

Abstract
Few nucleoside-derived natural products have been identified from animals, despite the ubiquity of nucleosides in living organisms. Here, we use a combination of synthesis and the emerging electron microscopy technique microcrystal electron diffraction to determine the structures of several N3-(β-glucopyranosyl)uric acid derivatives in Caenorhabditis elegans. These noncanonical gluconucleosides further integrate an ascaroside moiety, for which we present a shortened synthetic route. The production of a phosphorylated gluconucleoside is influenced by evolutionarily conserved insulin signaling.
Graphical Abstract

The nematodes Caenorhabditis elegans and Pristionchus pacificus are simple but powerful model systems for human physiology and biochemistry.1–4 Over the past decade, phenotypic screens of mutant collections and comparative metabolomic studies have led to the discovery of an extensive network of small molecule signals that plays a central role in the life history of C. elegans, regulating, e.g., aging, behavior, and development.5,6 These signaling molecules are derived from combinatorial assembly of building blocks from all major primary metabolic pathways, including nucleoside metabolism, demonstrating its importance beyond the biosynthesis of the canonical nucleosides in DNA and the different types of RNA.
The first example for this family of unusual nucleoside derivatives is the signaling molecule npar#1 (1), which incorporates a xyloadenosine and a glycoside of the dideoxysugar paratose (Figure 1a).1 npar#1 (1) and other ascaroside derivatives play a central role in regulating development in P. pacificus and C. elegans, respectively.5,6 Comparative metabolomics of peroxisomal β-oxidation (pβo) mutants and wild-type C. elegans7 revealed several additional nucleoside derivatives, including nuclas#33 (2), incorporating a 6-threonylcarbamoyl-adenosine (t6A) moiety esterified at its 3′-O-ribosyl position to ascaroside ascr#3, as well as gluconucleoside, pugl#1 (3), featuring an N9 anomeric linkage of 2-methylthio-N6-isopentenyladenine to glucose.
Figure 1.

(a) Unusual purine nucleoside and gluconucleosides from C. elegans and other nematodes. (b) Structures of uglas#1 and uglas#11 proposed on the basis of MS2 analysis and analogy to other glucosides and gluconucleosides in C. elegans,7 with dashed bonds referring to tentative assignments. N3-(Ribofuranosyl)uric acid (6) has been reported from bovine blood.8–11 Amounts of uglas#1 and uglas#11 relative to ascr#1 (7) were quantified in the wild type and mutants of the insulin/IGF-1 receptor daf-2 (also see Figure S1). ***p < 0.001.
Our attention was drawn to several other gluconucleoside derivatives from the pβo mutant screen that appeared to incorporate uric acid. These metabolites, uglas#1 (4), and a phosphorylated analogue named uglas#11 (5) were proposed to feature an N9-linked uric acid moiety, in analogy to gluconucleoside pugl#1 (3). Although uric acid is ubiquitous as a product of purine degradation, few natural small molecules or nucleoside derivatives incorporating uric acid have been described. The only known uric acid incorporating nucleoside, proposed to represent N3-(ribofuranosyl)uric acid (6), was isolated in the 1960s from bovine blood and hypothesized to function as a soluble carrier of uric acid.8–11 Uric acid serves as a major antioxidant capturing reactive oxygen species that are involved in diverse signaling pathways, regulating, e.g., immune function and aging.12 Moderate levels of uric acid supplementation were recently found to increase the life span in C. elegans, possibly by activating DAF-16/FOXO downstream of insulin/IGF-1 signaling.13 Analyzing the levels of uric acid-derived metabolites in long-lived mutants of the insulin/IGF-1 receptor daf-2,14,15 we found that production of uglas#11 is upregulated ~10-fold specifically in the daf-2(e1368) allele, but not in the daf-2(m577) allele, relative to wild-type worms, whereas abundances of the putative building blocks glucosyl uric acid and ascr#1 (7) as well as that of other ascarosides were not significantly increased in daf-2 mutants (Figure 1b and Figure S1).
