

Abstract
Halopyridines are key building blocks for synthesizing pharmaceuticals, agrochemicals, and ligands for metal complexes, but strategies to selectively halogenate pyridine C–H precursors are lacking. We designed a set of heterocyclic phosphines that are installed at the 4-position of pyridines as phosphonium salts and then displaced with halide nucleophiles. A broad range of unactivated pyridines can be halogenated, and the method is viable for late-stage halogenation of complex pharmaceuticals. Computational studies indicate that C–halogen bond formation occurs via an SNAr pathway, and phosphine elimination is the rate-determining step. Steric interactions during C–P bond cleavage account for differences in reactivity between 2- and 3-substituted pyridines.
Graphical Abstract
INTRODUCTION
Haloarenes are fundamental building block compounds in which the carbon–halogen bond enables access to an array of derivatives with precise regiocontrol (eq 1).1 Furthermore, haloarenes are inherently valuable in functional molecules and frequently occur in pharmaceuticals and agrochemicals.2 Halogenation methods are historically important in synthetic chemistry; numerous seminal advances in synthetic methodology use the carbon–halogen bond as a platform, and haloarene synthesis by electrophilic aromatic substitution (EAS) reactions is central to understanding aromatic reactivity.3 In EAS processes, reaction of the arene π-system with an electrophilic halogen source forms the carbon– halogen bond. However, this reactivity principle typically favors halogenation of electron-rich and electron-neutral aromatics. Electron-deficient π-systems, such as pyridines, are electronically mismatched toward EAS processes; their halogenation reactions require harsh conditions and are significantly more limited in scope.4 Developing broadly applicable pyridine halogenation methods will address current limitations in accessing essential synthetic halopyridine intermediates and biologically relevant molecules.5
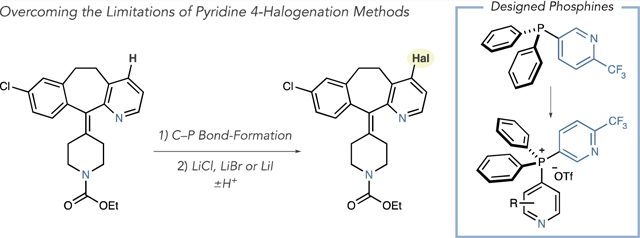
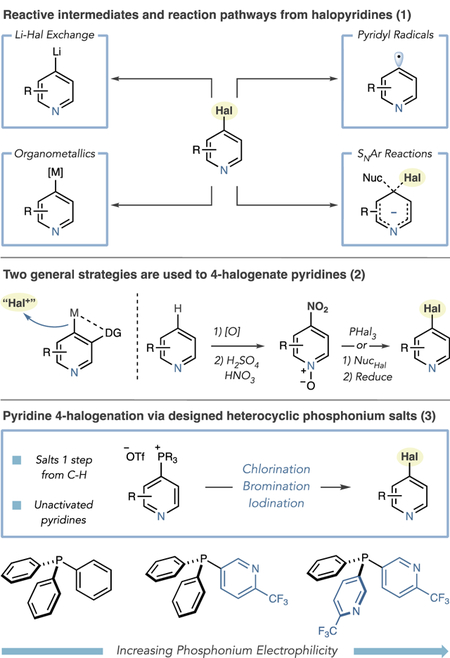
Positional selectivity is a useful way to classify pyridine halogenation reactions. EAS processes are 3-selective and often require strong mineral acids as solvents or Lewis acid promotion with elevated temperatures and elemental halides.6 Lower temperatures and alternate electrophiles can be used to halogenate pyridines, but electron-donating groups are typically required.7 2-Selective halogenation reactions use pyridine N-oxides, and Hartwig reported that AgF2 directly 2-fluorinates pyridines.8,9 Two strategies are generally used to halogenate pyridines at the 4-position (eq 2). First, metalation-trapping sequences exploit directing groups such as carbonyls and halides.10 Second, sequences that convert pyridines into N-oxides are followed by 4-selective nitration. Halopyridines are then formed directly by treatment with PHal3 or P(O)Hal3 reagents, or by displacing the nitro group with a nucleophilic halide and then reducing the N-oxide.11,12 Requiring preinstalled functional groups, strong bases, oxidants, and highly acidic media limits the applicability of these approaches.13 As a result, there are considerably fewer commercial 4-halopyridines than other isomers, and those available can be prohibitively expensive. Our goal was to develop a general strategy to halogenate pyridines at the 4-position that tolerates a range of functional groups as well as steric and electronic variance.14 Herein, we present a two-step approach that hinges on designing heterocyclic phosphine reagents (eq 3). The process uses metal halides, or halogen acids, to displace electrophilic phosphonium ions, applies to other azines, and functions on complex substrates including late-stage halogenation of pharmaceuticals.
 |
RESULTS AND DISCUSSION
Phosphonium salts can be selectively formed at the 4-position of pyridines and displaced by nucleophiles.15 We envisioned that nucleophilic halides could displace the phosphonium group and considered two mechanistic pathways at the outset: halide addition to the phosphonium ion to form a P(V) intermediate followed by ligand coupling or an SNAr pathway with PPh3 as a leaving group.16 As there are no reports of C–Hal bond formation via phosphorus ligand–ligand coupling reactions, we strongly preferred an SNAr mechanism, and in either case, we suspected the halide countercation would play an important role in coordinating to the pyridine N-atom.
We tested a set of nucleophilic chloride sources with isomeric salts 1 and 2 (Scheme 1A). Despite investigating a range of reaction conditions, only low yields of 3 and 4 could be obtained using HCl in dioxane at 80 °C. Given that these PPh3-derived phosphonium salts did not react efficiently with chloride nucleophiles, we considered that more electrophilic analogs were required. Therefore, we implemented a set of criteria to prepare more reactive phosphonium salts, as shown in Scheme 1B. First, introducing a pyridyl ligand would increase the electrophilicity of the resulting phosphonium salt, where two pyridines, rather than one, could be activated by Lewis or Brønsted acids.17 Second, we altered the C–P bond substitution pattern in the pyridine component to ensure the pyridine of interest was selectively chlorinated; both ligand-coupling processes and SNAr reactions are unfavorable at the 3-position of pyridines.16–18 Third, installing a 2-CF3 group would prevent reaction with Tf2O during the salt-forming stage and ensure C–P bond formation occurs on the pyridine of interest, rather than on the phosphine reagent.19 Importantly, preparing phosphine I is straightforward in one step from diphenylphosphine and 2-trifluoromethyl-5-bromopyridine (Scheme 1C).
Scheme 1. Design of Heteroarylphosphinesa,b.
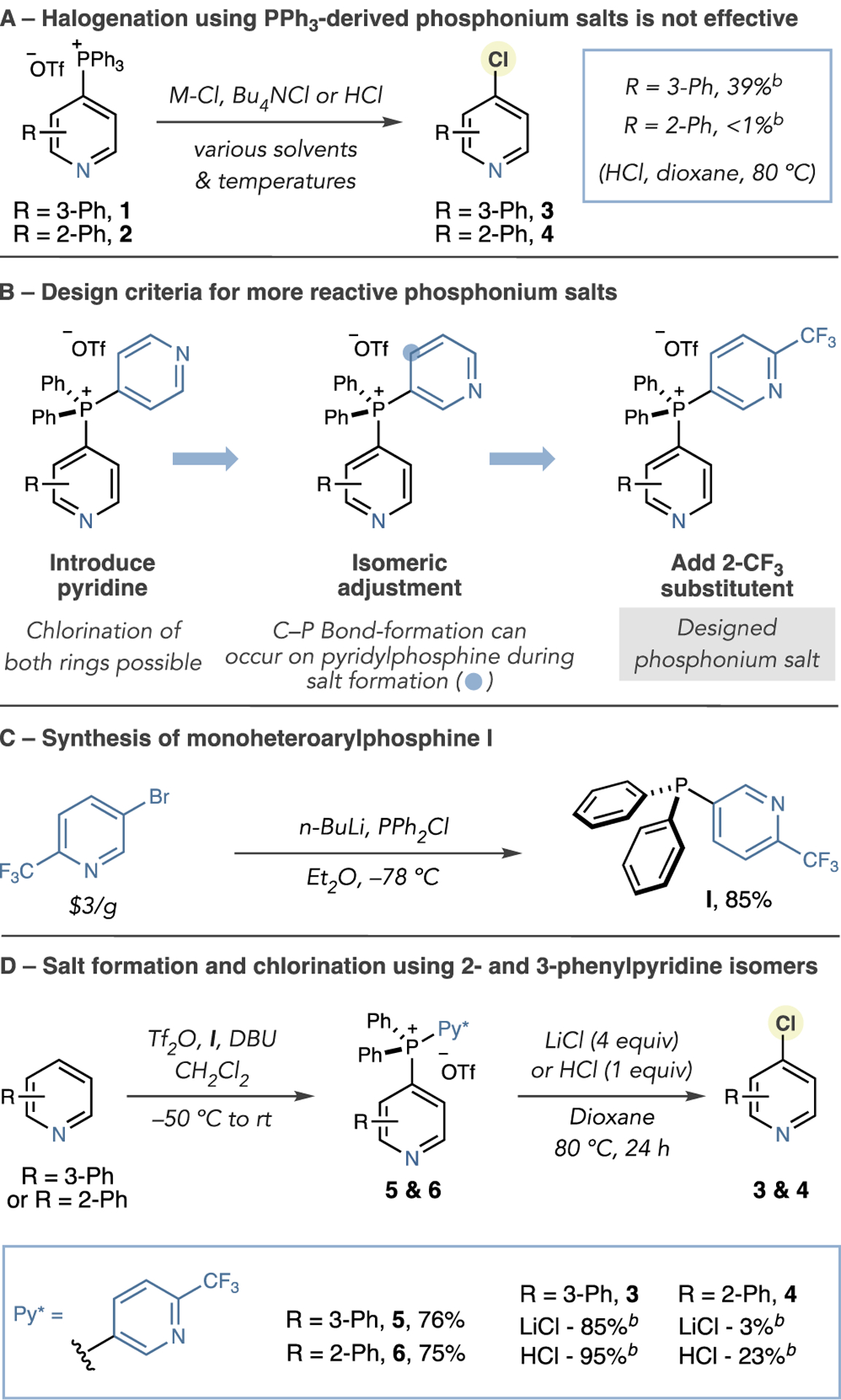
aIsolated yields shown (unless otherwise stated). bYields calculated by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
To test the hypothesis that more electrophilic phosphonium salts are viable for chlorination, we selected 2-phenylpyridine and 3-phenylpyridine as test substrates (Scheme 1D). We synthesized the corresponding phosphonium salts 5 and 6 in good yields and then subjected them to a range of metal chlorides or HCl and examined a range of reaction parameters (see the Supporting Information (SI) for full details). The results showed that 3-substituted isomer 3 was obtained in high yields using LiCl or HCl, but significantly lower amounts of the 2-substituted isomer 4 formed. Notably, we did not detect any chlorination of the 2-CF3 pyridine group in the crude reaction mixtures. Our hypothesis that phosphonium electrophilicity can influence reactivity appeared valid; however, as 2-substituted salt 6 was less reactive, we suspected that steric destabilization from the 3-phenyl substituent in 5 was also a significant factor. Therefore, to chlorinate 2-substituted pyridines, we speculated that more electrophilic phosphonium salts were required (Scheme 2A) and synthesized modified phosphine II, possessing two pyridyl groups (Scheme 2B). In line with this approach, salt 7 was prepared in good yield, and heating in dioxane at 80 °C with 4 equiv of LiCl, or 1 equiv of HCl, efficiently formed chlorinated product 4 (Scheme 2C). An increase in electrophilicity of phosphonium salts is predicted computationally at both the 4-position and the P-atom in the order of salts derived from PPh3, I, and II (Figure S2). A one-pot salt-formation–halogenation reaction is possible, but significant decreases in yield and reversion of the phosphonium salt to the parent C–H compound are observed (Table S6).
