

Abstract
Laminin-α2 related congenital muscular dystrophy (LAMA2-CMD) is a fatal muscle disease caused by mutations in the LAMA2 gene. Laminin-α2 is critical for the formation of laminin-211 and -221 heterotrimers in the muscle basal lamina. LAMA2-CMD patients exhibit hypotonia from birth and progressive muscle loss that results in developmental delay, confinement to a wheelchair, respiratory insufficiency and premature death. There is currently no cure or effective treatment for LAMA2-CMD. Several studies have shown laminin-111 can serve as an effective protein-replacement therapy for LAMA2-CMD. Studies have demonstrated early treatment with laminin-111 protein results in an increase in life expectancy and improvements in muscle pathology and function. Since LAMA2-CMD patients are often diagnosed after advanced disease, it is unclear if laminin-111 protein therapy at an advanced stage of the disease can have beneficial outcomes. In this study, we tested the efficacy of laminin-111 protein therapy after disease onset in a mouse model of LAMA2-CMD. Our results showed laminin-111 treatment after muscle disease onset increased life expectancy, promoted muscle growth and increased muscle stiffness. Together these studies indicate laminin-111 protein therapy either early or late in the disease process could serve as an effective protein replacement therapy for LAMA2-CMD.
Introduction
Congenital muscular dystrophy (CMD) is defined as a group of rare neuromuscular diseases with a prevalence estimated at 0.5/100 000 people. Laminin-α2 related CMD (LAMA2-CMD) accounts for a quarter of those cases (1). LAMA2-CMD is caused by mutations in the LAMA2 gene resulting in loss-of-function truncation or complete loss of laminin-α2 protein (2). The α2 chain is essential for the assembly of heterotrimers laminin-211 and laminin-221, which are the predominant laminin isoforms in adult skeletal muscle. Laminin-211 and -221 are ~900 kDa proteins located in the basal lamina and provide a critical link between the muscle cell and extracellular matrix (ECM) (3).
Loss-of-function mutations in the laminin-α2 chain which causes LAMA2-CMD results in a severe, congenital onset phenotype that has devastating clinical outcomes. Severe LAMA2-CMD presents hypotonia from birth and developmental delays (4). Other prominent clinical features include progressive muscle weakness, contractures, scoliosis and respiratory insufficiency, which often results in premature death (5). Further complications include seizures, cardiac arrhythmias, neuropathy and brain abnormalities (4,6). Currently, there is no approved or effective treatment for this fatal muscle disease.
Studies in mice show that treatment with laminin-111 protein, a laminin isoform expressed during embryonic development and adult kidney (7), reduces muscle pathology (8,9) and dramatically increases life expectancy in a mouse model of LAMA2-CMD when administered at two weeks of age (10). However, since most LAMA2-CMD patients are diagnosed after significant myopathy, it is unclear whether laminin-111 protein can be as effective in patients at a later stage in disease.
In this study, we investigated if laminin-111 protein therapy could be effective at preventing disease progression after muscle disease in a mouse model of LAMA2-CMD. The dyW−/− mice show mild to no myopathy at 2 weeks of age, while at 4 weeks of age they show hindlimb paralysis, mild kyphosis and smaller size (6,11). We show that laminin-111 treatment when started at 4 weeks of age, i.e. after disease onset, increased the life expectancy of dyW−/− mice compared with control animals. We show laminin-111 protein therapy at this age increased extensor digitorum longus (EDL) stiffness in response to a contraction-injury protocol. Finally, we show laminin-111 treatment promoted an increase in satellite cells and improved muscle regeneration. Together these results support the idea that laminin-111 could serve as an effective protein replacement therapy after disease onset in a mouse model of LAMA2-CMD.
Results
Laminin-111 protein localizes to the muscle sarcolemma after disease onset in a mouse model of LAMA2-CMD
To determine if laminin-111 treatment after disease onset resulted in myomatrix localization of the exogenously delivered protein, female dyW−/− mice were treated with weekly intraperitoneal (i.p.) doses of 10 mg/kg/week of Englebreth-Holm-Swarm (EHS) laminin-111 or PBS from 4 to 8 weeks of age (Fig. 1A). At 8 weeks of age, mice were humanely euthanized and muscle tissue harvested.