The proposed structure of uglas#1 (4) includes uric acid, glucose, and an ascaroside moiety; however, the connectivity of the different moieties could not be elucidated on the basis of MS2 spectra alone and thus remained undetermined as samples that are large enough for NMR spectroscopic characterization could not be isolated.7 To determine connectivity and obtain samples for exploring biological function, we pursued a synthetic approach that would provide access to different structural isomers. Previous work showed that under thermodynamic control, guanine glucosylation using the silyl Hilbert–Johnson method favors N9 attachment, as in RNA and DNA, for 2′-O-benzoylated substrates.16 However, when applied to the case of uric acid (8) and protected glucose (9), we obtained a mixture of three regioisomers. 1H–13C HMBC spectra of the three isomers in conjunction with chemical shift data of uric acid17 suggested that the major product represented the N3 regioisomer (10, 39%), whereas the two minor products were assigned as the N1-substituted (11, 9%) and likely N7-substituted (12, 4%) regioisomers. N9 glucosylation was not observed (Figure 2a). However, not all expected HMBC correlations could be observed in the spectra of the N1 and N7 regioisomers, due to the broad line shapes of some of the NH protons (Figure 2a and Figures S2–S4). Aminolysis of the major N3 isomer (10) as well as combined N1 and N7 isomers (11 and 12) produced samples of the three glucosyl uric acid isomers. Comparison with C. elegans exo-metabolome samples using HPLC-HRMS indicated that N3-linked isomer 13 (named gluric#1) is abundantly produced by C. elegans (Figure 2b).
Figure 2.
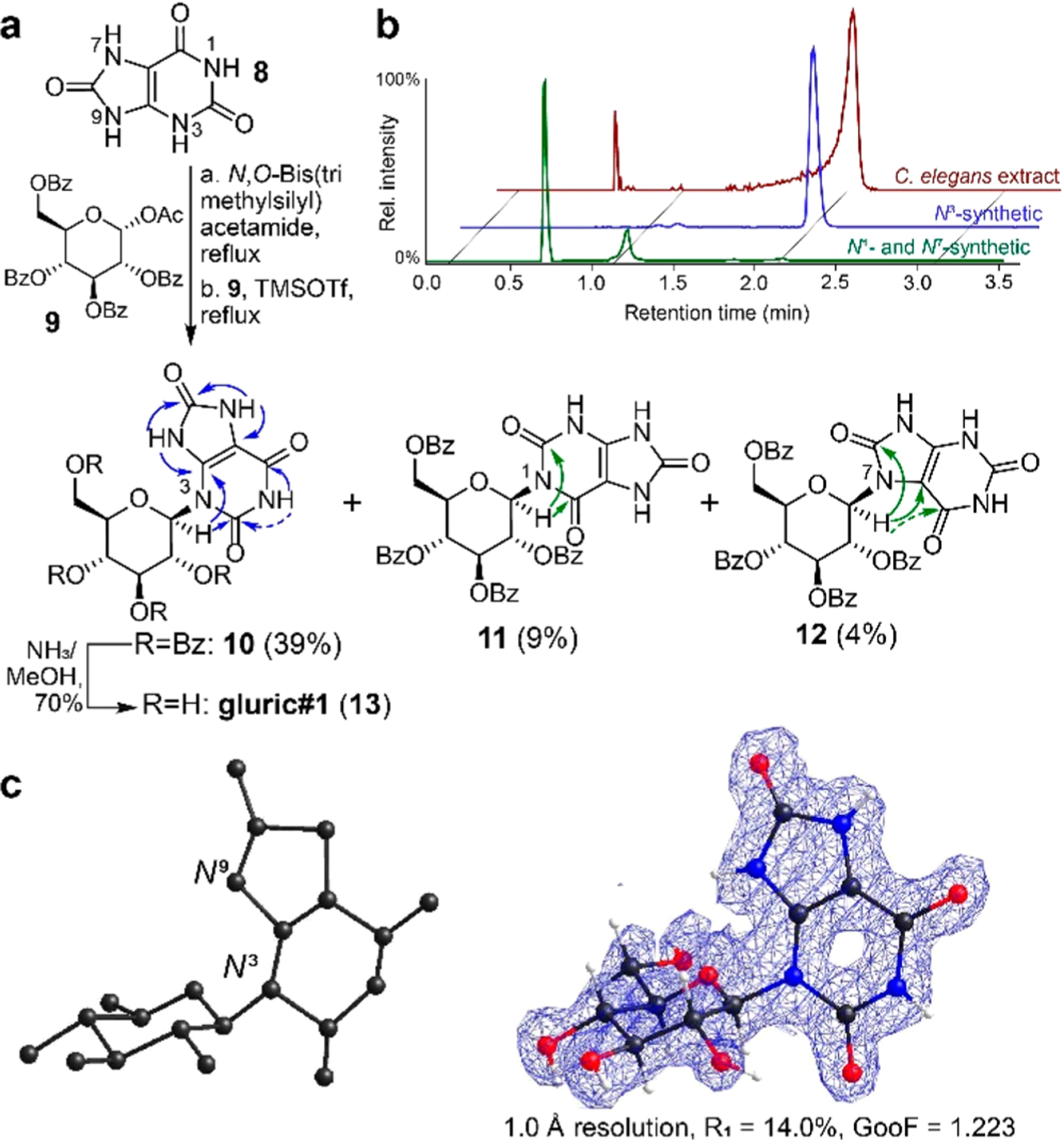
(a) Silyl Hilbert–Johnson glucosylation of uric acid (8) favors N3 substitution (10) with the N1 (11) and N7 (12) isomers as minor products. Arrows indicate relevant HMBC correlations; weak correlations are shown as dashed lines (also see Figures S2–S4). (b) Aminolysis of 10–12 and subsequent comparison with C. elegans exo-metabolome by HPLC-HRMS revealed the N3-β-isomer (13, gluric#1) as the major glucosyluric acid metabolite in C. elegans. (c) Initial MicroED structural solution (left) and final refined structure with overlaid density (right).
Given that not all regioisomers 10–12 provided complete HMBC spectra and 1H line shapes of gluric#1 (13) were too broad to obtain additional HMBC data, we sought to unambiguously confirm our structural assignment for 13 using microcrystal electron diffraction (MicroED). MicroED is an emerging cryo-electron microscopy (CryoEM) technique that has recently been demonstrated to be a powerful tool for small molecule structure elucidation but has remained largely unproven in natural product