

Abstract
Background
Chromosome conformation capture-based methods, especially Hi-C, enable scientists to detect genome-wide chromatin interactions and study the spatial organization of chromatin, which plays important roles in gene expression regulation, DNA replication and repair etc. Thus, developing computational methods to unravel patterns behind the data becomes critical. Existing computational methods focus on intrachromosomal interactions and ignore interchromosomal interactions partly because there is no prior knowledge for interchromosomal interactions and the frequency of interchromosomal interactions is much lower while the search space is much larger. With the development of single-cell technologies, the advent of single-cell Hi-C makes interrogating the spatial structure of chromatin at single-cell resolution possible. It also brings a new type of frequency information, the number of single cells with chromatin interactions between two disjoint chromosome regions.
Results
Considering the lack of computational methods on interchromosomal interactions and the unsurprisingly frequent intrachromosomal interactions along the diagonal of a chromatin contact map, we propose a computational method dedicated to analyzing interchromosomal interactions of single-cell Hi-C with this new frequency information. To the best of our knowledge, our proposed tool is the first to identify regions with statistically frequent interchromosomal interactions at single-cell resolution. We demonstrate that the tool utilizing networks and binomial statistical tests can identify interesting structural regions through visualization, comparison and enrichment analysis and it also supports different configurations to provide users with flexibility.
Conclusions
It will be a useful tool for analyzing single-cell Hi-C interchromosomal interactions.
Keywords: Single-cell Hi-C, Interchromosomal interactions, Networks, Statistically frequent
Background
Stretching the DNA in a human cell, it would be about two meters long, but how can it fit into a tiny space of about 6 microns across? DNA of cells of different tissues (e.g. neural cells and heart cells) are essentially the same, but why do these cells function disparately and what factors turn the genes’ on and off and result in the disparities? To gain insights into these questions, advances in chromosome conformation capture-based technologies have provided researchers a great opportunity to study the higher-order spatial organization of chromatin. A popular method is chromosome conformation capture with high-throughput sequencing (Hi-C), in which genomes are cross-linked with formaldehyde, fragmented with enzymes, randomly ligated in proximity and finally sequenced by next-generation sequencing platforms. After raw reads are processed by bioinformatics pipelines, a genome-wide contact map of a collection of cells is generated and it reveals intrachromosomal interactions and interchromosomal interactions. Intrachromosomal interactions refer to the valid ligations between DNA fragments of the same chromosome and interchromosomal interactions refer to the valid ligations between DNA fragments of different chromosomes. Intrachromosomal interactions are the majority of chromatin interactions in Hi-C experiments and their interaction frequencies are genomic distance dependent [1]. Interchromosomal interactions are two orders of magnitude weaker than intrachromosomal interactions [2] and interchromosomal interactions contain a higher proportion of noise than intrachromosomal interactions [3].
As the popularity of the Hi-C approach grows, large amounts of data have been generated and significant endeavors are devoted to developing computational methods and tools. These computational methods and tools can be coarsely divided into two categories, Hi-C data processing and downstream analysis. For the first category, there are some existing tools used to generate valid chromatin interactions from raw sequencing reads [4–12]. They follow similar processing steps and may adopt different sequence alignment strategies (pre-truncation, iterative and trimming), filtering criteria (read-level, read-pair level, strand and distance) and normalization methods (explicit-factor correction, matrix balancing and joint correction). Besides, there are some computational tools to exam the quality of Hi-C data by measuring the reproducibility of Hi-C replicates [10, 13–15]. For the second category, there are several major analysis tasks to gain insights into the spatial structure and function of chromatin. A/B compartments which correspond to open and closed chromatin can be identified by using Principle Component Analysis on transformed chromatin contact maps [16]. Megabase-sized Topologically Associating Domains (TADs) can be discovered by using a Hidden Markov Model with a directionality index [17]. There are other methods available to detect TADs [18–23]. As TADs are defined as continuous chromosomal loci, these methods only take intrachromosomal interactions into consideration. Statistically significant long-range chromatin interactions are extracted from Hi-C data. As there is no prior knowledge about interchromosomal interactions, computational methods focus on intrachromosomal interactions because the frequency of interactions between two intrachromosomal loci heavily depends on the genomic distance between the loci. Some methods identify statistically significant chromatin interactions by fitting the frequencies of intrachromosomal interactions with certain distributions, such as power-law [16], double-exponential [24] and negative binomial [25]. Instead of assuming a certain distribution, a nonparametric method [26] identifies statistically significant chromatin interactions by estimating the genomic distance-dependence relationship with splines. Furthermore, there is a method [19] extracting significant chromatin interactions as calling peaks in a chromatin contact map within the surrounding two-dimensional region. Hi-C data are also used to construct three-dimensional models of chromatin structure. Some methods [24, 27–33] try to learn a consensus chromatin structure of a collection of cells. Some methods [34–39] are intended to learn a set of chromatin structures representative of the observed chromatin interaction data. Besides the above downstream analysis tasks, there are some computational methods to carry out differential analysis on Hi-C data [40, 41] and multiple two-dimensional visualization tools exist [42–45]. For a comprehensive list of computational tools on Hi-C data, please check out the Omictools website [46] on high-throughput chromosome conformation capture data analysis software tools.
There are substantial computational methods and tools for downstream analysis of Hi-C data, however, most of them focus on intrachromosomal interactions and little attention is paid to interchromosomal interactions. Partly because there is no prior knowledge such as the strong genomic distance-dependence relationship between frequency of intrachromosomal interactions and the genomic distance. In addition, the frequency of the interchromosomal interactions is much lower than intrachromosomal interactions while their search space is much larger (bin pairs across chromosomes VS bin pairs within chromosomes). To the best of our knowledge, there are few computational studies that are dedicated to bulk Hi-C interchromosomal interactions. One study presents an investigation on human and mouse interchromosomal contacts and provides insights into mammalian chromatin organization [17]. A recent work develops a computational method based on an autoencoder and a multilayer perceptron classifier to impute high-resolution interchromosomal interactions [47]. Another paper presents two computational methods to estimate the transcription factors enriched in the interchromosomal interactions in yeast [48].
