

Abstract
The human multidrug transporter P-glycoprotein (Pgp or ABCB1) sets up pharmacological barriers to many clinically important drugs, a therapeutic remedy for which has yet to be formulated. For the rational design of mechanism-based inhibitors (or modulators), it is necessary to map the potential sites for modulator interaction and understand their modes of communication with the other functional domains of Pgp. In this study, combining directed mutagenesis with homology modeling, we provide evidence of two modulator-specific sites at the lipid protein interface of Pgp. Targeting 21 variant positions in the COOH-terminal transmembrane (TM) regions, we find residues M948 (in TM11) and F983, M986, V988, and Q990 (all four in TM12) critically involved in substrate-site modulation by a thioxanthene-based allosteric modulator cis-(Z)-flupentixol. Interestingly, for ATP-site modulation by the same modulator, only two (M948 and Q990) of those four residues appear indispensable, together with two additional residues, T837 and I864 in TM9 and TM10, respectively, suggesting independent modes of communication linking the allosteric site with the substrate binding and ATPase domains. None of the seven residues identified prove to be critical for modulation of the substrate or ATP sites by Pgp modulators that are transported by the pump, such as cyclosporin A or verapamil, indicating their specificity for cis-(Z)-flupentixol. On the other hand, ATP-site modulation by verapamil proves to be highly sensitive to replacement at positions F716 (in TM7) and I765 (in TM8), and to a more moderate extent at I764 and L772 (both in TM8). Homology modeling based on the known crystal structures of the bacterial multidrug transporter SAV1866 and the mouse Pgp homologue maps the identified residues primarily at the lipid–protein interface of Pgp, in two spatially distinct modulator-specific clusters. The two modulatory sites demonstrate negative synergism in influencing ATP hydrolysis, consolidating their spatial distinctness. Because Pgp is known to recruit drug molecules directly from the lipid bilayer, identification of modulatory sites at the lipid–protein interface and at the same time outside the conventional central drug binding cavity is mechanistically revealing.
Graphical Abstract
The human multidrug transporter (Pgp or ABCB1) is an ATP-driven pump that transports structurally unrelated hydrophobic compounds, which include many anticancer and antimicrobial agents.1,2 It is a 1280-amino acid protein with 40% sequence identity between its NH2-and COOH-terminal halves that are connected by an 80-amino acid linker region.3 Each half contains a hydrophobic region that folds into six membrane-spanning α-helices and a hydrophilic cytosolic domain.4–7 The transmembrane (TM) helices from the two halves closely associate to form a single dug-translocating pathway.8,9 Similarly, the two cytosolic regions, also known as the nucleotide binding domains (NBDs), interact intimately to form two ATP binding and hydrolysis sites.10 The binding and hydrolysis of ATP are functionally coupled to outward translocation of drug substrates.11,12
The most intriguing aspect of Pgp function is its ability to recognize and transport a broad spectrum of structurally unrelated compounds, which include several anticancer and antimicrobial agents.1,2 Apart from its transport substrates, Pgp also interacts with a battery of lipophilic compounds with modulatory potential, which can be exploited for reversal of drug resistance and improved bioavailability of therapeutic agents.13 Like substrates, modulators also are of diverse chemical origin, indicating an equally complex mode of interaction with Pgp.14–16 Although several Pgp modulators have been identified, none of the compounds succeed in overcoming clinical drug resistance, necessitating a mechanism-based development of new agents with improved efficacy. To achieve that goal, a detailed mapping of the modulator interaction sites in Pgp and understanding their modes of inactivation remain absolutely essential.
Experimental evidence suggests recruitment of drug molecules by Pgp directly from the lipid bilayer17–20 and an active involvement of TM regions in the recruitment process.21 Susceptibility to chemical modification of specifically engineered cysteine residues in a cysteine-less human Pgp and its protection by transport substrates mapped amino acid residues in the vicinity of the putative drug interaction sites and delineated the central drug binding pocket22 involved in drug recognition and transport. Photoaffinity labeling combined with directed mutagenesis has identified two symmetrical portals at the interface of the two transmembrane domains (TMDs),23,24 which could represent a major exit for drug molecules from or entrance for drug molecules into the central cavity.25 Apart from these studies, independent efforts, including our own, demonstrate allosteric modes of action for many Pgp modulators and the presence of modulatory sites distinct from the sites of substrate recognition, with a heterotropic effect on substrate binding.26–29 However, the identity of those allosteric sites and their spatial relationship with the central drug binding cavity are yet to be defined.
In this study, combining directed mutagenesis with molecular modeling, we identify two modulator-selective sites at the lipid—protein interface of Pgp, through which essential steps like ATP hydrolysis and substrate binding are functionally modulated.
EXPERIMENTAL PROCEDURES
Chemicals and Cell Lines.
All chemicals, unless otherwise mentioned, were purchased from Sigma (St. Louis, MO). cis-(Z)-Flupentixol was purchased from Research Biochemicals International or Sigma. [125I]IAAP (2200 Ci/mmol) was from NEN PerkinElmer Life and Analytical Sciences (Boston, MA). The QuickChange Site-Directed Mutagenesis Kit was obtained from Stratagene (La Jolla, CA), and the oligonucleotides were custom synthesized by Sigma. The Sf9 insect cells, the MAX Efficiency DH10Bac Competent Cells, and the Bac-to-Bac Baculovirus Expression Systems were purchased from Invitrogen Life Technologies. The Sf-900 II SFM cell insect cell culture media and penicillin-streptomycin were purchased from Invitrogen. Pgp-specific polyclonal antibody 4007 was a generous gift from M. M. Gottesman of the Laboratory of Cell Biology, National Cancer Institute, National Institutes of Health. Nucleotide sequencing of the recombinant cDNA constructs was conducted in the Uniformed Services University Bio Instrumentation Core facility.
Directed Mutagenesis.
The MDR1 cDNA was amplified via polymerase chain reaction (PCR) from plasmid pTM1-MDR1-(H6)30 with a BamHI site inserted at the 5’-end using the oligonucleotide 5’-CACGATAATACCGGATCCATGGATCTTGAAG-3’ and cloned into the BamHI- and XhoI-cut pFastBac plasmid supplied by Invitrogen, creating plasmid pKM2-MDR1-WT, containing the wild-type human MDR1 cDNA. Single-alanine replacements were introduced into the wild-type backbone using the QuickChange mutagenesis kit from Stratagene and appropriate oligonucleotide primers. The desired nucleotide replacements were confirmed by Big-Dye version 3.1 (BD Sciences) nucleotide sequencing of the MDR1 open reading frame in recombinant plasmids pKM2-MDR1- V715A, -F716A, -I719A, -G723A, -I764A, -I765A, -I768A, -F770A, -L772A, -T837A, -I840A, -I864A, -I867A, -A871F, -T945A, -M948A, -F983A, -M986A, -V988A, -Q990A, and -V991A.
Generation of Baculovirus Constructs with Alanine-Substituted Recombinant Pgps.
Recombinant baculoviruses encoding mutant Pgps were generated using the standard laboratory protocol supplied with the Invitrogen Bac-to-Bac Baculovirus Expression System. In brief, DH10Bac bacterial cells harboring a helper virus were cotransformed either with plasmid pKM2-MDR1-WT (containing wild-type Pgp) or with each of the 21 plasmids carrying recombinant Pgp with single-alanine substitutions. The DH10Bac cells containing the recombinant bacmids expressing wild type Pgp or mutant Pgps were purified by blue-white selection on Blue-gal plates. The recombinant bacmids were isolated from the positive clones and checked for the right insertion by PCR amplification of the inserted fragment using pUC/M13-specific primers (forward, 5’-CCCAGTCACGACGTTGTAAAACG-3’; pUC/ M13 reverse, 5’-AGCGGATAACAATTTCACACAGG-3’). Following confirmation, monolayers of Sf9 insect cells were grown to 75% confluency and transfected with the recombinant bacmids using Cellfectin reagent according to the recommended protocol of Invitrogen. Transfected cells were allowed to grow at 27 °C for 72 h, and recombinant viruses secreted into the medium were collected by separating the cells through centrifugation at 500g and 4 °C for 10 min. The viral supernatant was filtered through a nitrocellulose filter with a pore size of 0.25 μm to remove any residual cells or cell debris. The viral titer was then amplified through multiple rounds (four to six) of infection of Sf9 cells grown using a method similar to that for transfection with bacmids, until a final titer of ~107 pfu/mL was obtained. Amplified viruses were filtered and stored at 4 °C for future use.
Baculovirus-Mediated Expression of Recombinant Pgps in Sf9 Insect Cells.
Wild-type and recombinant Pgps were expressed in Sf9 insect cells using the baculoviral constructs described above. Sf9 cells were cultured at 27 °C in monolayers to 75% confluency in Sf-900 II SFM medium supplemented with 0.5% penicillin-streptomycin prior to infection. Infections with respective recombinant baculoviruses at 10 pfu/cell were conducted in a minimal volume (approximately 1 mL/1 million cells) for 2 h with brief and gentle rocking every 15 min. The infected cells were diluted with Sf-900 II SFM medium and allowed to grow for 72 h at 27 °C, after which cells were harvested by centrifugation at 500g for 10 min at 4 °C. Harvested cells were washed twice by centrifugation and resuspension with ice-cold phosphate-buffered saline (PBS) containing 1% aprotinin. Washed cells were finally resuspended in homogenization buffer [50 mM Tris-HCl (pH 7.5), 50 mM mannitol, 2 mM EGTA, 1 mM DTT, 1 mM AEBSF, and 1% aprotinin] and stored at —80 °C for future use.
Preparation of the Total Membrane from Sf9 Cells Expressing Wild-Type and Recombinant Pgps.
Crude membranes were prepared according to the method of Dey et al.26 Membranes, in 100 μL aliquots, were frozen on dry ice and stored at —70 °C until they were used. Protein concentrations were measured by a modified Lowry protocol31 using BSA as a standard.
Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS–PAGE) and Immunoblot Analysis.
Electrophoresis and immunoblot analysis were performed as described previously.32 Immunodetection was conducted using human Pgp-specific antiserum 4007, originally developed against a COOH-terminal fragment of the protein.33
Measuring Drug-Stimulated ATP Hydrolysis by Pgp.
ATP hydrolysis by wild-type Pgp and the alanine-substituted recombinant Pgps in isolated membrane vesicles from insect cells was assessed by measuring the vanadate-sensitive release of inorganic phosphate from MgATP in the presence or absence of 0.3 mM sodium orthovanadate, following a colorimetric assay originally developed by Sarkadi et al.,34 with minor modifications.26 ATP hydrolysis data were expressed as fold stimulation of the basal activity present in the absence of any modulators. The kinetic analysis of the data was conducted using nonlinear fit V0 = (Vmax[S])/(Km + [S]), where V0 is the fold stimulation, Vmax the maximal fold stimulation, [S] the concentration of modulatory compounds, and Km the concentration of modulators for half-maximal stimulation. Data were generated in triplicate, and the averages of all three data sets were used for analysis. Standard errors are shown as error bars in the figures.
[125I]IAAP Binding to Wild-Type Pgp and Recombinant Pgps.
