KX, Pu D, Xiao Q. Angiotensin-(1-7) administration attenuates Alzheimer’s disease-like neuropathology in rats with streptozotocin-induced diabetes via mas receptor activation. Neuroscience. 2017;346:267–277. doi: 10.1016/j.neuroscience.2017.01.027. [DOI] [PubMed] [Google Scholar]
- Chevallier NL, Soriano S, Kang DE, Masliah E, Hu G, Koo EH. Perturbed neurogenesis in the adult hippocampus associated with presenilin-1 A246E mutation. Am J Pathol. 2005;167(1):151–159. doi: 10.1016/S0002-9440(10)62962-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu WC, Ho WC, Lin MH, Lee HH, Yeh YC, Wang JD, Chen PC, Health Data Analysis in Taiwan (hDATa) Research Group Angiotension receptor blockers reduce the risk of dementia. J Hypertens. 2014;32(4):938–947. doi: 10.1097/HJH.0000000000000086. [DOI] [PubMed] [Google Scholar]
- Cifuentes D, Poittevin M, Dere E, Broquères-You D, Bonnin P, Benessiano J, Pocard M, Mariani J, Kubis N, Merkulova-Rainon T, Lévy BI. Hypertension accelerates the progression of Alzheimer-like pathology in a mouse model of the disease. Hypertension. 2015;65(1):218–224. doi: 10.1161/HYPERTENSIONAHA.114.04139. [DOI] [PubMed] [Google Scholar]
- Claassen JA. New cardiovascular targets to prevent late onset Alzheimer disease. Eur J Pharmacol. 2015;763(Pt A):131–134. doi: 10.1016/j.ejphar.2015.05.022. [DOI] [PubMed] [Google Scholar]
- Culman J, von Heyer C, Piepenburg B, Rascher W, Unger T. Effects of systemic treatment with irbesartan and losartan on central responses to angiotensin II in conscious, normotensive rats. Eur J Pharmacol. 1999;367(2–3):255–265. doi: 10.1016/s0014-2999(98)00983-2. [DOI] [PubMed] [Google Scholar]
- Dandapat A, Hu CP, Chen J, Liu Y, Khan JA, Remeo F, Carey RM, Hermonat PL, Mehta JL. Over-expression of angiotensin II type 2 receptor (agtr2) decreases collagen accumulation in atherosclerotic plaque. Biochem Biophys Res Commun. 2008;366(4):871–877. doi: 10.1016/j.bbrc.2007.11.061. [DOI] [PubMed] [Google Scholar]
- Danielyan L, Klein R, Hanson LR, Buadze M, Schwab M, Gleiter CH, Frey WH., II Protective effects of intranasal losartan in the APP/PS1 transgenic mouse model of Alzheimer disease. Rejuvenation Res. 2010;13(2–3):195–201. doi: 10.1089/rej.2009.0944. [DOI] [PubMed] [Google Scholar]
- de la Torre JC. Vascular risk factor detection and control may prevent Alzheimer’s disease. Ageing Res Rev. 2010;9(3):218–225. doi: 10.1016/j.arr.2010.04.002. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Karran E. The cellular phase of Alzheimer’s disease. Cell. 2016;164(4):603–615. doi: 10.1016/j.cell.2015.12.056. [DOI] [PubMed] [Google Scholar]
- Duron E, Hanon O. Vascular risk factors, cognitive decline, and dementia. Vasc Health Risk Manag. 2008;4(2):363–381. doi: 10.2147/vhrm.s1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esiri MM, Nagy Z, Smith MZ, Barnetson L, Smith AD. Cerebrovascular disease and threshold for dementia in the early stages of Alzheimer’s disease. Lancet. 1999;354(9182):919–920. doi: 10.1016/S0140-6736(99)02355-7. [DOI] [PubMed] [Google Scholar]
- Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64(6):575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- Feng Y, Xia H, Santos RA, Speth R, Lazartigues E. Angiotensin-converting enzyme 2: a new target for neurogenic hypertension. Exp Physiol. 2010;95(5):601–606. doi: 10.1113/expphysiol.2009.047407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrington L, Palmer LE, Love S, Horsburgh KJ, Kelly PA, Kehoe PG. Angiotensin II-inhibition: effect on Alzheimer’s pathology in the aged triple transgenic mouse. Am J Transl Res. 2012;4(2):151–164. [PMC free article] [PubMed] [Google Scholar]