With the development of single-cell technologies, some single-cell Hi-C (scHi-C) approaches [49–51] are invented and therefore we can examine chromatin interactions at single-cell resolution. They also bring a new type of frequency information, the number of single cells with chromatin interactions between two disjoint chromosome regions. Generally these chromosome regions are defined by dividing chromosomes into equal-sized bins according to a resolution specified by users. Considering the lack of computational methods on interchromosomal interactions and the obvious pattern of intrachromosomal interactions along the diagonal of a chromatin contact map, we propose a computational method dedicated to analyzing interchromosomal interactions of single-cell Hi-C with this new frequency information. The fundamental difference between our research and previous research on interchromosomal interactions is our research is based on the new frequency information observed from each cell among all cells profiled. Since a bulk Hi-C experiment pools cells together at the very beginning so it can’t discern whether a chromosomal interaction is shared by single cells or not. Therefore, computational methods on bulk Hi-C experiments don’t consider the new frequency information at single-cell level, which is not available in bulk Hi-C experiments. In addition, when dealing with frequent interchromosomal interactions our method takes multiple contact maps as its inputs while computational methods on bulk Hi-C take one contact map as their inputs. What is more, to the best of our knowledge there is no tool available for frequent interchromosomal interactions. Specifically, we develop a computational tool to identify regions with statistically frequent interchromosomal interactions and make it accessible to the public. We believe that the regions associated with statistically frequent interchromosomal interactions under the single-cell context may be helpful for new hypotheses and functionally important therefore deserve more attention. Finally, frequent pattern mining is a longstanding topic in data mining research [52].
Our contributions may be stated as follows:
We propose a computational method to identify regions associated with statistically frequent interchromosomal interactions at single-cell resolution.
To the best of our knowledge, we are the first to implement a tool to serve the purpose and make it open to the public. To accommodate different scHi-C experiments, the tool is flexible on configurations.
We demonstrate that using our proposed tool on two real scHi-C data sets, it can identify interesting structural regions.
The rest of paper is organized as follows. The “Method” delineates our proposed method in detail. The “Data” introduces two scHi-C data sets as our inputs. The “Results and discussion” demonstrates that our proposed tool’s usability on identifying interesting regions and flexibility of configurations. The “Conclusion” sections concludes that the tool will be useful for analyzing scHi-C interchromosomal interactions.
Method
In Fig. 1, the workflow of our proposed tool is illustrated and it includes three steps, network construction, statistical measurement calculation and region selection. The inputs of our tool are chromatin interactions of single cells, which are represented in heatmaps and can be easily generated with scHi-C processing pipelines such as NueProcess [53]. The outputs of our tool are identified regions, whose interchromosomal interactions are statistically frequent, along with frequencies and p-values. They are provided to help users refine identified regions with some frequency or p-value cutoff.
Fig. 1.
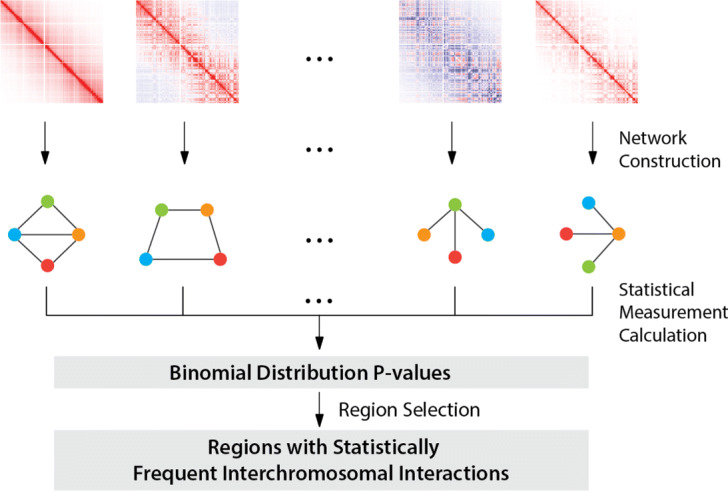
Workflow of the proposed method based on networks and statistical tests
First, we construct a network by using interchromosomal interactions for each cell respectively. Due to low read coverages of scHi-C experiments and the more complex chromosomal structures of larger mammalian genomes, i.e. homo sapiens and mus musculus, chromosomes are divided into equal-sized bins to accumulate sufficient signals. Each bin is represented as a node with an index, and if there is an interchromosomal interaction whose two ends fall within two bins then the corresponding two nodes are connected with an edge. Instead of counting the number of interchromosomal interactions between bins, we are more concerned about their presence or absence because of the scarcity and variability of interchromosomal interactions in single cells. Therefore, an unweighted network is constructed for each cell.
Second, we develop a measurement to quantify how statistically frequent for an edge to be detected among single cells. To avoid an overestimation of this measurement and therefore reduce false positives, we first remove nodes without any intrachromosomal and interchromosomal interactions among all cells to narrow down the search space of edges, which originally is all node pairs of different chromosomes. Assume the number of edges in the edge search space is M, the number of single cells is N, and the number of interchromosomal interactions for cell i is represented as ni. Then represents the probability for cell i to have an edge between two nodes of different chromosomes. If a given edge is observed in t cells, we can use the following equations to calculate its p-value.
| 1 |
| 2 |
| 3 |
Similar to previous research [27, 54, 55], in Eq. 1 the binomial distribution is applied to estimate the p-value that reflects how likely it is for an edge to be observed in at least a given number of cells among all single cells. The rationality behind the selection of the binomial distribution is assuming whether there is an edge between two nodes of different chromosomes is a Bernoulli trial, the binomial distribution can capture edges that appear so frequent in multiple single cells that they reach statistical significance among all single cells. These frequent edges can only be detected in scHi-C experiments instead of bulk Hi-C experiments because subtle single-cell level information is pooled in bulk Hi-C experiments. Equation 2 is used to quantify the probability of an edge with all cells considered, which is determined by a function in Eq. 3. Users can configure the selection of these functions through a parameter. For scHi-C experiments with larger genomes or low sequencing depths, it is recommended to use max to select regions with highly statistically frequent interchromosomal interactions; therefore fewer regions would be selected. To the contrary, min is applied to select more regions. For scHi-C experiments with smaller genomes or high sequencing depths, min increases the odds for some regions to be selected while max may find nothing. mean is a balance between max and min, so the number of identified regions falls between them.