Figure 1.

Exogenous laminin-111 protein localizes to the myomatrix of dyW−/− mice. (A) DyW−/− mice were treated with weekly i.p. injections of 10 mg/kg EHS laminin-111 or PBS from 4 to 8 weeks of age. After the last treatment, preclinical outcome measures were undertaken. (B) Laminin-α2 was only detected in dyW+/− muscle section and not dyW−/− animals treated with PBS or laminin-111. Laminin-α1 was only detected in dyW−/− mice treated with laminin-111 protein. Primary antibody-negative control for laminin-α2 and laminin-α1 immunostaining. Scale bar 100 μm.
To confirm that dyW−/− mice lacked laminin-α2 protein, tibialis anterior (TA) muscle sections were subjected to immunofluorescence to detect the laminin-α2 chain. Muscle from heterozygous mice (dyW+/−) served as a positive control. As expected, the dyW+/− mice showed laminin-α2 localized to the muscle basal lamina. In contrast, dyW−/− treated with PBS or laminin-111 lacked the laminin-α2 chain indicating loss of laminin-211/221 in the basal lamina (Fig. 1B). These results confirm that the dyW−/− mice in this study lacked laminin-211/221 in the muscle basal lamina.
To determine if laminin-111 treatment after disease onset resulted in localization to the myomatrix of dyW−/− mice, TA muscle sections were subjected to immunofluorescence to detect the laminin-α1 chain. Since laminin-111 is only found in developing embryonic skeletal muscle and not in adult skeletal muscle, we did not observe any laminin-α1 immunofluorescence in the muscle basal lamina of muscle sections from dyW+/− or dyW−/− mice treated with vehicle (Fig. 1B). In contrast, we observed laminin-α1 immunofluorescence in the basal lamina of dyW−/− treated with laminin-111 after disease onset (Fig. 1B). These results show that i.p. injections of EHS laminin-111 after disease onset resulted in localization of the exogenous laminin-111 protein to the muscle ECM of dyW−/− mice.
Laminin-111 treatment after disease onset increased life expectancy of a mouse model of LAMA2-CMD
LAMA2-CMD patients experience progressive muscle disease starting from birth including weight loss and reduced life expectancy (5). A previous study in the dyW−/− mouse model of LAMA2-CMD reported treatment with 10 mg/kg/week EHS starting at 2 weeks of age increased average survival of dyW−/− mice by 3.5-fold (10).
Since most patients with LAMA2-CMD already have significant muscle disease by the time they are diagnosed, we wanted to determine the efficacy of laminin-111 protein replacement after disease onset. In this study, female dyW−/− mice were treated with 10 mg/kg/week EHS laminin-111 or PBS vehicle delivered via i.p. injections starting at 4 weeks of age. At 2 weeks of age, the dyW−/− myopathy is relatively mild, while at 4 weeks of age mice present with significant muscle pathology as well as hindlimb paralysis and kyphosis.
Our results showed that treatment with EHS laminin-111 increased the life expectancy 2-fold, from 15 weeks of age to 30 weeks of age. The oldest dyW−/− mouse treated with laminin-111 starting at 4 weeks of age survived to 69 weeks (Fig. 2A). All dyW−/− mice treated with vehicle died before 29 weeks of age. We observed no significant differences in body weights between laminin-111 and vehicle-treated mice. However, after 29 weeks, laminin-111 treated mice showed a 1 g increase in average body weight that indicated stabilization of body weight. In contrast, PBS treated mice that survived continued to lose body weight (Fig. 2B). These results indicate treatment with laminin-111 after disease onset increased the survival and maintained the body weight of the dyW−/− mouse model of LAMA2-CMD.
Figure 2.

Laminin-111 treatment after disease onset increased the life expectancy of the dyW−/− mouse model of LAMA2-CMD. (A) Survival analysis of laminin-111 versus PBS treated dyW−/− mice starting at 4 weeks of age. (B) Weekly body weight measurements of dyW mice treated with laminin-111 or PBS starting at 4 weeks of age.