Photoaffinity labeling of crude membranes with the Pgp substrate [125I]IAAP was conducted according to the method of Dey et al.26 Radioactivity associated with the Pgp band was quantified directly by PhosphorImager analysis and captured on X-ray film for documentation.
Sequence Alignment of the Mammalian Pgps To Select Target Residues for Alanine Substitution.
The amino acid sequences for human MDR1 (P08183), mouse MDR1A (NM_011076.1) and MDR1B (NM_011075.1), beagle MDR1 (AF045016.1), rat MDR1 (NM_012623.2), and hamster MDR1 (P21448) were obtained from GenBank. The multiple-amino acid sequence alignment was conducted using the Internet-based general purpose multiple-sequence alignment program ClustalW2, licensed by the European Bioinformatics Institute (EBI) of the European Molecular Biology Laboratory.
Homology Modeling of Human Pgp Based on Crystal Structures of Sav1866 and Mouse Pgp.
For the N-terminal NBD, Pgp was found to be significantly identical in sequence with the crystal structure of Sav1866 in the Protein Data Bank (entry 2HYD). The initial monomeric NBD model was generated by replacing the residues on the Sav1866 model with those of the aligned Pgp sequence. Structure-based sequence alignment allows insertions and deletions to be moved to loop regions without interfering with the modeling of secondary structure elements.
To model the N-terminal transmembrane domain (TMD), TM helical segments were first predicted with various computer programs such as SOSUI,3 PHD,36 HMMTOP,37 and TMHMM,38 from which a consensus of six TM helices emerged. The boundary of each predicted helix was adjusted to conform to a set of empirical rules derived from known membrane protein structures.39,40 Adjustments were made to the model whenever residues in the model had stereochemistry violations.
The same procedure was used to build the C-terminal half of Pgp. The dimeric model was then generated by superposition of the monomers onto the Sav1866 dimer in the modeling program O and subsequently refined in CNS41 to optimize van der Waals interactions and to conform to the standard stereochemistry values of bond angles, bond lengths, and dihedral angles.
Lipophilic Index Calculation.
Lipophilic scores (LIPS) for predicted TM helices were calculated on the basis of the empirical hydrophobic scales of individual amino acids.42
RESULTS
Modulation of Pgp-Substrate Interaction Induced by Allosteric Modulator cis-(Z)-Flupentixol Is Sensitive to Replacement of Specific Variant Residues with Alanine.
Modulators of Pgp either directly compete for the substrate site(s) or indirectly modulate substrate binding through allosteric interaction, either of which can be studied using a radiolabeled photoaffinity compound, [125I]-iodoarylazidoprazosin ([125I]IAAP).29 Unlike most other modulators, such as cyclosporine A that directly blocks substrate binding, the thioxanthene-based allosteric modulator cis-(Z)-flupentixol prevents substrate dissociation, resulting in a dramatic increase in the level of Pgp-[125I]IAAP interaction.29,43 To identify the molecular determinants of this allosteric effect, we generated single-alanine substitutions at 21 variant positions in the COOH-terminal TM regions (Figure 1) and studied their impact on the stimulatory potential of cis-(Z)-flupentixol on [125I]IAAP binding (Figure 2A). Multiple-sequence alignment of closely related mammalian homologues revealed that 60% of the amino acid residues in the TM regions of Pgp remained strictly conserved during evolution, suggesting their indispensability in maintaining the native structure and physiological function of the protein. It is reasonable to propose that the primary selective force behind evolution of the invariant residues in human Pgp has been preservation of transport function, whereas interaction of modulatory drugs is relatively incidental. Modulators interact with Pgp either by physically mimicking transport substrates or through structural complementarity to allosteric sites with modulatory function. Therefore, it is less likely that the key components of allosteric modulator interaction, as opposed to those of substrate recognition, remain confined mainly within invariant (strictly conserved) residues. To identify residues selectively involved in allosteric modulation and at the same time preserve the normal function of the protein, we cautiously restricted our target residues within amino acid positions that were not strictly conserved.
Figure 1.
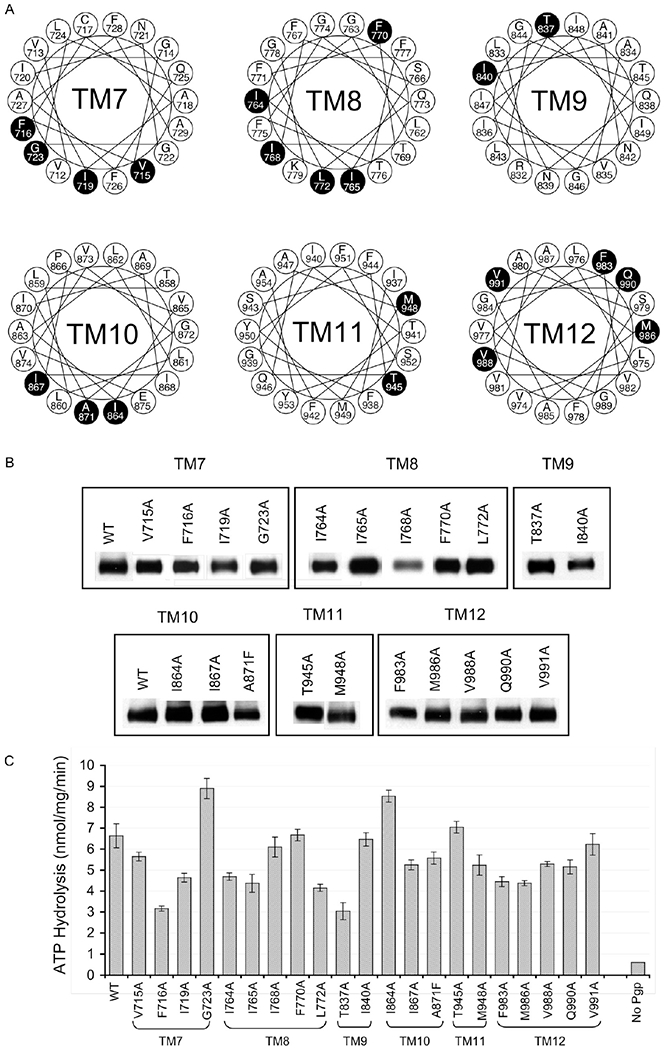
Generation of recombinant Pgps with single-alanine substitutions. (A) Amino acid residues targeted for single-amino acid substitution. The putative TM regions of the COOH-terminal half of Pgp (eight) are presented as helical wheels, to show the 21 variant residues targeted (in filled circles) for single-amino acid substitution. Human MDR1 and MDR2, beagle MDR1, mouse MDR1a, MDR1b, and MDR2, Chinese hamster PGP1, PGP2, and PGP3, and rat MDR1 and MDR2 were aligned as described in Experimental Procedures. If the 14 naturally occurring alanines are excluded, 37 variant residues were identified, of which six of eight nonconserved, four of six semiconserved, and 10 of 23 conserved changes are targeted by alanine substitution. Of the 14 alanines in variant positions, only one (A871) was targeted for substitution with phenylalanine. (B) Expression of recombinant human Pgp in insect cells using the baculovirus-mediated expression system. Isolated membranes (10 μg/well) from insect cells expressing either wild-type human Pgp or the Pgp alanine mutants were subjected to SDS-PAGE, electro-transferred to a nitrocellulose membrane, and probed with Pgp-specific antibody 4007.33 Signals were generated using an HRP-conjugated goat anti-rabbit secondary antibody and ECL reagents and captured on X-ray film. The image is a representative of three individual membrane preparations and subsequent immunoblotting. (C) Basal ATPase activity of Pgp mutants. The basal rate of ATP hydrolysis of the 21 Pgp mutants in insect cell membrane preparation was measured using a vanadate-sensitive phosphate release assay as described in Experimental Procedures and compared with that of the insect cell membranes containing the wild-type Pgp and membranes without Pgp expression. Data are averages of three different experiments and expressed as nanomoles per milligram of membrane protein per minute.
Figure 2.
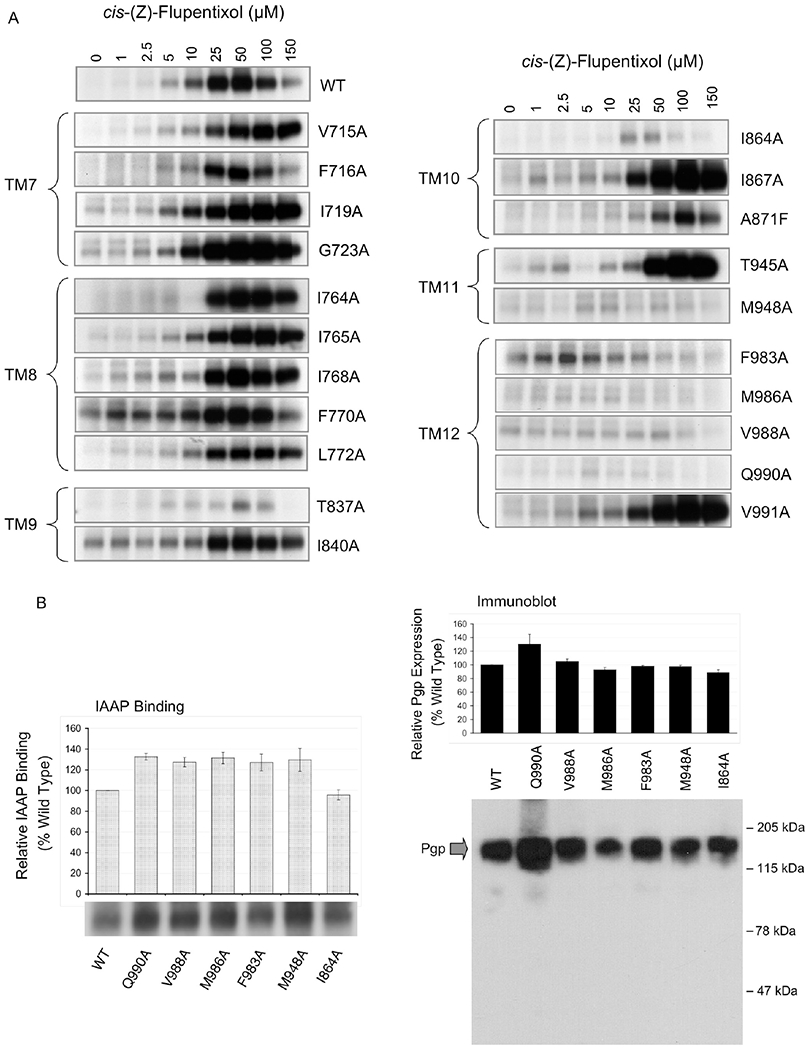
Effect of alanine substitutions on cis-(Z)-flupentixol-mediated stimulation of [125I]IAAP binding. (A) Stimulation of binding of [125I]IAAP to Pgp mutants. Wild-type Pgp and Pgp alanine mutants in isolated membranes (10 μg of total protein) of insect cells were incubated with and photoaffinity labeled with 5 nM [125I]IAAP in the presence or absence of varying concentrations of Pgp modulator cis-(Z)-flupentixol. Labeled membranes (2.5 μg per lane) were subjected to SDS-PAGE, and radioactivity associated with Pgp was captured on X-ray film. The relevant portion of the gel showing radioactivity associated with Pgp is included in the figure. There was no other specific radioactive band observed in the gel other than that corresponding to Pgp. (B) Basal binding of [125I]IAAP to cis-(Z)-flupentixol-specific Pgp mutants. Wild-type Pgp and the cis-(Z)-flupentixol-specific Pgp mutants in isolated membranes (10 μg of total protein) were photoaffinity labeled with 5 nM [125I]IAAP in the absence of any modulators. Labeled membranes (2.5 μg per lane) were subjected to SDS-PAGE, and radioactivity associated with Pgp was captured on X-ray film (left panel image) as well as quantified using a PhosphorImager (left panel graph). Aliquots (1 μg) of the same samples were immunodetected with Pgp-specific polyclonal antibody 4007 (right panel image), and the amount of Pgp in each band was determined by densitometric analysis (right panel graph).