At last, p-values are adjusted by the Bonferroni correction and a user provided p-value cutoff, e.g. 0.05, is applied to select regions associated with statistically frequent interchromosomal interactions.
Data
To demonstrate that our proposed tool can be used to identify interesting structural regions, we use data from two existing scHi-C studies as our input data sets.
The first study [56] investigated the cell-cycle dynamics of chromosomal organization at single-cell resolution. The authors processed single F1 hybrid 129 × Castaneus mouse embryonic stem cells (mESCs) grown in 2i media using 1.5 million reads per cell on average. They analyzed 1,171 cells with fluorescence-activiated cell sorting, which labeled these cells to different cell-cycle phases based on levels of the DNA replication marker geminin and DNA content. Among them, 280 cells with a prefix of 1CDX1 were labeled as G1 phase; 303 cells with a prefix of 1CDX2 were labeled as Early-S phase; 262 cells with a prefix of 1CDX3 were labeled as Mid-S phase; 326 cells with a prefix of 1CDX4 were labeled as Late-S phase. We treat cells of different cell-cycle phases separately and feed them as inputs of our tool respectively. Therefore we identify regions with statistically frequent interchromosomal interactions for different cell-cycle phases.
The second one [50] developed a single-nucleus Hi-C protocol which provides >10-fold more contacts per cell than the previous method [49] to investigate chromatin organization at oocyte-to-zygote transition in mice. There are 40 transcriptionally active oocytes labeled as non-surrounded nucleolus (NSN), 76 transcriptionally inactive oocytes labeled as surrounded nucleolus (SN), 30 maternal nuclei from zygotes and 24 paternal nuclei from zygotes. Maternal and paternal nuclei are extracted from predominantly G1 phase zygotes.
Results and discussion
Both data sets have single cells/nuclei of four conditions, therefore we run the proposed tool on single cells/nuclei of each condition respectively. Since the genomes used in the two experiments are large and sequencing read coverages are low, to accumulate sufficient interchromosomal interactions in a bin, we set the bin size to 500 kilobases (kb), which is also used in other existing studies [55, 57]. We first show that our tool can identify regions with statistically frequent interchromosomal interactions, then demonstrate that our tool is flexible to different configurations, which support sliding windows for region diversity, different functions to estimate the probability of having an edge between two nodes thereby providing adaptability of identified regions, and a configuration of different bin sizes e.g. 500kb VS 1 megabases (Mb).
Usability of identifying interesting regions
To demonstrate the usability of our proposed method, we first display identified regions in visualization, then compare the identified regions and at last carry out enrichment analysis with other genomics features such as CTCF binding sites and enhancers etc.
Identification of statistically frequent regions
In Fig. 2, identified regions associated with statistically frequent interchromosomal interactions among single cells of the cell-cycle data set are visualized in Circos [58]. The max function is configured for our method. The banded ideograms are mouse chromosomes (1-19, X and Y) and the black lines between them are interchromosomal interactions and the ends of these lines correspond to identified regions in chromosomes. Figure 2a shows the results of single cells of G1 phase; Fig. 2b shows the results of single cells of Early-S phase; Fig. 2c shows the results of single cells of Mid-S phase; and Fig. 2d shows the results of single cells of Late-S phase.
Fig. 2.
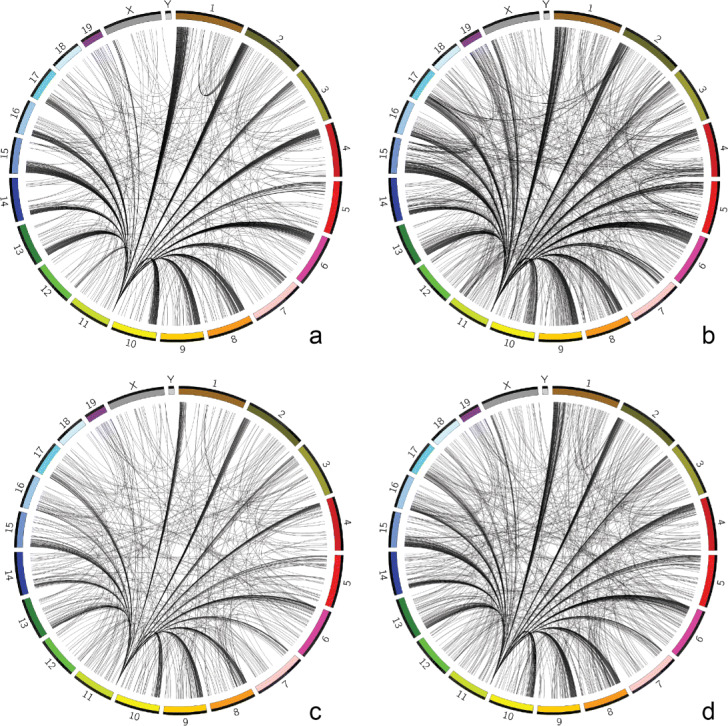
Identified regions of the cell-cycle data set. Visualizing genome-wide identified regions and their interchromosomal interactions of the cell-cycle data set with an adjusted p-value cutoff of 0.05 in Circos plots. a single cells of G1 phase; b single cells of Early-S phase; c single cells of Mid-S phase; d single cells of Late-S phase
Among all four Circos plots, there is an apparent common hub in chromosome 11 (between 3Mb and 3.5Mb) whose interchromosomal interactions are highly enriched. The finding of this hub is corroborated by previous research with bulk Hi-C experiments to study interchromosomal contact networks in mammalian genomes [55]. They also discovered this hub in the mouse genome. Our finding confirms the hub’s existence at single-cell level and rules out the possibility that its existence is solely contributed by very few cells with a large amount of interchromosomal interactions in the region. In addition, these four Circos plots are similar but not exactly the same, which means single cells of different cell phases share some interchromosomal interactions but also have some variabilities on interchromosomal interactions.