applications.18,19 Synthetic gluric#1 (13), isolated by reversed-phase flash chromatography as a white powder, was deposited on a Quantifoil holey-carbon TEM grid. Initial electron micrographs of the samples showed numerous microcrystalline domains, including prisms 1–3 μm long. Continuous rotation selected area diffraction data were collected from 104 crystals. Merging of four data sets provided a high-resolution (1.0 Å) and high-completeness direct method solution (R1 = 14.0%). Importantly, without refinement or any user input (besides the molecular formula), the initial structural solution obtained from SHELXD20 clearly confirms N3 glucosylation, despite lacking some atoms on the sugar fragment (Figure 2c and Figure S5). Anisotropic refinement of this structure allowed for placement of all atoms of gluric#1 (13), providing for unambiguous confirmation of the structure.
The preference for N3 glucosylation in our synthesis is consistent with calculated π-electron densities at purine nitrogen atoms21 and previously reported N3 glucosylation and ribosylation of uric acid using a silver-based silyl Hilbert–Johnson approach.22 These results further suggested that uglas#1 and uglas#11, like gluric#1 (13), also represent N3-β-linked gluconucleosides, in contrast to the original proposal7 and canonical N9-linked purine nucleosides in DNA and RNA, and in agreement with the connectivity in the previously reported N3-(ribofuranosyl)uric acid (6).8–11
For the synthesis of the proposed 2,4-dideoxyglycoside (ascaroside) moiety in uglas#1 and uglas#11, we employed a new, shorter route to access the key intermediate, dibenzoylated ascarylose (16).23 Methylation of α-L-rhamnose (14) followed by tosylation using p-TsCl and Bu2SnCl2 under basic conditions regioselectively afforded a 3-O-tosylated intermediate24,25 which was reduced upon treatment with NaH and LiAlH4, affording 1-O-methyl ascarylose, which was subsequently benzoylated with BzCl, furnishing 15. Optimized reaction conditions prevented the formation of a branched-chain deoxyfuranose side product derived from ring contraction, which had been reported for similar deoxygenation reactions (see Methods).26 Demethylation of 15 afforded target precursor 16, which was converted into dibenzylated ascaroside 17 as previously described.27
To determine the position of attachment of the ascaroside side chain to N3-(β-glucopyranosyl)uric acid (13), we first pursued a nonselective approach. Steglich esterification of dibenzylated ascr#1 (17)27 and gluric#1 (13) provided a mixture of three dibenzylated uglas#1 isomers, containing the 6′-O-substituted isomer (18, 40%) as the major component, which was separated from two minor isomers derived from acylation of secondary alcohols (Figure 3a).