Laminin-111 increases stiffness in the EDL muscle at a late stage of disease in a mouse model of LAMA2-CMD
LAMA2-CMD patients and dyW−/− mice exhibit severe progressive muscle weakness, and in the case of the mouse model the weakness is apparent by approximately 4 weeks of age. Hindlimb paralysis is especially evident in these animals rendering dyW−/− mice immobile by 10 weeks of age. We next determined whether laminin-111 treatment protected laminin-α2 deficient muscles from contraction-induced muscle injury (12). In this study, our treatment groups were compared with dyW+/− control mice, which demonstrate no myopathy or defects in muscle regeneration (13).
To test this hypothesis, we isolated the EDL and soleus muscles from the hindlimbs of laminin-111 and PBS treated dyW−/− mice, as well as dyW+/− control animals. Contractility assays were performed as previously described (14). Single twitch (1 Hz) and tetanus (150 Hz) contractions were performed in triplicate, followed by force frequency stimuli from 1 to 180 Hz. Table 1 summarizes the body weights of the treatment groups, as well as EDL and soleus muscle measurements used to normalize contractility data. We observed no differences in isometric contraction values for the twitch, tetani or force frequency responses between laminin-111 and PBS treated dyW−/− mice for either EDL or soleus muscles. However, the dyW+/− control group showed significantly higher strength for all measurements (Supplementary Material, Fig. S1A–F).
Table 1.
Summary of mouse muscle measurements
| EDL | Soleus | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Mouse | Treatment | N-number | Mass (g) | Mass (mg) | L o (mm) | CSA (mm2) | Mass (mg) | L o (mm) | CSA (mm2) |
| DyW+/− | NT | 5 | 20.20 ± 1.3 | 8.74 ± 0.45 | 11.60 ± 0.3 | 0.71 ± 0.03 | 6.08 ± 0.33 | 9.76 ± 0.40 | 0.59 ± 0.03 |
| DyW−/− | PBS | 5 | 13.14 ± 4.9 | 4.96 ± 0.54 | 10.02 ± 0.3 | 0.46 ± 0.03 | 4.46 ± 0.74 | 9.06 ± 0.31 | 0.45 ± 0.06 |
| DyW−/− | Laminin-111 | 5 | 12.90 ± 1.0 | 4.94 ± 0.39 | 10.32 ± 0.3 | 0.44 ± 0.02 | 4.26 ± 0.40 | 9.58 ± 0.27 | 0.42 ± 0.04 |
The contraction-induced injury protocol included a stimulus regimen of five fused tetanic contractions at 80 Hz for 500 ms, followed by an eccentric contraction at 0.5 Lo/s to a maximum stretch of 10% Lo (Lo, optimal muscle length). This regimen generates high peak stress that can lead to extensive injury in dystrophic and wild-type muscle (15). Both isometric and eccentric peaks were quantified and graphed for each treatment group and muscle. Each muscle was challenged with five stretches, one every 4 min. Figure 3A shows an overlay of stretch 1 where each peak is identified for clarity. Figure 3B and C shows the isometric peaks of all stretches in EDL and soleus, respectively. Results showed that laminin-111 had no effect on the isometric peak contraction compared with PBS treated dyW−/− muscles, but the control group dyW+/− was significantly higher in both muscle types (Fig. 3B and C; N = 5, P-value < 0.0001***, <0.05*).
Figure 3.

Laminin-111 treatment after disease onset increased the stiffness of the EDL muscle. (A) Normalized force profiles of EDL stimulated muscle from PBS and laminin-111 treated dyW−/− and dyW+/− mice at stretch 1. (B) EDL and (C) soleus isometric peak measurements at 1–5 stretches stimulations in PBS and laminin-111 treated dyW−/− and dyW+/− (N = 5, P-value < 0.0001***, <0.05*). (D) EDL and (E) soleus eccentric peak values at 1–5 stretches stimulations in PBS and laminin-111 treated dyW and dyW+/− (N = 5, P-value < 0.05#). (F) EDL and (G) soleus stiffness measurements at 1–5 stretches in PBS and laminin-111 treated dyW and dyW+/− (N = 5, P-value < 0.023*, < 0.05#, < 0.001##). Two way ANOVA was used to determine changes between time-point curves*, and Bonferroni post-test was used to determine changes between time-points#.