Scheme 2. Chlorination of 2-Substituted Pyridinesa,b.
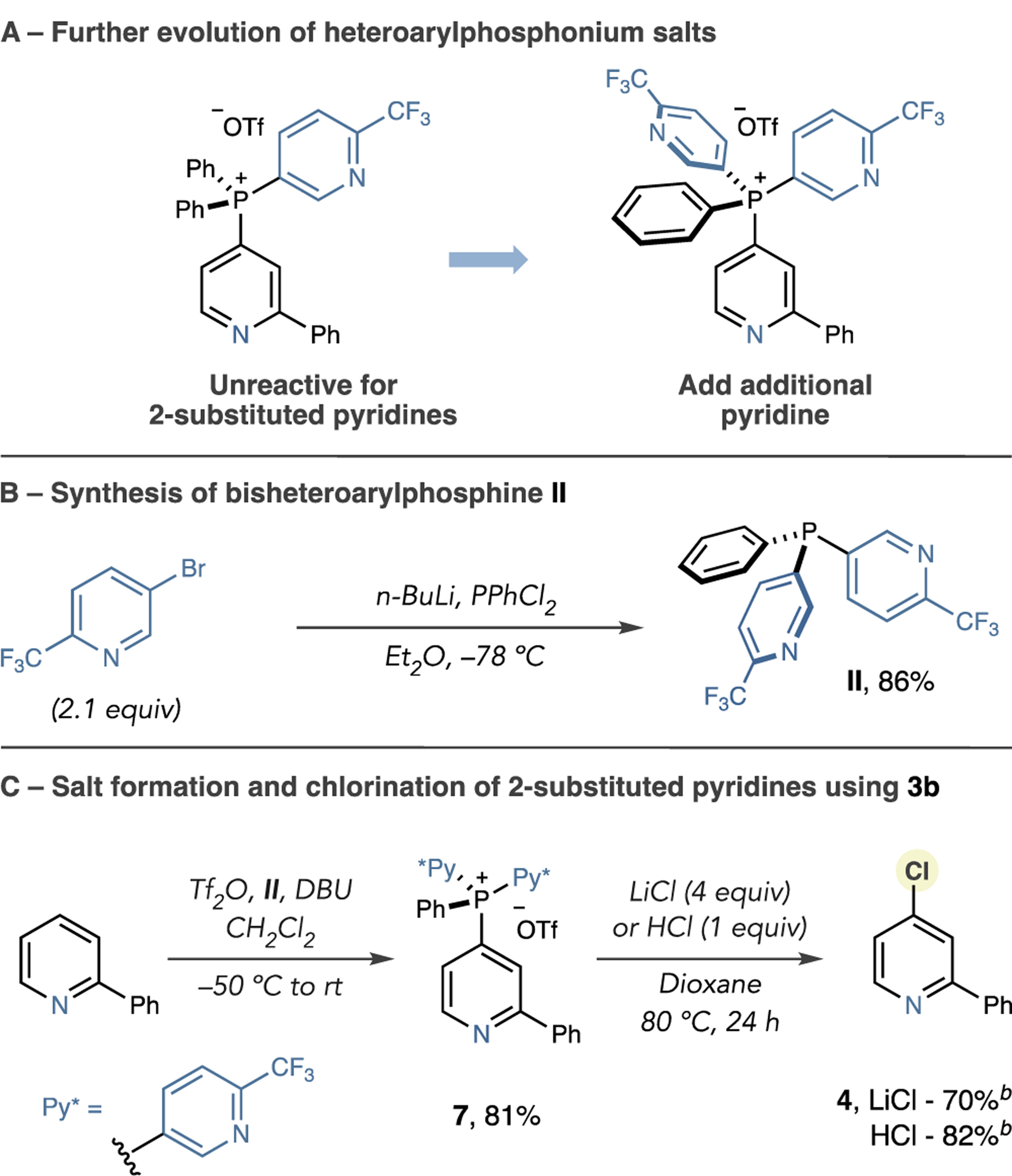
aIsolated yields shown (unless otherwise stated). bYield calculated by 1H NMR or GC analysis using 1,3,5-trimethoxybenzene as an internal standard.
A 3,5-disubstituted pyridine presented further opportunity to examine the effects of steric destabilization on the of reactivity phosphonium salts with chloride nucleophiles (Scheme 3). Based on the observations in Scheme 1, the significant steric hindrance in these systems was expected to result in more facile chlorination. Forming salt 8 proved challenging using phosphine I, although the subsequent chlorination reaction was effective (9). In contrast, we obtained PPh3-derived salt 10 in a much higher yield, and 9 formed in comparable yield. The greater steric destabilization present in these systems outweighs the requirement for electron-deficient phosphoniums such that designed phosphines I and II are replaceable with PPh3 for the reactions of 3,5-disubstituted substrates.
Scheme 3. Chlorination of 3,5-Disubstituted Pyridinesa,b,c.
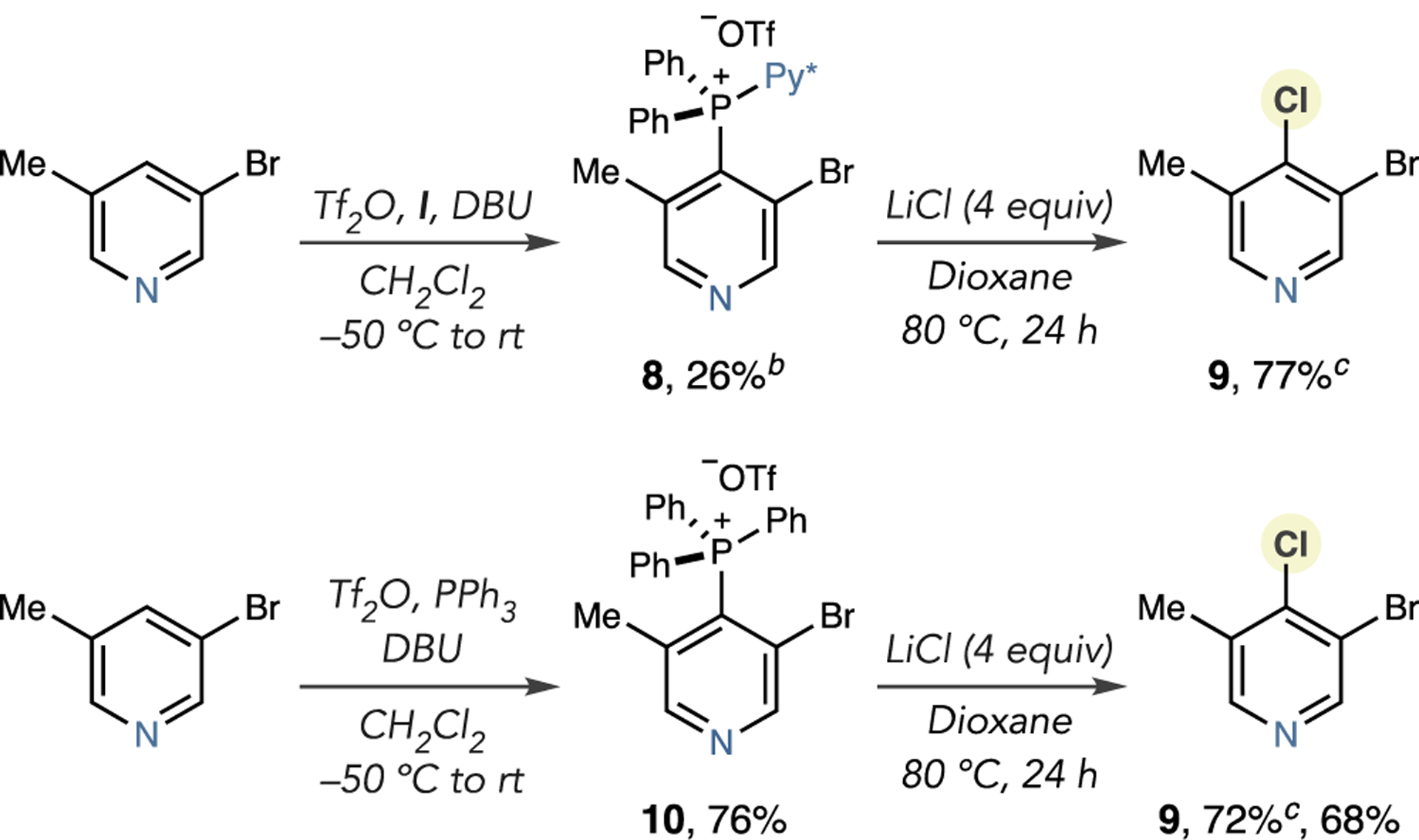
aIsolated yields shown (unless otherwise stated). bSalt isolated with 5% of an unknown impurity. cYields calculated by 1H NMR using 1,3,5-trimethoxybenzene as an internal standard.