In Fig. 3, identified regions associated with statistically frequent interchromosomal interactions among single cells/nuclei of the oocyte-to-zygote data set are visualized. Figure 3a shows the results of single oocytes labeled as NSN; Fig. 3b shows the results of single oocytes labeled as SN; Fig. 3c shows the results of single maternal nuclei from zygotes; and Fig. 3d shows the results of single paternal nuclei from zygotes. Our tool reports much fewer regions on this data set and there is no hub. The absence of the hub may be partly because of cell discrepancies on cell types and cell cycles. To be more specific, in the second research, oocytes and maternal/paternal nuclei from zygotes only contain a single set of chromosomes. However, for the chromosome 11 from 3Mb to 3.5Mb, there are comparatively more interchromosomal interactions among all four Circos plots. Additionally, a similar interchromosomal interaction pattern is observed: there are some shared interchromosomal interactions but there are also some variabilities at single-cell resolution.
Fig. 3.
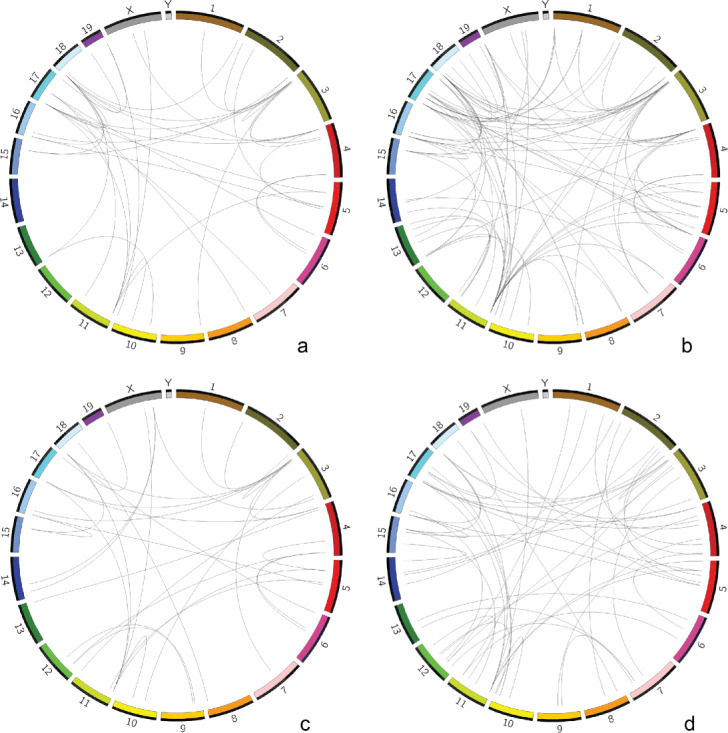
Identified regions of the oocyte-to-zygote data set. Visualizing genome-wide identified regions and their interchromosomal interactions of the oocyte-to-zygote data set with an adjusted p-value cutoff of 0.05 in Circos plots. a single oocytes labeled as NSN; b single oocytes labeled as SN; c maternal nuclei from zygotes; d paternal nuclei from zygotes
Pairwise comparisons of identified regions
For the cell-cycle data set, we compare the identified regions from single cells of different phases and examine the similarity and dissimilarity. In Table 1, single cells of different phases share a lot of common regions. There are some unique regions in each phased single cells. All pairs have more common regions than unique regions except the comparison between Early-S and Mid-S. Because the number of common regions is limited by the identified regions from single cells at Mid-S phase and single cells at Early-S phase report the most identified regions.
Table 1.
Pairwise comparisons of the cell-cycle data set
| Comparison | Common | Unique in former | Unique in latter |
|---|---|---|---|
| G1 VS Early-S | 757 | 219 | 569 |
| G1 VS Mid-S | 526 | 450 | 198 |
| G1 VS Late-S | 708 | 268 | 335 |
| Early-S VS Mid-S | 595 | 731 | 129 |
| Early-S VS Late-S | 767 | 559 | 276 |
| Mid-S VS Late-S | 597 | 127 | 446 |
We also compare the identified regions from single cells of the oocyte-to-zygote data set. In Table 2, single cells of different conditions share some regions and there are more unique regions than common regions. This phenomenon seems inconsistent with what we have observed in the cell-cycle data set. But it does make sense and reflects the different types of single cells/nuclei used in their experiments. When identified regions from oocytes labeled NSN are compared with the ones from other cells/nuclei, the oocytes labeled SN share the most common regions because both of them are the same type of cells and their common regions are limited by the identified regions from oocytes labeled NSN; single maternal nuclei share more regions than single paternal nuclei because oocytes and single maternal nuclei are both from females while single paternal nuclei are from males. The same reason can also be applied to explain why oocytes labeled SN share more common regions with single maternal nuclei than single paternal nuclei. At last, single maternal nuclei and single paternal nuclei share the fewest common regions because some are from females and the others are from males.
Table 2.
Pairwise comparisons of the oocyte-to-zygote data set
| Comparison | Common | Unique in former | Unique in latter |
|---|---|---|---|
| NSN VS SN | 35 | 2 | 49 |
| NSN VS maternal | 18 | 19 | 15 |
| NSN VS paternal | 15 | 22 | 36 |
| SN VS maternal | 21 | 63 | 12 |
| SN VS paternal | 19 | 65 | 32 |
| maternal VS paternal | 13 | 20 | 38 |
Enrichment analysis of identified regions
To improve the interpretation of identified regions, we carry out enrichment analysis of identified regions with genomic features, which are available in the cell-cycle data set. As there are too many identified regions in the data set, we select top ranked regions/nodes according to the numbers of statistically frequent unweighted edges with a cutoff (≥3 except ≥4 for single cells at Early-S phase because there are too many top regions). Therefore we obtain 16 regions for single cells at G1 phase, 37 regions for single cells at Early-S phase, 34 regions for single cells at Mid-S phase and 47 regions for single cells at Late-S phase. Genomic features of mESC cell line are downloaded from this paper [59] and they are CTCF binding sites, enhancer sites, H3K4me3 peaks, H3K27ac peaks and Pol II peaks.