Figure 3.

(a) Nonselective synthesis of uglas#1 isomers. (b) HPLC-HRMS ion chromatograms showing that synthetic 2′-O regioisomer 21 co-eluted with the major natural uglas#1 isomer in C. elegans metabolome extracts. In addition, the natural extracts contain small amounts of the 3′-O and 6′-O isomers, 22 and 23, respectively.
The 1H NMR spectroscopic signals of one of the two minor isomers exhibited extreme line broadening of the glucose-attached protons, likely due to slow interconversion of different conformers, which prevented full NMR spectroscopic characterization of this compound (Figure S6). However, dqfCOSY spectra processed to emphasize quickly decaying signals demonstrated 2′-O-acylation (19, 15%). The other minor isomer was identified as the 3′-O isomer (20, 8%), whereas a 4′-O-acylated isomer was not observed. Subsequent Pd/C-catalyzed hydrogenation of the 6′-O-substituted isomer (18) and a 2:1 mixture of the 2′-O (19) and 3′-O (20) isomers provided three uglas#1 isomers whose retention times and MS2 spectra were compared to that of natural uglas#1. We found that the synthetic 2′-O isomer 21 co-eluted with C. elegans metabolite uglas#17 and produced virtually identical MS2 spectra (Figure S7). In addition, we detected small amounts of the 3′-O (22) and 6′-O (23) isomers, named uglas#12 and uglas#14, respectively, in C. elegans endo-metabolome samples (Figure 3b and Figure S8).
Next, we pursued selective synthesis of the major natural isomer, 2′-O-acylated 21. gluric#1 (13) was protected as the 4′,6′-O-benzylidene derivative (Scheme 1) and then coupled with dibenzylated ascr#1 (17) via Steglich esterification, which produced primarily 2′-O-acylated isomer 24, in addition to a trace of 3′-O isomer 25 (>25:1), in overall 64% yield. Subsequent hydrogenation gave uglas#1 (21) in 83% yield. The much better line shapes of the 1H NMR spectra of the benzylidene derivative 24 allowed to further confirm our earlier assignment of uglas#1 as the 2′-O-acylated isomer (Figure S9).
Scheme 1.
