

Chromosomal resistance to metronidazole has emerged in clinical Clostridioides difficile isolates, but the genetic mechanisms remain unclear. This is further hindered by the inability to generate spontaneous metronidazole-resistant mutants in the lab to interpret genetic variations in clinical isolates. We therefore constructed a mismatch repair mutator in nontoxigenic ATCC 700057 to survey the mutational landscape for de novo resistance mechanisms. In separate experimental evolutions, the mutator adopted a deterministic path to resistance, with truncation of the ferrous iron transporter FeoB1 as a first-step mechanism of low-level resistance.
KEYWORDS: drug resistance evolution, redox stress, iron metabolism
ABSTRACT
Chromosomal resistance to metronidazole has emerged in clinical Clostridioides difficile isolates, but the genetic mechanisms remain unclear. This is further hindered by the inability to generate spontaneous metronidazole-resistant mutants in the lab to interpret genetic variations in clinical isolates. We therefore constructed a mismatch repair mutator in nontoxigenic ATCC 700057 to survey the mutational landscape for de novo resistance mechanisms. In separate experimental evolutions, the mutator adopted a deterministic path to resistance, with truncation of the ferrous iron transporter FeoB1 as a first-step mechanism of low-level resistance. Deletion of feoB1 in ATCC 700057 reduced the intracellular iron content, appearing to shift cells toward flavodoxin-mediated oxidoreductase reactions, which are less favorable for metronidazole’s cellular action. Higher-level resistance evolved from sequential acquisition of mutations to catalytic domains of pyruvate-ferredoxin/flavodoxin oxidoreductase (PFOR; encoded by nifJ), a synonymous codon change to putative xdh (xanthine dehydrogenase; encoded by CD630_31770), likely affecting mRNA stability, and last, frameshift and point mutations that inactivated the iron-sulfur cluster regulator (IscR). Gene silencing of nifJ, xdh, or iscR with catalytically dead Cas9 revealed that resistance involving these genes occurred only when feoB1 was inactivated; i.e., resistance was seen only in the feoB1 deletion mutant and not in the isogenic wild-type (WT) parent. Interestingly, metronidazole resistance in C. difficile infection (CDI)-associated strains carrying mutations in nifJ was reduced upon gene complementation. This observation supports the idea that mutation in PFOR is one mechanism of metronidazole resistance in clinical strains. Our findings indicate that metronidazole resistance in C. difficile is complex, involving multigenetic mechanisms that could intersect with iron-dependent and oxidoreductive metabolic pathways.
INTRODUCTION
Clostridioides difficile infection (CDI) is a leading cause of diarrhea in hospitalized patients in developed countries. Since 2003, the emergence and spread of epidemic strains have significantly increased the incidence and severity of CDI. The public health burden of epidemic C. difficile is evident in the United States from the total estimated in-hospital deaths, combined from health care- and community-associated CDIs, of 30,600 and 20,500 deaths in 2011 and 2017, respectively (1).
Metronidazole is a 5-nitroimidazole prodrug that has been the preferred treatment for mild to moderate CDI because of its potent anti-anaerobic activity and low cost (2). Following cellular uptake in anaerobes, the nitro group of metronidazole is reduced by electrons arising from cofactors like ferredoxin and flavodoxin in reactions catalyzed by oxidoreductases, such as pyruvate-ferredoxin/flavodoxin oxidoreductase (PFOR) (3, 4). This produces an unstable nitroimidazole anion that may be converted to reactive nitroso and hydroxylamine intermediates, which react with DNA, proteins, and non-protein thiols to form adducts (5).
The efficacy of metronidazole has decreased with the emergence of epidemic strains. Therefore, in the 2017 treatment guidelines of the Infectious Diseases Society of America/Society for Healthcare Epidemiology of America (IDSA/SHEA), metronidazole is no longer a first-line drug for adult CDI (6). However, there is a need for scientific evidence to explain why the efficacy of metronidazole has decreased since its prescribing is likely to continue until the new guidelines become standard practice. The decline in metronidazole efficacy appears to correlate with the emergence and spread of resistant strains of different ribotype backgrounds; metronidazole resistance is defined by a EUCAST breakpoint of >2 μg/ml (7–9). For example, Thorpe et al. reported that ∼8% of U.S. isolates (2011 to 2016) are resistant to metronidazole (8), while the rate in Europe ranged from 0.1% to 0.5% for isolates collected between 2011 and 2014 (7).
The first attempt to characterize the genetic basis for chromosomal metronidazole resistance in a clinical strain was by Lynch et al. (10). After the strain was isolated on metronidazole-containing agar, it was then passaged in vitro in the presence of drug and contained mutations in PFOR, the ferric uptake regulator (Fur), and the oxygen-independent coproporphyrinogen III oxidase (HemN), among other changes. In another study, Moura et al. evaluated a nontoxigenic metronidazole-resistant C. difficile strain showing that it adjusted metabolic pathways that are linked to PFOR activity (11). Most recently, Boekhoud et al. (12) reported that C. difficile clinical isolates carried a conjugative plasmid (pCD-METRO) conferring resistance to metronidazole. However, pCD-METRO was not present in several metronidazole-resistant CDI-associated ribotypes, indicating that pCD-METRO is not a universal mechanism of resistance. Moreover, it is still unknown which genetic determinants on pCD-METRO are directly responsible for resistance. Taken together, it appears that C. difficile could have multiple ways to evolve metronidazole resistance, but these mechanisms remain unclear or require genetic and biochemical validation in naive hosts.
In contrast to mechanisms in C. difficile, there are multiple examples of mechanisms used by bacteria and protozoa to resist the action of metronidazole. For example, Helicobacter pylori clinical isolates often display metronidazole resistance by mutating the NADPH nitroreductases RdxA and FrxA that activate metronidazole (13). Other resistance mechanisms in H. pylori include mutations to the Fur protein that enhances levels of superoxide dismutase (SOD) to detoxify nitroimidazole reactive species (14); the homeostatic regulator HsrA also coordinates derepression of SOD and other global oxidative stress responses against metronidazole (15). Metronidazole-resistant Bacteroides fragilis may produce a nitroimidazole reductase (NimA) that converts metronidazole to a nontoxic amine, overexpress recA (encoding DNA recombination/repair protein RecA) to repair DNA damaged by metronidazole, or decrease drug activation by disrupting its ferrous iron transporter protein (FeoAB) (16–19). In Giardia lamblia, Entamoeba histolytica, and Trichomonas vaginalis, metronidazole resistance often correlates with these protozoans attenuating enzymatic pathways that activate metronidazole (3, 20). For example, decreased production and activity of PFOR enzyme causes metronidazole resistance in G. lamblia (21). Other mechanisms to detoxify metronidazole also occur in E. histolytica, such as decreased expression of ferredoxin and flavin reductase, which affect electron transfer to metronidazole, and upregulation of antioxidants (peroxiredoxin or SOD) (22, 23). Collectively, microbial resistance to metronidazole mainly involves altering drug activation, detoxifying its reactive species, or repairing cellular damage caused by the drug.