Consistent with our previous finding, cis-(Z)-flupentixol induced a concentration-dependent increase in the level of binding of [125I]IAAP to wild-type Pgp, which was progressively alleviated at concentrations higher than 50 μM. The reduction in the level of stimulation at higher concentrations can be attributed to the presence of a secondary site of interaction for cis-(Z)-flupentixol, with inhibitory potential, or to a nonspecific membrane effect of high modulator concentrations. This is reminiscent of the bell-shaped curves observed in the stimulation of ATP hydrolysis by many Pgp-interacting drugs.26,44 Most of the Pgp mutants that retained flupentixol-mediated stimulation of [125I]IAAP binding, in our study, demonstrated a similar decrease in the level of stimulation at higher concentrations of the modulator. For identifying residues that allosterically modulate substrate binding, we exclusively focused on substitutions that affect the stimulatory component of modulation.
As one can see in Figure 2A, none of the substitutions in TM7 (G723A, I719A, F716A, and V715A) or TM8 (L772A, F770A, I768A, I765A, and I764A) was critical for stimulation of [125I]IAAP binding by cis-(Z)-flupentixol. On the other hand, replacement of residues M948 (of TM11) and F983, V988, M986, and Q990 (of TM12) abrogated the stimulatory effect, suggesting that these residues were crucial in mediating the modulatory action of cis-(Z)-flupentixol. Although alanine substitution of T837 (in TM9) and I864 (in TM10) also had a recognizable effect, a >2-fold residual stimulation in each case suggested that the residues were not indispensable for the stimulatory action. A comparable level of basal binding of [125I]IAAP to wild-type Pgp and Pgp mutants M948A, F983A, V988A, M986A, and Q990A (Figure 2B) rules out the possibility that the reduced level of stimulation in the mutants is due to loss of [125I]IAAP binding and not stimulation per se.
To investigate whether M948, Q990, F983, M986, and V988 are part of a general module involved in the regulation of substrate binding or are molecular components specific to cis-(Z)-flupentixol, we studied the effect of a structurally unrelated Pgp modulator, cyclosporin A, on binding of [125I]IAAP to all 21 mutants. Cyclosporin A, a transport substrate for Pgp, is known to block [125I]IAAP binding presumably through direct competition.29 Results showed no comparable effect on the inhibitory potential of cyclosporin A by any of the substitutions, with more than 75% inhibition observed in each case at a modulator concentration as low as 5 μM (Figure 3). Although a moderate effect was observed in Pgp mutants I867A, A871F, and V988A, none of the replacements proved to be absolutely critical for inhibition by cyclosporin A (Figure 3). These results established the specificity of the identified residues toward the allosteric modulator cis-(Z)-flupentixol and ruled out any nonspecific defect affecting modulator interaction in general.
Figure 3.
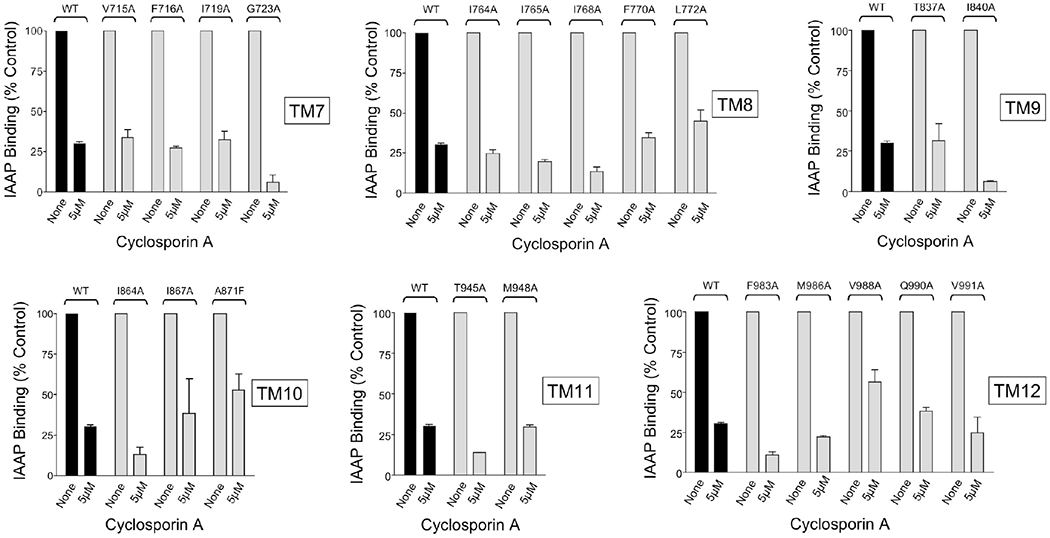
Inhibition of binding of [125I]IAAP to Pgp mutants by cyclosporin A. Wild-type Pgp and Pgp alanine mutants in isolated membranes (10 μg of total protein) of insect cells were photoaffinity labeled with 5 nM [125I]IAAP in the presence of absence of varying concentrations of Pgp modulator cyclosporin A. Labeled membranes (2.5 μg per lane) were subjected to SDS-PAGE, and radioactivity associated with Pgp was quantified using a PhosphorImager. For wild-type Pgp and for each Pgp alanine mutant, the data were expressed as the percentage of the amount of [125I]IAAP bound in the absence of cyclosporin A. The data are averages of two independent experiments.
Allosteric Determinants of ATP-Site Stimulation Are Nonidentical but Overlapping with Those of Substrate-Site Modulation.
Many transport substrates and modulators that are transported by Pgp stimulate the rate of basal ATP hydrolysis by the pump.45,46 Interestingly, cis-(Z)-flupentixol, although not transported, effectively stimulates hydrolysis of ATP by Pgp.47,48 Although the mechanistic significance of this futile cycle is yet to be understood, it underscores the allosteric nature of the modulator. To determine whether ATP-site modulation by cis-(Z)-flupentixol utilizes the same molecular mediators required for modulation of substrate binding, we studied the basal rate of ATP hydrolysis (Figure 1C) by all 21 mutants and its stimulation in the presence of the modulator (Figure 4). Compared to the membrane vesicles prepared from cells without Pgp expression, all the mutants showed detectable levels of basal hydrolysis ranging from 3 to 9 nmol mg of (membrane protein)−1 min−1 (Figure 1C), suggesting that none of the substitutions abrogate ATP-site function. As expected, cis-(Z)-flupentixol stimulated ATP hydrolysis by wild-type Pgp in a concentration-dependent manner, which was completely inhibited by the transition-state analogue sodium orthovanadate (data not shown). However, in the mutants, a wide range of effects varying from no noticeable alteration to almost complete loss of stimulation was observed (Figure 4). To recognize residues that were absolutely critical for stimulation, we focused primarily on those replacements that mimicked the inhibitory effect of 0.3 mM sodium orthovanadate (data not shown) and demonstrate <2-fold stimulation over their basal activity (Table1).
Figure 4.
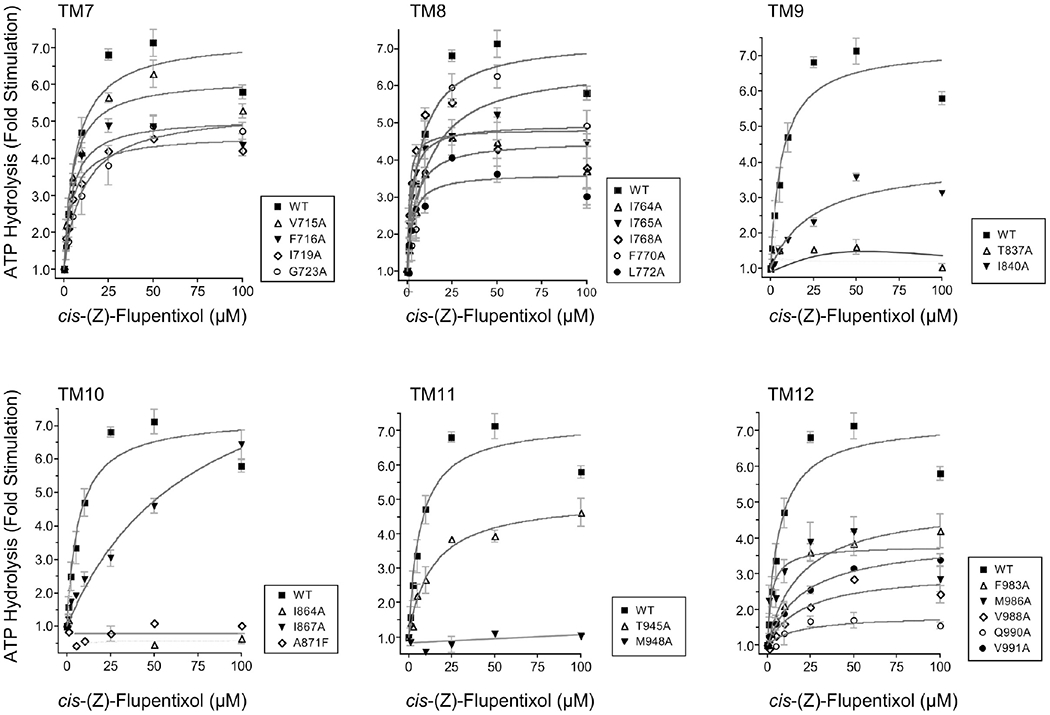
Effect of the single-amino acid substitutions on cis-(Z)-flupentixol-stimulated ATP hydrolysis. The vanadate-sensitive ATP hydrolysis by the wild type and alanine-substituted Pgps was measured in the presence of varying concentrations (0–100 μM) of Pgp modulator cis-(Z)-flupentixol. For each TM helix, the cis-(Z)-flupentixol-stimulated ATPase activity of Pgp alanine mutants were compared to that of wild-type Pgp. The data were expressed as fold stimulation of the basal activity as a function of cis-(Z)-flupentixol concentration. Each point represents the mean value of the data obtained from three independent experiments. Data were analyzed by nonlinear regression using GraphPad PRISM 4.
Table 1.