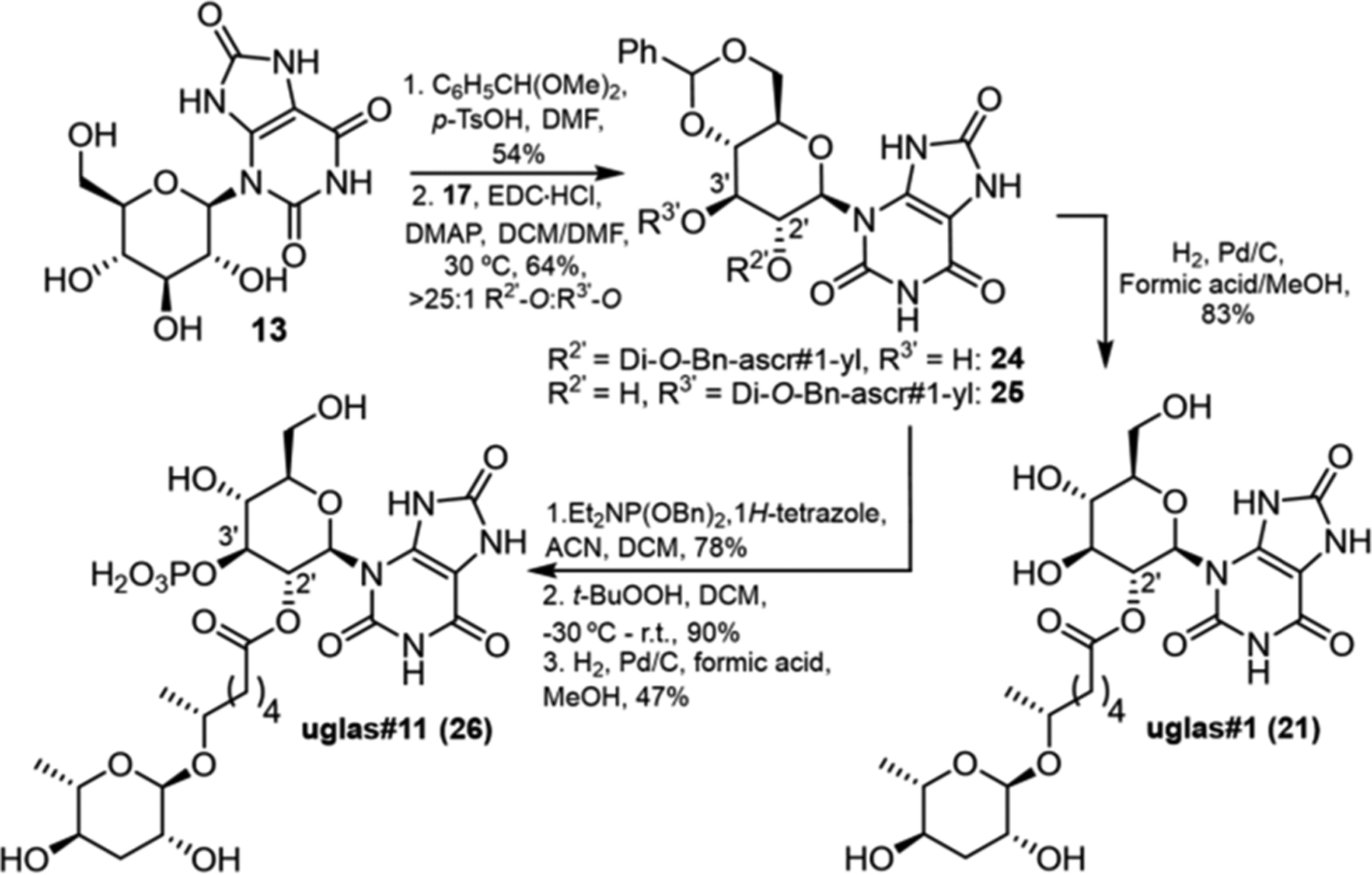
Selective Synthesis of uglas#1 and uglas#11
To determine the position of the phosphate moiety in uglas#11 (26) (Scheme 1), we considered the structure of iglu#2 (27), a glucosylated indole derivative from C. elegans, for which 3′-O-phosphorylation had been demonstrated (Figure 4).28 Phosphitylation of 24 using dibenzyl N,N-diethylphosphoramidite and 1H-tetrazole, followed by oxidation with t-butyl hydroperoxide selectively yielded the protected 3′-O-phosphate in 70% yield over two steps. Subsequent deprotection of benzyl and benzylidene moieties via Pd/C-catalyzed hydrogenation furnished uglas#11 (26), whose HPLC retention times and MS2 spectra were indistinguishable from those of natural uglas#11 in the C. elegans metabolome (Figures S10 and S11). Using synthetic uglas#11 as a standard, we determined that uglas#11 concentrations range from ~25 nM in the wild type to ~250 nM in daf-2(e1368) worms (Figures S1 and S12).
Figure 4.

Examples of modular glucosyl metabolites/ascaroside O-glycoside esters produced by C. elegans with the indicated variation in substitution specificity.