After identifying a set of phosphines, we explored the substrate scope of the pyridines and related azines amenable to the chlorination process (Table 1). Based on the substitution pattern, we matched pyridines with one of three phosphines I, II, and PPh3. For pyridines possessing a 3- or 5-substituent (but not both), monoheterocyclic phosphine I is most appropriate. From Scheme 1, both HCl and LiCl are effective chlorination reagents, but we proceeded with LiCl because of the likelihood of a broader substrate scope and compatibility with acid-sensitive groups, such as Boc-protected amines. As a typical case, chloropyridine 11 was obtained in 56% yield using LiCl, without evidence of Boc-deprotection. Using HCl as a reagent, the corresponding secondary amine was observed in the reaction mixture using LCMS analysis. The chlorination step tolerated 3-substituents such as pyrazoles, alkynes, and other substituted pyridines (12–15). The two-step process also chlorinated 2,3- and 2,5-disubstituted pyridines in moderate to good yields for each stage (16–22). In this set, chlorinating a 2-cyanopyridine was unsuccessful using LiCl or HCl, and the starting phosphonium salt was largely unreacted (19). On the other hand, 2-chloro substituents can be present, although the reaction requires 72 h to reach completion (20). We hypothesized that the cyano group prevents pyridine activation by the Lewis or Brønsted acid. Phosphine I is also a suitable reagent for quinolines and isoquinolines for which we obtained isomeric chlorinated products 23–26 with complete control of regioselectivity. Diazines 27–29 were successfully chlorinated, as were fused triazines 30 and 31.
Table 1.
Chlorination of Pyridine, Quinoline and Diazine Building Blocksa,b,c,d,e,f
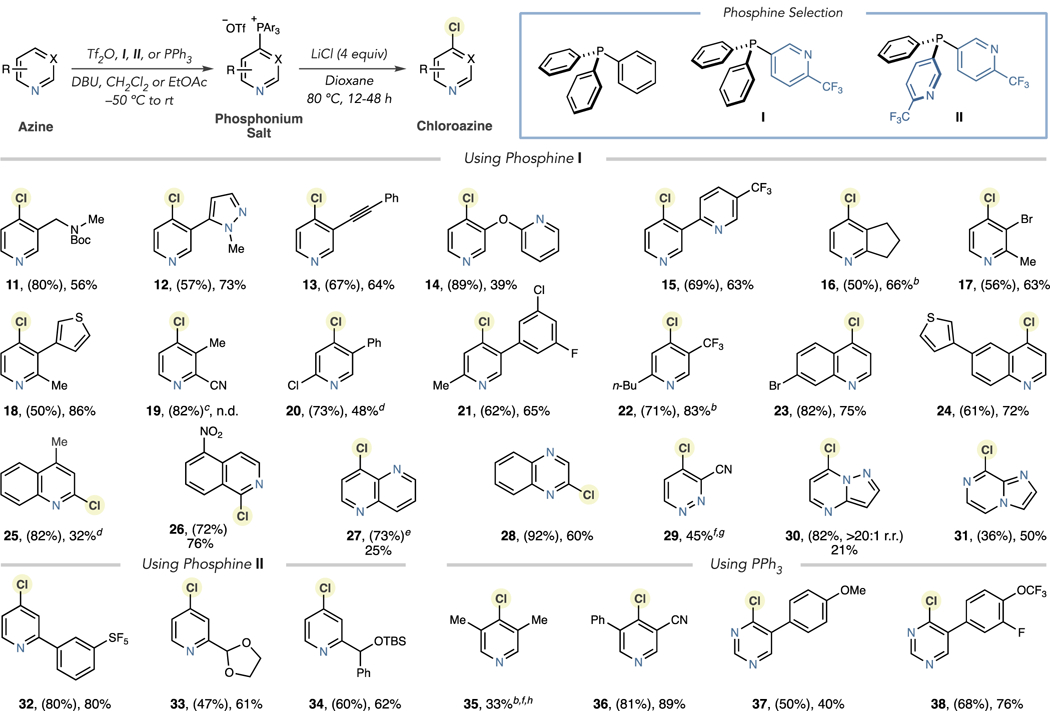
|
Isolated yields of single regioisomers (unless stated) shown with yields of phosphonium salts in parentheses.
Yields calculated by 1H NMR using 1,3,5-trimethoxybenzene or triphenylmethane as an internal standard.
Isolated as an inseparable 17:1 mixture with an impurity.
Run for 72 h.
Isolated as an inseparable 7:1 mixture with an impurity.
Chlorination was performed directly on the crude phosphonium salt and yield for two steps is reported.
Run for 5 h.
Run using HCl in 1,4-dioxane (1.0 equiv) instead of LiCl.
Phosphine II and PPh3 were then used to chlorinate 2- and 3,5-substituted pyridines. Using the former, we obtained an SF5-aryl derivative 32 without difficulty. The acid-sensitive groups in chlorides 33 and 34 were again preserved using LiCl; we observed TBS deprotection by LCMS analysis using HCl as a chloride source. Forming chlorides 35 and 36 is viable using PPh3-derived salts, and as pyrimidines undergo facile SNAr reactions, we proposed that this attribute would also enable chlorination using PPh3 as a reagent. Using this approach, we obtained aryl-substituted chloropyrimidines 37 and 38 in moderate yields.
Next, we developed protocols to install halides other than chlorides using phosphonium salt 7 as a test substrate (Scheme 4). For bromination, low yields of product 39 were obtained using LiBr, KBr, or Bu4NBr as nucleophiles at 80 °C. However, when we combined 4 equiv of LiBr with 1 equiv of TfOH, bromination occurred in good yield, presumably because protonation generates a more reactive pyridinium salt. Using the analogous iodide salts, either no reaction or low yields of pyridyl iodide 40 were observed at 80 °C; heating the reactions at 120 °C and prolonging the reaction times to 48 h did result in iodination, and again, combining LiI with TfOH was optimal. Using these conditions, we chose a selection of substrates from Table 1 to examine bromination and iodination (Scheme 4). The reaction conditions translated well to halogenate a 2-aryl-SF5 derivative (42 and 43). Phosphonium salts derived from II and PPh3 also required acid for bromination and iodination with products 45–48 obtained in moderate to good yields. When we examined fluoride nucleophiles or HF sources, phosphonium salts predominately cleaved to the parent C–H compounds and no fluorinated products formed using these protocols. Efforts are currently ongoing in our laboratory to improve this fluorination process.
Scheme 4. Pyridine Bromination and Iodinationa,b.
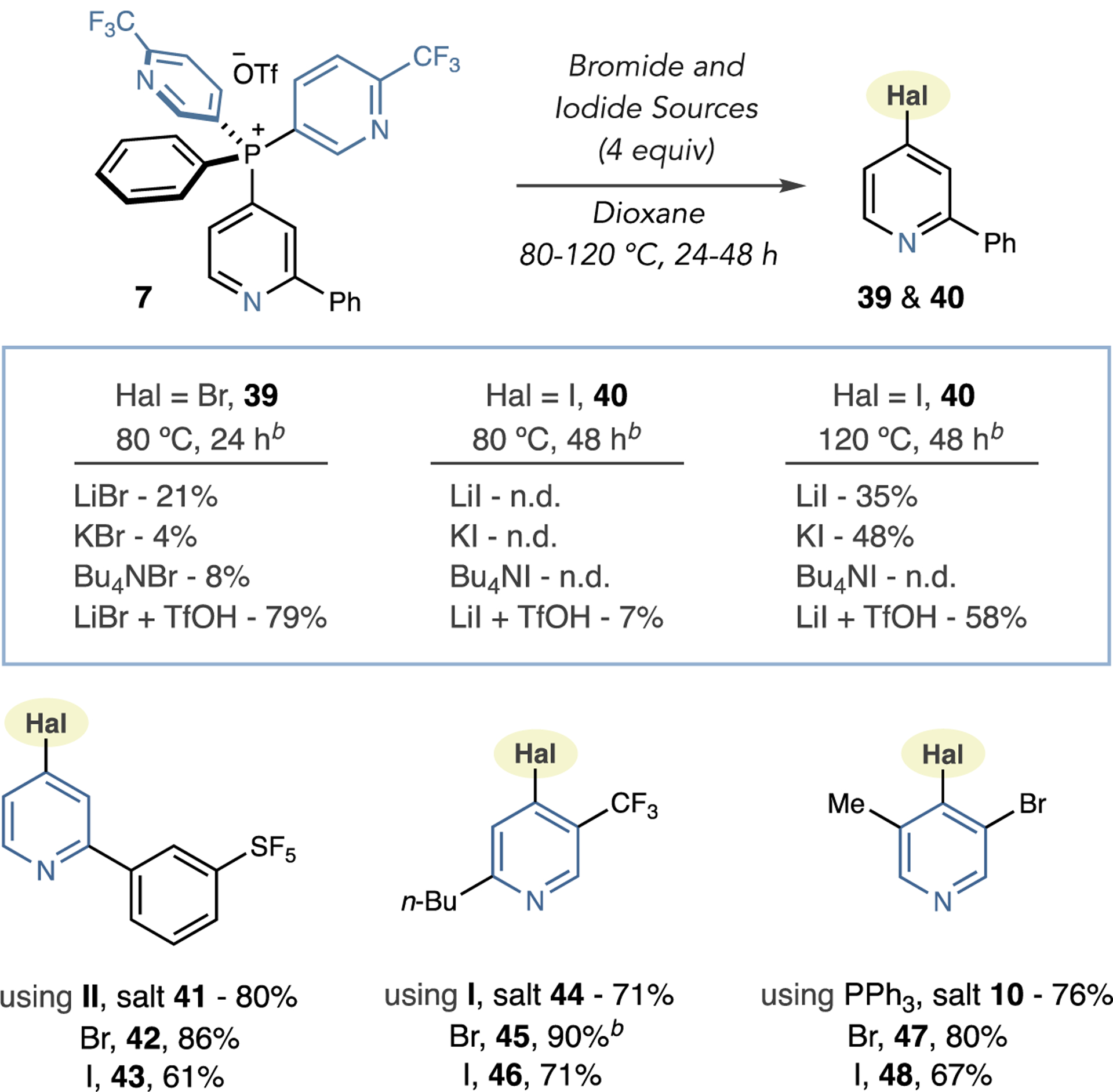
aIsolated yields. bYields by 1H NMR or GC using 1,3,5-trimethoxybenzene triphenylmethane as internal standards.