Stimulation of ATP Hydrolysis by ris-(Z)-Flupentixol and Verapamil
| cis-(Z)-flupentixol-mediated stimulation | verapamil-mediated stimulation | |||||
|---|---|---|---|---|---|---|
| maximal stimulation (x-fold basal) | concentration for half-maximal stimulation μM) | remark | maximal stimulation (x-fold basal) | concentration for half-maximal stimulation μM) | remark | |
| WT | 7.3 ± 0.4 | 6.8 ± 1.5 | 5.6 ± 0.19 | 6.6 ± 1.04 | ||
| V715A | 6.2 ± 0.2 | 5.0 ± 0.9 | 6.4 ± 0.37 | 16.3 ± 3.3 | ||
| V716A | 5.1 ± 0.2 | 4.7 ± 1.1 | NDb | NDb | a | |
| I719A | 4.6 ± 0.08 | 4.7 ± 0.4 | 5.8 ± 0.26 | 6.5 ± 1.31 | ||
| G723A | 5.3 ± 0.3 | 10.93 ± 2.7 | 5.4 ± 0.1 | 11.6 ± 0.88 | ||
| I764A | 4.5 ± 0.4 | 4.1 ± 2 | 3.2 ± 0.26 | 27.9 ± 8.4 | ||
| I765A | 4.96 ± 0.2 | 2.3 ± 0.47 | NDb | NDb | a | |
| I768A | 4.8 ± 0.2 | 1.2 ± 0.43 | 4.8 ± 0.19 | 8.5 ± 1.4 | ||
| F770A | 6.6 ± 0.6 | 10.9 ± 3.9 | 8.6 ± 0.59 | 13.2 ± 3.2 | ||
| L772A | 3.6 ± 0.2 | 2.96 ± 0.9 | 3.2 ± 0.11 | 9.5 ± 1.6 | ||
| T837A | 1.1 ± 0.06 | NDb | a | 2.6 ± 0.1 | 3.1 ± 0.9 | |
| I840A | 4.0 ± 0.3 | 25.6 ± 6.5 | 6.9 ± 0.2 | 13.5 ± 0.5 | ||
| I864A | 0.6 ± 0.05 | NDb | a | 4.1 ± 0.25 | 0.9 ± 1.04 | |
| I867A | 10.2 ± 1.3 | 72.3 ± 18.5 | 10.2 ± 0.52 | 30.7 ± 3.1 | ||
| A871F | NDb | NDb | NDb | NDb | a | |
| T945A | 5.0 ± 0.3 | 13.7 ± 3.1 | 6.4 ± 0.15 | 6.9 ± 0.71 | ||
| M948A | NDb | NDb | a | 6.8 ± 1.5 | 44.4 ± 12.4 | |
| F983A | 4.95 ± 0.36 | 19.59 ± 5.0 | 6.1 ± 0.38 | 6.6 ± 1.8 | ||
| M986A | 3.8 ± 0.3 | 2.6 ± 1.5 | 7.7 ± 0.76 | 30.0 ± 8.6 | ||
| V988A | 3.0 ± 0.2 | 21.4 ± 7.6 | 6.5 ± 0.31 | 16.9 ± 2.9 | ||
| Q990A | 1.8 ± 0.4 | 18.9 ± 2.6 | a | 7.7 ± 0.58 | 12.9 ± 3.7 | |
| V991A | 3.9 ± 0.2 | 21.1 ± 5.0 | 5.9 ± 0.59 | 21.9 ± 7.2 | ||
Mutants with <2-fold maximal stimulation (<25% of the wild-type level).
Activity too low to be analyzed.
Similar to our observation for substrate binding, none of the residues from TM7 (G723, I719, F716, and V715) or TM8 (L772, F770, I768, I765, and I764) was found to be essential for stimulation of ATP hydrolysis by cis-(Z)-flupentixol (Figure 4). On the other hand, replacement of T837 (of TM9), I864 (of TM10), M986 (of TM11), and Q990 (of TM12) severely affected the stimulatory potential of the modulator. It is worth mentioning that two of these four residues, M986 and Q990, were equally critical for stimulation of [125I]IAAP binding by cis-(Z)-flupentixol, which suggested a nonidentical but overlapping footprint for the two modulatory activities induced by the same allosteric modulator.
Residues Involved in ATP-Site Stimulation by Verapamil Are Distinctly Different from Those by Allosteric Modulator cis-(Z)-Flupentixol.
To determine the specificity of the residues identified as critical components of ATP-site modulation by the allosteric modulator cis-(Z)-flupentixol, we studied the stimulatory potential of a structurally as well as functionally unrelated Pgp modulator, verapamil, in all 21 mutants. Verapamil, a calcium channel blocker that is efficiently transported by Pgp,49,50 is known to inhibit transport of other substrates. As indicated in Figure 5 and Table 1), alanine substitution at only two of 21 positions, F716 (of TM7) and I765 (of TM8), disrupted stimulation by verapamil. Although a moderate loss of stimulation was observed in mutants L772A and I764A, a >2-fold residual activity in each case suggested that neither of the two residues was indispensable. Interestingly, none of the replacements affecting cis-(Z)-flupentixol-mediated stimulation (Figure 4) had any significant effect on stimulation by verapamil (Figure 5), further suggesting the specificity of the identified residues toward a particular modulator. This, however, was with the exception of mutant A871F, which replaced a naturally occurring alanine with a phenylalanine. The more general effect evident in A871F most likely represented a local structural perturbation caused by replacement of the methyl side chain of alanine with a bulkier benzyl functional group of phenylalanine. It was also noticeable that alanine replacement at position T837, which incurred a complete loss of stimulation by cis-(Z)-flupentixol, had a moderate effect on stimulation by verapamil too, which was marginally 2-fold of the basal activity.
Figure 5.
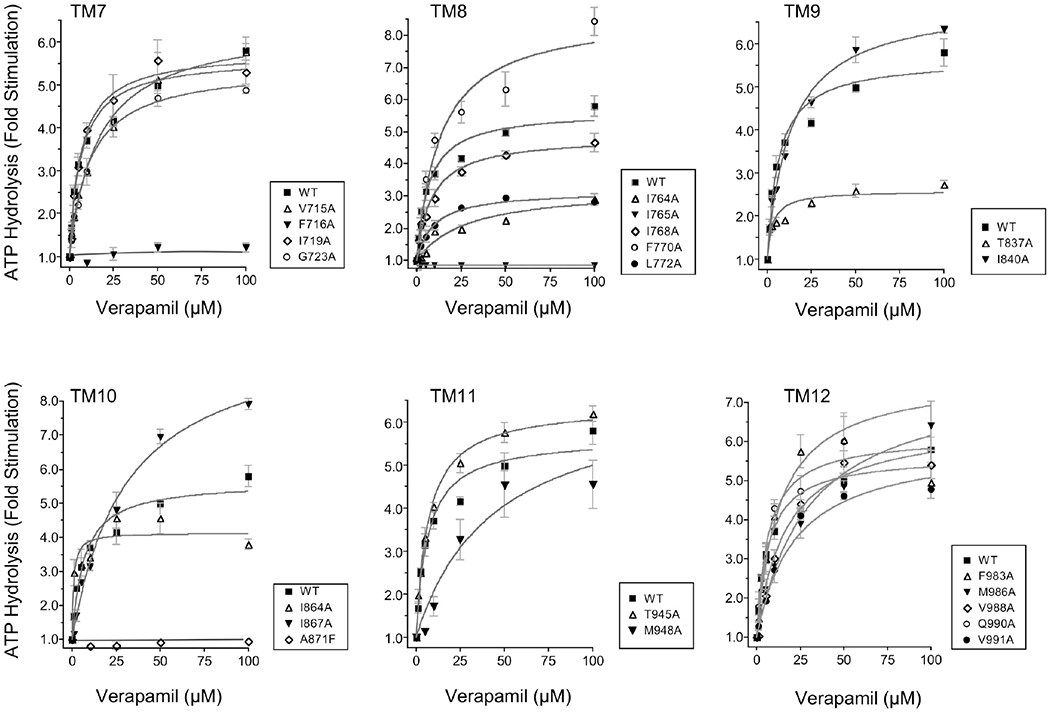
Verapamil-mediated stimulation of ATP hydrolysis in the alanine-substituted Pgp mutants. Vanadate-sensitive ATP hydrolysis by wild-type Pgp and recombinant Pgps with single-amino acid substitutions was assessed in the presence of varying concentrations (0–100 μM) of Pgp modulator verapamil. For each TM helix, the verapamil-stimulated ATPase activities of Pgp alanine mutants were compared to that of wild-type Pgp. The data are expressed as fold stimulation of the basal activity as a function of verapamil concentration, and each point represents the mean value of three independent experiments, with standard errors shown as error bars. Data were analyzed by nonlinear regression using GraphPad PRISM 4 for the determination of Vmax and Km.
Known Crystal Structures of Closely Related ABC Transporters Allow Homology Modeling of Human Pgp.
To visualize where the mutated residues are located in the three-dimensional environment of the protein, we performed a molecular modeling study of human Pgp. We used the structure of ABC exporter Sav1866 (Protein Data Bank entry 2HYD) from the bacterium Staphylococcus aureus51 as a template to model Pgp in a nucleotide-bound state. Sav1866 is 27% identical and 42% similar to Pgp in overall sequence, based on an optimal alignment of the two sequences. The degree of sequence identity or similarity in the NBD regions is considerably greater than in the TMD, and this degree of sequence similarity permits homology models of both NH2-and COOH-terminal NBDs to be built directly on the basis of the Sav1866 crystal structure, which was crystallized in an NBD-closed conformation in the presence of ADP (ADP-bound model). Molecular modeling is the process of integration of existing knowledge and therefore represents a powerful tool for generating hypotheses for further experimental verification. However, atomic models generated by this process are not crystal structures and thus contain errors. The accuracy of a model is largely dependent on sequence similarity and the quality of the template. Although the human Pgp model for the ADP-bound form is based on the high-resolution crystal structure of bacterial transporter Sav1866, its accuracy in the TMD may suffer because of the low level of sequence similarity. The accuracy of the Pgp model in the nucleotide-free state may also be compromised because of the low-resolution template. Nevertheless, these models can still provide a three-dimensional context for our biochemical results and provide guidance for new experiments.