- Gebre AK, Altaye BM, Atey TM, Tuem KB, Berhe DF. Targeting renin-angiotensin system against Alzheimer’s disease. Front Pharmacol. 2018;9:440. doi: 10.3389/fphar.2018.00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol (1985) 2006;100(1):328–335. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer’s disease. Science. 2006;314(5800):777–781. doi: 10.1126/science.1132814. [DOI] [PubMed] [Google Scholar]
- Goel R, Bhat SA, Hanif K, Nath C, Shukla R. Angiotensin II receptor blockers attenuate lipopolysaccharide-induced memory impairment by modulation of NF-kappaB-mediated BDNF/CREB expression and apoptosis in spontaneously hypertensive rats. Mol Neurobiol. 2018;55(2):1725–1739. doi: 10.1007/s12035-017-0450-5. [DOI] [PubMed] [Google Scholar]
- Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, Sellke FW, Seshadri S, American Heart Association Stroke Council, Council on Epidemiology and Prevention, Council on Cardiovascular Nursing, Council on Cardiovascular Radiology and Intervention, and Council on Cardiovascular Surgery and Anesthesia Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(9):2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe MJ, Heinsen H, Amaro E Jr, Grinberg LT, Teipel SJ. Cognitive correlates of basal forebrain atrophy and associated cortical hypometabolism in mild cognitive impairment. Cereb Cortex. 2016;26(6):2411–2426. [DOI] [PMC free article] [PubMed]
- Haberl RL, Decker PJ, Einhaupl KM. Angiotensin degradation products mediate endothelium-dependent dilation of rabbit brain arterioles. Circ Res. 1991;68(6):1621–1627. doi: 10.1161/01.res.68.6.1621. [DOI] [PubMed] [Google Scholar]
- Hamel E, Nicolakakis N, Aboulkassim T, Ongali B, Tong XK. Oxidative stress and cerebrovascular dysfunction in mouse models of Alzheimer’s disease. Exp Physiol. 2008;93(1):116–120. doi: 10.1113/expphysiol.2007.038729. [DOI] [PubMed] [Google Scholar]
- Hayashi K, Suzuki H, Saruta T. Segmental differences in angiotensin receptor subtypes in interlobular artery of hydronephrotic rat kidneys. Am J Phys. 1993;265(6 Pt 2):F881–F885. doi: 10.1152/ajprenal.1993.265.6.F881. [DOI] [PubMed] [Google Scholar]
- Henrion D, Kubis N, Levy BI. Physiological and pathophysiological functions of the AT(2) subtype receptor of angiotensin II: from large arteries to the microcirculation. Hypertension. 2001;38(5):1150–1157. doi: 10.1161/hy1101.096109. [DOI] [PubMed] [Google Scholar]
- Ho JK, Nation DA, Alzheimer’s Disease Neuroimaging Initiative Memory is preserved in older adults taking AT1 receptor blockers. Alzheimers Res Ther. 2017;9(1):33. doi: 10.1186/s13195-017-0255-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JAR. Long-term effects of Abeta42 immunisation in Alzheimer’s disease: follow-up of a randomised, placebo-controlled phase I trial. Lancet. 2008;372(9634):216–223. doi: 10.1016/S0140-6736(08)61075-2. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nat Rev Neurosci. 2004;5(5):347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- Iadecola C. The pathobiology of vascular dementia. Neuron. 2013;80(4):844–866. doi: 10.1016/j.neuron.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iturria-Medina Y, et al. Early role of vascular dysregulation on late-onset Alzheimer’s disease based on multifactorial data-driven analysis. Nat Commun. 2016;7:11934. doi: 10.1038/ncomms11934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson L, Eldahshan W, Fagan SC, Ergul A. Within the brain: the renin angiotensin system. Int J Mol Sci. 2018;19(3):876. [DOI] [PMC free article] [PubMed]