Diversifying complex pyridine-containing structures is valuable for medicinal chemistry, and selective halogenation represents a means to access multiple analogs by subsequently transforming the C–Hal bond. We first tested compounds representative of drug fragments or lead compounds (Table 2). Using phosphine I, a precursor to the antihistamine Bepotastine was chlorinated and brominated (49 and 50). Halogenation of two isomeric ester-containing structures proceeded in good overall yields for the two-step process, and ester C–O bonds were not cleaved during the process (51–53). Site-selective halogenation is a valuable attribute of this protocol; we obtained bis-pyridyl halides 54–56 with exclusive selectivity favoring the pyridine without 2- or 6-substitution in each case. Table 2 also shows late-stage halogenation of pyridine-containing pharmaceuticals. The 2-substituted pyridines in Bisacodyl and a Vismodegib derivative were chlorinated to form 57 and 58 using phosphine II. Monoheterocyclic phosphine I was used to generate a variety of halide derivatives of Etoricoxib, Loratadine, Nicoboxil, and Abiraterone Acetate, with exclusive 4-selectivity in all cases (59–66). With the 4-position in the pesticide Quinoxyfen blocked, the 2-position of the quinoline was chlorinated (67). Finally, the two-step process was effective at chlorinating the quinoxaline core within a protected version of Varenicline in moderate yield (68).
Table 2.
Halogenation of Pyridine-Containing Fragments and Pharmaceuticalsa,b,c,d
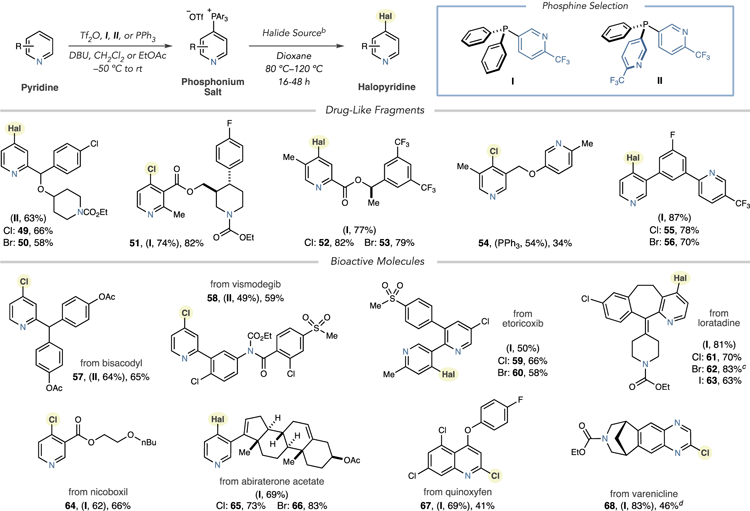
|
Isolated yields of single regioisomers (unless stated) shown with yields of phosphonium salts in parentheses from I, II, or PPh3.
Chlorination: LiCl (4.0 equiv), 80 °C. Bromination: LiBr (4.0 equiv), TfOH (1.0 equiv), 80 °C. Iodination: LiI (4.0 equiv), TfOH (1.0 equiv), 120 °C.
Run without TfOH.
Run for 72 h.
To further emphasize that diverse libraries of analogs could be generated using this halogenation strategy, we tested our previously reported site-selective switching protocol on an MK-1064 precursor (Scheme 5).20 Using phosphine PPh3 and DBU as a base, salt formation and subsequent chlorination occurred on the 2,6-unsubstituted ring to form chloride 69, in line with the kinetically preferred reaction with Tf2O. Although the yield of the subsequent chlorination was low, only one isomer formed. Applying the base-switch protocol, using NMe2Cy as well as 2 equiv of Tf2O and II, allowed us to synthesize isomeric pyridyl chloride 70 with excellent control of regioselectivity and site selectivity. This result aligns with the rationale where the phosphine adds to the Tf-activated 2- and 3,5-disubstituted pyridine rings to form dearomatized adducts. Steric interactions between the 3- and 5-substituents and the trialkylamine base prevent rearomatization, whereas these effects are absent in the 2-substituted pyridine. Numerous transformations then apply to 69 and 70 to synthesize libraries of isomeric compounds.
Scheme 5. Site-Selective Chlorinationa.
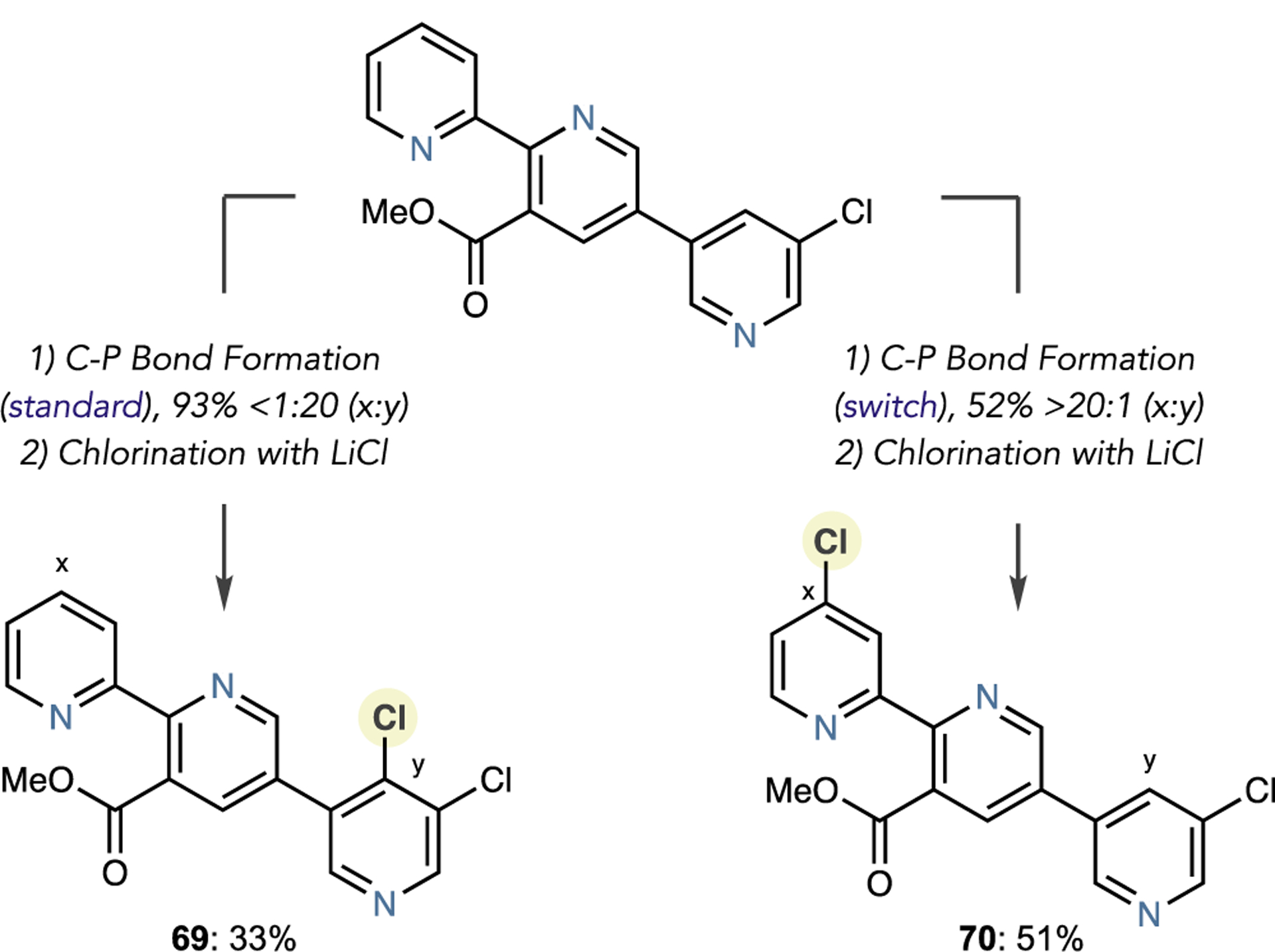
aIsolated yields are shown. Standard C–P bond formation: Heterocycle (1.0 equiv), Tf2O (1.0 equiv), PPh3 (1.1 equiv), DBU, (1.0 equiv), CH2Cl2. Switch C–P bond formation: Heterocycle (1.0 equiv), Tf2O (2.0 equiv), Phosphine II (2.0 equiv), NMe2Cy (2.0 equiv), CH2Cl2.