There have been two critical barriers to knowledge of metronidazole resistance in C. difficile. First, metronidazole-resistant mutants are described as being unstable, as evident from inconsistent metronidazole susceptibility profiles of strains and the related poor performance of the Etest method for metronidazole (10, 24). Second, there is an inability to select laboratory mutants to allow for controlled genetic studies (25). To address these challenges, from a laboratory perspective, we constructed a mutator tool by deleting the DNA mismatch repair system in a nontoxigenic C. difficile strain. Mutators are employed for accelerated evolution to study mutation accumulation in bacteria (26, 27). Using this concept, we investigated the genomic landscape of C. difficile for de novo resistance mechanisms that are evolutionarily feasible, albeit in vitro. Validated in vitro mechanisms were then used to interpret genetic changes in CDI-associated strains, showing that mutated PFOR is likely to contribute to metronidazole resistance in clinical strains.
RESULTS AND DISCUSSION
Hypermutator construction by deletion of DNA MMR genes.
The MutSL mismatch repair (MMR) proteins are involved in correcting replicative errors (28), whereby MutS identifies mispaired or unpaired bases and recruits the endonuclease MutL that initiates the removal and repair of misincorporated bases (28). C. difficile was found to carry adjacent mutS and mutL genes (CD630_19770 and CD630_19760, respectively), in addition to the mutS homolog mutS2 (CD630_07090). To construct a C. difficile mutator, we chose the strain ATCC 700057 (29) because it is nonpathogenic, lacking the toxin genes tcdA and tcdB (30), and is widely used for antibiotic susceptibility testing (24). We individually deleted mutS, mutL, mutS2, and the entire mutSL operon by allelic exchange using pMTL-SC7215; deletions were confirmed by PCR (31). Mutability was assessed based on the frequencies in which spontaneous mutants were isolated to the antibiotics rifaximin and fidaxomicin at 4× their agar MICs (0.5 and 0.25 μg/ml, respectively). As shown in Fig. 1A mutation frequencies were enhanced upon deletion of mutS or mutL, but deletion of mutS2 had no effect. Deletion of mutSL caused the highest increase (>80-fold) in mutability with respect to wild-type (WT) ATCC 700057; i.e., mutation frequencies were 10−6 for 700057 ΔmutSL versus 10−8 for the WT ATCC 700057, and this strain was significantly more mutable than a ΔmutS or ΔmutL strain, as determined by unpaired t tests (see Table S1 in the supplemental material). Mutability was reversed when 700057 ΔmutSL was complemented with WT mutSL (Fig. S1). Thus, 700057 ΔmutSL (i.e., mutator) was used to select the metronidazole-resistant mutants described below.
FIG 1.

Evolution of metronidazole (MTZ) resistance using a mismatch repair (MMR)-deficient mutator. After deletion of various MMR genes (mutS, mutSL, mutL, or mutS2) in WT ATCC 700057, the derivatives were tested for a change in mutation frequencies (MF), the ability of the ΔmutSL mutator to evolve resistance by serial passage on agars with metronidazole, and mutations associated with metronidazole resistance in the ΔmutSL mutator. (A) Fold change in mutation frequencies for rifaximin and fidaxomicin relative to levels of the WT ATCC 700057. Values are means ± standard deviations from three biological replicates. (B) Two independent experimental evolutions with the ΔmutSL mutator resulted in metronidazole-resistant mutants in contrast to results for the WT. (C) The order in which mutations were accumulated in the two evolution experiments suggests that there is a deterministic path to resistance. Numbers along the blue arrow in panel C represent passage numbers.
Experimental evolution of metronidazole resistance.
The agar MIC of metronidazole against the WT and mutator was 0.25 μg/ml. However, spontaneous mutants could not be selected by plating >10 overnight cultures of each strain onto agars containing 2× and 4× MICs (data not shown). Both strains were therefore serially passaged on agars containing various concentrations of metronidazole (0.125 to 16 μg/ml); each passage was incubated for up to 3 days to obtain growth. In the first experimental evolution, by the ninth passage, the mutator evolved resistance as its population was inhibited by 2 μg/ml (Fig. 1B). By the 15th passage, the population was inhibited at 8 μg/ml. In contrast, stable mutants did not arise from the WT ATCC 700057, even with up to 15 serial passages in the presence of the drug (Fig. 1B). This is consistent with prior reports of an inability to isolate in vitro metronidazole-resistant mutants of C. difficile (25). To further comprehend the evolutionary path to resistance, we conducted a separate evolution experiment with the mutator and identified that, by the 6th and 17th passages, the population was inhibited by 2 and 16 μg/ml of drug, respectively (Fig. 1B and C).
Identification of genetic changes associated with de novo metronidazole resistance.
As expected, the mutator accumulated insertions, substitutions, and deletions across the genome and showed no genomic site specificity. Functional gene classification showed that mutations occurred to iron transporters, iron-sulfur proteins, oxidoreductases, carbohydrate metabolism, fatty acid metabolism, cell surface/division proteins, and other genes (Fig. S2). Table S2 lists the entire set of mutations that were identified in the sequenced endpoint mutants. However, to study mutations associated with resistance, we focused on proteins involved in cellular redox and iron homeostasis that may affect the activation of metronidazole.
(i) Identification of genetic changes in evolution experiment 1.
From cultures at the endpoint of evolution experiment 1 (passage 15), we isolated and sequenced the genomes of three mutant colonies, designated JWD-1 (MIC = 8 μg/ml), JWD-2 (MIC = 32 μg/ml), and JWD-3 (MIC = 32 μg/ml). They all carried a frameshift deletion (Glu38fs) in FeoB1, encoded by CD630_14790, a substitution in PFOR (Gln803Arg) encoded by nifJ (CD630_26820), and a synonymous change (GAG2070GAA, where underlining indicates the change) in a putative xanthine dehydrogenase xdh, encoded by CD630_31770. JWD-2 and JWD-3 also carried unique changes of Lys51fs and Val76Ala, respectively, in the iron-sulfur cluster regulator (IscR), encoded by CD630_12780. These mutations in IscR may explain why JWD-2 and JWD-3 were 4-fold more resistant to metronidazole than JWD-1, which lacked changes to the regulator. We identified the order in which mutations arose (Fig. 1C) by determining agar MICs and by Sanger sequencing of three individual mutant colonies each from passages 10 and 13. Mutants from passage 10 had metronidazole MICs of 2 μg/ml and carried the above variations in PFOR and FeoB1, while mutants (MICs of 8 μg/ml) from passage 13 also carried the synonymous change in xdh.