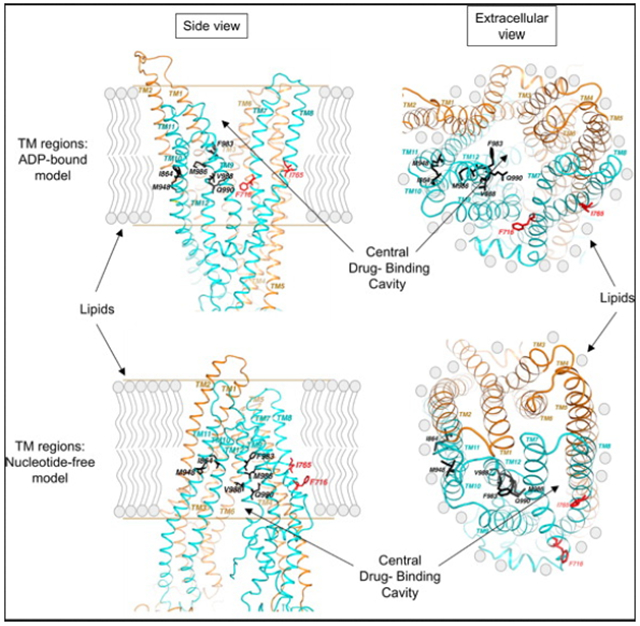
Because the degrees of sequence similarity for the TMD regions between Pgp and Sav1866 are considerably lower and a correct homology model is largely dependent on a correct sequence alignment, the TMD models constructed on the basis of the initial sequence alignment required incorporation of structural knowledge known to integral membrane proteins and knowledge-based modeling, making the modeling process for the two TMDs an iterative one. A survey of known membrane protein structures that were determined at high resolution showed that lipid-facing residues of a TM helix are very hydrophobic and often nonconserved.42 Thus, the approach we employed in aligning Pgp sequence in the TM helices with the sequence of those of Sav1866 was to calculate the LIPS score, which is a product of lipophilicity and Shannon sequence entropy, for each position on a TM helix to ascertain the correct assignment of residues to transmembrane helices.42 For each alignment, residues mapped to a TM helix were grouped into three categories: lipid-facing, interior cavity-facing, and hydrophobic core. The LIPS score for each residue was calculated. These scores were combined for each category of residues. The highest combined score for lipid-facing residues often indicates an optimal sequence alignment for the TM helix. The final Pgp model with ADP bound in a NBD-closed state was built by combining the two halves together based on the dimeric Sav1866 structure.51 A representation of the resulting Pgp model is given in Figure 6A–C. The model lacks the 32 NH2-terminal residues and four COOH-terminal residues; it also is missing the entire linker region of 61 residues between G632 and I692. The model is considered to be in a conformation with the substrate binding pocket of the TMDs accessible to extracellular space and the NBDs tightly associated with bound ADP molecules. Additionally, the structure of mouse Pgp6 gave us the opportunity to construct a human Pgp model in an NBD-open state (also termed the nucleotide-free conformation) (Figure 6D–F). Because the sequences from the two species are 87% identical, it permitted direct homology modeling for both NBDs and TMDs.
Figure 6.
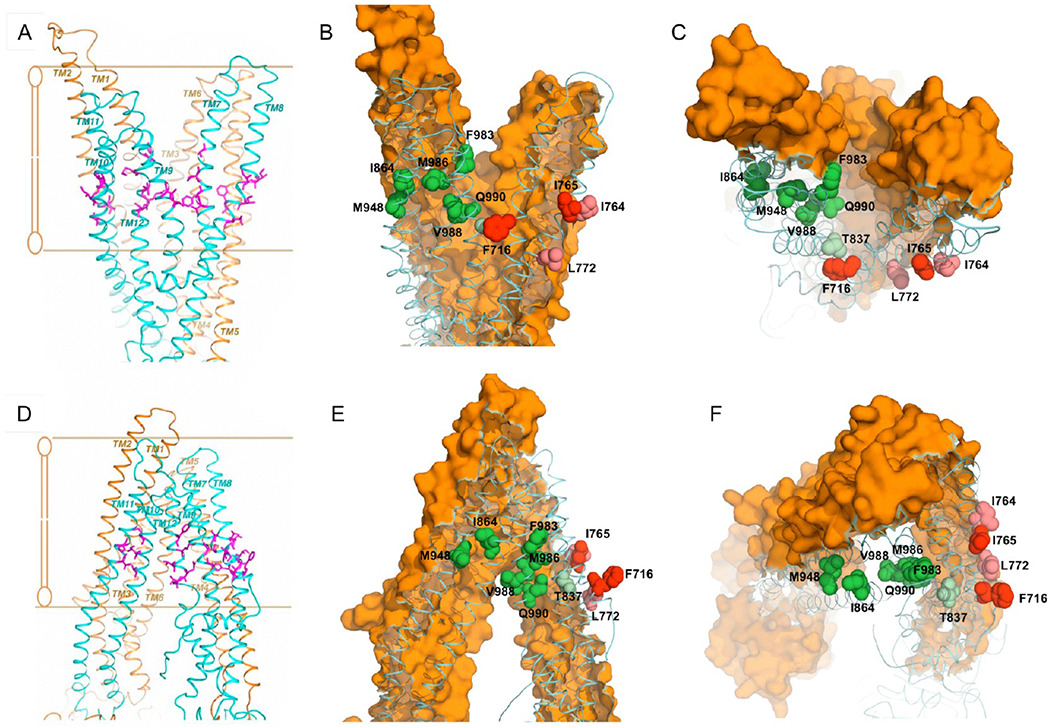
Spatial distribution of targeted residues and residues critical for modulation of human Pgp in the nucleotide-free and ADP-bound conformations. The transmembrane regions of the ADP-bound (A–C) and nucleotide-free (D–F) Pgp models are depicted in isolation from the nucleotide binding domains. (A and D) The amino acid residues subjected to alanine substitution are colored purple and viewed parallel to the plane of the lipid bilayer. (B, C, E, and F) Amino acid residues with a modulator-specific contribution are colored different shades of green [for cis-(Z)-flupentixol] and red or pink (for verapamil), for both ADP-bound (B and C) and nucleotide-free (E and F) conformations, viewed parallel (B and E) and perpendicular (C and F) to the plane of the lipid bilayer from the extracellular side. The NH2-terminal TM helices are colored orange (surface contour), while the COOH-terminal helices are colored blue (ribbon). The lipid-embedded portion of Pgp is highlighted in panels A and D with parallel lines.
Residues Critical for the Functional Modulation of Pgp Form Modulator-Specific Clusters at the Lipid-Protein Interface.
When the mutated residues in TM7 (V715, F716, I719, and G723) and TM8 (I764, I765, I768, F770, and L772) were mapped onto the three-dimensional ADP-bound model, it became clear that they were located in the membrane-buried region of the structure near the inner leaflet of the bilayer (Figure 6A,D). However, the residues with a critical role in verapamil-mediated stimulation of ATP hydrolysis, F716 and I765, both faced the lipid bilayer (Figures 6B,C and 7B,C), suggesting that verapamil might interact with these residues on the outer surface of Pgp to stimulate ATPase activity. Consistent with that, residues L772 and I764 (of TM8), which when replaced with alanines had a moderate but recognizable effect on stimulation by verapamil (Figure 5), were found to be located in the vicinity of the F716–1765 pair, with a similar lipid-facing orientation (Figures 6B,C and 7B,C). The nucleotide-free model of Pgp displayed the same lipid-facing distribution for all four residues (Figures 6E,F and 7A). It is worth mentioning that residue F716 was positioned in the ADP-bound structure at the split of the two bundles of TM helices, which might be part of a substrate exit or entry pathway (Figure 7B,C). Because verapamil is a transport substrate of Pgp, this transition of F716 to a spatial orientation closer to the split could be related to its subsequent transfer to the central drug binding cavity.
Figure 7.
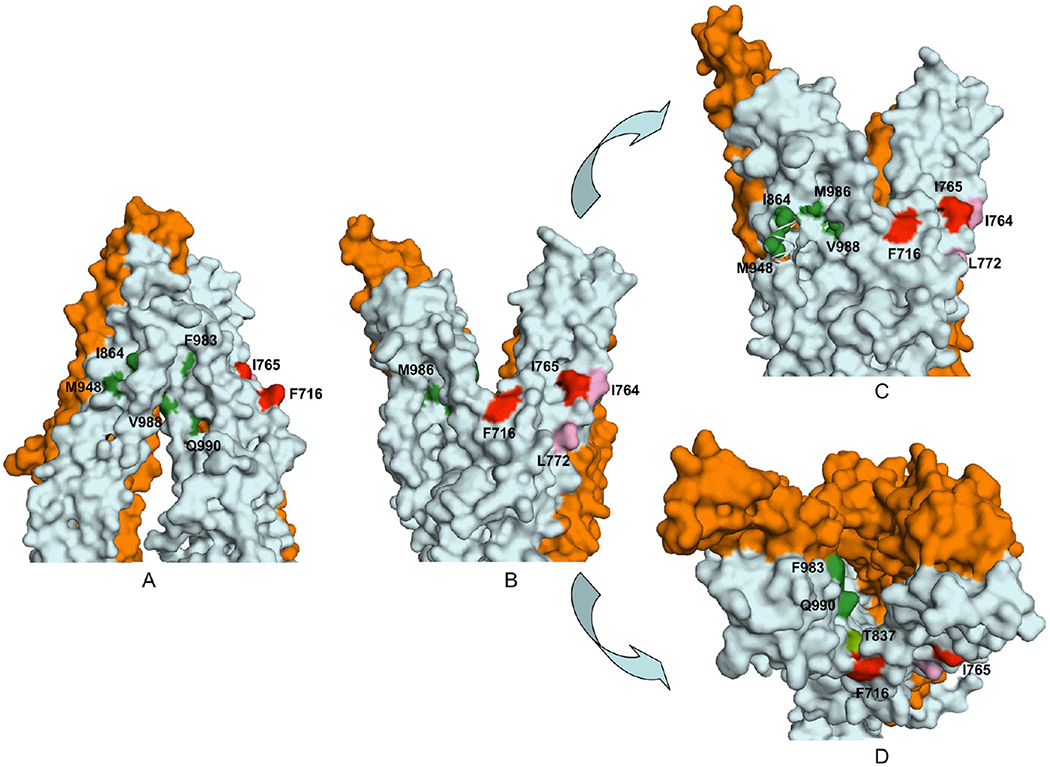
Surface contour diagram depicting the portal and the lipid-facing side pocket in relation to the modulator-specific residues. (A) Close-up view from parallel to the plane of the lipid bilayer of the TM regions, in the nucleotide-free state of Pgp, depicting the portal formed by a split between TM12 and TM10 and −11. (B) Same as panel A, except in the ADP-bound conformation showing formation of a side pocket with modulator-specific residues located inside it. (C) ADP-bound conformation with an ~15° clockwise rotation around the axis perpendicular to the plane of the lipid bilayer. The model shows part of TM10 removed to better visualize the modulator-specific residues located inside the side pocket. (D) View of panel B from the extracellular side of the membrane perpendicular to the plane of the lipid bilayer. The amino acid residues critical for ris-(Z)-flupentixol interaction (T837, I864, M948, F983, V988, M986, and Q990) are colored different shades of green and for verapamil red (F716 and I765) or pink (I764 and L772).
Mutations in TM10 (I864, I867, and A871), TM11 (T945 and M948), and TM12 (F983, M986, V988, Q990, and V991) were similarly mapped to the three-dimensional models and found to be located also in the membrane-buried portion of Pgp, but relatively more toward the outer leaflet of the bilayer (Figure 6B). In that ADP-bound form, TM10, TM11, and TM12 grouped together, creating a side pocket (Figure 7B,C) that was in direct contact with the membrane lipid bilayer. Four of the seven residues essential for interaction with cis-(Z)-flupentixol, I864, M948, M986, and V988, were found to be located within this lipid-facing side pocket (Figure 7C), whereas the other three, Q990, F983, and T837, although part of the same cluster, were oriented toward the central cavity (Figure 7D). In the nucleotide-free model, TM10 and TM11 remained separated from TM12, turning that side pocket into a portal (Figure 7A), through which substrates could gain access to the central cavity of Pgp. The formation of the portal splits the flupentixol-specific cluster, distancing the I864-M948 pair from residues M986 and V988 in TM12 (Figure 6B,E). Reorientation of TM12 associated with the phenomenon leaves all cis-(Z)-flupentixol-specific residues in a lipid-facing orientation except for residues M986 and T837 (Figure 7A).
The Modulator-Specific Sites Demonstrate Negative Synergism in ATP-Site Stimulation.
