

Atherosclerosis is a chronic disease involving multiple cells in the arterial wall, including monocytes/macrophages, vascular smooth muscle cells (VSMC), and endothelial cells that are activated by immune responses1. It is thus clear that inflammation is a critical player in the pathogenesis of atherosclerosis and its complications, including stroke and myocardial infarction. Inflammatory processes contribute to all stages of the disease, including plaque stability and thrombosis. Targeting inflammation for atherosclerosis treatment is a major clinical endeavor, and, despite the success of statins, there is still an unmet need for additional therapies. Furthermore, atherosclerosis has strong genetic susceptibility, with genome-wide association studies (GWAS) identifying several risk loci. However, the specific causal variants, their target genes, cell types affected, and mechanisms of action in atherosclerosis are not very clear.
The histone deacetylase 9 (HDAC9) has been implicated in lipid metabolism, progression of atherosclerosis, and macrophage polarization via alterations in histone acetylation at target genes2,3. GWAS revealed several genetic variants in the HDAC9 locus are associated with multiple vascular diseases, including myocardial infarction, stroke, atherosclerotic aortic calcification, and peripheral artery disease4–8. HDAC9 risk alleles are associated with higher expression of HDAC9 in human monocytes/macrophages and human atherosclerotic plaques. Enhanced HDAC9 expression also increases vascular calcification and decreases contractility of human aortic VSMCs4,7,8. However, the molecular mechanisms by which HDAC9 promotes vascular inflammation and atherosclerosis are unclear.
In this issue of Circulation Research, Asare et al9 used a combination of in vitro assays and in vivo genetic mouse models, including Hdac9 deficient (Hdac9−/−) mice, as well as therapeutic interventions to investigate mechanisms underlying HDAC9 involvement in atherosclerosis and vascular inflammation. Bone marrow transplantation experiments were used to evaluate the effects of hematopoietic deficiency of Hdac9. Apoe−/− mice that received bone marrow from Hdac9−/− mice showed smaller and fewer atherosclerotic lesions as compared to those that received WT (Hdac9+/+) bone marrow. Furthermore, advanced lesions from Hdac9−/− mice had features of greater plaque stability with increased fibrous cap thickness relative to WT. To investigate this further, the authors treated bone marrow-derived macrophages (BMDMs) from WT or Hdac9−/− mice with the inflammatory cytokine TNF-α Hdac9−/− BMDMs showed decreased expression of proinflammatory cytokines and chemokines in response to TNF-α, as compared to WT BMDMs. These results support the proinflammatory role of HDAC9 in BMDMs and plaque vulnerability.
To further determine the mechanisms by which HDAC9 regulates inflammation, the authors postulated that transcription factor nuclear factor-κB (NF-κB), a major regulator of inflammation, might be a target of HDAC910. Co-immunoprecipitation experiments in HEK293 cells revealed that HDAC9 interacts with IKKα and IKKβ, both key upstream kinases that activate NF-κB. Phosphorylation of the NF-κB transcriptionally active p65 subunit by IKK results in p65 nuclear translocation and binding to its target gene promoters. Functionally, the observed interactions between HDAC9, IKKα, and IKKβ resulted in the deacetylation of IKKα and IKKβ, leading to their increased activity as demonstrated by higher p65 phosphorylation (Fig. 1). Leukocyte recruitment to inflamed vascular endothelium is a key feature of atherosclerosis. Some of the results showing HDAC9 mediated NF-κB activation were also observed in human umbilical vein endothelial cells (HUVECs). Loss-of-function studies in HUVECs showed that HDAC9 depletion decreased p65 phosphorylation and its nuclear localization, and reduced TNF-α-induced expression of key proinflammatory NF-κB target genes. Although these data strongly support the proinflammatory role of HDAC9 acting as a positive effector of NF-κB activation, the experiments were performed in HEK293 and HUVEC cells. As the in vivo experiments suggested a compelling role for myeloid cell-derived HDAC9 in the observed phenotypes, the authors could have also performed some of these biochemical experiments in monocyte/macrophages, and even with VSMC that modulate plaque stability. Furthermore, examining the nuclear localization of HDAC9 in TNF-α treated cells may help distinguish the cytosolic versus nuclear functions of HDAC9.
Figure 1. HDAC9 actions in atherosclerosis.