(ii) Identification of genetic changes in evolution experiment 2.
JWD-4 (MIC = 64 μg/ml), an endpoint mutant (passage 17) of experiment 2, was isolated, and the genome was sequenced. Similar to the above endpoint mutants, JWD-4 contained disruptions to FeoB1 (Lys40fs), PFOR (Pro32Leu), and IscR (Lys51fs) but also acquired a Gly270Asp substitution in xanthine permease (CD630_20910). The order in which mutations arose (Fig. 1C) was also determined from agar MIC tests and Sanger sequencing of a minimum of three colonies per time point. This revealed that FeoB1 was disrupted by passage 4 (MIC = 1 μg/ml) and that disruptions occurred in PFOR by passage 7 (MIC = 2 μg/ml), in xanthine permease by passage 14 (MIC = 16 μg/ml), and in IscR by passage 17 (MIC = 64 μg/ml). The occurrence of similar genetic changes in the two independent serial passage experiments suggests that there was a deterministic path to metronidazole resistance.
Explanation of de novo genetic variations associated with metronidazole resistance.
(i) FeoB1 participation. Under anaerobic conditions, iron mostly exists in the ferrous (Fe2+) form. In C. difficile, FeoB1 is predicted to be the main iron transporter as it is the most upregulated iron transporter under low-iron conditions and in vivo in hamsters (32, 33). In vitro, the homologs FeoB2 and FeoB3 are thought to be less responsive to changes in iron (32). Thus, the loss of FeoB1 may have lowered the supply of iron to iron carrier proteins that mediate electron transfer to metronidazole (34). In support of our findings, deletion of feoAB in B. fragilis conferred a 10-fold decrease in metronidazole activity (18).
(ii) Oxidoreductases PFOR and XDH participation. PFOR catalyzes the oxidation of pyruvate to acetyl-coenzyme A (CoA), while XDH (CD630_31770; annotated as a molybdenum-binding xanthine dehydrogenase) catalyzes the oxidation of purines. Sequence alignment of PFOR homologs of C. difficile (CD630_26820), Desulfovibrio africanus (Desaf_2186), and Moorella thermoacetica (GenBank accession number Q2RMD6) (similarity of 58.7% and 62.7%, respectively) indicated that the Pro32Leu substitution occurred adjacent to the critical threonine-31 site that forms the catalytic domain for pyruvate binding. The Gln803Arg substitution occurred in domain VI upstream of cysteine-815 that binds the proximal [4Fe-4S] cluster (35, 36) (Fig. S3). These substitutions likely affected the enzyme’s catalytic activity, involving electron transfer to electron carrier proteins. To evaluate the synonymous change in xdh, we analyzed its secondary structure using CLC Genomics Workbench, version 12.0, which is based upon the RNA folding algorithm Mfold (37), to gauge potential effects on RNA translation. Our analysis of xdh, shown in Fig. S4, shows that GAG2070GAA is predicted to introduce an unstable stem-loop, with a positive free energy change (ΔG of 3.8 kcal/mol for the mutant compared to a ΔG of −25.9 kcal/mol for the WT), which is unfavorable for translation (38). With regard to xanthine permease, the role of this transporter in metronidazole resistance is presently unclear although we speculate that the Gly270Asp substitution might affect the supply of xanthine to XDH.
(iii) IscR participation. Iron-sulfur [Fe-S] clusters are essential to the biochemistry of several proteins that conduct electron transfer reactions (39, 40). In pathogenic bacteria the assembly and incorporation of [Fe-S] clusters are mostly controlled by the isc and suf operons, of which isc is the housekeeping system; furthermore, C. difficile lacks the suf operon (41). Holo-IscR binds DNA as a homodimer, but damage to its [4Fe-4S] by reactive free radicals or iron starvation increases cellular levels of apo-IscR, triggering defensive mechanisms, including antioxidants such as cysteine and non-protein thiols (42). The Lys51fs variation, in strains JWD-2 and JWD-4, disabled the function of IscR, by forming an aberrant protein lacking cysteines-92, -98, and -104 that are required to bind [4Fe-4S] (43). To characterize the effect of the Val76Ala substitution in IscR in JWD-3, we analyzed the crystal structure of dimeric IscR bound to DNA from Escherichia coli (PDB accession number 4CHU) using UCSF Chimera (44). The IscR proteins from C. difficile ATCC 700057 and E. coli K-12 are closely related, with ∼41% identical amino acids, and valine-76 is a conserved amino acid. In the dimeric structure (Fig. S5), valine-76 on chain A occurs with other lipophilic residues in a dimer interface where it hydrophobically interacts with leucine-113 on chain B. It is plausible that the less hydrophobic alanine-76 reduces hydrophobic steric packing for stabilizing the dimeric DNA binding conformation of IscR.
FeoB1-mediated cellular changes associated with metronidazole resistance.
(i) Deleting feoB1 affects resistance and iron content. Since feoB1 was inactivated early in the serial evolution (Fig. 1C), we first examined the role of this gene by deleting it in ATCC 700057. The deletion mutant (700057ΔfeoB1) grew in the presence of 0.5 μg/ml of metronidazole (broth MIC = 1 μg/ml), whereas the WT strain grew with 0.125 μg/ml of drug (broth MIC of 0.25 μg/ml) (Fig. 2A). Susceptibility was restored by complementing the ΔfeoB1 strain with WT feoB1, expressed from its own promoter in pMTL84151 (Fig. 2A). There was ∼21% lower iron content in the mutant (0.0753 ± 0.002 ppm) than in the WT (0.0956 ± 0.004 ppm), as determined by inductively coupled plasma optical emission spectrometry (ICP-OES). Consistent with the reduced intracellular iron content, 700057 ΔfeoB1 also showed increased transcription of fur (3.9-fold ± 0.17-fold) and the ferrichrome ABC transporter subunit (fhuB) (5.13-fold ± 0.25-fold) compared to the WT level in the absence of drug (Fig. 2B); this was significant (P < 0.0005), as determined by an unpaired t test. In the presence of the drug, the ΔfeoB1 mutant showed 10.50-fold ± 0.96-fold (fur) and 9.16-fold ± 0.25-fold (fhuB) higher gene expression levels than the WT (P < 0.0005) (Fig. 2B).
FIG 2.