For the above selected regions of each phase, the numbers of genomic features are counted respectively. Then we ranomly select the same number of regions and count the numbers of genomic features falling into these randomly selected regions respectively. We carry out this randomization strategy 50,000 times and therefore we obtain empirical background samples for each genomic feature. We calculate the z-score for each genomic feature. In Table 3, most of genomic features are enriched (≥1.97, which corresponds to 0.05 in p-value) except enhancer. What is more important, for single cells at Early-S phase, all the genomic features are highly enriched. (When ≥3 is used as the cutoff, the results become more enriched.) H3K4me3 and H3K27ac are active gene transcirption marks. Pol II plays very important roles in gene transcription. An enhancer increases the likelihood of gene transcription. CTCF plays important rols in chromatin structure and insolates the spread of heterochromatin. Early-S phase corresponds to the commencement of DNA replication. These genomic features seems working coordinately to facilitate the initialization of DNA replication.
Table 3.
Identified Regions’ Enrichment Analysis of the cell-cycle data set
| Input | CTCF | enhancer | H3K4me3 | H3K27ac | Pol II |
|---|---|---|---|---|---|
| G1 | 2.82 | 1.05 | 1.75 | 2.63 | 2.48 |
| Early-S | 10.86 | 9.81 | 12.48 | 12.05 | 12.58 |
| Mid-S | 2.81 | 1.48 | 3.08 | 2.74 | 3.64 |
| Late-S | 3.37 | 1.74 | 4.33 | 4.36 | 5.05 |
Flexibility of configurations
To make our tool flexible to accommodate different scHi-C experiments, we support different configurations, which include sliding windows for region diversity, edge probability functions for adjustability of identified regions and different bin sizes.
Configuration of sliding windows
By default, our tool divides chromosomes into bins from the first bases of chromosomes to the last ones, which limits the starting and ending positions of regions. To overcome this limitation, our tool supports a sliding window strategy by moving bins toward the last bases certain bases (e.g. 100kb). It lets users decide where their regions’ starting and ending positions through a parameter. In Table 4, we adopt four sliding windows of sizes of 100kb, 200kb, 300kb and 400kb and compare the identified regions with the ones by default (no sliding window). If identified regions mediated by some interchromosomal interactions from the no sliding window condition overlap with identified regions from a sliding window condition at both ends, we treat these regions as common identified regions; otherwise they are different. Therefore, we can calculate the common identified regions between no sliding window and sliding windows. In Table 4, we conclude that most identifed regions between no sliding window and sliding windows are common because some shared interchromosomal interactions fall into these regions. But as these common regions’ starting and ending positions are different, our tool diversifies the identified regions to users. What is more interesting is the single cells at Early-S phase share the fewest identified regions between no sliding window and sliding windows of different sizes. As DNA synthesis commences at Early-S phase, interchromosomal interactions may vary or involve in DNA synthesis initialization activites more at this phase than other phases. In Table 5 of the oocyte-to-zygote data set, we can reach the same conclusion that most identified regions are common between no sliding window and sliding windows of different sizes and meanwhile there are some different regions.
Table 4.
Overlapping identified regions of the cell-cycle data set with no sliding window and sliding windows of different sizes
| Input Data | 100kb | 200kb | 300kb | 400kb |
|---|---|---|---|---|
| G1 | 92.11% | 92.01% | 92.01% | 95.49% |
| Early-S | 85.52% | 86.05% | 86.73% | 89.22% |
| Mid-S | 90.33% | 89.92% | 91.16% | 93.65% |
| Late-S | 93.19% | 91.08% | 91.66% | 94.44% |
Table 5.
Overlapping identified regions of the oocyte-to-zygote data set with no sliding window and sliding windows of different sizes
| Input Data | 100kb | 200kb | 300kb | 400kb |
|---|---|---|---|---|
| oocyte NSN | 100% | 92.01% | 92.01% | 95.49% |
| oocyte SN | 86.90% | 89.29% | 89.29% | 92.86% |
| pronucleus maternal | 93.94% | 93.94% | 90.91% | 100% |
| pronucleus paternal | 94.12% | 90.20% | 90.20% | 92.16% |
Configuration of edge probability functions
Our proposed tool supports three functions, max, mean and min, to estimate the probability of an edge between two nodes of different chromosomes, therefore improving adjustability of identified regions. In Table 6 of the cell-cycle data set and Table 7 of the oocyte-to-zygote data set, our tool configured with the max function identifies the fewest regions; our tool configured with the min function identifies the most regions and our tool configured with the mean funciton falls between them. This is because if we fix other variables except p in Eq. 1, a large p entails a large p-value and a small p entails a small p-value. As we have explained in the second to last paragrpah of Method, users can select these functions according to the sizes of genomes and sequencing depths used in their experiments. Therefore, our proposed tool provides adaptability of identified regions.
Table 6.
Number of identified regions of the cell-cycle data set with edge probability functions
| Input Data | max | mean | min |
|---|---|---|---|
| G1 | 976 | 1651 | 2133 |
| Early-S | 1326 | 2579 | 7714 |
| Mid-S | 724 | 1833 | 2991 |
| Late-S | 1043 | 1999 | 6058 |
Table 7.