It became apparent after mapping the modulator-specific residues in the three-dimensional model of Pgp that the interaction sites for cis-(Z)-flupentixol and verapamil were spatially distinct from each other. To understand the interrelationship between these two sites, we studied the combined effect of both modulators on activation of the ATP site. As one can see in Figure 8 (top left), although both modulators stimulated ATP hydrolysis independently, the verapamil-stimulated ATP hydrolysis in wild-type Pgp was effectively antagonized by cis-(Z)-flupentixol in a concentration-dependent manner, with a 50% reduction of the level of stimulation observed at a cis-(Z)-flupentixol concentration of 50 μM. On the other hand, 50 μM cis-(Z)-flupentixol, by itself, had a stimulatory effect on the basal ATPase activity, which was similarly blocked by increasing the concentration of verapamil (Figure 8). This suggested a negative synergism between the stimulatory signals generated by the two modulators. To positively establish that the observed synergism is an outcome of specific interactions of the modulators at their respective sites identified in our study, the effects of both cis-(Z)-flupentixol-specific and verapamil-specific mutants on the phenomenon were tested. As one can see in the top panels (middle and right) of Figure 8, the negative synergistic effect of cis-(Z)-flupentixol on verapamil-stimulated ATP hydrolysis was considerably compromised in I864A and Q990A, Pgp mutants defective with respect to ATP-site stimulation by cis-(Z)-flupentixol. Reciprocally, the inhibitory effect of verapamil on cis-(Z)-flupentixol-mediated stimulation of the ATP site was obliterated in the verapamil-specific Pgp mutant I765A (Figure 8, bottom right). The data suggest that although the two sites are spatially distinct, their communication with the ATP sites cannot process in parallel to generate an additive effect.
Figure 8.
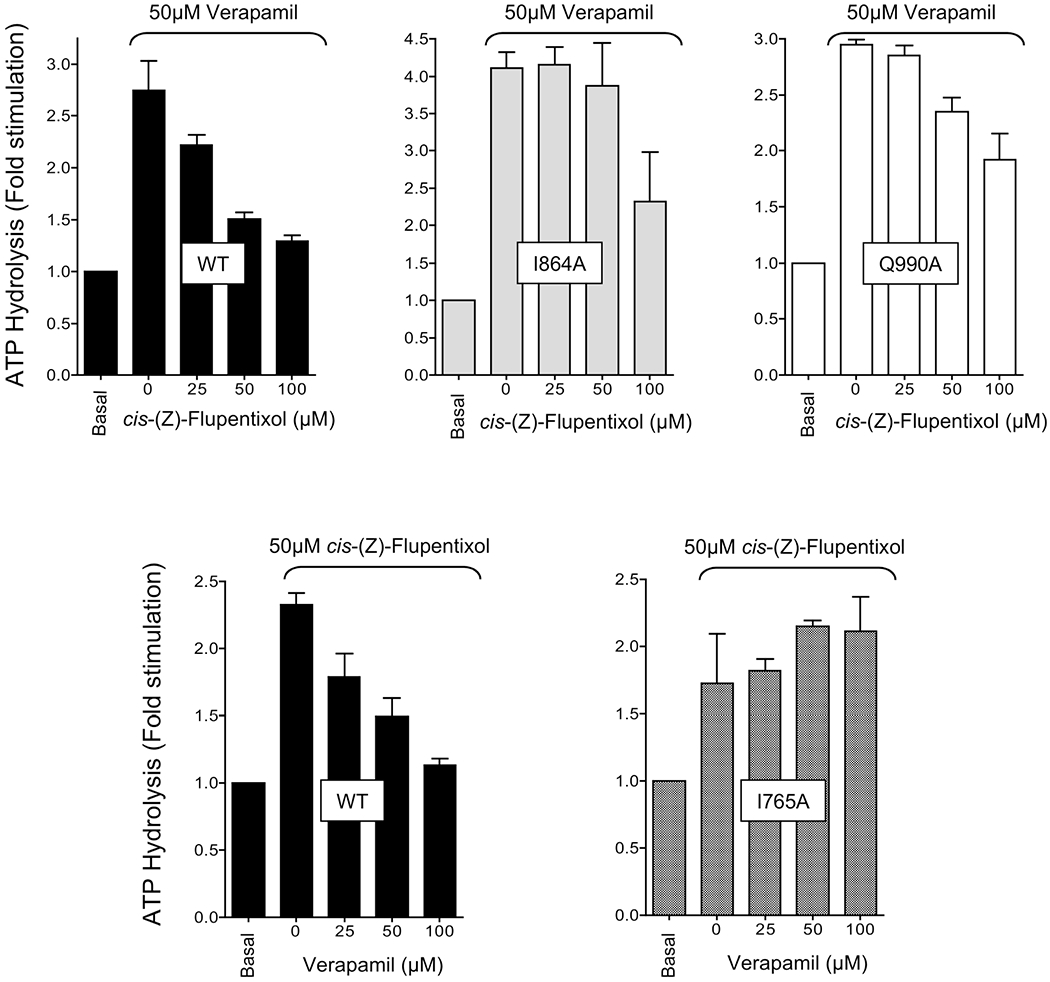
Negative synergism between the stimulatory effects of ds-(Z)-flupentixol and verapamil on ATP hydrolysis. ATP hydrolysis by the wild type and Pgp mutants I864A, Q990A, and I765A, in isolated membrane vesicles, was conducted either after preincubation with 50 ^M verapamil (top) or with 50 ^M ds-(Z)-flupentixol (bottom) followed by incubation with increasing concentrations of ds-(Z)-flupentixol (top) or verapamil (bottom), using the vanadate-sensitive phosphate release assay. Data are expressed as fold stimulation of the basal ATPase activity observed for respective Pgp mutants or the wild type. Each set represents averages of three independent experiments.
DISCUSSION
Human Pgp interacts with an inordinately broad spectrum of structurally unrelated compounds, some of which are recognized as transport substrates and the others act as functional modulators. Although biochemical analysis22 and structural studies6 reveal the presence of a central drug binding cavity within the membrane-embedded region acting as a polyspecific drug recognition module, the facts that Pgp-interacting drugs are directly recruited from the lipid bilayer17–20 and several of them act allosterically26–29 opened up the possibility of additional sites outside the central cavity. In agreement with that, recent reports have pointed out the existence of “gates” or “portals” at the TMD interfaces allowing drug molecules to enter and exit the central cavity with appreciable degrees of physicochemical specificity.23–25,52 However, the interaction sites for modulators acting allosterically remained to be mapped. In this study, combining alanine substitution mutagenesis with molecular modeling, we identify two spatially distinct modulatory sites at the lipid—protein interface of Pgp, outside the central drug binding cavity and distinct from the sites identified at the TMD interfaces, that are involved in the functional regulation of ATP hydrolysis and substrate binding.
The versatility in multidrug recognition by Pgp has been attributed to multiple, but overlapping, drug interaction sites within the drug translocating pathway (including the central drug binding cavity and at the TMD interfaces) with induced-fit recognition capability.22,53 However, the fact that certain pharmacological agents modulate Pgp function yet are not transported54 instigated our hunt for allosteric site(s). Among the Pgp modulators with allosteric modes of action, thioxanthene derivative cis-(Z)-flupentixol has been relatively well characterized, and its mechanistic distinctness with modulators that are transport substrates, such as verapamil and cyclosporin A, is well established;29,43,48,54,55 however, the molecular basis of their different modes of action remained to be resolved.
By targeting only 21 variant positions in the COOH-terminal TM regions of Pgp (Figure 1), we identify five residues (M948, F983, M986, V988, and Q990) that play a crucial role in substrate-site modulation by cis-(Z)-flupentixol (Figure 2). These residues cluster within a 10 Å radius of each other, outside the central drug binding cavity (Figure 6), demarcating the region as a potential site of interaction for the allosteric modulator. Because none of those residues prove to be critical for substrate ([125I]IAAP) binding (Figure 2B) or its modulation by cyclosporin A (Figure 3), a modulator that is efficiently transported by Pgp,29,56 it reemphasizes the allosteric nature of the site.
Interestingly, two of the residues found to be crucial for substrate-site modulation, M948 and Q990, also contribute critically to the modulation of ATP hydrolysis by the same modulator (Figure 4), which emphatically labels the cluster as a major interaction site for cis-(Z)-flupentixol. This is further supported by the fact that the two additional residues found to be essential for ATP-site modulation, I864 and T837 (Figure 4), map either within or in the vicinity of the cluster (Figure 6). It is debatable, however, whether the cluster represents an actual footprint of the interacting modulator or is a congregation of residues critical for transmitting modulatory changes. Because replacement of these residues dramatically reduces the maximal level of stimulation (Vmax), it is more likely that their contribution extends beyond being physical contacts for binding. We cautiously propose that allosteric communications with the ATP and substrate sites require one or more intermolecular (modulator—Pgp) recognition events and intramolecular (within Pgp) rearrangements that are sensitive to side chain replacements at these locations. Because M948 and Q990 prove to be equally important for ATP- and substrate-site modulation (Figures 2 and 4), their side chains could be part of a core component responsible for modulator recognition.
It is noteworthy that, although the molecular signatures for ATP-site and substrate-site modulation by cis-(Z)-flupentixol overlap, they are not identical. Substitutions F983A, M986A, and V988A completely abrogate stimulation of substrate binding (Figure 2) with only modest effects on ATP-site stimulation (Figure 4), which indicates that the two modulatory events are not tightly coupled to each other. Similarly, a reciprocal effect was evident in T837A and I864A, where a 3—4-fold stimulation of substrate binding was retained (Figure 2) even though ATP-site stimulation was almost abrogated (Figure 4). This defines the cis-(Z)-flupentixol-specific cluster as a point from which allosteric communication diverges toward the ATP binding domain and the substrate binding site. In our opinion, the distinction between the molecular players involved in ATP- and substrate-site modulation by the same Pgp modulator is a significant step toward understanding the molecular mechanism of allosteric communication between the different functional domains of Pgp.
In the same context, the spatial distinctness of the cis-(Z)-flupentixol-specific cluster from the central drug binding cavity is particularly significant. This perhaps explains why cis-(Z)-flupentixol does not compete with [125I]IAAP binding. [125I]IAAP is a transport substrate of Pgp29 and is likely to bind at the central drug binding cavity. Photoaffinity labeling with [125I]IAAP and identification of the labeled peptides have identified a major site of [125I]IAAP interaction in the COOH-terminal TM region of Pgp, which spans residues 758—80057 encompassing TM8. Interestingly, none of the five TM8 residues targeted in our study proved to be critical for the modulation of cis-(Z)-flupentixol interaction (Figures 2A and 4), supporting the allosteric nature of the interaction of flupentixol with Pgp.