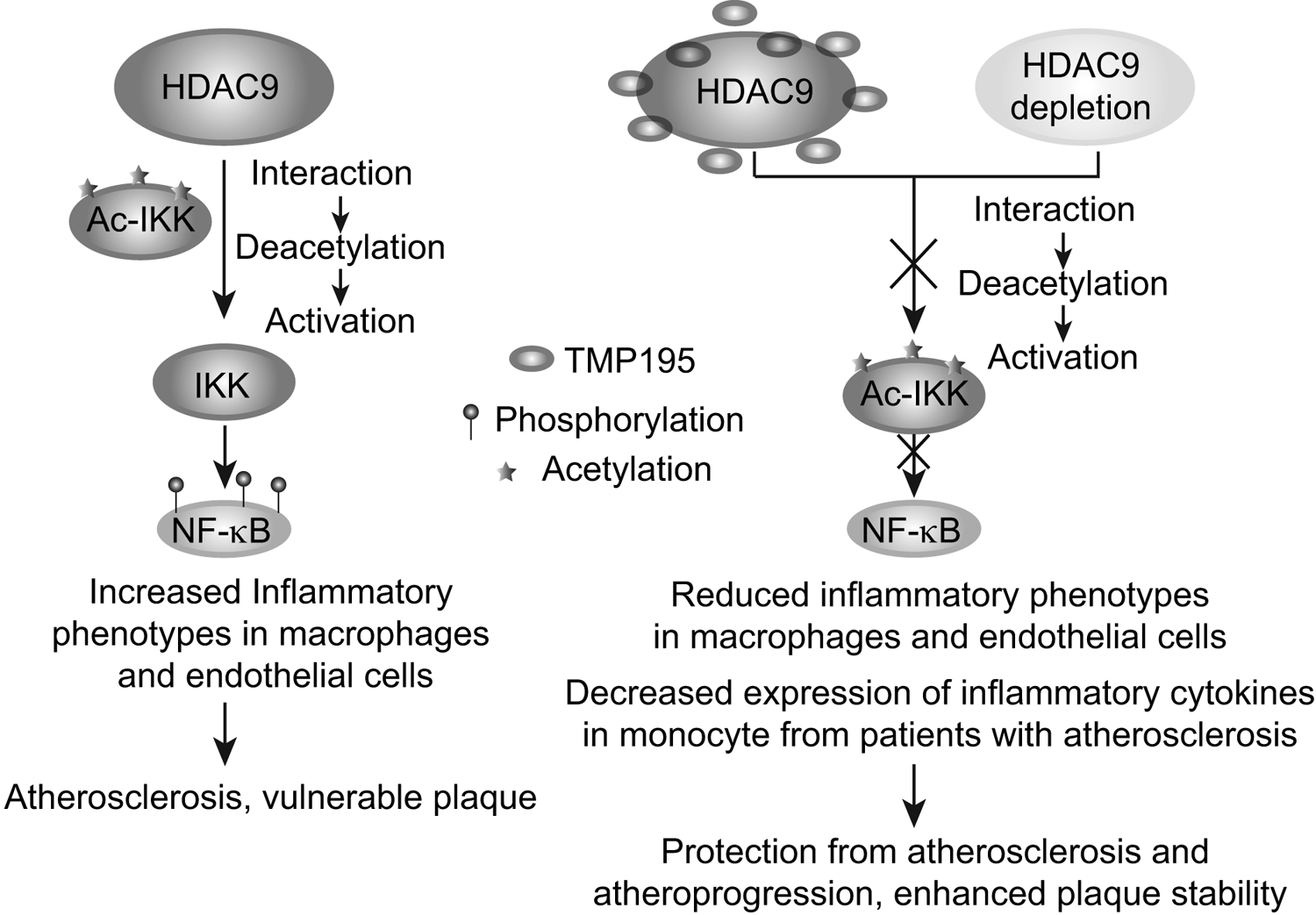
HDAC9 risk alleles can increase HDAC9 expression. HDAC9 interacts with IKK kinases, leading to their deacetylation and activation. IKK phosphorylates and activates NF-κB p65 leading to increased inflammatory phenotypes in macrophages and endothelial cells associated with atherosclerosis and vulnerable plaque. HDAC9 deletion or pharmacological inhibition by TMP195 inhibits such inflammatory phenotypes and ameliorates atherosclerosis and enhances plaque stability in mice.