Taken together, our results show that connectivity of the building blocks in the uglas family of metabolites deviates from that found in canonical nucleosides and previously identified C. elegans metabolites. Furthermore, we report a much-simplified route for the synthesis of protected ascarylose (16), which will greatly facilitate synthesis of diverse ascaroside-derived signaling molecules.
Although NMR spectroscopic techniques provide sufficient proof for the structures of a majority of newly discovered natural products, the case presented here highlights the challenges that can arise from broad line shapes or the absence of non-exchangeable protons near heteroaromatic cores. Moreover, growing crystals large enough for single-crystal X-ray crystallography is frequently not feasible. MicroED addresses these challenges, here providing unambiguous corroboration of N3 substitution of 13, which otherwise could only be inferred indirectly from solution-state NMR spectra of a precursor, 10. The ability to obtain crystal structures from microcrystals of natural products (provided as simple powders without crystallization) will likely facilitate identification of a wide range of novel metabolites.
Our data are in agreement with earlier reports that natural glycosylated uric acid derivatives represent the N3-linked isomers (e.g., in bovine blood),8–11 in contrast to many other nucleoside derivatives described from animals, and also contrasting with the structure of pugl#1 (3), an N9-linked gluconucleoside from C. elegans. Notably, the N3-glucosyluric acid moiety has not been previously characterized from natural sources, despite the ubiquity of the building blocks uric acid and glucose. The most abundant members of the uglas family, uglas#1 (21) and uglas#11 (26), feature 2′-O attachment of the ascaroside side chain, whereas all previously identified ascarosyl glucosides are attached at position 1′ (e.g., glas#10, 28) or 6′ (e.g., iglas#1, 29).7,29 These findings suggest that uglas#1 (21) and related modular metabolites are the products of highly specific biosynthetic pathways. Notably, upregulation of uglas#11 (26) production was observed only in the daf-2(e1368) mutant, whereas this was not observed for daf-2(m577), which carries a mutation in a different location of the DAF-2 ligand binding domain. Ongoing studies investigate potential roles of uglas#1 and uglas#11 and related metabolites for phenotypes associated with perturbation of insulin signaling.14
Supplementary Material
ACKNOWLEDGMENTS
The authors thank David Kiemle (ESF Syracuse) for help with the acquisition of NMR spectra and Russell N. Burkhardt and Bennett W. Fox (Boyce Thompson Institute) for assistance with mass spectrometry. This work was partly supported by the National Institutes of Health (R35 GM131877 to F.C.S. and T32GM008500 to B.J.C.) and the Howard Hughes Medical Institute. The authors also thank the Packard Foundation (fellowship to H.M.N.) and Bristol Myers Squibb (Unrestricted Grant in Synthetic Organic Chemistry to H.M.N.) for generous support.
Footnotes
The authors declare the following competing financial interest(s): F.C.S. is a founder of Ascribe Bioscience Inc.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.0c02038.
Experimental procedures, supporting figures, supporting tables, and NMR spectra (PDF)
Accession Codes
CCDC 2020283 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Contributor Information
Brian J. Curtis, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Lee Joon Kim, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Chester J. J. Wrobel, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
James M. Eagan, Ascribe Bioscience, Ithaca, New York 14853, United States.
Rubin A. Smith, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Jessica E. Burch, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States
Henry H. Le, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States
Alexander B. Artyukhin, Chemistry Department, College of Environmental Science and Forestry, State University of New York, Syracuse, New York 13210, United States
Hosea M. Nelson, Department of Chemistry and Biochemistry, University of California, Los Angeles, California 90095, United States.
Frank C. Schroeder, Boyce Thompson Institute and Department of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, United States.