On the other hand, the specificity of the cis-(Z)-flupentixol-selective residues, identified in this study, is remarkable; none of them proved to be essential for modulation by verapamil or cyclosporin A (Figures 3 and 5), modulators that are Pgp transport substrates. Instead, verapamil-mediated stimulation was sensitive to changes at positions F716 and I765 (Figure 5), neither of which mapped within the cis-(Z)-flupentixol-specific side pocket (Figure 7) or proved to be essential for modulation by cis-(Z)-flupentixol (Figures 2 and 4). The fact that the residues involved in ATP-site modulation by the two types of modulators are not identical and remain spatially separated from each other strongly argues against a unifying mechanism orchestrating TMD—NBD crosstalk. However, the negative synergism between the action of cis-(Z)-flupentixol and verapamil in influencing ATP hydrolysis (Figure 8) suggests that, although the sites for the two modulators are spatially distinct from each other, the stimulatory signals must eventually converge prior to activation of the ATP site. Independent crosstalk with the NBD by the two modulatory sites would have resulted in an additive effect of the two stimulatory drugs. The fact that when one modulator antagonizes the other it reduces the level of stimulation to a level lower than that achieved individually by the respective modulators while acting alone (Figure 8, top left and bottom left) conclusively rules out the possibility of any physical competition between them for interaction with Pgp.
Given the fact that cyclosporin A is a dodecapeptide, it is noteworthy that none of the 21 targeted residues proved to be essential for cyclosporin A-mediated inhibition of [125I]IAAP binding. The noninvolvement of the COOH-terminal TM residues in modulation by cyclosporin A is, however, consistent with the fact that the only cyclosporin A interaction deficient mutation identified to date is a deletion at position F335, which is in the NH2-terminal TM region.58 Although photolabeling with a diazirine derivative of cyclosporin A claimed the peptide fragment spanning residues 953—1007 as the major site of labeling,59 the lack of sequence information about the peptide did not conclusively rule out other possibilities. Using a completely different approach involving thiol-reactive derivatives and specifically engineered cysteine residues in a cysteine-less Pgp, binding sites for verapamil and rhodamine have been mapped primarily within the central drug binding cavity of Pgp.8,60,61 In contrast, the verapamil-specific residues, identified in this study, mapped outside the central drug binding cavity and oriented toward the lipid face under both conformational states (Figures 6 and 7) of Pgp. We propose that F716 and 1765 could be part of a module involved in the specificity check during the recruitment of verapamil from the lipid bilayer, whereas those identified by D. Clarke’s group constitute the site of subsequent interaction critical for stimulation of ATP hydrolysis.
It is remarkable that both modulators, cis-(Z)-flupentixol and verapamil, have interaction sites at the lipid—protein interface of Pgp. The importance of the lipid—protein interface in the functional regulation of membrane transporters and channels is gradually unfolding. Integral membrane proteins, at their outer surface, intimately interact with the bilayer through annular lipids, in both specific and nonspecific manners.62 Specific interactions between protein surface residues and annular lipids often play determining roles in protein structure and function63 and in interaction with pharmacological agents.64 The antimalarial compound quinacrine noncompetitively binds to the nicotinic acetylcholine receptor65 through its lipid—protein interface66,67 and regulates channel activity.68 Similar interaction sites for general anesthetics have been identified at the lipid—protein interface of the synaptosomal Ca2+-ATPase.69 Although it has been long established that Pgp recruits its drug substrates and modulators directly from the lipid bilayer and membrane lipid composition largely dictates Pgp function,70 experimental data and structural studies primarily highlighted the central drug binding cavity and portals at the TMD interfaces as major sites for substrate and modulator interaction6,22 Against the backdrop of this widely accepted notion, our mutagenesis study revealing the lipid—protein interface as a critical component of modulator interaction and modulator selectivity opens up new possibilities. A lipid-facing drug binding cavity with modulatory properties has been identified in the structure of the glycine α-1 receptor,71 and a physically separated allosteric site for NMDA antagonist dizoclipine has been described in the acetylcholine receptor;72 however, to the best of our knowledge, the spatial identity of such sites has never been reported for any multidrug transporter or any ATP-binding cassette transporters. Given the susceptibility of multidrug transporters to functional modulation by membrane lipid composition as well as membrane soluble xenobiotics, recognition of modulatory sites at the lipid—protein interface of Pgp is mechanistically unraveling.
ACKNOWLEDGMENTS
We thank Dr. Michael Gottesman for his critical assessment of the manuscript and the generous gift of Pgp antibody 4007 and Dr. Roopa Biswas and Dr. Ernest Maynard for their valuable input on the manuscript.
Funding
The work was supported by U.S. Public Flealth Services Grant GM067926 and Uniformed Services University Grant C071FU.
ABBREVIATIONS
- TM
transmembrane
- NBD
nucleotide binding domain
- ABC
ATP-binding cassette
- [125I]IAAP
[125I]iodoarylazidoprazosin
Footnotes
The authors declare no competing financial interest.
REFERENCES
- (1).Gottesman ΜM, and Pastan I (1988) The multidrug-transporter: A double-edged sword. /. Biol. Chem 263, 12163–12166. [PubMed] [Google Scholar]
- (2).Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, and Gottesman ΜM (1999) Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu. Rev. Pharmacol. Toxicol 39, 361–398. [DOI] [PubMed] [Google Scholar]
- (3).Chen C. j., Chin JE, Ueda K, Clark DP, Pastan I, Gottesman ΜM, and Roninson IB (1986) Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 47, 381–389. [DOI] [PubMed] [Google Scholar]
- (4).Gottesman ΜM, and Pastan I (1993) Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochem 62, 385–427. [DOI] [PubMed] [Google Scholar]
- (5).Zolnerciks JK, Wooding C, and Linton KJ (2007) Evidence for a Sav1866-like architecture for the human multidrug transporter P-glycoprotein. FASEB J. 21, 3937–3948. [DOI] [PubMed] [Google Scholar]
- (6).Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, and Chang G (2009) Structure of P-glycoprotein reveals a molecular basis for polyspecific drug binding. Science 323, 1718–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gottesman ΜM, Ambudkar SV, and Xia D (2009) Structure of a multidrug transporter. Nat Biotechnol. 27, 546–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Loo TW, and Clarke DM (2001) Defining the drug-binding site in the human multidrug resistance P- glycoprotein using a methanethiosulfonate analog of verapamil, MTS-verapamil. J. Biol. Chem 276, 14972–14979. [DOI] [PubMed] [Google Scholar]
- (9).Loo TW, and Clarke DM (2001) Determining the dimensions of the drug-binding domain of human P- glycoprotein using thiol cross-linking compounds as molecular rulers. J. Biol. Chem 276, 36877–36880. [DOI] [PubMed] [Google Scholar]
- (10).Loo TW, and Clarke DM (1994) Reconstitution of drug-stimulated ATPase activity following co-expression of each half of human P-glycoprotein as separate polypeptides. J. Biol. Chem 269, 7750–7755. [PubMed] [Google Scholar]
- (11).Horio M, Gottesman ΜM, and Pastan I (1988) ATP-dependent transport of vinblastine in vesicles from human multidrug-resistant cells. Proc. Natl. Acad. Sci. U.S.A 85, 3580–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ramachandra M, Ambudkar SV, Chen D, Hrycyna CA, Dey S, Gottesman ΜM, and Pastan I (1998) Human P- glycoprotein exhibits reduced affinity for substrates during a catalytic transition state. Biochemistry 37, 5010–5019. [DOI] [PubMed] [Google Scholar]
- (13).Scala S, Akhmed N, Rao US, Pauli K, Lan LB, Dickstein B, Lee JS, Elgemeie GH, Stein WD, and Bates SE (1997) P- glycoprotein substrates and antagonists cluster into two distinct groups. Mol. Pharmacol 51, 1024–1033. [DOI] [PubMed] [Google Scholar]
- (14).Pearce HL, Winter MA, and Beck WT (1990) Structural characteristics of compounds that modulate P-glycoprotein-associated multidrug resistance. Adv. Enzyme Regul 30, 357–373. [DOI] [PubMed] [Google Scholar]
- (15).Hait WN, and Aftab DT (1992) Rational design and pre-clinical pharmacology of drugs for reversing multidrug resistance. Biochem. Pharmacol 43, 103–107. [DOI] [PubMed] [Google Scholar]
- (16).Klopman G, Srivastava S, Kolossvary I, Epand RF, Ahmed N, and Epand RM (1992) Structure-activity study and design of multidrug-resistant reversal compounds by a computer automated structure evaluation methodology. Cancer Res. 52, 4121–4129. [PubMed] [Google Scholar]
- (17).Raviv Y, Pollard ΗB, Bruggemann EP, Pastan I, and Gottesman ΜM (1990) Photosensitized labeling of a functional multidrug transporter in living drug-resistant tumor cells. J. Biol. Chem 265, 3975–3980. [PubMed] [Google Scholar]
- (18).de Graaf D, Sharma RC, Mechetner EB, Schimke RT, and Roninson IB (1996) P-glycoprotein confers methotrexate resistance in 3T6 cells with deficient carrier-mediated methotrexate uptake. Proc. Natl. Acad. Sci. U.S.A 93, 1238–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shapiro AB, and Ling V (1997) Extraction of Hoechst 33342 from the cytoplasmic leaflet of the plasma membranes by P- glycoprotein. Eur. J. Biochem 250, 122–129. [DOI] [PubMed] [Google Scholar]
- (20).Shapiro AB, and Ling V (1998) Transport of LDS-751 from the cytoplasmic leaflet of the plasma membrane by the rhodamine-123-selective site of P-glycoprotein. Eur. J. Biochem 254, 181–188. [DOI] [PubMed] [Google Scholar]
- (21).Loo TW, and Clarke DM (1999) The transmembrane domains of the human multidrug resistance P-glycoprotein are sufficient to mediate drug binding and trafficking to the cell surface. J. Biol. Chem 274, 24759–24765. [DOI] [PubMed] [Google Scholar]
- (22).Loo TW, and Clarke DM (2008) Mutational analysis of ABC proteins. Arch. Biochem. Biophys 476, 51–64. [DOI] [PubMed] [Google Scholar]
- (23).Parveen Z, Stockner T, Bentele C, Pferschy S, Kraupp M, Freissmuth M, Ecker GF, and Chiba P (2011) Molecular dissection of dual pseudosymmetric solute translocation pathways in human P-glycoprotein. Mol. Pharmacol 79, 443–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Pleban K, Kopp S, Csaszar E, Peer M, Hrebicek T, Rizzi A, Ecker GF, and Chiba P (2005) P-glycoprotein substrate binding domains are located at the transmembrane domain/transmembrane domain interfaces: A combined photoaffinity labeling-protein homology modeling approach. Mol. Pharmacol 67, 365–374. [DOI] [PubMed] [Google Scholar]