Number of identified regions of the oocyte-to-zygote data set with edge probability functions
| Input Data | max | mean | min |
|---|---|---|---|
| oocyte NSN | 37 | 79 | 199 |
| occyte SN | 84 | 229 | 1846 |
| pronucleus maternal | 33 | 50 | 268 |
| pronucleus paternal | 51 | 51 | 274 |
Configuration of bin sizes
Finally, our tool also supports different bin sizes. As scHi-C experiments have low read coverages and scarce interchromosomal interactions, we need to use large bin sizes to accumulate sufficient interchromosomal interactions in a bin. We run our tool with bin_size=1Mb on the two data sets and compare the identified regions with the ones of bin_size=500kb. We find that the identified regions of bin_size=500kb and bin_size=1Mb are quite similar for most single cells except the Early-S phased single cells in the cell-cycle data set. In Fig. 4b of bin_size=1Mb, the hub of the chromosome 11 at 3Mb becomes less obvious as it is overshadowed by enrichment of other interchromosomal interactions because of the increased bin size and single cells of this particular cell phase. Therefore, different bin sizes may affect the identified regions.
Fig. 4.

Comparing identified regions of Early-S phased single cells with different bin sizes. a bin_size=500kb b bin_size=1Mb
Conclusion
In this paper, we introduce a computational method to identify regions associated with statistically frequent interchromosomal interactions at single-cell resolution and implement it as an open source tool, which is the first serving the purpose to the best of our knowledge. Its workflow includes network construction, binomial statistical measurement calculation and region selection. We demonstrate its usability on two existing scHi-C data. On the cell-cycle data set, the tool discovers a hub in the mouse chromosome 11 from 3Mb to 3.5Mb, which is endorsed by a previous study on interchromosomal contact networks with bulk Hi-C experiments. On the oocyte-to-zygote data set, there is no apparent hub at the region, but comparatively interchromosomal interactions are enriched. Identified regions’ pairwise comparisons show that our method identifies common regions between different data sets and also reflects the true dissimilarity such as different cell types. Identified regions’ enrichment analysis helps improve the interpretation of top ranked identified regions and these genomic features are highly enriched for single cells at Early-S phase, which implies our top ranked regions may be functionally important. We also exhibit our proposed tool’s flexibility on configurations, which support sliding windows for diverse regions, edge probability functions for adjustable regions and different bin sizes. Overall, it will be a useful tool for analyzing scHi-C interchromosomal interactions.
Due to low sequencing depths of scHi-C experiments and the paucity of interchromosomal interactions, identifying high resolution regions of several kilobases (e.g. 8kb) is extremely difficult. Our tool can run with this resolution but due to the limitation of scHi-C data, it can’t identify any regions passing the statistical tests. We will try to mitigate this problem by imputing high-resolution interchromosomal interactions with data of other experiments such as interchromosomal interactions from bulk Hi-C experiments. In addition, further research is needed to improve the signal-to-noise ratio for scHi-C experiments.
Acknowledgements
Some experiments were conducted on NDSU Center for Computationally Assisted Science and Technology (CCAST).
About this supplement
This article has been published as part of BMC Bioinformatics Volume 21 Supplement 14, 2020: Selected original articles from the Sixth International Workshop on Computational Network Biology: Modeling, Analysis, and Control (CNB-MAC 2019): bioinformatics. The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-21-supplement-14.
Abbreviations
- Hi-C
chromosome conformation capture with high-throughput sequencing
- TAD
topologically associating domain
- scHi-C
single-cell chromosome conformation capture with high-throughput sequencing
- kb
kilobases
- Mb
megabases
- mESC
mouse embryonic stem cell
- NSN
non-surrounded nucleolus
- SN
surrounded nucleolus
Authors’ contributions
Conceptualization: LL; methodology: LL; software: CB; figures: CB and LL; investigation: CB and LL; writing: LL; funding acquisition: LL. All author(s) read and approved the final manuscript.
Funding
This research was supported by Seed Award from ND EPSCoR (FAR0030613) and NDSU Startup Award to LL. Publications costs are funded by NDSU Startup Award to LL.
Availability of data and materials
For the implementaion details of our tool, please check out it at https://github.com/bignetworks2019/Inter-chromosomal-interactions. Currently it supports the following genomes, mm9, mm10, hg18 and hg19. It can be easily extended to other organisms.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Lajoie BR, Dekker J, Kaplan N. The hitchhiker’s guide to hi-c analysis: practical guidelines. Methods. 2015;72:65–75. doi: 10.1016/j.ymeth.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarnataro S, Chiariello AM, Esposito A, Prisco A, Nicodemi M. Structure of the human chromosome interaction network. PloS ONE. 2017;12(11):0188201. doi: 10.1371/journal.pone.0188201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lin D, Bonora G, Yardımcı GG, Noble WS. Computational methods for analyzing and modeling genome structure and organization. Wiley Interdiscip Rev Syst Biol Med. 2019;11(1):1435. doi: 10.1002/wsbm.1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wingett S, Ewels P, Furlan-Magaril M, Nagano T, Schoenfelder S, Fraser P, Andrews S. Hicup: pipeline for mapping and processing hi-c data. F1000Research. 2015;4:1310. doi: 10.12688/f1000research.7334.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Imakaev M, Fudenberg G, McCord RP, Naumova N, Goloborodko A, Lajoie BR, Dekker J, Mirny LA. Iterative correction of hi-c data reveals hallmarks of chromosome organization. Nat Methods. 2012;9(10):999. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Castellano G, Le Dily F, Pulido AH, Beato M, Roma G. Hic-inspector: a toolkit for high-throughput chromosome capture data. bioRxiv. 2015. 10.1101/020636.