COMPUTATIONAL STUDIES
We turned to quantum chemical calculations to model the mechanism of carbon–halogen bond formation, using density functional theory (DFT)21 with the SMD solvation model (1,4-dioxane)22 to study these reactions. Results are presented at the ωB97X-D/def2-QZVPP//ωB97X-D/def2-SVP level of theory. The presence of anionic nucleophiles and hypervalent P(V) species present potential challenges for computation,23 and benchmarking studies were carried out. The use of larger basis sets and additional diffuse basis functions during geometry optimizations and single-point energy calculations, including Def2-TZVPP, Def2-TZVPPD, and Def2-QZVPPD, were examined, and very similar results were obtained with these different protocols (see the Studies using larger basis sets with diffuse functions section of the SI). We selected 3-Ph and 2-Ph phosphonium salts 5 and 6, respectively, as the substrates for the computational study. We included the triflate counteranion in all calculations unless otherwise stated, since the calculated energy of the dissociated ions in dioxane is considerably larger than that of the ion pair (19.0 kcal/mol). However, the overall mechanism is qualitatively unaffected by omitting this counterion (Figure S7).
First, we address the question of whether hypervalent P(V) intermediates are formed prior to C–X bond formation. Figure 1 shows the calculated Wiberg Bond Orders (WBOs)24 and bond distances of P–X bonds for the optimized geometries of 6 with the different halides used experimentally. Chloro-, bromo-, and iodophosphoranes do not form stable pentavalent geometries and instead prefer phosphonium halide ion-pair structures. Fluorophosphoranes, on the other hand, form stable P(V) intermediate structures as illustrated in Figure 1B. These DFT results are consistent with tabulated P–X bond strengths (P–Br: 267, P–Cl 289, P–F 439 kJ/mol).25 Excluding fluorination, these results implicate the direct attack of the halide at carbon (i.e., an SNAr pathway) rather than via phosphorus ligand-coupling.
Figure 1.
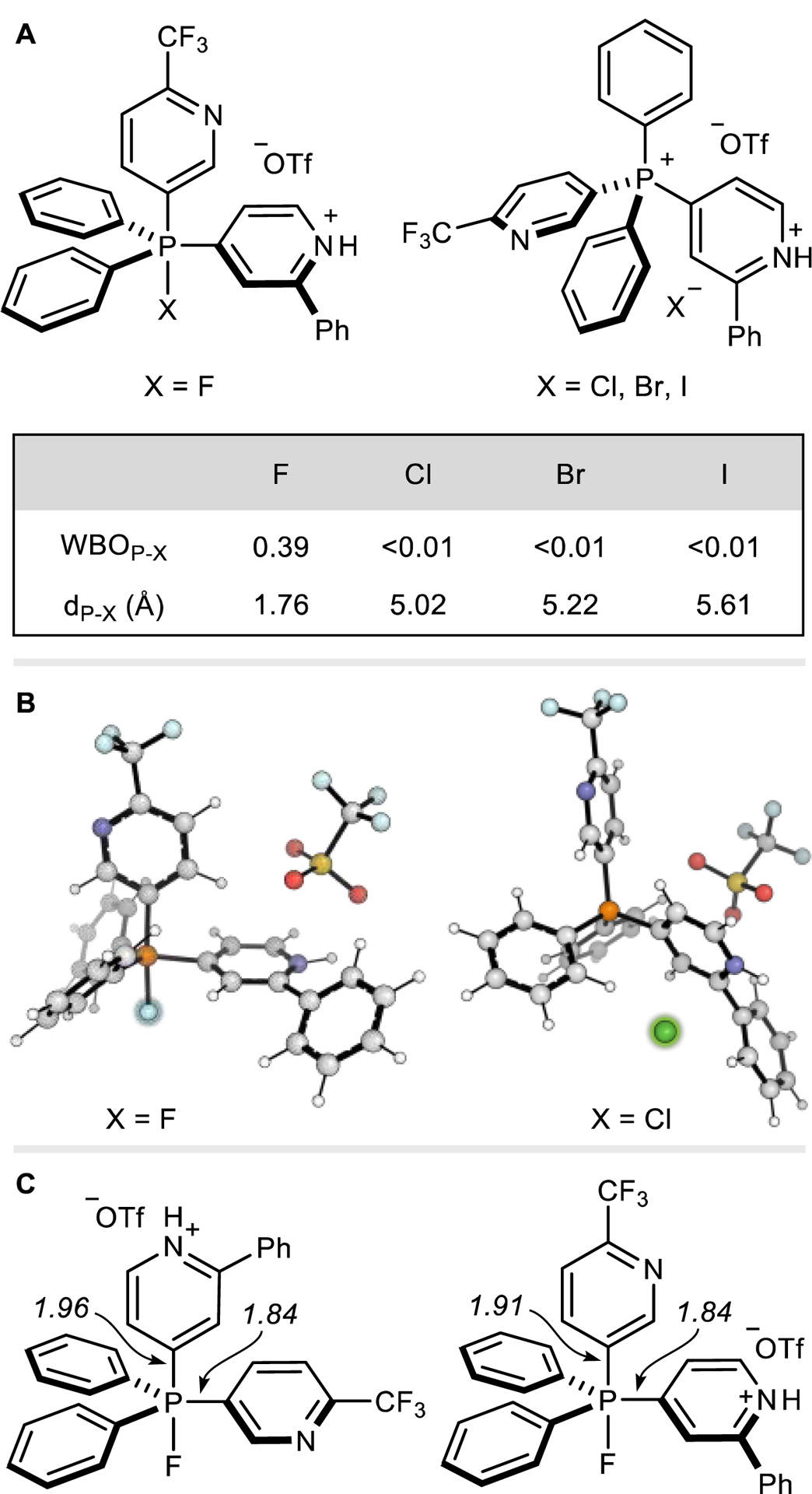
(A) WBO and distances of P–X bonds of the different species formed with phosphonium salts and different halogen anions. (B) Representation of the most stable conformers when using F and Cl atoms. (C) Lengths of axial and equatorial P–C bonds, in Å, in multiple conformations of the fluorophosphorane studied.
Figure 2A shows the overall Gibbs energy profile for the formation of chloropyridines 3 and 4. The computed transition structures (TSs) indicate an SNAr-type mechanism takes place.26 A relatively flat potential energy surface exists, across which two discrete steps occur: addition of chloride at the 4-position of an activated pyridinium (here modeled by HCl protonation) in TS-I, and the subsequent cleavage of the C–P bond in TS-II. The intervening Meisenheimer complex, Int-III, is a stable intermediate structure, although this lies very close in energy with the first TS. The second TS, forming products 3 and 4 and phosphine byproduct, lies highest in energy and is the rate-limiting step. Figure 2B highlights the critical role of a Brønsted or Lewis acid additive (either HCl or LiCl), as the activation barrier is raised prohibitively high (42.5 kcal/mol for salt 6) in their absence, via a concerted mechanism.27 The calculations also clarify why reagents such as Bu4NCl are not effective due to the relatively weak Lewis acidity of the ammonium counterions with pyridine Lewis bases. The differential reactivity of 2- and 3-substituted pyridines results from steric interactions that are enhanced as the phosphine departs in TS-II (Figure 2C). Compared to TS-I, the phosphine lies further out of the aromatic plane, bringing it closer of the ring substituents. As seen in the NCI plots, the P-aryl groups make contact with the 3-Ph substituent of salt 5 while these interactions are less significant with the more remote 2-Ph group in salt 6. The computed ΔΔG‡ of 2.8 kcal/mol is indicative of around a 50-fold increase in reactivity of 5 over 6. Computations suggest that this energy difference is due to sterics, rather than arising from differences in pyridine electronics, since similarly sized but electronically distinct groups are predicted to behave in the same way (Figure S6). This finding reinforces the role of sterics over electronic effects, and we believe the hypothesis is generalizable to a reasonable extent based on the substrates examined in this study.
Figure 2.