- Jellinger KA. The enigma of mixed dementia. Alzheimers Dement. 2007;3(1):40–53. doi: 10.1016/j.jalz.2006.09.002. [DOI] [PubMed] [Google Scholar]
- Jiang T, Gao L, Shi J, Lu J, Wang Y, Zhang Y. Angiotensin-(1-7) modulates renin-angiotensin system associated with reducing oxidative stress and attenuating neuronal apoptosis in the brain of hypertensive rats. Pharmacol Res. 2013;67(1):84–93. doi: 10.1016/j.phrs.2012.10.014. [DOI] [PubMed] [Google Scholar]
- Jiang T, Yu JT, Zhu XC, Zhang QQ, Tan MS, Cao L, Wang HF, Lu J, Gao Q, Zhang YD, Tan L. Angiotensin-(1-7) induces cerebral ischaemic tolerance by promoting brain angiogenesis in a Mas/eNOS-dependent pathway. Br J Pharmacol. 2014;171(18):4222–4232. doi: 10.1111/bph.12770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, Zhang YD, Zhou JS, Zhu XC, Tian YY, Zhao HD, Lu H, Gao Q, Tan L, Yu JT. Angiotensin-(1-7) is reduced and inversely correlates with tau hyperphosphorylation in animal models of Alzheimer’s disease. Mol Neurobiol. 2016;53(4):2489–2497. doi: 10.1007/s12035-015-9260-9. [DOI] [PubMed] [Google Scholar]
- Jing F, Mogi M, Sakata A, Iwanami J, Tsukuda K, Ohshima K, Min LJ, Steckelings UM, Unger T, Dahlöf B, Horiuchi M. Direct stimulation of angiotensin II type 2 receptor enhances spatial memory. J Cereb Blood Flow Metab. 2012;32(2):248–255. doi: 10.1038/jcbfm.2011.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaria RN. Cerebral vessels in ageing and Alzheimer’s disease. Pharmacol Ther. 1996;72(3):193–214. doi: 10.1016/s0163-7258(96)00116-7. [DOI] [PubMed] [Google Scholar]
- Khachaturian AS, Zandi PP, Lyketsos CG, Hayden KM, Skoog I, Norton MC, Tschanz JT, Mayer LS, Welsh-Bohmer KA, Breitner JC. Antihypertensive medication use and incident Alzheimer disease: the Cache County study. Arch Neurol. 2006;63(5):686–692. doi: 10.1001/archneur.63.5.noc60013. [DOI] [PubMed] [Google Scholar]
- Kisler K, Nelson AR, Montagne A, Zlokovic BV. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci. 2017;18(7):419–434. doi: 10.1038/nrn.2017.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramar EA, et al. Role of nitric oxide in angiotensin IV-induced increases in cerebral blood flow. Regul Pept. 1998;74(2–3):185–192. doi: 10.1016/s0167-0115(98)00039-1. [DOI] [PubMed] [Google Scholar]
- Kramar EA, et al. The effects of angiotensin IV analogs on long-term potentiation within the CA1 region of the hippocampus in vitro. Brain Res. 2001;897(1–2):114–121. doi: 10.1016/s0006-8993(01)02100-x. [DOI] [PubMed] [Google Scholar]
- Labandeira-Garcia JL, Rodríguez-Perez AI, Garrido-Gil P, Rodriguez-Pallares J, Lanciego JL, Guerra MJ. Brain renin-angiotensin system and microglial polarization: implications for aging and neurodegeneration. Front Aging Neurosci. 2017;9:129. doi: 10.3389/fnagi.2017.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane CA, et al. Associations between vascular risk across adulthood and brain pathology in late life: evidence from a British Birth Cohort. JAMA Neurol. 2019;18(10):942–952. [DOI] [PMC free article] [PubMed]