(A) Effect of feoB1 deletion on growth with metronidazole (MTZ). The ΔfeoB1 mutant grew better with metronidazole, as shown from optical density (absorbance, Abs) readings (Abs600nm), whereby the mutant was inhibited at 1 μg/ml, while the WT and complemented ΔfeoB1 mutant strains were inhibited at 0.25 μg/ml. (B and C) Transcription of iron response genes in the ΔfeoB1 mutant with respect to the level in the WT in the absence and presence of metronidazole. Increased expression of fur and fhuB (3.9-fold ± 0.17-fold and 5.13-fold ± 0.25-fold, respectively; ***, P < 0.0005) without drug supports iron limitation in an feoB1 mutant. Expression of fur and fhuB increased when cells were exposed to metronidazole (10.50-fold ± 0.96-fold and 9.16-fold ± 0.25-fold, respectively; ***, P < 0.0005). Deletion of feoB1 caused downregulation of ferredoxin (−2.46-fold ± 0.09-fold) and upregulation of flavodoxin (2.04-fold ± 0.06-fold) relative to WT levels in the absence of metronidazole. These genes were further differentially expressed upon exposure to metronidazole; a reduction in ferredoxin expression (−7.51-fold ± 0.21-fold) and an increase in flavodoxin expression (6.860-fold ± 0.43-fold; ***, P < 0.0005) were observed. (D) Intracellular concentration of dimetridazole (DMZ) after 1 h in the ΔfeoB1 and WT strains suggests less drug accumulation (by ∼33.63%) in the mutant (*, P < 0.05). Statistical significance was determined by unpaired t tests in GraphPad Prism, version 8.0.
(ii) Effect on expression of cofactors that activate metronidazole. Since the loss of feoB1 reduced iron content in cells, we reasoned that this may diminish metronidazole activation by limiting iron-dependent electron carrier proteins such as ferredoxins. Ferredoxin and flavodoxin are small redox proteins that accept and transfer electrons to metronidazole from oxidoreductases, such as PFOR. Transcriptional analysis revealed that in the absence of drug, 700057 ΔfeoB1 downregulated (−2.46-fold ± 0.09-fold) ferredoxin (fdx, i.e., CD630_06271) and upregulated (2.04-fold ± 0.06-fold) flavodoxin (fldx, i.e., CD630_19990) relative to the WT levels (Fig. 2C). When 700057 ΔfeoB1 was exposed to metronidazole, there was further downregulation of fdx (−7.51-fold ± 0.21-fold) and upregulation of fldx (6.860-fold ± 0.43-fold) (Fig. 2C), which was significant (P < 0.0001), as determined by an unpaired t test. Other ferredoxin and flavodoxin homologs (Fig. S6) were expressed at lower levels, supporting published studies that CD630_06271 and CD630_19990 are the most responsive homologs of ferredoxin and flavodoxin during iron stress in C. difficile (32, 33). Ferredoxins are better at transferring electrons to metronidazole due to carriage of an iron-sulfur cluster that has a lower redox potential than flavodoxin, which carries flavin mononucleotide (45); but the exact homolog(s) of ferredoxin that activates metronidazole is unknown in C. difficile. Overall, these results indicate that loss of feoB1 led to reduced iron content, prompting a shift to flavodoxin-mediated oxidoreductase reactions that enable cells to resist metronidazole.
(iii) Effect on cellular accumulation of nitroimidazole. Since the above results suggest that the loss of feoB1 might decrease metronidazole activation, we quantified the intracellular concentration of dimetridazole, a related nitroimidazole (19). Dimetridazole was used due to the commercial availability of 5-amino-1,2-dimethylimidazole, a likely end product of dimetridazole activation (19) that could be used as a standard in liquid chromatography-tandem mass spectrometry (LC-MS/MS). Against dimetridazole, 700057 ΔfeoB1 showed the same level of resistance to metronidazole (i.e., broth MIC of 1 μg/ml). Concentrated cells were exposed to 1 mM dimetridazole for 1 h, and intracellular concentrations were measured in cell lysates. In 700057 ΔfeoB1, dimetridazole was present at 7.31 ± 0.53 μM (Fig. 2D), which represents about 34% lower intracellular drug accumulation than that in the WT (11.07 ± 1.26 μM). The difference in drug uptake was statistically significant (P < 0.05), as determined by an unpaired t test. The accumulation of metronidazole and other nitroimidazoles into bacteria is thought to occur in a concentration-dependent manner, whereby as the drug is activated in cells, more drug is taken up (46). Thus, the observed lower intracellular accumulation of dimetridazole in 700057 ΔfeoB1 might indicate less drug activation, which could be expected with a shift to flavodoxin-mediated metabolism. However, we could not detect the amine end product, which would have provided a measure of drug activation.
Resistance mediated by nifJ, xdh, and iscR appears to require the loss of feoB1.
To assess the roles played by nifJ, xdh, and iscR in metronidazole resistance, we silenced these genes using a xylose-inducible CRISPR interference vector. In this approach, fusion of antisense nucleotides to the guide RNA of Cas9 blocks gene transcription (47), and this is a more facile approach than deleting genes in C. difficile. As a positive control, an antisense sequence to feoB1 was included to measure the effect of gene silencing on growth with metronidazole. Even though gene silencing reduced transcript levels by ∼10-fold (Fig. 3A), resistance was detected only in the strain expressing the antisense to feoB1 (broth MIC of 1 μg/ml); the strain carrying the empty vector was inhibited by 0.25 μg/ml (Fig. 3B). Given that feoB1 mutants arose early in the passages (Fig. 1C), we questioned whether the expression of resistance involving nifJ, xdh, or iscR required the loss of feoB1. As shown in Fig. 3C, the above-mentioned antisense sequences enhanced survival of 700057 ΔfeoB1, shifting broth MICs to 8 μg/ml. Thus, the loss of feoB1 early in the serial evolution might have influenced the direction of evolution of resistance toward genes that, when disrupted, took advantage of lower iron content (i.e., combinatorial or epistatic resistance).
FIG 3.

Effect of silencing nifJ, iscR, or xdh on resistance in the ΔfeoB1 mutant and WT strain. (A) In the WT strain, depletion of mRNA for feoB1, nifJ, iscR, and xdh occurs when antisense sequences (AS) are induced with xylose (2%, wt/vol) (A), but when the WT was grown in various concentrations of metronidazole, resistance was seen only with depletion of feoB1 (i.e., growth at 0.5 μg/ml and MIC of 1 μg/ml). The MIC for strains expressing other antisense sequences was 0.25 μg/ml. (C) Conversely, in the ΔfeoB1 mutant, expression of any of the three antisense sequences caused resistance (i.e., MICs of 8 μg/ml versus 1 μg/ml against the ΔfeoB1 mutant).