However, the eccentric peaks showed differences with laminin-111 treatment in the EDL of dyW−/− mice. At stretches 1 and 2, the PBS-treated EDL eccentric peak was significantly lower compared with the EDL in dyW+/− control, while the laminin-111 treated EDL showed increased values closer to the heterozygous control (Fig. 3D; N = 5, P-value < 0.05). The soleus, on the other hand, did not show significant differences between treatments in dyW−/− groups, nor between the dyW+/− control group (Fig. 3E; N = 5).
Using Petrof’s protocol, stiffness was defined as the change between the difference between isometric and eccentric peaks (Δ Normalized force) and the difference in length during contraction or 10% Lo (Δ Muscle length; Eq. (1), Stiffness equation).
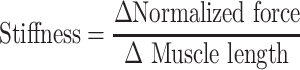 |
(1) |
Using this calculation, we found a significant increase in the stiffness of laminin-111 treated EDL muscle compared with PBS treated dyW−/− and dyW+/− muscles (N = 5, P-value <0.023*). More specifically, at stretches 2, 3 and 4, laminin-111 treated dyW−/− EDL muscles were significantly greater compared with dyW+/− muscles (N = 5, P-value < 0.05#, < 0.001###). We found no differences between PBS treated dyW−/− and dyW+/− muscles. The soleus muscle did not show any changes between laminin-111, PBS treated dyW−/− or dyW+/− mice for either eccentric peak or stiffness (N = 5).
Laminin-111 after disease onset improved muscle repair and regeneration in a LAMA2-CMD mouse model
LAMA2-CMD patients have muscle that exhibit myofiber size variation and hypotrophic muscle fibers (16). Studies have shown that treatment with EHS laminin-111 improves muscle regeneration and repair in wild-type muscle (17), mouse and dog model of DMD (8,18) as well as mouse model of LAMA2-CMD (9,10).
To test whether laminin-111 treatment improved muscle repair after disease onset, we quantified centrally located nuclei (CLN) fibers and used the Feret diameter to quantify myofiber size. We found that percent of CLN fibers was not changed between laminin-111 versus PBS-treated muscles (Fig. 4B; N = 5 and 6, respectively, P-value of 0.28). However, the histogram of CLN-fiber diameters showed a significant shift toward larger myofiber diameters from a mean of 27.32 μm in PBS treatment to 33.29 μm in laminin-111 treated muscles (Fig. 4C; N = 5 and 6, respectively, P-value of <0.0001). Additionally, total fiber diameter showed a significant increase in diameters indicating hypertrophy from 26.79 μm in PBS to 32.65 μm in laminin-111 treated groups (N = 5 and 6, respectively, P-value of <0.0001) (Fig. 4D). These results suggest that the presence of laminin-111 in the muscle ECM aided in on-going muscle repair yielding an increase in average myofiber size.
Figure 4.

Laminin-111 treatment after disease onset promoted an increase in myofiber size. (A) Muscle fibers (white) and nuclei (blue) were imaged in laminin-111 and PBS treated sections of TA. CLN fibers were determined by the nuclear position in the center of fibers (yellow arrows). Scale bar 100 μm. (B) Percent of CLN were quantified in each treatment group. Laminin-111 treated muscle showed no significant changes in CLN compared with PBS treated (N = 5 and 6, respectively). (C) Myofiber diameters were quantified in CLN fibers showing an increase in myofiber size in laminin-111 versus PBS treated dyW mice groups (N = 6 and 5, respectively, P-value of <0.0001). (D) Total fiber diameters also indicated increased myofiber size in laminin-111 versus PBS treated groups (N = 6 and 5, respectively, P-value of <0.0001).
Next, we tested if laminin-111 protein therapy after disease onset improved the regenerative capacity of muscle by counting the number of myofibers in the TA muscle positive for embryonic Myosin Heavy Chain (eMHC) (Fig. 5A). We found an increase in eMHC positive myofibers from 7.51% in PBS treated animals to 11.06% in laminin-111 treated mice (N = 5, P-value of 0.001) (Fig. 5B). Additionally, we quantified levels of the satellite cell specific transcription factor paired box 7 (Pax7) transcript factor (Fig. 5C). Our results showed a 1.6-fold increase in Pax7-positive cells from 0.84 cells per field of view in PBS treated animals to 1.36 cells per field of view in laminin-111 treated muscles (Fig. 5D) (N = 5 and 6, respectively, P-value of 0.049). These results indicate that treatment with laminin-111 promoted an increase the regenerative capacity of laminin-α2 deficient muscle after disease onset in dyW−/− mice.