- Langbaum JB, Chen K, Lee W, Reschke C, Bandy D, Fleisher AS, Alexander GE, Foster NL, Weiner MW, Koeppe RA, Jagust WJ, Reiman EM, Alzheimer’s Disease Neuroimaging Initiative Categorical and correlational analyses of baseline fluorodeoxyglucose positron emission tomography images from the Alzheimer’s disease neuroimaging initiative (ADNI) Neuroimage. 2009;45(4):1107–1116. doi: 10.1016/j.neuroimage.2008.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Chai SY, Mendelsohn FAO, Morris MJ, Allen AM. Potentiation of cholinergic transmission in the rat hippocampus by angiotensin IV and LVV-hemorphin-7. Neuropharmacology. 2001;40(4):618–623. doi: 10.1016/s0028-3908(00)00188-x. [DOI] [PubMed] [Google Scholar]
- Lennon MJ, Makkar SR, Crawford JD, Sachdev PS. Midlife hypertension and Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2019;71(1):307–316. doi: 10.3233/JAD-190474. [DOI] [PubMed] [Google Scholar]
- Li H, Guo Q, Inoue T, Polito VA, Tabuchi K, Hammer RE, Pautler RG, Taffet GE, Zheng H. Vascular and parenchymal amyloid pathology in an Alzheimer disease knock-in mouse model: interplay with cerebral blood flow. Mol Neurodegener. 2014;9:28. doi: 10.1186/1750-1326-9-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Wang YJ, Zhang M, Xu ZQ, Gao CY, Fang CQ, Yan JC, Zhou HD, On behalf of the Chongqing Ageing Study Group Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology. 2011;76(17):1485–1491. doi: 10.1212/WNL.0b013e318217e7a4. [DOI] [PubMed] [Google Scholar]
- Li M, Tejada T, Lambert JP, Nicholson CK, Yahiro E, Ambai VT, Ali SF, Bradley EW, Graham RM, Dell'Italia LJ, Calvert JW, Naqvi N. Angiotensin type 2-receptor (AT2R) activation induces hypotension in apolipoprotein E-deficient mice by activating peroxisome proliferator-activated receptor-gamma. Am J Cardiovasc Dis. 2016;6(3):118–128. [PMC free article] [PubMed] [Google Scholar]
- Li NC, Lee A, Whitmer RA, Kivipelto M, Lawler E, Kazis LE, Wolozin B. Use of angiotensin receptor blockers and risk of dementia in a predominantly male population: prospective cohort analysis. BMJ. 2010;340:b5465. doi: 10.1136/bmj.b5465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Liu S, Tanabe C, Maeda T, Zou K, Komano H. Differential effects of angiotensin II receptor blockers on Abeta generation. Neurosci Lett. 2014;567:51–56. doi: 10.1016/j.neulet.2014.03.030. [DOI] [PubMed] [Google Scholar]
- Lu J, Wu L, Jiang T, Wang Y, Zhao H, Gao Q, Pan Y, Tian Y, Zhang Y. Angiotensin AT2 receptor stimulation inhibits activation of NADPH oxidase and ameliorates oxidative stress in rotenone model of Parkinson’s disease in CATH.a cells. Neurotoxicol Teratol. 2015;47:16–24. doi: 10.1016/j.ntt.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65(4):545–551. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matavelli LC, Siragy HM. AT2 receptor activities and pathophysiological implications. J Cardiovasc Pharmacol. 2015;65(3):226–232. doi: 10.1097/FJC.0000000000000208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurer K, Volk S, Gerbaldo H. Auguste D and Alzheimer’s disease. Lancet. 1997;349(9064):1546–1549. doi: 10.1016/S0140-6736(96)10203-8. [DOI] [PubMed] [Google Scholar]
- McCoy AT, Benoist CC, Wright JW, Kawas LH, Bule-Ghogare JM, Zhu M, Appleyard SM, Wayman GA, Harding JW. Evaluation of metabolically stabilized angiotensin IV analogs as procognitive/antidementia agents. J Pharmacol Exp Ther. 2013;344(1):141–154. doi: 10.1124/jpet.112.199497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecca AP, Regenhardt RW, O’Connor TE, Joseph JP, Raizada MK, Katovich MJ, Sumners C. Cerebroprotection by angiotensin-(1-7) in endothelin-1-induced ischaemic stroke. Exp Physiol. 2011;96(10):1084–1096. doi: 10.1113/expphysiol.2011.058578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MC, Foster C, Brunner HR, Liu L. A systematic comparison of the properties of clinically used angiotensin II type 1 receptor antagonists. Pharmacol Rev. 2013;65(2):809–848. doi: 10.1124/pr.112.007278. [DOI] [PubMed] [Google Scholar]