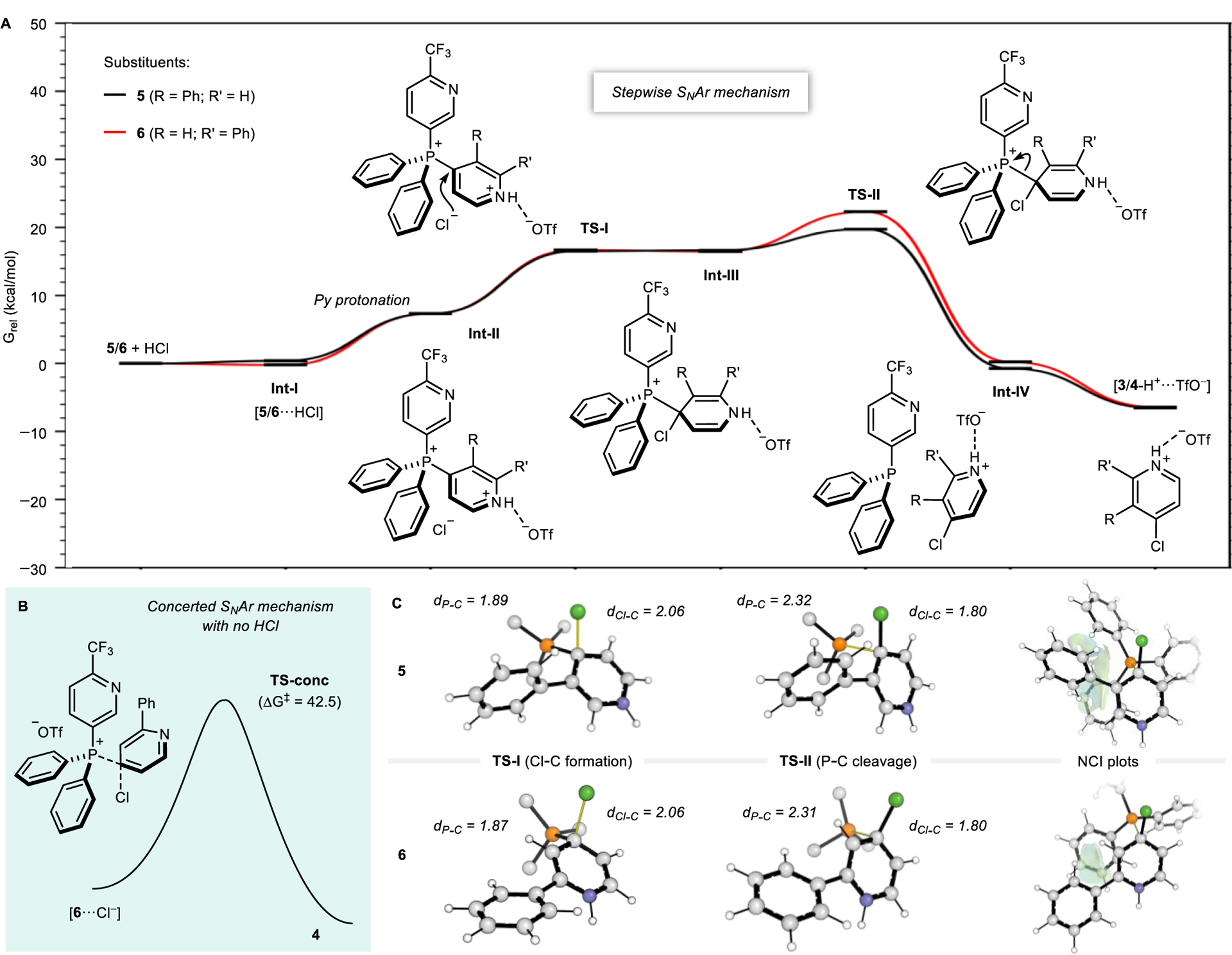
(A) Boltzmann weighted relative G in kcal/mol during the formation of products 3 (3-Ph substituent, black line) and 4 (2-Ph substituent, red line) at 80 °C (ωB97X-D/Def2-QZVPP//ωB97X-D/Def2-SVP, SMD with 1,4-dioxane). (B) Reaction coordinate to form 4 when using Cl– instead of HCl. ΔG‡ is shown in kcal/mol. (C) Representations of the most favorable TS-I and TS-II steps with P–C and Cl–C bond distances (in Å) when using 5 and 6, along with NCIPlot32 surfaces of the corresponding TS-II steps. Bonds involved in the TSs are represented as yellow lines. Phosphorus substituents and the TfO– counteranion are omitted for clarity in some cases.
The computed activation barrier obtained in 1,4-dioxane (20–22 kcal/mol) would be indicative of room temperature sreactivity,28 whereas experimentally we required heating to 80 °C. We attribute this in part to the heterogeneous nature of the reaction, and potential solubility effects, that were not modeled computationally. Qualitative conclusions concerning the C– Hal bond-forming process and the differences in reactivity between 2- and 3-substituted pyridines, which are consistent with experiment, are unaffected by these differences.29,30
Finally, we considered the failure of phosphonium salt fluorination to occur under these the same conditions, and the return of the parent C–H bond as the major outcome. As described above, a mechanistic switch vs other halogens is expected, involving the intermediacy of a P(V) fluorophosphorane. In the optimized geometry of this intermediate, the axial P–Cpyr bond is lengthened, and therefore weakened, considerably relative to its equatorial counterpart (1.96 and 1.91 vs 1.84 Å, Figure 1C), and can potentially decompose in the same manner as when pyridine phosphonium salts react with carbonates, hydroxides, and alkoxides.31 Elongation of the axial P–Cpyr bond can occur relatively easily (computationally, stretching by 0.2 Å costs just 3.9 kcal/mol), such that reaction of this pyridyl group with an external proton source at the 4-position is possible; we have not identified the nature of the proton source at this point in our studies and have observed the same result with rigorously dried reaction reagents and solvents.
CONCLUSIONS
In summary, we have developed a set of designed phosphine reagents that enable 4-selective halogenation of pyridines. The key design element was to incorporate electron-deficient pyridine ligands on the phosphine reagents so that the corresponding phosphonium salts are more reactive toward halide nucleophiles. Pyridines with a variety of substitution patterns and variations in steric and electronic properties are amenable to this two-step strategy, which is also effective for late-stage halogenation of complex pharmaceuticals. Computational studies indicate that C–Hal bond formation occurs via a stepwise SNAr pathway that requires N-activation of the pyridyl group. Phosphine elimination is the rate-determining step. Steric interactions between the departing phosphine and pyridyl substituents are most pronounced during C–P bond cleavage and account for differences in reactivity between 2-and 3-substituted pyridines. Given the deficiency in existing methods to produce these halogenated products, we anticipate the protocol will be useful in medicinal chemistry. Current efforts are focusing on elucidating the mechanism of the carbon–halogen bond-forming step and will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by The National Institutes of Health (NIGMS) under Award Number R01 GM124094. Further support came from startup funds from Colorado State University and partial support from the National Science Foundation under Grant No. (1753087). We acknowledge the RMACC Summit supercomputer, supported by the NSF (ACI-1532235 and ACI1532236), and the Extreme Science and Engineering Discovery Environment (XSEDE) allocations TG-CHE180056 and TG-CHE190111.
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04674.
Experimental procedures, spectral data and details of the computational methods (PDF)
Molecular coordinates and thermochemistry data (ZIP)
REFERENCES
- (1).(a) Crawley ML; Trost BM Applications of transition metal catalysis in drug discovery and development an industrial perspective; Wiley: Hoboken, NJ, 2012. [Google Scholar]; (b) Bunnett JF; Zahler RE Aromatic Nucleophilic Substitution Reactions. Chem. Rev 1951, 49, 273–412. [Google Scholar]; (c) Bailey WF; Patricia JJ The Mechanism of the Lithium -Halogen Interchange Reaction: A Review of the Literature. J. Organomet. Chem 1988, 352, 1–46 [Google Scholar]; (d) Seath CP; Vogt DB; Xu Z; Boyington AJ; Jui NT Radical Hydroarylation of Functionalized Olefins and Mechanistic Investigation of Photo-catalytic Pyridyl Radical Reactions. J. Am. Chem. Soc 2018, 140, 15525–15534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) McGrath NA; Brichacek M; Njardarson JT A Graphical Journey of Innovative Organic Architectures That Have Improved Our Lives. J. Chem. Educ 2010, 87, 1348–1349. [Google Scholar]; (b) Wilcken R; Zimmermann MO; Lange A; Joerger AC; Boeckler FM Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology. J. Med. Chem 2013, 56, 1363–1388. [DOI] [PubMed] [Google Scholar]; (c) Andriska V; György M; Miklós N Pesticide chemistry; Elsevier: Amsterdam, 1988. [Google Scholar]; (d) Jeschke P The Unique Role of Halogen Substituents in the Design of Modern Agrochemicals. Pest Manage. Sci 2010, 66, 10–27. [DOI] [PubMed] [Google Scholar]; (e) Jeschke P Latest Generation of Halogen-Containing Pesticides. Pest Manage. Sci 2017, 73, 1053–1066. [DOI] [PubMed] [Google Scholar]
- (3).(a) Olah GA Aromatic Substitution. XXVIII. Mechanism of Electrophilic Aromatic Substitutions. Acc. Chem. Res 1971, 4, 240–248. [Google Scholar]; (b) Galabov B; Nalbantova D; von Schleyer PR; Schaefer HF Electrophilic Aromatic Substitution: New Insights into an Old Class of Reactions. Acc. Chem. Res 2016, 49, 1191–1199. [DOI] [PubMed] [Google Scholar]
- (4).Joule JA; Mills K Heterocyclic Chemistry, 4th ed.; Blackwell: Malden, MA, 2000. [Google Scholar]
- (5).(a) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals: Mini-perspective. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]; (b) Murakami K; Yamada S; Kaneda T; Itami K C-H Functionalization of Azines. Chem. Rev 2017, 117, 9302–9332. [DOI] [PubMed] [Google Scholar]; (c) Baumann M; Baxendale IR An Overview of the Synthetic Routes to the Best Selling Drugs Containing 6-Membered Heterocycles. Beilstein J. Org. Chem 2013, 9, 2265–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Grimmett MR Halogenation of Heterocycles: II. Six- and Seven-Membered Rings. Adv. Heterocycl. Chem 1993, 58, 271–329. [Google Scholar]
- (6).(a) den Hertog HJ; Van der Does L; Landheer CA Bromination of Pyridine in Fuming Sulphuric Acid. Recl. Trav. Chim. Pays-Bas 1962, 81, 864–870. [Google Scholar]; (b) van der Does L; den Hertog HJ Bromination of Methylpyridines in Fuming Sulfuric Acid. Recl. Trav. Chim. Pays-Bas 1965, 84, 951–964. [Google Scholar]; (c) Lokhov RE; Lokhova SS; Gaidarova NM; Belen’kii LI Bromination of Pyridine in the Presence of Some Lewis Acids. Chem. Heterocycl. Compd 1981, 17, 923–926. [Google Scholar]; (d) Pearson DE; Hargrove WW; Chow JKT; Suthers BR The Swamping Catalyst Effect. III. The Halogenation of Pyridine and Picolines. J. Org. Chem 1961, 26, 789–792. [Google Scholar]
- (7).(a) Rodriguez RA; Pan C-M; Yabe Y; Kawamata Y; Eastgate MD; Baran PS Palau’chlor: A Practical and Reactive Chlorinating Reagent. J. Am. Chem. Soc 2014, 136, 6908–6911. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fosu SC; Hambira CM; Chen AD; Fuchs JR; Nagib DA Site-Selective C-H Functionalization of (Hetero)Arenes via Transient, Non-Symmetric Iodanes. Chem 2019, 5, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bagal SK; Andrews M; Bechle BM; Bian J; Bilsland J; Blakemore DC; Braganza JF; Bungay PJ; Corbett MS; Cronin CN; Cui JJ; Dias R; Flanagan NJ; Greasley SE; Grimley R; James K; Johnson E; Kitching L; Kraus ML; McAlpine I; Nagata A; Ninkovic S; Omoto K; Scales S; Skerratt SE; Sun J; Tran-Dubé M; Waldron GJ; Wang F; Warmus JS Discovery of Potent, Selective, and Peripherally Restricted Pan-Trk Kinase Inhibitors for the Treatment of Pain. J. Med. Chem 2018, 61, 6779–6800. [DOI] [PubMed] [Google Scholar]; (d) Di Lello P; Pastor R; Murray JM; Blake RA; Cohen F; Crawford TD; Drobnick J; Drummond J; Kategaya L; Kleinheinz T; Maurer T; Rougé L; Zhao X; Wertz I; Ndubaku C; Tsui V Discovery of Small-Molecule Inhibitors of Ubiquitin Specific Protease 7 (USP7) Using Integrated NMR and in Silico Techniques. J. Med. Chem 2017, 60, 10056–10070. [DOI] [PubMed] [Google Scholar]; (e) Liang Y; Lin F; Adeli Y; Jin R; Jiao N Efficient Electrocatalysis for the Preparation of (Hetero)Aryl Chlorides and Vinyl Chloride with 1,2-Dichloroethane. Angew. Chem., Int. Ed 2019, 58, 4566–4570. [DOI] [PubMed] [Google Scholar]; (f) Wang M; Zhang Y; Wang T; Wang C; Xue D; Xiao J Story of an Age-Old Reagent: An Electrophilic Chlorination of Arenes and Heterocycles by 1-Chloro-1,2-Benziodoxol-3-One. Org. Lett 2016, 18, 1976–1979. [DOI] [PubMed] [Google Scholar]