- 7.Hwang Y-C, Lin C-F, Valladares O, Malamon J, Kuksa PP, Zheng Q, Gregory BD, Wang L-S. Hippie: a high-throughput identification pipeline for promoter interacting enhancer elements. Bioinformatics. 2014;31(8):1290–2. doi: 10.1093/bioinformatics/btu801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmid MW, Grob S, Grossniklaus U. Hicdat: a fast and easy-to-use hi-c data analysis tool. BMC Bioinformatics. 2015;16(1):277. doi: 10.1186/s12859-015-0678-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Servant N, Varoquaux N, Lajoie BR, Viara E, Chen C-J, Vert J-P, Heard E, Dekker J, Barillot E. Hic-pro: an optimized and flexible pipeline for hi-c data processing. Genome Biol. 2015;16(1):259. doi: 10.1186/s13059-015-0831-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sauria ME, Phillips-Cremins JE, Corces VG, Taylor J. Hifive: a tool suite for easy and efficient hic and 5c data analysis. Genome Biol. 2015;16(1):237. doi: 10.1186/s13059-015-0806-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Durand NC, Shamim MS, Machol I, Rao SS, Huntley MH, Lander ES, Aiden EL. Juicer provides a one-click system for analyzing loop-resolution hi-c experiments. Cell Syst. 2016;3(1):95–98. doi: 10.1016/j.cels.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lazaris C, Kelly S, Ntziachristos P, Aifantis I, Tsirigos A. Hic-bench: comprehensive and reproducible hi-c data analysis designed for parameter exploration and benchmarking. BMC Genomics. 2017;18(1):22. doi: 10.1186/s12864-016-3387-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang T, Zhang F, Yardımcı GG, Song F, Hardison RC, Noble WS, Yue F, Li Q. Hicrep: assessing the reproducibility of hi-c data using a stratum-adjusted correlation coefficient. Genome Res. 2017;27(11):1939–49. doi: 10.1101/gr.220640.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ursu O, Boley N, Taranova M, Wang YR, Yardimci GG, Stafford Noble W, Kundaje A. Genomedisco: A concordance score for chromosome conformation capture experiments using random walks on contact map graphs. Bioinformatics. 2018;34(16):2701–7. doi: 10.1093/bioinformatics/bty164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan K-K, Yardımcı GG, Yan C, Noble WS, Gerstein M. Hic-spector: a matrix library for spectral and reproducibility analysis of hi-c contact maps. Bioinformatics. 2017;33(14):2199–201. doi: 10.1093/bioinformatics/btx152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lieberman-Aiden E, Van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326(5950):289–93. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485(7398):376. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Filippova D, Patro R, Duggal G, Kingsford C. Identification of alternative topological domains in chromatin. Algoritm Mol Biol. 2014;9(1):14. doi: 10.1186/1748-7188-9-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, et al. A 3d map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159(7):1665–80. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lévy-Leduc C, Delattre M, Mary-Huard T, Robin S. Two-dimensional segmentation for analyzing hi-c data. Bioinformatics. 2014;30(17):386–92. doi: 10.1093/bioinformatics/btu443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Serra F, Baù D, Goodstadt M, Castillo D, Filion GJ, Marti-Renom MA. Automatic analysis and 3d-modelling of hi-c data using tadbit reveals structural features of the fly chromatin colors. PLoS Comput Biol. 2017;13(7):1005665. doi: 10.1371/journal.pcbi.1005665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weinreb C, Raphael BJ. Identification of hierarchical chromatin domains. Bioinformatics. 2015;32(11):1601–9. doi: 10.1093/bioinformatics/btv485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shin H, Shi Y, Dai C, Tjong H, Gong K, Alber F, Zhou XJ. Topdom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Res. 2015;44(7):70. doi: 10.1093/nar/gkv1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanizawa H, Iwasaki O, Tanaka A, Capizzi JR, Wickramasinghe P, Lee M, Fu Z, Noma K. -i. Mapping of long-range associations throughout the fission yeast genome reveals global genome organization linked to transcriptional regulation. Nucleic Acids Res. 2010;38(22):8164–77. doi: 10.1093/nar/gkq955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen C-A, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013;503(7475):290. doi: 10.1038/nature12644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ay F, Bailey TL, Noble WS. Statistical confidence estimation for hi-c data reveals regulatory chromatin contacts. Genome Res. 2014;24(6):999–1011. doi: 10.1101/gr.160374.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, Noble WS. A three-dimensional model of the yeast genome. Nature. 2010;465(7296):363. doi: 10.1038/nature08973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ay F, Bunnik EM, Varoquaux N, Bol SM, Prudhomme J, Vert J-P, Noble WS, Le Roch KG. Three-dimensional modeling of the p. falciparum genome during the erythrocytic cycle reveals a strong connection between genome architecture and gene expression. Genome Res. 2014;24(6):974–88. doi: 10.1101/gr.169417.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Varoquaux N, Ay F, Noble WS, Vert J-P. A statistical approach for inferring the 3d structure of the genome. Bioinformatics. 2014;30(12):26–33. doi: 10.1093/bioinformatics/btu268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Z, Li G, Toh K-C, Sung W-K. 3d chromosome modeling with semi-definite programming and hi-c data. J Comput Biol. 2013;20(11):831–46. doi: 10.1089/cmb.2013.0076. [DOI] [PubMed] [Google Scholar]
- 31.Ben-Elazar S, Yakhini Z, Yanai I. Spatial localization of co-regulated genes exceeds genomic gene clustering in the saccharomyces cerevisiae genome. Nucleic Acids Res. 2013;41(4):2191–201. doi: 10.1093/nar/gks1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baù D, Sanyal A, Lajoie BR, Capriotti E, Byron M, Lawrence JB, Dekker J, Marti-Renom MA. The three-dimensional folding of the α-globin gene domain reveals formation of chromatin globules. Nat Struct Mol Biol. 2011;18(1):107. doi: 10.1038/nsmb.1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lesne A, Riposo J, Roger P, Cournac A, Mozziconacci J. 3d genome reconstruction from chromosomal contacts. Nat Methods. 2014;11(11):1141. doi: 10.1038/nmeth.3104. [DOI] [PubMed] [Google Scholar]