- Mogi M, Li JM, Tsukuda K, Iwanami J, Min LJ, Sakata A, Fujita T, Iwai M, Horiuchi M. Telmisartan prevented cognitive decline partly due to PPAR-gamma activation. Biochem Biophys Res Commun. 2008;375(3):446–449. doi: 10.1016/j.bbrc.2008.08.032. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Lo EH, Iadecola C. The science of stroke: mechanisms in search of treatments. Neuron. 2010;67(2):181–198. doi: 10.1016/j.neuron.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of abeta 1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20(11):4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasser M, Clere N, Botelle L, Javellaud J, Oudart N, Faure S, Achard JM. Opposite effects of angiotensins receptors type 2 and type 4 on streptozotocin induced diabetes vascular alterations in mice. Cardiovasc Diabetol. 2014;13:40. doi: 10.1186/1475-2840-13-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naveri L, Stromberg C, Saavedra JM. Angiotensin IV reverses the acute cerebral blood flow reduction after experimental subarachnoid hemorrhage in the rat. J Cereb Blood Flow Metab. 1994;14(6):1096–1099. doi: 10.1038/jcbfm.1994.143. [DOI] [PubMed] [Google Scholar]
- Nicolakakis N, Aboulkassim T, Ongali B, Lecrux C, Fernandes P, Rosa-Neto P, Tong XK, Hamel E. Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci. 2008;28(37):9287–9296. doi: 10.1523/JNEUROSCI.3348-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura Y, Ito T, Saavedra JM. Angiotensin II AT(1) blockade normalizes cerebrovascular autoregulation and reduces cerebral ischemia in spontaneously hypertensive rats. Stroke. 2000;31(10):2478–2486. doi: 10.1161/01.str.31.10.2478. [DOI] [PubMed] [Google Scholar]
- Oba R, Igarashi A, Kamata M, Nagata K, Takano S, Nakagawa H. The N-terminal active Centre of human angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide. Eur J Neurosci. 2005;21(3):733–740. doi: 10.1111/j.1460-9568.2005.03912.x. [DOI] [PubMed] [Google Scholar]
- O’Brien JT, Markus HS. Vascular risk factors and Alzheimer’s disease. BMC Med. 2014;12:218. doi: 10.1186/s12916-014-0218-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongali B, Nicolakakis N, Tong XK, Aboulkassim T, Papadopoulos P, Rosa-Neto P, Lecrux C, Imboden H, Hamel E. Angiotensin II type 1 receptor blocker losartan prevents and rescues cerebrovascular, neuropathological and cognitive deficits in an Alzheimer’s disease model. Neurobiol Dis. 2014;68:126–136. doi: 10.1016/j.nbd.2014.04.018. [DOI] [PubMed] [Google Scholar]
- Oscanoa TJ, Amado J, Vidal X, Romero-Ortuno R. Angiotensin-receptor blockers (ARBs) and risk of Alzheimer’s disease: a meta-analysis. Curr Clin Pharmacol. 2020. [DOI] [PubMed]
- Paris JJ, Eans SO, Mizrachi E, Reilley KJ, Ganno ML, McLaughlin JP. Central administration of angiotensin IV rapidly enhances novel object recognition among mice. Neuropharmacology. 2013;70:247–253. doi: 10.1016/j.neuropharm.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park BM, Cha SA, Lee SH, Kim SH. Angiotensin IV protects cardiac reperfusion injury by inhibiting apoptosis and inflammation via AT4R in rats. Peptides. 2016;79:66–74. doi: 10.1016/j.peptides.2016.03.017. [DOI] [PubMed] [Google Scholar]
- Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, Iadecola C. NADPH oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid β peptide. J Neurosci. 2005;25(7):1769–1777. doi: 10.1523/JNEUROSCI.5207-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park MH, Kim H, Lim J, Ahn JS, Koh JY. Angiotensin II potentiates zinc-induced cortical neuronal death by acting on angiotensin II type 2 receptor. Mol Brain. 2013;6:50. doi: 10.1186/1756-6606-6-50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pase MP, Satizabal CL, Seshadri S. Role of improved vascular health in the declining incidence of dementia. Stroke. 2017;48(7):2013–2020. doi: 10.1161/STROKEAHA.117.013369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel JM, et al. Angiotensin IV receptor-mediated activation of lung endothelial NOS is associated with vasorelaxation. Am J Phys. 1998;275(6 Pt 1):L1061–L1068. doi: 10.1152/ajplung.1998.275.6.L1061. [DOI] [PubMed] [Google Scholar]
- Poirier J, Bertrand P, Poirier J, Kogan S, Gauthier S, Poirier J, Gauthier S, Davignon J, Bouthillier D, Davignon J. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet. 1993;342(8873):697–699. doi: 10.1016/0140-6736(93)91705-q. [DOI] [PubMed] [Google Scholar]
- Prusty SK, Sahu PK, Subudhi BB. Angiotensin mediated oxidative stress and neuroprotective potential of antioxidants and AT1 receptor blockers. Mini Rev Med Chem. 2017;17(6):518–528. doi: 10.2174/1389557516666161025094539. [DOI] [PubMed] [Google Scholar]
- Rompe F, Artuc M, Hallberg A, Alterman M, Ströder K, Thöne-Reineke C, Reichenbach A, Schacherl J, Dahlöf B̈, Bader M, Alenina N, Schwaninger M, Zuberbier T, Funke-Kaiser H, Schmidt C, Schunck WH, Unger T, Steckelings UM. Direct angiotensin II type 2 receptor stimulation acts anti-inflammatory through epoxyeicosatrienoic acid and inhibition of nuclear factor kappaB. Hypertension. 2010;55(4):924–931. doi: 10.1161/HYPERTENSIONAHA.109.147843. [DOI] [PubMed] [Google Scholar]
- Rossi GP. Losartan metabolite EXP3179: an AT1-receptor-independent treatment strategy for patients with the metabolic syndrome? Hypertension. 2009;54(4):710–712. doi: 10.1161/HYPERTENSIONAHA.109.138883. [DOI] [PubMed] [Google Scholar]
- Royea J, Zhang L, Tong XK, Hamel E. Angiotensin IV receptors mediate the cognitive and cerebrovascular benefits of losartan in a mouse model of Alzheimer’s disease. J Neurosci. 2017;37(22):5562–5573. doi: 10.1523/JNEUROSCI.0329-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royea J, et al. AT2R’s (angiotensin II type 2 receptor’s) role in cognitive and cerebrovascular deficits in a mouse model of Alzheimer disease. Hypertension. 2020a; HYPERTENSIONAHA11914431. [DOI] [PubMed]
- Royea J, Martinot P, Hamel E. Memory and cerebrovascular deficits recovered following angiotensin IV intervention in a mouse model of Alzheimer’s disease. Neurobiol Dis. 2020;134:104644. doi: 10.1016/j.nbd.2019.104644. [DOI] [PubMed] [Google Scholar]
- Rygiel K. Can angiotensin-converting enzyme inhibitors impact cognitive decline in early stages of Alzheimer’s disease? An overview of research evidence in the elderly patient population. J Postgrad Med. 2016;62(4):242–248. doi: 10.4103/0022-3859.188553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson AK, Irvine JC, Shihata WA, Dragoljevic D, Lumsden N, Huet O, Barnes T, Unger T, Steckelings UM, Jennings GL, Widdop RE, Chin-Dusting JPF. Compound 21, a selective agonist of angiotensin AT2 receptors, prevents endothelial inflammation and leukocyte adhesion in vitro and in vivo. Br J Pharmacol. 2016;173(4):729–740. doi: 10.1111/bph.13063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savoia C, Ebrahimian T, He Y, Gratton JP, Schiffrin EL, Touyz RM. Angiotensin II/AT2 receptor-induced vasodilation in stroke-prone spontaneously hypertensive rats involves nitric oxide and cGMP-dependent protein kinase. J Hypertens. 2006;24(12):2417–2422. doi: 10.1097/01.hjh.0000251902.85675.7e. [DOI] [PubMed] [Google Scholar]
- Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400(6740):173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69(24):2197–2204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- Serrano-Pozo A, Frosch MP, Masliah E, Hyman BT. Neuropathological alterations in Alzheimer disease. Cold Spring Harb Perspect Med. 2011;1(1):a006189. doi: 10.1101/cshperspect.a006189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simic G, et al. Volume and number of neurons of the human hippocampal formation in normal aging and Alzheimer’s disease. J Comp Neurol. 1997;379(4):482–494. doi: 10.1002/(sici)1096-9861(19970324)379:4<482::aid-cne2>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Siragy HM, Carey RM. The subtype-2 (AT2) angiotensin receptor regulates renal cyclic guanosine 3′,5′-monophosphate and AT1 receptor-mediated prostaglandin E2 production in conscious rats. J Clin Invest. 1996;97(8):1978–1982. doi: 10.1172/JCI118630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skoog I. Vascular aspects in Alzheimer’s disease. J Neural Transm Suppl. 2000;59:37–43. doi: 10.1007/978-3-7091-6781-6_6. [DOI] [PubMed] [Google Scholar]
- Skoog I, Gustafson D. Update on hypertension and Alzheimer’s disease. Neurol Res. 2006;28(6):605–611. doi: 10.1179/016164106X130506. [DOI] [PubMed] [Google Scholar]
- Sonnen JA, Larson EB, Crane PK, Haneuse S, Li G, Schellenberg GD, Craft S, Leverenz JB, Montine TJ. Pathological correlates of dementia in a longitudinal, population-based sample of aging. Ann Neurol. 2007;62(4):406–413. doi: 10.1002/ana.21208. [DOI] [PubMed] [Google Scholar]
- Stromberg C, Naveri L, Saavedra JM. Nonpeptide angiotensin AT1 and AT2 receptor ligands modulate the upper limit of cerebral blood flow autoregulation in rats. J Cereb Blood Flow Metab. 1993;13(2):298–303. doi: 10.1038/jcbfm.1993.37. [DOI] [PubMed] [Google Scholar]
- Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–150. doi: 10.1038/nrneurol.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Sato N, Takeuchi D, Kurinami H, Shinohara M, Niisato K, Kano M, Ogihara T, Rakugi H, Morishita R. Angiotensin receptor blocker prevented beta-amyloid-induced cognitive impairment associated with recovery of neurovascular coupling. Hypertension. 2009;54(6):1345–1352. doi: 10.1161/HYPERTENSIONAHA.109.138586. [DOI] [PubMed] [Google Scholar]
- Thomas WG, Mendelsohn FA. Angiotensin receptors: form and function and distribution. Int J Biochem Cell Biol. 2003;35(6):774–779. doi: 10.1016/s1357-2725(02)00263-7. [DOI] [PubMed] [Google Scholar]
- Tong XK, Nicolakakis N, Fernandes P, Ongali B, Brouillette J, Quirion R, Hamel E. Simvastatin improves cerebrovascular function and counters soluble amyloid-beta, inflammation and oxidative stress in aged APP mice. Neurobiol Dis. 2009;35(3):406–414. doi: 10.1016/j.nbd.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Torika N, Asraf K, Cohen H, Fleisher-Berkovich S. Intranasal telmisartan ameliorates brain pathology in five familial Alzheimer’s disease mice. Brain Behav Immun. 2017;64:80–90. doi: 10.1016/j.bbi.2017.04.001. [DOI] [PubMed] [Google Scholar]