Figure 5.

Laminin-111 treatment after disease onset increased muscle regeneration and satellite cells number in the dyW−/− mouse model of LAMA2-CMD. (A) Immunofluorescence to detect eMHC (green) positive myofibers in TA muscle sections. Scale bar 100 μm. (B) Quantification of eMHC positive fibers in the TA showed a significant increase in laminin-111 versus PBS treated animals (N = 5, P-value of 0.001). (C) Immunofluorescence to detect Pax7 in muscle satellite cells (yellow arrows). (D) Quantification of Pax7 positive cells revealed a significant increase in satellite cells in the TA muscle of laminin-111 treated animals compared with vehicle (N = 6 and 5, respectively, P-value of 0.049).
Discussion
LAMA2-CMD is a severe genetic disease that causes muscle wasting from birth. Early treatment with laminin-111 protein has been shown to prevent myopathy and extend lifespan in a mouse model of LAMA2-CMD. However, most patients are diagnosed later in life after significant muscle disease and it is currently unclear if laminin-111 protein therapy could be beneficial in these LAMA2-CMD patients. In this study, we aimed to test the hypothesis that laminin-111 protein treatment after disease onset can stall or reverse muscle disease in a mouse model of LAMA2-CMD. The answer to this question would better define the treatment period in which laminin-111 would be effective as a therapeutic in LAMA2-CMD. To test this hypothesis, we treated the dyW−/− mouse model of LAMA2-CMD starting at 4 weeks of age, when mice show significant muscle disease, and analyzed preclinical outcome measures at 8 weeks of age.
Our results showed that i.p. treatment with 10 mg/kg/week laminin-111 protein resulted in the localization of the biologic to the muscle basal lamina of dyW−/− mice. These results demonstrated that laminin-111 protein can move through the blood stream and integrate into the muscle ECM in laminin-211/221 deficient mice.
LAMA2-CMD patients and dyW−/− animals exhibit weight loss and premature death. We showed that treatment with laminin-111 protein after disease onset increased life expectancy of dyW−/− mice by 2-fold compared with vehicle-treated animals. In addition, laminin-111 treatment stabilized the body weight of dyW−/− animals. These studies demonstrate laminin-111 can serve as an effective protein-replacement therapy after significant muscle disease. Previously, we showed that when treatment is started at 2 weeks of age, laminin-111 treated dyW−/− increased lifespan by a 3.5-fold (10). At 2 weeks of age, the dyW−/− mouse exhibit less myopathy. These results suggest that laminin-111 is more effective when used early in LAMA2-CMD disease progress, but that treatment is still effective after disease onset and can improve outcome measures including life expectancy and body weight.
To determine if laminin-111 treatment after disease onset improved muscle function, we measured ex vivo muscle contractility and quantified isometric muscle contraction and contraction-induced injury following published studies (12). The dyW−/− mouse model exhibits severe demyelinating neuropathy in which muscles of the hindlimbs are severely affected. We showed no change in the isometric contractility in the soleus and EDL between laminin-111 and vehicle-treated animals. As expected, the wild-type control group was significantly stronger compared with laminin-111 and vehicle dyW−/− treatment groups. Furthermore, isometric and eccentric contractions in the EDL from dyW mice showed 2-fold lower values compared with dyW+/−, while the soleus showed 1.5-fold lower contractility values. However, the EDL showed stronger contraction compared with the soleus in the dyW+/− control groups. These results indicate that the soleus muscle may be less severely affected by LAMA2-CMD compared with the EDL muscle.