Certain HDAC inhibitors are already in clinical use for certain cancers11. They have also been tested in experimental models of CVD with varied outcomes. One group showed that the non-selective HDAC inhibitor trichostatin A (which inhibits most HDACs, class I, IIa, IIb) increased atherosclerotic lesion size3, while others reported anti-atherogenic function in macrophages. A highlight of the current study is that the authors examined the therapeutic potential of TMP195, a selective class IIa HDAC inhibitor, on both initiation of atherosclerosis as well as later stages. Apoe−/− mice fed with high-fat diet were administered TMP195 and evaluated for atherosclerotic phenotypes. Two-photon microscopy revealed TMP195 treated Apoe−/− mice had reduced lesion formation and decreased endothelial expression of adhesion molecules compared to vehicle control. Moreover, intravital microscopy showed TMP195 decreased myeloid cell rolling and adhesion to arteries, supporting its anti-inflammatory, atheroprotective role. TMP195 also increased IKKβ acetylation and decreased TNFα-induced p65 phosphorylation in BMDMs, which coincided with attenuated NF-κB-mediated proinflammatory gene expression. These data support the notion that TMP195 can phenocopy/mimic the effect of HDAC9 deletion. Further, RNA-seq revealed that TMP195 inhibits inflammatory phenotypes in TNFα-induced BMDMs. Of note, authors also performed RNA-seq in cells treated with the IKKβ inhibitor TPCA-1 and found 86% of shared genes between TMP195 and TPCA-1 treatment, suggesting TMP195 exerts its anti-inflammatory effects, at least in part, via inhibitory effects on IKKβ.
Finally, the authors performed critical experiments in a mouse model with established atherosclerosis to determine whether TMP195 also reduces atheroprogression and confers plaque stability. This intervention is important to evaluate the therapeutic potential of targeting HDAC9 with TMP195 because most treatments are administered only to CVD patients having existing lesions. TMP195 was administered to western diet-fed Apoe−/− mice after they developed lesions. Relative to control, TMP195 treatment decreased lesion size and invasion of monocytes and neutrophils into the atherosclerotic lesions, suggesting its protective role against atheroprogression. TMP195 also depicted protection at later stages of atherosclerosis, attenuating the size of advanced lesions, while concordantly promoting a more stable plaque phenotype (Fig. 1). Together, the data suggest TMP195 confers protective effects in early and advanced atherosclerosis in mice, highlighting translational significance. To determine human relevance, the authors treated monocytes isolated from patients with established atherosclerosis with TMP195, which led to decreased production of inflammatory cytokines and chemokines, along with reduced p65 phosphorylation.
This study further underscores the value of investigating GWAS candidate hits for the treatment of related CVDs. Notably, the authors examined the effect of TMP195 on both early and late atherosclerosis, mimicking the clinical situation. However, more studies are needed to determine the specificity and off-target effects of TMP195 and compare with other selective HDAC inhibitors. The vascular and myeloid cell-type specific effects of TMP195 related to atherosclerosis and stroke have not been assessed.
The authors focused primarily on the non-histone protein deacetylase role of HDAC9 with IKK proteins being the target. HDAC9 can also have nuclear roles by deacetylating histones at promoters of key target genes leading to their repression. Such epigenetic mechanisms were reported by Cao et al2, who showed Hdac9−/− mice depicted protection from atherosclerosis. These protective effects were attributed to increased histone H3K9 acetylation at the promoters of ABCA1 and ABCG1, factors involved in macrophage reverse cholesterol transport, with resultant increases in their expression and improvement in lipid homeostasis. Such targets/epigenetic mechanisms were not assessed in the current study. Apart from histone-modifying enzymes like HDACs, other regulatory elements such as enhancers are also implicated in CVDs, and small molecule inhibitors targeting them show protective effects12,13. Future studies could assess whether TMP195 exerts its anti-atherogenic and anti-inflammatory effects through mechanisms besides those described by Asare et al., including those affecting epigenetic processes and regulatory elements.