- (8).(a) Fier PS; Hartwig JF Selective C-H Fluorination of Pyridines and Diazines Inspired by a Classic Amination Reaction. Science 2013, 342, 956–960. [DOI] [PubMed] [Google Scholar]; (b) Fier PS; Hartwig JF Synthesis and Late-Stage Functionalization of Complex Molecules through C-H Fluorination and Nucleophilic Aromatic Substitution. J. Am. Chem. Soc 2014, 136, 10139–10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Wengryniuk SE; Weickgenannt A; Reiher C; Strotman NA; Chen K; Eastgate MD; Baran PS Regioselective Bromination of Fused Heterocyclic N-Oxides. Org. Lett 2013, 15, 792–795. [DOI] [PubMed] [Google Scholar]; (b) Trécourt F; Gervais B; Mongin O; Le Gal C; Mongin F; Quéguiner G First Syntheses of Caerulomycin E and Collismycins A and C. A New Synthesis of Caerulomycin A. J. Org. Chem 1998, 63, 2892–2897. [Google Scholar]; (c) Chen Y; Huang J; Hwang T-L; Chen MJ; Tedrow JS; Farrell RP; Bio MM; Cui S Highly Regioselective Halogenation of Pyridine N-Oxide: Practical Access to 2-Halo-Substituted Pyridines. Org. Lett 2015, 17, 2948–2951. [DOI] [PubMed] [Google Scholar]; (d) Yamanaka H; Araki T; Sakamoto T Site-Selectivity in the Reaction of 3-Substituted Pyridine 1-Oxides with Phosphoryl Chloride. Chem. Pharm. Bull 1988, 36, 2244–2247. [Google Scholar]
- (10).For selected reviews and examples, see:; (a) Manolikakes SM; Barl NM; Sämann C; Knochel P Regioselective Functionalization of Pyridines Using a Directed Metalation or a Halogen/Metal Exchange. Z. Naturforsch., B: J. Chem. Sci 2013, 68b, 411–422. [Google Scholar]; (b) El-Hiti GA; Hegazy AS; Alshammari MB; Masmali A Directed Lithiation of Simple Aromatics and Heterocycles for Synthesis of Substituted Derivatives. ARKIVOC 2015, iv, 19–47. [Google Scholar]; (c) El-Hiti GA; Smith K; Hegazy AS Directed Lithiation and Substitution of Pyridine Derivatives. Heterocycles 2015, 91, 479–504. [Google Scholar]; (d) Jaric M; Haag BA; Unsinn A; Karaghiosoff K; Knochel P Highly Selective Metalations of Pyridines and Related Heterocycles Using New Frustrated Lewis Pairs or Tmp-Zinc and Tmp-Magnesium Bases with BF3·OEt2. Angew. Chem., Int. Ed 2010, 49, 5451–5455. [DOI] [PubMed] [Google Scholar]; (e) Pomel V; Rovera JC; Godard A; Marsais F; Quéguiner G Synthesis of New Pyridine Intermediates as Precursors for the Elaboration of Streptonigrin Analogues by the Metalation-Cross-Coupling Strategy. J. Heterocycl. Chem 1996, 33, 1995–2005. [Google Scholar]; (f) Shi G; Takagishi S; Schlosser M Metalated Fluoropyridines and Fluoroquinolines as Reactive Intermediates: New Ways for Their Regioselective Generation. Tetrahedron 1994, 50, 1129–1134. [Google Scholar]; (g) Gu YG; Bayburt EK Synthesis of 4-Alkyl-3,5-Dibromo-, 3-Bromo-4,5-Dialkyl- and 3,4,5-Trialkylpyridines via Sequential Metalation and Metal-Halogen Exchange of 3,5-Dibromopyridine. Tetrahedron Lett. 1996, 37, 2565–2568. [Google Scholar]; (h) Pollet P; Turck A; Plé N; Quéguiner G Synthesis of Chiral Diazine and Pyridine Sulfoxides. Asymmetric Induction by Chiral Sulfoxides in an “Aromatic Ortho-Directed Metalation-Reaction with Electrophiles Sequence”. Diazines. 24. J. Org. Chem 1999, 64, 4512–4515. [Google Scholar]
- (11).For select bromination examples, see:; (a) Ashimori A; Ono T; Uchida T; Ohtaki Y; Fukaya C; Watanabe M; Yokoyama K Novel 1,4-Dihydropyridine Calcium Antagonists. I.: Synthesis and Hypotensive Activity of 4-(Substituted Pyridyl)-1, 4-Dihydropyridine Derivatives. Chem. Pharm. Bull 1990, 38, 2446–2458. [DOI] [PubMed] [Google Scholar]; (b) Diemer V; Chaumeil H; Defoin A; Fort A; Boeglin A; Carré C Syntheses of Sterically Hindered Zwitterionic Pyridinium Phenolates as Model Compounds in Nonlinear Optics. Eur. J. Org. Chem 2008, 2008, 1767–1776. [Google Scholar]; (c) Neumann U; Vögtle F 4,4′-Donor-Substituierte Und 6,6′-Difunktionalisierte 2,2′-Bipyridine. Chem. Ber 1989, 122, 589–591. [Google Scholar]; (d) Baron A; Herrero C; Quaranta A; Charlot M-F; Leibl W; Vauzeilles B; Aukauloo A Click Chemistry on a Ruthenium Polypyridine Complex. An Efficient and Versatile Synthetic Route for the Synthesis of Photoactive Modular Assemblies. Inorg. Chem 2012, 51, 5985–5987. [DOI] [PubMed] [Google Scholar]
- (12).For select chlorination examples, see:; (a) Pavlik JW; Vongnakorn T; Tantayanon S Synthesis and Spectroscopic Properties of Some Di- and Trideuterated Methylpyridines. J. Heterocycl. Chem 2009, 46, 213–216. [Google Scholar]; (b) Felts AS; Rodriguez AL; Blobaum AL; Morrison RD; Bates BS; Thompson Gray A; Rook JM; Tantawy MN; Byers FW; Chang S; Venable DF; Luscombe VB; Tamagnan GD; Niswender CM; Daniels JS; Jones CK; Conn PJ; Lindsley CW; Emmitte KA Discovery of N-(5-Fluoropyridin-2-Yl)-6-Methyl-4-(Pyrimidin-5-Yloxy)Picolinamide (VU0424238): A Novel Negative Allosteric Modulator of Metabotropic Glutamate Receptor Subtype 5 Selected for Clinical Evaluation. J. Med. Chem 2017, 60, 5072–5085. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Blank B; DiTullio NW; Deviny L; Roberts JT; Magnani A; Billig M; Saunders HL Synthesis and Hypoglycemic Activity of 4-Substituted 3-Mercaptopicolinic Acids. J. Med. Chem 1977, 20, 1572–1577. [DOI] [PubMed] [Google Scholar]
- (13).For examples of other 4-selective halogenation reactions on pyridines, see:; (a) Hamana M; Saito H Gamma-Bromination of Quinoline and Pyridine N-Oxides. Heterocycles 1979, 12, 475–479. [Google Scholar]; (b) Hwang SH; Wecksler AT; Zhang G; Morisseau C; Nguyen LV; Fu SH; Hammock BD Synthesis and Biological Evaluation of Sorafenib- and Regorafenib-like SEH Inhibitors. Bioorg. Med. Chem. Lett 2013, 23, 3732–3737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).For examples of recent 4-selective reactions on pyridines, see:; (a) Yang L; Semba K; Nakao Y Para-Selective C-H Borylation of (Hetero)Arenes by Cooperative Iridium/Aluminum Catalysis. Angew. Chem., Int. Ed 2017, 56, 4853–4857. [DOI] [PubMed] [Google Scholar]; (b) Gu Y; Shen Y; Zarate C; Martin R A Mild and Direct Site-Selective sp2 C-H Silylation of (Poly)Azines. J. Am. Chem. Soc 2019, 141, 127–132. [DOI] [PubMed] [Google Scholar]; (c) Nagase M; Kuninobu Y; Kanai M 4-Position-Selective C-H Perfluoroalkylation and Perfluoroarylation of Six-Membered Heteroaromatic Compounds. J. Am. Chem. Soc 2016, 138, 6103–6106. [DOI] [PubMed] [Google Scholar]
- (15).(a) Hilton MC; Dolewski RD; McNally A Selective Functionalization of Pyridines via Heterocyclic Phosphonium Salts. J. Am. Chem. Soc 2016, 138, 13806–13809. [DOI] [PubMed] [Google Scholar]; (b) Anderson RG; Jett BM; McNally A Selective Formation of Heteroaryl Thioethers via a Phosphonium Ion Coupling Reaction. Tetrahedron 2018, 74, 3129–3136. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Anderson RG; Jett BM; McNally A A Unified Approach to Couple Aromatic Heteronucleophiles to Azines and Pharmaceuticals. Angew. Chem., Int. Ed 2018, 57, 12514–12518. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Patel C; Mohnike M; Hilton MC; McNally A A Strategy to Aminate Pyridines, Diazines, and Pharmaceuticals via Heterocyclic Phosphonium Salts. Org. Lett 2018, 20, 2607–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Finet J-P Ligand Coupling Reactions with Heteroaromatic Compounds, Vol. 18; Pergamon: 1998; chapter 4. [Google Scholar]
- (17).For alternative uses of these 4,4’-bisheteroaryl phosphonium salts, see:; Hilton MC; Zhang X; Boyle BT; Alegre-Requena JV; Paton RS; McNally A Heterobiaryl Synthesis by Contractive C-C Coupling via P(V) Intermediates. Science 2018, 362, 799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Eicher T; Hauptmann S; Speicher A The chemistry of heterocycles: structure, reactions, syntheses, and applications, 3rd ed.; Wiley-VCH: Weinheim, 2012; pp 345–380. [Google Scholar]
- (19).Without the trifluoromethyl substituent at the 2-position, triflic anhydride activation of the phosphine pyridine is competitive with activation of the substrate pyridine.