Contraction-induced injury was performed next to reveal defects in muscle mechanical function. Eccentric contraction is defined as the forced lengthening of the muscle during contraction, which results in the disruption of the contractile apparatus (19). Irrespective of treatment, neither the EDL nor soleus muscles from dyW−/− mice showed increased susceptibility to contraction-induced injury (i.e. there were no decreases in isometric peak force with successive contractions) compared with the dyW+/− controls. Additionally, using the isometric and eccentric peaks, we calculated stiffness in both EDL and soleus muscles. We found that there were no significant changes in stiffness between PBS-treated EDL and soleus from dyW−/− and the dyW+/− control groups. These results indicate there was no defect in muscle stiffness in this LAMA2-CMD mouse model. However, the EDL from laminin-111-treated dyW−/− showed an increase in muscle stiffness compared with PBS treated dyW−/− mice as well as dyW+/− controls, while the soleus showed no significant changes with treatment. These results suggest that systemic treatment with laminin-111 treatment after disease onset alters the mechanical properties of the ECM of EDL muscle increasing muscle stiffness. These results may also suggest that higher or more frequent doses of laminin-111 may be required after disease onset to improve the mechanical properties of laminin-α2 deficient skeletal muscle.
Next, we showed that laminin-111 treatment after disease onset improves the histopathology of dyW−/− muscle. We found that CLN-positive fibers exhibited increased myofiber diameters indicating these muscles are being repaired more effectively compared with control animals. Perhaps this indicates that laminin-111 treatment after disease onset allowed for stabilization or increased survival of myofibers undergoing muscle repair. Laminin-111 treated muscles also showed a 2-fold increase in number of regenerating fibers compared with PBS. These findings are consistent with previous studies in other models of muscular dystrophy where laminin-111 treatment increased muscle regeneration (9,18,20). This observation further supports the idea that laminin-111 treatment can stabilize the muscle and promote muscle regeneration in the dyW−/− mouse.
Increased muscle regeneration raises the concern that laminin-111 treatment could exhaust the satellite cell pool, especially in older dyW−/− mice. To address this question, we quantified number of satellite cells in treated versus control muscles. We found a significant increase in satellite cells in the laminin-111 treated animals indicating that treatment may induce apical satellite cell proliferation and support asymmetric satellite cell division to restore the satellite cell pool (21) contributing to muscle regeneration without exhausting the repair capacity of muscle. Laminin-111 has been shown to stimulate apical division of satellite cells in a wild-type in vitro model (22). The cellular effects of laminin-111 on satellite cells remain to be explored in laminin-α2 deficient muscle.
There is currently no approved or effective treatment for LAMA2-CMD. In this study, we show that treatment with EHS mouse laminin-111 is able slow symptomatic muscle disease in the dyW−/− mouse model of LAMA2-CMD. These studies support the idea that laminin-111 protein therapy is an exciting mutation-independent therapeutic that could be beneficial before and after muscle disease in patients with LAMA2-CMD.
Materials and Methods
Survival study
Female dyW mice were treated i.p. with 10 mg/kg/week laminin-111 or an equal volume of PBS starting at 4 weeks of age. Survival was recorded with either morbidity or mortality. Morbidity was defined as 10% weight loss in the span of one week unable to drink or feed, animals exhibiting severe kyphosis and/or hindlimb paralysis as defined within our approved IACUC protocol. The weights of all mice were measured weekly throughout the study.
Ex vivo muscle contractility assay
The EDL and soleus muscles from deeply anesthetized mice treated with laminin-111 or control animals were isolated and placed in an oxygenated physiological salt solution bath (pH 7.6 PSS buffer) at 30°C. Muscles were mounted between two platinum wire electrodes, clamped at one tendon and attached at the other tendon to a force transducer (14). Electrical stimulation, force and length changes were recorded under computer control as previously described (14,23). L0, or optimal length, was determined using a caliper before contractility protocols were performed. Following contractility assays, muscles were dry-blotted and weighed to determine wet muscle weight. Cross-sectional area was calculated using L0 and wet weight to normalize force measurements (23).