REFERENCES
- (1).Bose N; Ogawa A; von Reuss SH; Yim JJ; Ragsdale EJ; Sommer RJ; Schroeder FC Angew. Chem 2012, 124, 12606–12611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kaletta T; Hengartner MO Nat. Rev. Drug Discovery 2006, 5, 387–399. [DOI] [PubMed] [Google Scholar]
- (3).Bumbarger DJ; Riebesell M; Rödelsperger C; Sommer RJ Cell 2013, 152, 109–119. [DOI] [PubMed] [Google Scholar]
- (4).Bento G; Ogawa A; Sommer RJ Nature 2010, 466, 494–497. [DOI] [PubMed] [Google Scholar]
- (5).Von Reuss SH; Schroeder FC Nat. Prod. Rep 2015, 32, 994–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Butcher RA Curr. Opin. Chem. Biol 2019, 50, 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Artyukhin AB; Zhang YK; Akagi AE; Panda O; Sternberg PW; Schroeder FC J. Am. Chem. Soc 2018, 140, 2841–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Hatfield D; Rinehart RR; Forrest HS J. Chem. Soc 1963, 899–902. [Google Scholar]
- (9).Forrest HS; Hatfield D; Lagowski JM J. Chem. Soc 1961, 963–968. [Google Scholar]
- (10).Hatfield D; Forrest HS Biochim. Biophys. Acta 1962, 62, 185–187. [DOI] [PubMed] [Google Scholar]
- (11).Lohrmann R; Lagowski JM; Forrest HS J. Chem. Soc 1964, 451–459. [Google Scholar]
- (12).Balaban RS; Nemoto S; Finkel T Cell 2005, 120, 483–495. [DOI] [PubMed] [Google Scholar]
- (13).Wan QL; Fu X; Dai W; Yang J; Luo Z; Meng X; Liu X; Zhong R; Yang H; Zhou Q Aging 2020, 12, 2840–2856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Kenyon CJ Nature 2010, 464, 504–512. [DOI] [PubMed] [Google Scholar]
- (15).Patel DS; Garza-Garcia A; Nanji M; McElwee JJ; Ackerman D; Driscoll PC; Gems D Genetics 2008, 178, 931–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Garner P; Ramakanth SJ Org. Chem 1988, 53, 1294–1298. [Google Scholar]
- (17).Kahn K; Serfozo P; Tipton PA J. Am. Chem. Soc 1997, 119, 5435–5442. [Google Scholar]
- (18).Jones CG; Martynowycz MW; Hattne J; Fulton TJ; Stoltz BM; Rodriguez JA; Nelson HM; Gonen T ACS Cent. Sci 2018, 4, 1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ting CP; Funk MA; Halaby SL; Zhang Z; Gonen T; Van Der Donk WA Science 2019, 365, 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Sheldrick GM Acta Crystallogr., Sect. A: Found. Crystallogr 2008, A64, 112–122. [DOI] [PubMed] [Google Scholar]
- (21).Miyaki M; Shimizu B Chem. Pharm. Bull 1970, 18, 1446–1456. [Google Scholar]
- (22).Birkofer L; Ritter A Angew. Chem 1965, 77, 414–426. [Google Scholar]
- (23).Jeong PY; Jung M; Yim YH; Kim H; Park M; Hong E; Lee W; Kim YH; Kim K; Paik YK Nature 2005, 433, 541–545. [DOI] [PubMed] [Google Scholar]
- (24).Muramatsu W; Tanigawa S; Takemoto Y; Yoshimatsu H; Onomura O Chem. - Eur. J 2012, 18, 4850–4853. [DOI] [PubMed] [Google Scholar]
- (25).Martinelli MJ; Vaidyanathan R; Van Khau V Tetrahedron Lett. 2000, 41, 3773–3776. [Google Scholar]
- (26).Baer HH; Astles DJ; Chin H-C; Siemsen L Can. J. Chem 1985, 63, 432–439. [Google Scholar]
- (27).Zhang YK; Sanchez-Ayala MA; Sternberg PW; Srinivasan J; Schroeder FC Org. Lett 2017, 19, 2837–2840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Stupp GS; Von Reuss SH; Izrayelit Y; Ajredini R; Schroeder FC; Edison AS ACS Chem. Biol 2013, 8, 309–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Von Reuss SH; Bose N; Srinivasan J; Yim JJ; Judkins JC; Sternberg PW; Schroeder FC J. Am. Chem. Soc 2012, 134, 1817–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.