- (20).Dolewski RD; Fricke PJ; McNally A Site-Selective Switching Strategies to Functionalize Polyazines. J. Am. Chem. Soc 2018, 140, 8020–8026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).(a) Gaussian 16, Revision B.01; Gaussian, Inc.: Wallingford, CT, 2016. (full reference available in the SI). [Google Scholar]; (b) Luchini G; Alegre-Requena JV; Funes-Ardoiz I; Paton RS GoodVibes: Automated Thermochemistry for Heterogeneous Computational Chemistry Data. F1000Research 2020, 9, 291 For functional ωB97X-D references: [Google Scholar]; (c) Becke AD Density-functional thermochemistry. V. Systematic Optimization of Exchange-Correlation Functionals. J. Chem. Phys 1997, 107, 8554–8560. [Google Scholar]; (d) Chai J-D; Head-Gordon M Long-Range Corrected Hybrid Density Functionals with Damped Atom-Atom Dispersion Corrections. Phys. Chem. Chem. Phys 2008, 10, 6615–6620. For Def2-SVP and Def2-QZVPP basis set references: [DOI] [PubMed] [Google Scholar]; (e) Weigend F; Ahlrichs R Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]; (f) Weigend F Accurate Coulomb-Fitting Basis Sets for H to Rn. Phys. Chem. Chem. Phys 2006, 8, 1057–1065. [DOI] [PubMed] [Google Scholar]
- (22).For IEF-PCM and SMD references:; (a) Cancés E; Mennucci B; Tomasi J A New Integral Equation Formalism for the Polarizable Continuum Model: Theoretical Background and Applications to Isotropic and Anisotropic Dielectrics. J. Chem. Phys 1997, 107, 3032–3041. [Google Scholar]; (b) Mennucci B; Cancés E; Tomasi J Evaluation of Solvent Effects in Isotropic and Anisotropic Dielectrics and in Ionic Solutions with a Unified Integral Equation Method: Theoretical Bases, Computational Implementation, and Numerical Applications. J. Phys. Chem. B 1997, 101, 10506–10517. [Google Scholar]; (c) Mennucci B; Tomasi J Continuum Solvation Models: A New Approach to the Problem of Solute’s Charge Distribution and Cavity Boundaries. J. Chem. Phys 1997, 106, 5151–5158. [Google Scholar]; (d) Tomasi J; Mennucci TB; Cancés E The IEF Version of the PCM Solvation Method: An Overview of a New Method Addressed to Study Molecular Solutes at the QM Ab Initio Level. J. Mol. Struct.: THEOCHEM 1999, 464, 211–226. [Google Scholar]; (e) Scalmani G; Frisch MJ Continuous Surface Charge Polarizable Continuum Models of Solvation. I. General formalism. J. Chem. Phys 2010, 132, 114110. [DOI] [PubMed] [Google Scholar]; (f) Marenich AV; Cramer CJ; Truhlar DG Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [DOI] [PubMed] [Google Scholar]
- (23).Pupo G; Ibba F; Ascough DMH; Vicini AC; Ricci P; Christensen KE; Pfeifer L; Morphy JR; Brown JM; Paton RS; Gouverneur V Asymmetric Nucleophilic Fluorination Under Hydrogen Bonding Phase-Transfer Catalysis. Science 2018, 360, 638–642. [DOI] [PubMed] [Google Scholar]
- (24).Wiberg KB Application of the Pople-Santry-Segal CNDO Method to the Cyclopropylcarbinyl and Cyclobutyl Cation and to Bicyclobutane. Tetrahedron 1968, 24, 1083–1096. [Google Scholar]
- (25).Dean JA Lange’s Handbook of Chemistry; McGraw-Hill; p 4.41. [Google Scholar]
- (26).Bowler JT; Wong FM; Gronert S; Keeffe JR; Wu W Reactivity in the Nucleophilic Aromatic Substitution Reactions of Pyridinium Ions. Org. Biomol. Chem 2014, 12, 6175–6180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).(a) Błaziak K; Danikiewicz W; Mąkosza M How Does Nucleophilic Aromatic Substitution Really Proceed in Nitroarenes? Computational Prediction and Experimental Verification. J. Am. Chem. Soc 2016, 138, 7276–7281. [DOI] [PubMed] [Google Scholar]; (b) Kwan EE; Zeng Y; Besser HA; Jacobsen EN Concerted Nucleophilic Aromatic Substitutions. Nat. Chem 2018, 10, 917–923. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Rohrbach S; Smith AJ; Pang JH; Poole DL; Tuttle T; Chiba S; Murphy JA Concerted Nucleophilic Aromatic Substitution Reactions. Angew. Chem., Int. Ed 2019, 58, 16368–16388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).(a) Eyring H The Activated Complex in Chemical Reactions. J. Chem. Phys 1935, 3, 107–115. [Google Scholar]; (b) Evans MG; Polanyi M Some Applications of the Transition State Method to the Calculation of Reaction Velocities, especially in Solution. Trans. Faraday Soc 1935, 31, 875–894. [Google Scholar]
- (29).(a) Nørskov JK; Abild-Pedersen F; Studt F; Bligaard T Density Functional Theory in Surface Chemistry and Catalysis. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 937–943. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mao Y; Wang H-F; Hu P Theory and Applications of Surface Micro-Kinetics in the Rational Design of Catalysts Using Density Functional Theory Calculations. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2017, 7, No. e1321. [Google Scholar]; (c) Zhao Z-J; Li Z; Cui Y; Zhu H; Schneider WF; Delgass WN; Ribeiro F; Greeley J Importance of Metal-Oxide Interfaces in Heterogeneous Catalysis: A Combined DFT, Micro-kinetic, and Experimental Study of Water-Gas Shift on Au/MgO. J. Catal 2017, 345, 157–169. [Google Scholar]; (d) Chen ZW; Chen LX; Wen Z; Jiang Q Understanding Electro-Catalysis by Using Density Functional Theory. Phys. Chem. Chem. Phys 2019, 21, 23782–23802. [DOI] [PubMed] [Google Scholar]
- (30).There is always an intrinsic source of error when using DFT methods and the error changes from one method to the others. For example, even though in all the cases the conclusions are the same (i.e., 3-Ph leads to faster reactions than 2-Ph, the mechanism goes through a stepwise SNAr, etc.), we found differences of several kcal/mol in the overall reaction barriers when using different functionals and solvents (Tables S7 and S10).
- (31).Koniarczyk JL; Hesk D; Overgard A; Davies IW; McNally A A General Strategy for Site-Selective Incorporation of Deuterium and Tritium into Pyridines, Diazines and Pharmaceuticals. J. Am. Chem. Soc 2018, 140, 1990–1993. [DOI] [PubMed] [Google Scholar]
- (32).(a) Johnson ER; Keinan S; Mori-Sanchez P; Contreras-Garcia J; Cohen J; Yang AW Revealing Noncovalent Interactions. J. Am. Chem. Soc 2010, 132, 6498–6506. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Contreras-Garcia J; Johnson ER; Keinan S; Chaudret R; Piquemal J-P; Beratan DN; Yang W NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput 2011, 7, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.