To confirm that combinatorial resistance was more protective, we exposed cells to 8 μg/ml of metronidazole and analyzed effects on DNA integrity, non-protein thiols, and transcription of redox-responsive genes (48, 49). The combinatorial resistant strains showed more intact DNA, while DNA damage was seen in 700057 ΔfeoB1 and was more marked in the WT strain (Fig. 4A). Similarly, after cells were exposed to metronidazole, there was less impact on free non-protein thiols in the combinatorial resistant strains than in the untreated samples (Fig. 4B). A significant decrease in non-protein thiols was evident in the WT strain, but this was less in its isogenic feoB1 mutant, suggesting that there was a decline in the formation of covalent adducts that occurs with activated metronidazole. Combinatorial resistant strains also showed reduced transcription of the DNA damage response gene recA (CD630_13280) and redox stress-responsive genes (hybrid cluster protein, CD630_21680; thioredoxin A, CD630_30330) (Fig. 4C). Taken together, these results confirm the role of feoB1 in the evolution of a novel epistatic mechanism of metronidazole resistance. Epistatic mechanisms of antimicrobial resistance are also seen in fluoroquinolone-resistant E. coli, with double mutations to gyrase A and topoisomerase IV, which produce high-level resistance that is greater than predicted from individual mutations (50).
FIG 4.

Biochemical and transcriptional validation that higher-level resistance (MIC = 8 μg/ml) occurs when mRNA for nifJ, iscR, or xdh is depleted by antisense sequence (AS) in the ΔfeoB1 mutant. (A) Metronidazole (MTZ)-induced DNA damage was reduced in the ΔfeoB1 mutant by expression of antisense sequence compared to the level in the ΔfeoB1 mutant and in the WT carrying the empty vector. As a positive control for DNA fragmentation, the WT was exposed to hydrogen peroxide (0.5%, vol/vol). Captured are lanes from gel photos shown in Fig. S7 in the supplemental material. Resistance was also confirmed by less depletion of low-molecular-weight thiols that form adducts with MTZ (B) and by reduced expression of recA (DNA recombination/repair protein RecA, CD630_13280), hcp (hybrid cluster protein, CD630_21680), and trxA (thioredoxin A, CD630_30330) that respond to activated metronidazole (C). Statistical significance was determined by unpaired t tests in GraphPad Prism, version 8.0. ** and *** indicate P < 0.005 and P < 0.0005, respectively.
Further assessment of the function of IscR in resistance.
Because IscR has not been previously linked to metronidazole resistance, we further studied its role by focusing on pyruvate-lactate metabolism, which is altered by iscR deletion in Clostridium perfringens (41). Additionally, in B. fragilis with decreased PFOR activity, a shift to lactate fermentation is accompanied by reduced metronidazole activity (51). In 700057 ΔfeoB1, depletion of iscR mRNA further diminished the transcription of nifJ (−6.17-fold ± 0.29-fold), which was already marginally reduced (−2.67-fold ± 0.18-fold) by the loss of feoB1 in 700057 ΔfeoB1 (Fig. 5A), which is significant (P < 0.005, unpaired t test). The strain bearing the iscR antisense also exhibited an increase in pyruvate (1.33 ± 0.03 nM) and lactate (1.86 ± 0.08 nM) compared to levels in the parent 700057 ΔfeoB1 (0.98 ± 0.02 nM and 1.13 ± 0.045 nM, respectively) (Fig. 5B). This can be expected if downregulation of nifJ further reduced PFOR levels in cells, which would concomitantly decrease metronidazole activation. A reduction in PFOR activity may also decrease oxidation of pyruvate to acetyl-CoA, allowing pyruvate to be converted to lactate via lactate dehydrogenase (LDH), as in B. fragilis (51); nonetheless, we did not measure LDH activities, which would also be confirmatory. However, it appears that loss of iscR (Fig. 1C) affected iron-sulfur-dependent electron transfer reactions and synergized with both the loss of feoB1 and mutated oxidoreductase enzymes, which occurred at earlier evolutionary steps. To confirm the hypothesis that loss of iscR affected electron transfer reactions, we tested the susceptibility of cells to the electron acceptor plumbagin, a naphthoquinone that is reduced in reactions catalyzed by oxidoreductases (52). In Xanthomonas campestris, the loss of iscR led to plumbagin resistance (53). Our results show that logarithmic growth of WT ATCC 700057 was completely inhibited by 0.5 μg/ml plumbagin (Fig. 5C). In contrast, growth of 700057ΔfeoB1 was inhibited at 4 μg/ml of the compound; resistance to plumbagin was also increased with depletion of iscR mRNA in 700057 ΔfeoB1 (i.e., broth MIC of 16 μg/ml). These findings are analogous to those for metronidazole.
FIG 5.

Analysis of how iscR inactivation promotes metronidazole (MTZ) resistance in the △feoB1 mutant. (A) Depletion of iscR mRNA in the △feoB1 mutant by antisense sequence (AS) caused downregulation of nifJ (P < 0.005, as determined by an unpaired t test in GraphPad Prism, version 8). This led to accumulation of pyruvate (∼35%) and lactate (∼65%) (B). All values are relative to levels in the control △feoB1 strain carrying an empty vector (EV). (C) Cross-resistance was seen with the electron acceptor plumbagin. Results suggest that loss of iscR globally affects iron-dependent oxidoreductase reactions, including that by PFOR.
Role of PFOR in metronidazole resistance in clinical strains.