- 34.Rousseau M, Fraser J, Ferraiuolo MA, Dostie J, Blanchette M. Three-dimensional modeling of chromatin structure from interaction frequency data using markov chain monte carlo sampling. BMC Bioinformatics. 2011;12(1):414. doi: 10.1186/1471-2105-12-414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giorgetti L, Galupa R, Nora EP, Piolot T, Lam F, Dekker J, Tiana G, Heard E. Predictive polymer modeling reveals coupled fluctuations in chromosome conformation and transcription. Cell. 2014;157(4):950–63. doi: 10.1016/j.cell.2014.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hu M, Deng K, Qin Z, Dixon J, Selvaraj S, Fang J, Ren B, Liu JS. Bayesian inference of spatial organizations of chromosomes. PLoS Comput Biol. 2013;9(1):1002893. doi: 10.1371/journal.pcbi.1002893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang S, Xu J, Zeng J. Inferential modeling of 3d chromatin structure. Nucleic Acids Res. 2015;43(8):54–54. doi: 10.1093/nar/gkv100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Peng C, Fu L-Y, Dong P-F, Deng Z-L, Li J-X, Wang X-T, Zhang H-Y. The sequencing bias relaxed characteristics of hi-c derived data and implications for chromatin 3d modeling. Nucleic Acids Res. 2013;41(19):183. doi: 10.1093/nar/gkt745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trieu T, Cheng J. Large-scale reconstruction of 3d structures of human chromosomes from chromosomal contact data. Nucleic Acids Res. 2014;42(7):52. doi: 10.1093/nar/gkt1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lun AT, Smyth GK. diffhic: a bioconductor package to detect differential genomic interactions in hi-c data. BMC Bioinformatics. 2015;16(1):258. doi: 10.1186/s12859-015-0683-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu L, Ruan J. Utilizing networks for differential analysis of chromatin interactions. J Bioinforma Comput Biol. 2017;15(06):1740008. doi: 10.1142/S021972001740008X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhou X, Lowdon RF, Li D, Lawson HA, Madden PA, Costello JF, Wang T. Exploring long-range genome interactions using the washu epigenome browser. Nat Methods. 2013;10(5):375. doi: 10.1038/nmeth.2440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paulsen J, Sandve GK, Gundersen S, Lien TG, Trengereid K, Hovig E. Hibrowse: multi-purpose statistical analysis of genome-wide chromatin 3d organization. Bioinformatics. 2014;30(11):1620–2. doi: 10.1093/bioinformatics/btu082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Durand NC, Robinson JT, Shamim MS, Machol I, Mesirov JP, Lander ES, Aiden EL. Juicebox provides a visualization system for hi-c contact maps with unlimited zoom. Cell Syst. 2016;3(1):99–101. doi: 10.1016/j.cels.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang Y, Song F, Zhang B, Zhang L, Xu J, Kuang D, Li D, Choudhary MN, Li Y, Hu M, et al. The 3d genome browser: a web-based browser for visualizing 3d genome organization and long-range chromatin interactions. Genome Biol. 2018;19(1):151. doi: 10.1186/s13059-018-1519-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Henry VJ, Bandrowski AE, Pepin A-S, Gonzalez BJ, Desfeux A. Omictools: an informative directory for multi-omic data analysis. Database. 2014;2014:069. doi: 10.1093/database/bau069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xiong K, Ma J. Revealing hi-c subcompartments by imputing inter-chromosomal chromatin interactions. Nat Commun. 2019;10(1):5069. doi: 10.1038/s41467-019-12954-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dai Y, Li C, Pei G, Dong X, Ding G, Zhao Z, Li Y, Jia P. Multiple transcription factors contribute to inter-chromosomal interaction in yeast. BMC Syst Biol. 2018;12(8):140. doi: 10.1186/s12918-018-0643-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nagano T, Lubling Y, Stevens TJ, Schoenfelder S, Yaffe E, Dean W, Laue ED, Tanay A, Fraser P. Single-cell hi-c reveals cell-to-cell variability in chromosome structure. Nature. 2013;502(7469):59. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Flyamer IM, Gassler J, Imakaev M, Brandão HB, Ulianov SV, Abdennur N, Razin SV, Mirny LA, Tachibana-Konwalski K. Single-nucleus hi-c reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature. 2017;544(7648):110. doi: 10.1038/nature21711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ramani V, Deng X, Qiu R, Gunderson KL, Steemers FJ, Disteche CM, Noble WS, Duan Z, Shendure J. Massively multiplex single-cell hi-c. Nat Methods. 2017;14(3):263. doi: 10.1038/nmeth.4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Han J, Cheng H, Xin D, Yan X. Frequent pattern mining: current status and future directions. Data Min Knowl Disc. 2007;15(1):55–86. [Google Scholar]
- 53.Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, Leeb M, Wohlfahrt KJ, Boucher W, O’Shaughnessy-Kirwan A, et al. 3d structures of individual mammalian genomes studied by single-cell hi-c. Nature. 2017;544(7648):59. doi: 10.1038/nature21429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kruse K, Sewitz S, Babu MM. A complex network framework for unbiased statistical analyses of dna–dna contact maps. Nucleic Acids Res. 2012;41(2):701–10. doi: 10.1093/nar/gks1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kaufmann S, Fuchs C, Gonik M, Khrameeva EE, Mironov AA, Frishman D. Inter-chromosomal contact networks provide insights into mammalian chromatin organization. PloS ONE. 2015;10(5):0126125. doi: 10.1371/journal.pone.0126125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nagano T, Lubling Y, Várnai C, Dudley C, Leung W, Baran Y, Cohen NM, Wingett S, Fraser P, Tanay A. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 2017;547(7661):61. doi: 10.1038/nature23001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu J, Lin D, Yardımcı GG, Noble WS. Unsupervised embedding of single-cell hi-c data. Bioinformatics. 2018;34(13):96–104. doi: 10.1093/bioinformatics/bty285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19(9):1639–45. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen Y, Yue F, McCleary DF, Ye Z, Edsall L, Kuan S, Wagner U, Dixon J, Lee L, Lobanenkov VV, et al. A map of the cis-regulatory sequences in the mouse genome. Nature. 2012;488(7409):116. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
For the implementaion details of our tool, please check out it at https://github.com/bignetworks2019/Inter-chromosomal-interactions. Currently it supports the following genomes, mm9, mm10, hg18 and hg19. It can be easily extended to other organisms.