Immunohistochemistry
The TA muscles from laminin-111 treated and control animals were harvested post last treatment and embedded in optimum cutting temperature (OCT) medium (Fisher Scientific, Waltham, Massachusetts) mix of OCT: 30% Sucrose (2:3 ratio) and slowly frozen down. Tissues were then cryosectioned at 10 μm thickness and stained with wheat-germ agglutinin (Vector Laboratories, Burlingame, California, 1:100) for 10 min, to visualize the fiber ECM. Tissue slides were then mounted in DAPI containing-Vectashield mounting media (Vector Laboratories, Burlingame, California) to visualize nuclei. Sections were permeabilized using 0.2% Triton in PBS for 30 min before blocking with Mouse on Mouse blocking reagent (Vector Laboratories, Burlingame, California) for 1 h. Primary antibodies were incubated on sections overnight: e-MHC (Developmental Studies Hybridroma Bank—DSHB, Iowa City, Iowa, BF-45, 1:40); Pax7 (DSHB, Iowa City, Iowa, BF-45, 1:20) anti-Laminin-α2 L0663 (Sigma, St. Louis, MO, 1:100) and MAB200 laminin-α1 antibody (1:1, gift from Dr Madeleine Durbeej). Slides were imaged using the Olympus Fluoview FV 1000 laser confocal microscope and analyzed using Image J-win32 software. Quantification of fiber size and central nucleation was quantified in whole section of the muscle. Regenerating fibers were quantified by taking five 20× frames. Pax7-positive cells were quantified by taking 5–10 40× frames.
Statistics
Statistical calculations were performed using GraphPath Prism 5 analysis software. Unpaired student’s t-test was used to calculate mean P-values between two groups. One-way ANOVA test was performed to assess significance between three groups. Two-Way ANOVA test was performed to determine statistical significance between time-point curves in three test groups. Bonferroni post-test was used to determine statistical significance between time-points. Means of experimental groups were considered statistically different when P-values < 0.05*, < 0.001** and < 0.0001***.
Supplementary Material
{kind=link}
Acknowledgements
The authors thank Dr Madeleine Durbeej for kindly providing the MAB200 anti-laminin-α1 antibody.
Conflict of Interest statement. The University of Nevada, Reno, has a patent on the therapeutic use of laminin-111 and its derivatives. This patent has been licensed to Prothelia Inc., Milford, MA. The University of Nevada, Reno has a small equity share in this company.
Author Contributions
D.J.B. conceptualization and study design; P.B-F. methodology, validation, formal analysis; P.B-F., K.B, M.D., B.W.C, A.O-S. investigation; D.J.B. resources; P.B-F., manuscript draft, P.B-F., D.J.B., R.W.G. editing the manuscript; D.J.B. project administration and funding acquisition.
Funding
National Institutes of Health/National Institutes of Arthritis and Muscoskeletal and Skin Diseases (R01AR064338-01A1, MDA628561, MDA238981 to D.J.B.); Mick Hitchcock Scholarship to P.B.F.
References
- 1. Graziano A., Bianco F., Moroni I., Messina S., Bruno C., Pegoraro E., Mora M., Astrea G., Magri F., Comi G.P. et al. (2015) Prevalence of congenital muscular dystrophy in Italy. Neurology, 84, 904–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kuang W., Xu H., Vachon P.H., Ling Liu F.L., Wewer U.M. and Engvall E. (1998) Merosin-deficient congenital muscular dystrophy. Partial genetic correction in two mouse models. Am. Soc. Clin. Investig., 102, 844–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Holmberg J. and Durbeej M. (2013) Laminin-211 in skeletal muscle function. Cell Adhes. Migr., 7, 111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Helbling-Leclerc A., Zhang X., Topaloglu H., Cruaud C., Tesson F., Weissenbach J., Tomé F.M., Schwartz K., Fardeau M. and Tryggvason K. (1995) Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat. Genet., 11, 216–218. [DOI] [PubMed] [Google Scholar]
- 5. Mohassel P., Reghan Foley A. and Bönnemann C.G. (2018) Extracellular matrix-driven congenital muscular dystrophies. Matrix Biol., 71–72, 188–204. [DOI] [PubMed] [Google Scholar]