Based on the results described above, we generated a preliminary model of chromosomally mediated metronidazole resistance (Fig. 6) to help interpret genetic changes seen in C. difficile clinical isolates, which lack pCD-METRO (12). To test the model, we analyzed the genomes of two metronidazole-resistant CDI-associated strains (491858 and 490054, ribotype 027) from an Israeli surveillance (9) and the resistant strain CD26A54_R reported by Lynch et al. (10). Agar MICs of metronidazole against 491858, 490054, and CD26A54_R were 8 to 16 μg/ml. Table S3 documents genetic differences in 491858 and 490054 compared to the genome of strain R20291, while those for CD26A54_R have been previously reported (10). Strains 491858 and 490054 both carried an Ala1018Val substitution in PFOR. Alignment with PFOR from D. africanus and M. thermoacetica (Fig. S4) revealed that alanine-1018 lies next to domain VI, which binds [4Fe-4S] and thiamine pyrophosphate (TPP). CD26A54_R carries a Gly423Glu substitution in domain III of PFOR (10), the binding site for the coenzyme A (CoA) substrate (35, 36); the mutation directly affects the conserved domain (423GLGSD427) that is thought to engage the pyrophosphate portion of CoA in the M. thermoacetica (Fig. S4) (36). In PFOR from M. thermoacetica, CoA is also stabilized by charge interaction with domain VI, namely, arginine-1016 (i.e., lysine-1023 in C. difficile). Hence, structural analysis is required to determine if the more non-polar valine-1018, instead of alanine, affects CoA binding in 491858 and 490054. To further evaluate PFOR, we complemented 491858 and CD26A54_R with the WT copy of nifJ expressed from its promoter in vector pMTL84151 and measured their agar MICs (Fig. 7). Complemented 491858 showed metronidazole agar MICs of 4 to 8 μg/ml (mean, 4.67 ± 1.50 μg/ml; n = 6 clones) and agar MICs of 8 to 16 μg/ml (13.33 ± 3.77 μg/ml; n = 6 clones) against the strain with the empty vector; these were significant (P < 0.005, unpaired t tests). Similarly, the MICs of complemented CD26A54_R were reduced (4 to 8 μg/ml; mean, 4.67 ± 1.50 μg/ml; n = 6 clones) compared to those of the empty vector control (8 to 16 μg/ml; mean, 10.66 ± 3.77 μg/ml; P < 0.05, unpaired t tests). However, complementation did not completely resensitize the strains to metronidazole, at least according to the EUCAST breakpoint of >2 μg/ml. This might be due to the presence of both WT and mutant copies of PFOR in the enzyme pool. It may also be due to differences in chromosomal and plasmid gene positions or to the fact that the strains contain other genetic variations that are predictive of metronidazole resistance (Table S3). For example, strain 491858 contained a Tyr130Ser substitution in a putative NimA-related 5-nitroimidazole reductase (CDR20291_1308). As mentioned above, Nim proteins reduce metronidazole to nontoxic metabolites in Bacteroides species (19). The Tyr130Ser substitution was recently reported in combination with HemN (Glu317Lys) in C. difficile strains that lacked pCD-METRO and had MICs of 2 to 4 μg/ml (54). Although CD26A54_R lacks mutation in the NimA homolog, it carries HemN (Tyr214fs) and Fur (Glu41Lys) mutations. The role of HemN in iron metabolism in C. difficile is unclear since it carries a partial set of genes for heme biosynthesis. Disruption of Fur might affect iron homeostasis, as reported in H. pylori (14). While 491858 and 490054 did not carry obvious disruptions to iron metabolism (Table S3), the additional mechanisms described above nevertheless support a model wherein metronidazole resistance in clinical strains could be multigenetic.
FIG 6.

A proposed model of in vitro resistance to metronidazole (MTZ) in C. difficile. (Left) In sensitive cells, metronidazole is activated by oxidoreductases (Ox-Red; e.g., pyruvate-ferredoxin/flavodoxin oxidoreductase [PFOR, nifJ]), resulting in free radical damage to DNA and protein damage. Activation then influences drug uptake. Susceptibility is influenced by iron uptake via transporters, mainly FeoB1 (the main iron transporter), and the electron carrier protein ferredoxin (Fdx) with low redox potential. (Right) In metronidazole-resistant cells, loss of FeoB1 decreases intracellular iron content, probably shifting cells from ferredoxin (Fdx)- to flavodoxin (Fldx)-mediated metabolism. Fldx is less effective in activating metronidazole. Loss of iscR synergizes with a defective feoB1 to further reduce metabolic activities that activate metronidazole.
FIG 7.
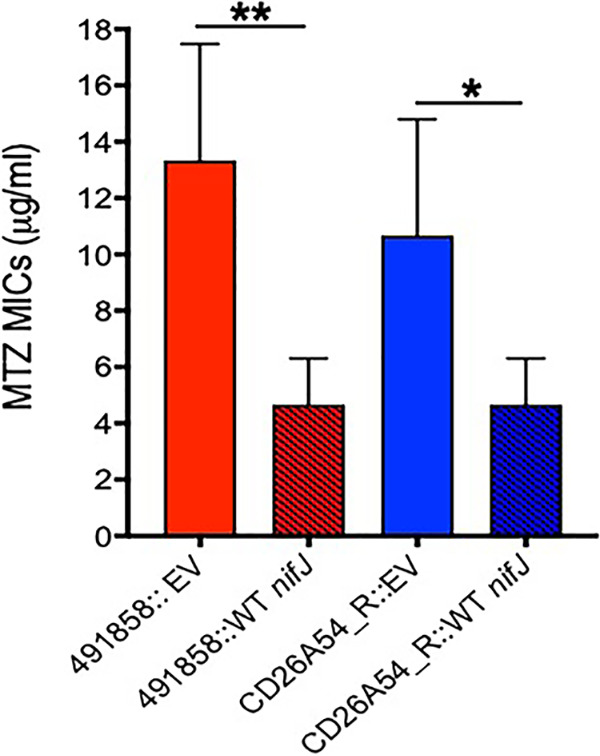
Role of PFOR in metronidazole resistance in clinical isolates. Two metronidazole (MTZ)-resistant CDI-associated strains (491858 and CD26A54_R) were complemented with the WT copy of nifJ. This caused a reduction in metronidazole agar MICs. MIC reductions were significant (**, P < 0.005; *, P < 0.05 for 491858 and CD26A54_R, respectively), as determined by unpaired t tests in GraphPad Prism, version 8.0. EV, empty vector. Strains 491858 and CD26A54_R carried Ala1018Val and Gly423Glu mutations, respectively, in PFOR.
Conclusions.
The involvement of mutations to multiple genes might partly explain why C. difficile was slow to evolve chromosomal resistance to metronidazole in spite of the drug being used since the 1980s. While mutations to feoB1 and oxidoreductases are known to individually cause metronidazole resistance in other organisms, to the best of our knowledge there are no reports of epistatic interactions between these mechanisms, as we observed from in vitro studies. FeoB1 is essential for colonization and virulence in other bacteria, and during CDI in mice, feoB1 is >200-fold upregulated, suggesting iron limitation in the gut (55). Thus, it is improbable that C. difficile will inactivate FeoB1 during metronidazole therapy in vivo. Indeed, the above-described clinical strains, or those reported elsewhere (54), do not carry mutations in FeoB1. In the host, C. difficile also encounters oxygen fluctuations as the environmental redox potential in the colon is increased following dysbiosis (56). This is evident from significant in vivo transcription of superoxide dismutase (CDR20291_1529) and antioxidant thiol proteins, suggesting that C. difficile responds to in vivo oxidative stress (57). Taken together, two possible scenarios are also likely to drive differences between in vitro and in vivo metronidazole resistance mechanisms. First, low-iron conditions in the colon might favor the use of iron-sparing cofactors like flavodoxin instead of ferredoxin, which is less effective for metronidazole activation, and as such could synergize with genetic changes in PFOR or other oxidoreductases to promote higher-level resistance. Second, resistance might also be elevated by mechanisms that enhance the expression of genes that confer tolerance to in vivo oxidative stress and metronidazole, such as superoxide dismutase. Resistance by such types of strains could be higher in vivo than in vitro, but this difference could be difficult to detect in iron-rich medium or anaerobic medium with antioxidants. These two scenarios are important because metronidazole achieves low concentrations only in the lumen (average of 9.3 μg/g in watery stool) (58), and even modest increases in resistance following mutations could enhance the survival of C. difficile during metronidazole therapy.