- (25).Crowley E, and Callaghan R (2010) Multidrug efflux pumps: Drug binding—gates or cavity? FEBS J. 277, 530–539. [DOI] [PubMed] [Google Scholar]
- (26).Dey S, Ramachandra M, Pastan I, Gottesman MM, and Ambudkar SV (1997) Evidence for two nonidentical drug-interaction sites in the human P-glycoprotein. Proc. Natl. Acad. Sci. U.S.A 94, 10594–10599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Pascaud C, Garrigos M, and Orlowski S (1998) Multidrug resistance transporter P-glycoprotein has distinct but interacting binding sites for cytotoxic drugs and reversing agents. Biochem. J 333, 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Martin C, Berridge G, Higgins CF, Mistry P, Charlton P, and Callaghan R (2000) Communication between multiple drug binding sites on P-glycoprotein. Mol. Pharmacol 58, 624–632. [DOI] [PubMed] [Google Scholar]
- (29).Maki N, Hafkemeyer P, and Dey S (2003) Alosteric modulation of human P-glycoprotein. Inhibition of transport by preventing substrate translocation and dissociation. J. Biol. Chem 278, 18132–18139. [DOI] [PubMed] [Google Scholar]
- (30).Ramachandra M, Ambudkar SV, Gottesman ΜM, Pastan I, and Hrycyna CA (1996) Functional characterization of a glycine 185-to-valine substitution in human P-glycoprotein by using a Vaccinia-based transient expression system. Mol. Biol. Cell 7, 1485–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Bailey JL (1967) Techniques in Protein Chemistry, pp 340–341, Elsevier, New York. [Google Scholar]
- (32).Germann UA, Chambers TC, Ambudkar SV, Licht T, Cardarelli CO, Pastan I, and Gottesman ΜM (1996) Characterization of phosphorylation-defective mutants of human P- glycoprotein expressed in mammalian cells. J. Biol. Chem 271, 1708–1716. [DOI] [PubMed] [Google Scholar]
- (33).Tanaka S, Currier SJ, Bruggemann EP, Ueda K, Germann UA, Pastan I, and Gottesman ΜM (1990) Use of recombinant P-glycoprotein fragments to produce antibodies to the multidrug transporter. Biochem. Biophys. Res. Commun 166, 180–186. [DOI] [PubMed] [Google Scholar]
- (34).Sarkadi B, Price EM, Boucher RC, Germann UA, and Scarborough GA (1992) Expression of the human multidrug resistance cDNA in insect cells generates a high activity drug-stimulated membrane ATPase. J. Biol. Chem 267, 4854–4858. [PubMed] [Google Scholar]
- (35).Hirokawa T, Boon-Chieng S, and Mitaku S (1998) SOSUI: Classification and secondary structure prediction system for membrane proteins. Bioinformatics 14, 378–379. [DOI] [PubMed] [Google Scholar]
- (36).Rost B (1996) PHD: Predicting one-dimensional protein structure by profile-based neural networks. Methods Enzymol. 266, 525–539. [DOI] [PubMed] [Google Scholar]
- (37).Tusnady GE, and Simon I (2001) The HMMTOP transmembrane topology prediction server. Bioinformatics 17, 849–850. [DOI] [PubMed] [Google Scholar]
- (38).Krogh A, Larsson B, von Heijne G, and Sonnhammer EL (2001) Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol 305, 567–580. [DOI] [PubMed] [Google Scholar]
- (39).Sipos L, and von Heijne G (1993) Predicting the topology of eukaryotic membrane proteins. Eur. J. Biochem 213, 1333–1340. [DOI] [PubMed] [Google Scholar]
- (40).von Heijne G (1994) Membrane proteins: From sequence to structure. Annu. Rev. Biophys. Biomol. Struct 23, 167–192. [DOI] [PubMed] [Google Scholar]
- (41).Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, Read RJ, Rice LM, Simonson T, and Warren GL (1998) Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 54, 905–921. [DOI] [PubMed] [Google Scholar]
- (42).Adamian L, and Liang J (2006) Prediction of transmembrane helix orientation in polytopic membrane proteins. BMC Struct. Biol 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Maki N, and Dey S (2006) Biochemical and pharmacological properties of an allosteric modulator site of the human P-glycoprotein (ABCBl). Biochem. Pharmacol 72, 145–155. [DOI] [PubMed] [Google Scholar]
- (44).Litman T, Zeuthen T, Skovsgaard T, and Stein WD(1997) Structure-activity relationships of P-glycoprotein interacting drugs: Kinetic characterization of their effects on ATPase activity. Biochim. Biophys. Acta 1361, 159–168. [DOI] [PubMed] [Google Scholar]
- (45).Ambudkar SV, Lelong IH, Zhang J, Cardarelli CO, Gottesman ΜM, and Pastan I (1992) Partial purification and reconstitution of the human multidrug-resistance pump: Characterization of the drug-stimulatable ATP hydrolysis. Proc. Natl. Acad. Sci. U.S.A 89, 8472–8476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Szabó K, Welker E, Bakos, Müller M, Roninson I, Váradi A, and Sarkadi B (1998) Drug-stimulated nucleotide trapping in the human multidrug transporter MDR1. Cooperation of the nucleotide binding domains./. Biol. Chem 273, 10132–10138. [DOI] [PubMed] [Google Scholar]
- (47).Dey S, Hafkemeyer P, Pastan I, and Gottesman MM(1999) A single amino acid residue contributes to distinct mechanisms of inhibition of the human multidrug transporter by stereoisomers of the dopamine receptor antagonist flupentixol. Biochemistry 38, 6630–6639. [DOI] [PubMed] [Google Scholar]
- (48).Maki N, Moitra K, Silver C, Ghosh P, Chattopadhyay A, and Dey S (2006) Modulator-Induced Interference in Functional Cross Talk between the Substrate and the ATP Sites of Human P- glycoprotein. Biochemistry 45, 2739–2751. [DOI] [PubMed] [Google Scholar]
- (49).Yusa K, and Tsuruo T (1989) Reversal mechanism of multidrug resistance by verapamil: Direct binding of verapamil to P- glycoprotein on specific sites and transport of verapamil outward across the plasma membrane of K562/ADM cells. Cancer Res 49, 5002–5006. [PubMed] [Google Scholar]
- (50).Omote H, and Al-Shawi MK (2002) A novel electron paramagnetic resonance approach to determine the mechanism of drug transport by P-glycoprotein. J. Biol. Chem 277, 45688–45694. [DOI] [PubMed] [Google Scholar]
- (51).Dawson RJ, and Locher KP (2007) Structure of the multidrug ABC transporter Sav1866 from Staphylococcus aureus in complex with AMP-PNP. FEBS Lett. 581, 935–938. [DOI] [PubMed] [Google Scholar]
- (52).Loo TW, and Clarke DM (2005) Do drug substrates enter the common drug-binding pocket of P-glycoprotein through “gates”? Biochem. Biophys. Res. Commun 329, 419–422. [DOI] [PubMed] [Google Scholar]
- (53).Loo TW, and Clarke DM (2005) Recent progress in understanding the mechanism of P-glycoprotein-mediated drug efflux. J. Membr. Biol 206, 173–185. [DOI] [PubMed] [Google Scholar]
- (54).Ghosh P, Moitra K, Maki N, and Dey S (2006) Allosteric modulation of the human P-glycoprotein involves conformational changes mimicking catalytic transition intermediates. Arch. Biochem. Biophys 450, 100–112. [DOI] [PubMed] [Google Scholar]
- (55).Maki N, Moitra K, Ghosh P, and Dey S (2006) Allosteric modulation bypasses the requirement for ATP hydrolysis in regenerating low affinity transition state conformation of human P- glycoprotein. J. Biol. Chem 281, 10769–10777. [DOI] [PubMed] [Google Scholar]
- (56).Saeki T, Ueda K, Tanigawara Y, Hori R, and Komano T (1993) Human P-Glycoprotein transports Cyclosporin-A and FK506. J. Biol. Chem 268, 6077–6080. [PubMed] [Google Scholar]
- (57).Isenberg B, Thole H, Tummler B, and Demmer A (2001) Identification and localization of three photobinding sites of iodoarylazidoprazosin in hamster P-glycoprotein. Eur. J. Biochem 268, 2629–2634. [DOI] [PubMed] [Google Scholar]
- (58).Chen G, Duran GE, Steger KA, Lacayo NJ, Jaffrezou JP, Dumontet C, and Sikic BI (1997) Multidrug-resistant human sarcoma cells with a mutant P-glycoprotein, altered phenotype, and resistance to cyclosporins. J. Biol. Chem 272, 5974–5982. [DOI] [PubMed] [Google Scholar]
- (59).Demeule M, Laplante A, Murphy GF, Wenger RM, and Beliveau R (1998) Identification of the cyclosporin-binding site in P- glycoprotein. Biochemistry 37, 18110–18118. [DOI] [PubMed] [Google Scholar]
- (60).Loo TW, Bartlett MC, and Clarke DM (2003) Methanethiosulfonate derivatives of rhodamine and verapamil activate human P-glycoprotein at different sites. J. Biol. Chem 278, 50136–50141. [DOI] [PubMed] [Google Scholar]
- (61).Loo TW, and Clarke DM (2002) Location of the rhodamine-binding site in the human multidrug resistance P- glycoprotein. J. Biol. Chem 277, 44332–44338. [DOI] [PubMed] [Google Scholar]
- (62).Ernst AM, Contreras FX, Brugger B, and Wieland F (2010) Determinants of specificity at the protein-lipid interface in membranes. FEBS Lett. 584, 1713–1720. [DOI] [PubMed] [Google Scholar]
- (63).Walcher S, Altschuh J, and Sandermann H Jr. (2001) The lipid/protein interface as xenobiotic target site: Kinetic analysis of the nicotinic acetylcholine receptor. J. Biol. Chem 276, 42191–42195. [DOI] [PubMed] [Google Scholar]
- (64).Barrantes FJ (2004) Structural basis for lipid modulation of nicotinic acetylcholine receptor function. Brain Res. Brain Res. Rev 47, 71–95. [DOI] [PubMed] [Google Scholar]
- (65).Arias HR, Valenzuela CF, and Johnson DA (1993) Quinacrine and ethidium bind to different loci on the Torpedo acetylcholine receptor. Biochemistry 32, 6237–6242. [DOI] [PubMed] [Google Scholar]
- (66).Li L, Lee YH, Pappone P, Palma A, and McNamee MG (1992) Site-specific mutations of nicotinic acetylcholine receptor at the lipid-protein interface dramatically alter ion channel gating. Biophys. J 62, 61–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Bouzat C, Roccamo AM, Garbus I, and Barrantes FJ (1998) Mutations at lipid-exposed residues of the acetylcholine receptor affect its gating kinetics. Mol. Pharmacol 54, 146–153. [DOI] [PubMed] [Google Scholar]
- (68).Valenzuela CF, Kerr JA, and Johnson DA (1992) Quinacrine binds to the lipid-protein interface of the Torpedo acetylcholine receptor: A fluorescence study. J. Biol. Chem 267, 8238–8244. [PubMed] [Google Scholar]
- (69).Pflugmacher D, and Sandermann H Jr. (1998) The lipid/ protein interface as a target site for general anesthetics: A multiple-site kinetic analysis of synaptosomal Ca2+-ATPase. Biochim. Biophys. Acta 1415, 174–180. [DOI] [PubMed] [Google Scholar]
- (70).Eckford PD, and Sharom FJ (2008) Interaction of the P- glycoprotein multidrug efflux pump with cholesterol: Effects on ATPase activity, drug binding and transport. Biochemistry 47, 13686–13698. [DOI] [PubMed] [Google Scholar]
- (71).Bertaccini EJ, Shapiro J, Brutlag DL, and Trudell JR (2005) Homology modeling of a human glycine a1 receptor reveals a plausible anesthetic binding site. J. Chem. Inf. Model 45, 128–135. [DOI] [PubMed] [Google Scholar]
- (72).Arias HR, McCardy EA, and Blanton MP (2001) Characterization of the dizocilpine binding site on the nicotinic acetylcholine receptor. Mol. Pharmacol 59, 1051–1060. [DOI] [PubMed] [Google Scholar]