- 6. Willmann R., Gordish-Dressman H., Meinen S., Rüegg M.A., Yu Q., Nagaraju K., Kumar A., Girgenrath M., Coffey C.B.M., Cruz V. et al. (2017) Improving reproducibility of phenotypic assessments in the DyW mouse model of laminin-α2 related congenital muscular dystrophy. J. Neuromuscul. Dis., 4, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Miner J.H., Patton B.L., Lentz S.I., Gilbert D.J., Snider W.D., Jenkins N.A., Copeland N.G. and Sanes J.R. (1997) The laminin α chains: expression, developmental transitions, and chromosomal locations of α1-5, identification of heterotrimeric laminins 8-11, and cloning of a novel α3 isoform. J. Cell Biol., 137, 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rooney J.E., Gurpur P.B., Yablonka-Reuveni Z. and Burkin D.J. (2009) Laminin-111 restores regenerative capacity in a mouse model for alpha 7 integrin congenital myopathy. Am J Pathol., 174, 256–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ry P.M., Minogue P., Hodges B.L. and Burkin D.J. (2014) Laminin-111 improves muscle repair in a mouse model of merosin-deficient congenital muscular dystrophy. Hum. Mol. Genet., 23, 383–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rooney J.E., Knapp J.R., Hodges B.L., Wuebbles R.D. and Burkin D.J. (2012) Laminin-111 protein therapy reduces muscle pathology and improves viability of a m ouse model of merosin-deficient congenital muscular dystrophy. Am. J. Pathol., 180, 1593–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Connolly A.M., Keeling R.M., Mehta S., Pestronk A., Sanes J.R. and Sanes J.R. (2001) Three mouse models of muscular dystrophy: the natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin alpha 2-deficient mice. Neuromuscul. Disord., 11, 703–712. [DOI] [PubMed] [Google Scholar]
- 12. Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M. and Sweeney H.L. (1993) Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. U. S. A., 90, 3710–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kuang W., Xu H., Vilquin J.T. and Engvall E. (1999) Activation of the lama 2 gene in muscle regeneration: abortive regeneration in laminin alpha 2-deficiency. Lab. Investig., 79, 1601–1613. [PubMed] [Google Scholar]
- 14. Sperringer J.E. and Grange R.W. (2016) In vitro assays to determine skeletal muscle physiologic function. Methods Mol. Biol., 1460. [DOI] [PubMed] [Google Scholar]
- 15. Warren G.L., Lowe D.A. and Armstrong R.B. (1999) Measurement tools used in the study of eccentric contraction-induced injury. Sport. Med., 27, 43–59. [DOI] [PubMed] [Google Scholar]
- 16. Løkken N., Born A.P., Duno M. and Vissing J. (2015) LAMA2-related myopathy: frequency among congenital and limb-girdle muscular dystrophies. Muscle Nerve, 52, 547–553. [DOI] [PubMed] [Google Scholar]
- 17. Pinto-Mariz F., Rodrigues Carvalho L., Prufer De Queiroz Campos Araujo A., De Mello W., Gonçalves Ribeiro M., Cunha M.D.C.S.A., Cabello P.H., Riederer I., Negroni E., Desguerre I. et al. (2015) CD49d is a disease progression biomarker and a potential target for immunotherapy in Duchenne muscular dystrophy. Skelet. Muscle, 5, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Barraza-Flores P., Fontelonga T.M., Wuebbles R.D., Hermann H.J., Nunes A.M., Kornegay J.N. and Burkin D.J. (2019) Laminin-111 protein therapy enhances muscle regeneration and repair in the GRMD dog model of Duchenne muscular dystrophy. Hum. Mol. Genet.. doi: 10.1093/hmg/ddz086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Moens P., Baatsen P.H.W.W. and Maréchal G. (1993) Increased susceptibility of EDL muscles from mdx mice to damage induced by contractions with stretch. J. Muscle Res. Cell Motil., 14, 446–451. [DOI] [PubMed] [Google Scholar]
- 20. Riederer I., Bonomo A.C., Mouly V. and Savino W. (2015) Laminin therapy for the promotion of muscle regeneration. FEBS Lett., 589, 3449–3453. [DOI] [PubMed] [Google Scholar]
- 21. Yin H., Price F. and Rudnicki M.A. (2013) Satellite cells and the muscle stem cell niche. Physiol. Rev., 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rayagiri S.S., Ranaldi D., Raven A., Mohamad Azhar N.I.F., Lefebvre O., Zammit P.S., Borycki A.-G.G., Farhana N.I., Azhar M., Lefebvre O. et al. (2018) Basal lamina remodeling at the skeletal muscle stem cell niche mediates stem cell self-renewal. Nat. Commun., 9, 1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grange R.W., Gainer T.G., Marschner K.M., Talmadge R.J. and Stull J.T. (2002) Fast-twitch skeletal muscles of dystrophic mouse pups are resistant to injury from acute mechanical stress. AJP Cell Physiol., 283, C1090–C 1101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.