In the aftermath of metronidazole not being recommended for adult CDI, there remain a number of outstanding questions concerning the apparent multigenetic and complex mechanisms of resistance. Certainly, it is important to establish which resistance mechanisms do or do not impose physiological costs as this factor might influence the future epidemiology of strains in the clinic. There is also a need to identify if strains carry resistance mechanisms that promote cross-resistance to host oxidative stress, which is a factor that could impact disease severity. Hence, studies are still warranted to assess the extent to which metronidazole resistance could shape C. difficile evolution, epidemiology, and pathophysiology.
MATERIALS AND METHODS
Strains and culture conditions.
Here, we studied nontoxigenic C. difficile ATCC 700057 and its derivative and ribotype 027 CDI-associated strains (491858, 490054, and CD26A54_R). Strains 491858 and 490054 were from an Israeli surveillance (9) and were previously studied for vancomycin resistance (59); CD26A54_R (10) was also previously reported. All strains were routinely grown in prereduced brain heart infusion (BHI) broth or agar at 37°C in a Whitley A35 anaerobic workstation (Don Whitley Scientific). Escherichia coli strains NEB 5-alpha, CA434, and SD46 were grown at 37°C in LB broth or agar. All strains and plasmids used in this study are listed in Table S3 in the supplemental material. d-Cycloserine (250 μg/ml), cefoxitin (8 μg/ml), and thiamphenicol (15 μg/ml) were used to selectively culture C. difficile-containing plasmids, whereas chloramphenicol (15 μg/ml) and ampicillin (50 μg/ml) or kanamycin (50 μg/ml) were used to grow E. coli SD46 and CA434, respectively. Experiments with open C. difficile cultures were performed in a class II, type A2 biological safety cabinet or in the A35 workstation; autoclaving and 10% (vol/vol) bleach were used for decontamination of cultures and work surfaces.
Antimicrobial susceptibility.
(i) Agar MICs. MICs of the metronidazole-resistant laboratory mutants, the clinical isolates, and their complemented counterparts were determined on BHI agar supplemented with hemin (5 μg/ml) and an inoculum of 105 CFU/ml. Agars contained doubling dilutions of metronidazole from 0.06 to 64 μg/ml.
(ii) Broth MICs and growth kinetics. Twofold serial dilutions of drugs were made in sterile prereduced BHI broth. Overnight-grown cultures were diluted 1:100 and grown to an optical density at 600 nm (OD600) of ∼0.3 before being added to the prereduced 96-well plates. The plates were incubated anaerobically for 24 h in a Synergy H1 microplate reader (BioTek), and OD600 values were automatically recorded. For induction of antisense nucleotides, 2% (wt/vol) xylose was added to the wells.
Determination of mutation frequencies.
Briefly, C. difficile strains were grown overnight in BHI broth, centrifuged, and concentrated 10-fold. Aliquots (0.1 ml) of each culture were plated onto prereduced BHI agar plates containing 0.32 μg/ml fidaxomicin or 0.5 μg/ml rifaximin, values representing 4× MICs of the drugs. Similarly, WT and mutator cultures (>10 each) were concentrated and plated onto agar plates containing 2× or 4× the metronidazole MIC (i.e., 0.5 or 1 μg/ml, respectively). After 48 h of incubation, the mutation frequencies were calculated as the number of resistant colonies divided by total viable counts.
Experimental evolution by serial passaging.
From an overnight culture on agar, C. difficile colonies were resuspended into 1 ml of BHI broth to produce an OD600 of ∼0.8 to 1.0. At each passage step, an aliquot of 0.01 ml (inoculum of 105 to 106 CFU/ml) was spread onto BHI agar containing 0.25× to 64× the MIC of metronidazole. After 48 to 72 h of incubation, colonies were isolated from the highest concentration permitting visible growth and were resuspended in fresh broth before the suspension was passaged with higher drug concentrations. After the passage was set up, the remaining bacterial suspension was stored in glycerol at −80°C.
Genetic manipulation of C. difficile.
The vector pMTL-SC7215 was used to delete target genes in C. difficile by allelic exchange (31). Allelic exchange vectors to delete mutS, mutS2, mutSL, mutL, and feoB1 were conjugated into C. difficile via E. coli donor strains (CA434 or SD46). Allelic exchange was conducted as described previously (31). Successful gene knockouts were confirmed by PCR. The strains lacking mutSL and feoB1 were complemented with the entire mutSL operon and feoB1, driven by their own promoters in the vector pMTL84151; clinical isolates were also complemented with a WT copy of nifJ, driven by its own promoter cloned in pMTL84151. Primers used in the study are described in Table S4.
Gene knockdown.
The xylose-inducible vector pXWpxyl-dcas9 was used to silence the transcription of feoB1, nifJ, iscR, and xdh, as previously described (47). The single guide RNAs (sgRNAs) (Table S5) targeting the above-mentioned genes were synthesized and cloned into the vector’s PmeI site by GenScript Biotech (New Jersey). Induction of genes in the antisense direction was performed with 2% (wt/vol) xylose.
Genome sequencing and analysis.
Whole-genome sequencing was done by paired-end sequencing at SeqMatics LLC (California) and c (Texas). CLC Genomics Workbench, version 12 (Qiagen), was used to de novo assemble WT ATCC 700057. The assembled genome of the WT was annotated using Rapid Annotation using Subsystem Technology (RAST) (49) and was used for mapping the mutator 700057 ΔmutSL and metronidazole-resistant mutants (JWD-1, JWD-2, JWD-3, and JWD-4). Sequence variations were identified using the quality-based variant detection tool in CLC Genomics Workbench with default parameters (≥10-fold coverage of the reference position and sequence variation of ≥35% of mapped reads). Mutations detected in the endpoint mutants, compared to the WT sequence, were screened against the 700057 ΔmutSL strain to remove shared variations to focus on those that arose during the evolutions. For confirmation, the variations in ≥90% of mapped reads were further analyzed manually. The coverage for each genomic location was calculated in the CLC software, and zero coverage regions were then identified using a customized Perl script. Sanger sequencing confirmed genetic changes. The clinical strains 491858 and 490054 were mapped to the genome of C. difficile R20291, and genetic variations were identified, exactly as we recently reported (59).