Several treatment modalities are available for atherosclerosis prevention, but fewer options for the vulnerable plaque. So, HDAC9 inhibitors might be one future treatment option for plaque vulnerability in advanced disease and/or exclusively for patients expressing the Hdac9 risk alleles in the era of personalized medicine. In summary, Asare et al. provide strong evidence that HDAC9 promotes vascular inflammation and plaque vulnerability via NF-κB activation, and that a class IIa HDAC inhibitor TMP195 is effective against atherosclerosis, atheroprogression, and plaque vulnerability (Fig. 1). This study offers a mechanistic explanation for the strong association of the HDAC9 locus with vascular disease.
Sources of Funding
We gratefully acknowledge funding from the National Institutes of Health (NIH): R01 DK065073 and R01 HL106089 (to RN)
Footnotes
Disclosures: None
References:
- 1.Libby P, Ridker PM, Hansson GK, Leducq Transatlantic Network on A. Inflammation in atherosclerosis: From pathophysiology to practice. J Am Coll Cardiol. 2009;54:2129–2138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cao Q, Rong S, Repa JJ, St Clair R, Parks JS, Mishra N. Histone deacetylase 9 represses cholesterol efflux and alternatively activated macrophages in atherosclerosis development. Arterioscler Thromb Vasc Biol. 2014;34:1871–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Choi JH, Nam KH, Kim J, Baek MW, Park JE, Park HY, Kwon HJ, Kwon OS, Kim DY, Oh GT. Trichostatin a exacerbates atherosclerosis in low density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2005;25:2404–2409 [DOI] [PubMed] [Google Scholar]
- 4.Malhotra R, Mauer AC, Lino Cardenas CL, Guo X, Yao J, Zhang X, Wunderer F, Smith AV, Wong Q, Pechlivanis S, et al. Hdac9 is implicated in atherosclerotic aortic calcification and affects vascular smooth muscle cell phenotype. Nat Genet. 2019;51:1580–1587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.International Stroke Genetics C, Wellcome Trust Case Control C, Bellenguez C, Bevan S, Gschwendtner A, Spencer CC, Burgess AI, Pirinen M, Jackson CA, , et al. Genome-wide association study identifies a variant in hdac9 associated with large vessel ischemic stroke. Nat Genet. 2012;44:328–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Malik R, Chauhan G, Traylor M, Sargurupremraj M, Okada Y, Mishra A, Rutten-Jacobs L, Giese AK, van der Laan SW, Gretarsdottir S, et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes. Nat Genet. 2018;50:524–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Markus HS, Makela KM, Bevan S, Raitoharju E, Oksala N, Bis JC, O’Donnell C, Hainsworth A, Lehtimaki T. Evidence hdac9 genetic variant associated with ischemic stroke increases risk via promoting carotid atherosclerosis. Stroke. 2013;44:1220–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Prestel M, Prell-Schicker C, Webb T, Malik R, Lindner B, Ziesch N, Rex-Haffner M, Roh S, Viturawong T, Lehm M, et al. The atherosclerosis risk variant rs2107595 mediates allele-specific transcriptional regulation of hdac9 via e2f3 and rb1. Stroke. 2019;50:2651–2660 [DOI] [PubMed] [Google Scholar]
- 9.Asare Y, Campbell-James TA, Bokov Y, Yu LL, Prestel M, El Bounkari O, Roth S, Megens RT, Straub T, Thomas K, et al. Histone deacetylase 9 activates ikk to regulate atherosclerotic plaque vulnerability. Circ Res. 2020; 127: xxx-xxx. [DOI] [PubMed] [Google Scholar]
- 10.de Winther MP, Kanters E, Kraal G, Hofker MH. Nuclear factor kappab signaling in atherogenesis. Arterioscler Thromb Vasc Biol. 2005;25:904–914 [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Seto E. Hdacs and hdac inhibitors in cancer development and therapy. Cold Spring Harb Perspect Med. 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brown JD, Lin CY, Duan Q, Griffin G, Federation A, Paranal RM, Bair S, Newton G, Lichtman A, Kung A, , et al. Nf-kappab directs dynamic super enhancer formation in inflammation and atherogenesis. Mol Cell. 2014;56:219–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Das S, Senapati P, Chen Z, Reddy MA, Ganguly R, Lanting L, Mandi V, Bansal A, Leung A, Zhang S,et al. Regulation of angiotensin ii actions by enhancers and super-enhancers in vascular smooth muscle cells. Nat Commun. 2017;8:1467. [DOI] [PMC free article] [PubMed] [Google Scholar]