- Trigiani LJ, Royea J, Lacalle-Aurioles M, Tong XK, Hamel E. Pleiotropic benefits of the angiotensin receptor blocker candesartan in a mouse model of Alzheimer disease. Hypertension. 2018;72(5):1217–1226. doi: 10.1161/HYPERTENSIONAHA.118.11775. [DOI] [PubMed] [Google Scholar]
- Tsukuda K, Mogi M, Iwanami J, Min LJ, Sakata A, Jing F, Iwai M, Horiuchi M. Cognitive deficit in amyloid-beta-injected mice was improved by pretreatment with a low dose of telmisartan partly because of peroxisome proliferator-activated receptor-gamma activation. Hypertension. 2009;54(4):782–787. doi: 10.1161/HYPERTENSIONAHA.109.136879. [DOI] [PubMed] [Google Scholar]
- Verret L, Jankowsky JL, Xu GM, Borchelt DR, Rampon C. Alzheimer’s-type amyloidosis in transgenic mice impairs survival of newborn neurons derived from adult hippocampal neurogenesis. J Neurosci. 2007;27(25):6771–6780. doi: 10.1523/JNEUROSCI.5564-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villapol S, Saavedra JM. Neuroprotective effects of angiotensin receptor blockers. Am J Hypertens. 2015;28(3):289–299. doi: 10.1093/ajh/hpu197. [DOI] [PubMed] [Google Scholar]
- Vinh A, Widdop RE, Drummond GR, Gaspari TA. Chronic angiotensin IV treatment reverses endothelial dysfunction in ApoE-deficient mice. Cardiovasc Res. 2008;77(1):178–187. doi: 10.1093/cvr/cvm021. [DOI] [PubMed] [Google Scholar]
- Wang J, Ho L, Chen L, Zhao Z, Zhao W, Qian X, Humala N, Seror I, Bartholomew S, Rosendorff C, Pasinetti GM. Valsartan lowers brain beta-amyloid protein levels and improves spatial learning in a mouse model of Alzheimer disease. J Clin Invest. 2007;117(11):3393–3402. doi: 10.1172/JCI31547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QG, Xue X, Yang Y, Gong PY, Jiang T, Zhang YD. Angiotensin IV suppresses inflammation in the brains of rats with chronic cerebral hypoperfusion. J Renin-Angiotensin-Aldosterone Syst. 2018;19(3):1470320318799587. doi: 10.1177/1470320318799587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayner MJ, Armstrong DL, Phelix CF, Wright JW, Harding JW. Angiotensin IV enhances LTP in rat dentate gyrus in vivo. Peptides. 2001;22(9):1403–1414. doi: 10.1016/s0196-9781(01)00475-2. [DOI] [PubMed] [Google Scholar]
- Widdop RE, Jones ES, Hannan RE, Gaspari TA. Angiotensin AT2 receptors: cardiovascular hope or hype? Br J Pharmacol. 2003;140(5):809–824. doi: 10.1038/sj.bjp.0705448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WL, Munn C, Ross RC, Harding JW, Wright JW. The role of the AT4 and cholinergic systems in the nucleus Basalis Magnocellularis (NBM): effects on spatial memory. Brain Res. 2009;1272:25–31. doi: 10.1016/j.brainres.2009.03.025. [DOI] [PubMed] [Google Scholar]
- Wisniewski T, Goni F. Immunotherapeutic approaches for Alzheimer’s disease. Neuron. 2015;85(6):1162–1176. doi: 10.1016/j.neuron.2014.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright JW, Harding JW. Brain renin-angiotensin--a new look at an old system. Prog Neurobiol. 2011;95(1):49–67. doi: 10.1016/j.pneurobio.2011.07.001. [DOI] [PubMed] [Google Scholar]
- Wright JW, Kawas LH, Harding JW. The development of small molecule angiotensin IV analogs to treat Alzheimer’s and Parkinson’s diseases. Prog Neurobiol. 2015;125:26–46. doi: 10.1016/j.pneurobio.2014.11.004. [DOI] [PubMed] [Google Scholar]
- Xie W, Zhu D, Ji L, Tian M, Xu C, Shi J. Angiotensin-(1-7) improves cognitive function in rats with chronic cerebral hypoperfusion. Brain Res. 2014;1573:44–53. doi: 10.1016/j.brainres.2014.05.019. [DOI] [PubMed] [Google Scholar]