Quantification of intracellular iron by ICP-OES.
Overnight-grown cells were subcultured (1:100) in fresh BHI broth and harvested after 6 h for ICP-OES analysis to determine the cellular iron levels. Samples were washed twice, centrifuged at 5,000 rpm, and air dried. Samples were first digested in a pressurized microwave at 250°C at an initial pressure of 4 × 106 Pa; at 250°C the pressure was about 8 × 106 Pa. Precisely weighed samples were loaded into the digestion tube, and 4 ml of 16N HNO3 was added. After digestion, concentrated HNO3 was evaporated at 100°C before addition of 5 ml of 2% HNO3 and heating at 120°C to redissolve the samples. After samples were weighed, iron content was measured with an Agilent 725 ICP-OES. Calibration curves were made using standard solutions of iron at concentrations of 5 ppb to 500 ppb.
LC-MS/MS analysis of cellular lysates.
Tenfold-concentrated, logarithmically growing cells (OD600 of ∼0.3) were exposed to 1 mM dimetridazole, and samples were harvested after 1 h of incubation. Cellular lysates (10 μl) were mixed with methanol (90 μl), vortexed, and centrifuged at 15,000 × g for 10 min. Supernatant (5 μl) was injected onto a UHPLC-Q Exactive MS system for analysis.
Analysis of DNA damage.
Samples were harvested as described above after exposure to 8 μg/ml metronidazole, and DNA damage was analyzed by alkaline agarose gel electrophoresis as described previously (60), except for the following changes. The lysis buffer contained lysozyme (500 μg/ml), and the agarose plugs were incubated at 37°C for 4 h. Hydrogen peroxide (0.5%, vol/vol) was used as a positive control to assess DNA fragmentation.
Quantification of low-molecular-weight thiols.
Samples harvested as described above were processed in 5% (wt/vol) trichloroacetic acid and later with Tris-HCl to remove proteins as previously described (48). Free-thiol content in the processed samples was then measured by fluorescence (emission, 510 nm; excitation, 390 nm) using a thiol fluorescent detection kit (Invitrogen), according to the manufacturer’s instructions. Free-thiol content in strains exposed to metronidazole was determined relative to levels in untreated samples.
Transcriptional analysis.
After the samples were harvested as described above, bacterial RNAprotect reagent (Qiagen) was added to cultures, and RNA was isolated using Qiagen’s RNeasy minikit. RNA was converted to cDNA by Moloney murine leukemia virus (M-MLV) reverse transcriptase (Quantabio) using qScript cDNA SuperMix. A qScript One-Step SYBR green quantitative reverse transcription-PCR (qRT-PCR) kit, 6-carboxy-X-rhodamine (ROX; Quantabio), and gene-specific primers were used to amplify genes in an Applied Biosystems ViiA7 real-time PCR machine. Transcript levels were calculated by the comparative threshold cycle (ΔΔCT) method, and data were normalized to 16S rRNA values.
Quantification of lactate and pyruvate.
Samples were harvested as described above; lactate and pyruvate were detected using a pyruvate colorimetric/fluorometric assay kit (Biovison) and a Lactate-Glo assay kit (Promega).
Data availability.
Genomic data for 491858, 490054, and CD26A54_R are available in GenBank under accession numbers SAMN12323916, SAMN12323915, and NZ_AMDM00000000, respectively.
Supplementary Material
ACKNOWLEDGMENTS
This work was in part funded by grants R56AI126881 and R01AI139261 to J.G.H. from the National Institute of Allergy and Infectious Diseases at the National Institutes of Health. The funders had no role in study design, in data collection and interpretation of the findings, or in the writing and submission of the manuscript.
ICP-OES analysis was conducted at the ICP lab, Department of Earth and Atmospheric Sciences, University of Houston, Texas. LC-MS/MS analysis was performed at NMR and Drug Metabolism Core, Advanced Technology Core, Baylor College of Medicine, Houston, Texas.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Guh AY, Mu Y, Winston LG, Johnston H, Olson D, Farley MM, Wilson LE, Holzbauer SM, Phipps EC, Dumyati GK, Beldavs ZG, Kainer MA, Karlsson M, Gerding DN, McDonald LC, Emerging Infections Program Clostridioides difficile Infection Working Group . 2020. Trends in U.S. burden of Clostridioides difficile Infection and Outcomes. N Engl J Med 382:1320–1330. doi: 10.1056/NEJMoa1910215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Peláez T, Alcalá L, Alonso R, Rodríguez-Créixems M, García-Lechuz JM, Bouza E. 2002. Reassessment of Clostridium difficile susceptibility to metronidazole and vancomycin. Antimicrob Agents Chemother 46:1647–1650. doi: 10.1128/aac.46.6.1647-1650.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Leitsch D, Burgess AG, Dunn LA, Krauer KG, Tan K, Duchene M, Upcroft P, Eckmann L, Upcroft JA. 2011. Pyruvate:ferredoxin oxidoreductase and thioredoxin reductase are involved in 5-nitroimidazole activation while flavin metabolism is linked to 5-nitroimidazole resistance in Giardia lamblia. J Antimicrob Chemother 66:1756–1765. doi: 10.1093/jac/dkr192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sisson G, Goodwin A, Raudonikiene A, Hughes NJ, Mukhopadhyay AK, Berg DE, Hoffman PS. 2002. Enzymes associated with reductive activation and action of nitazoxanide, nitrofurans, and metronidazole in Helicobacter pylori. Antimicrob Agents Chemother 46:2116–2123. doi: 10.1128/aac.46.7.2116-2123.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lloyd D, Pedersen JZ. 1985. Metronidazole radical anion generation in vivo in Trichomonas vaginalis: oxygen quenching is enhanced in a drug-resistant strain. J Gen Microbiol 131:87–92. doi: 10.1099/00221287-131-1-87. [DOI] [PubMed] [Google Scholar]
- 6.McDonald LC, Gerding DN, Johnson S, Bakken JS, Carroll KC, Coffin SE, Dubberke ER, Garey KW, Gould CV, Kelly C, Loo V, Shaklee Sammons J, Sandora TJ, Wilcox MH. 2018. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis 66:e1–e48. doi: 10.1093/cid/cix1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Freeman J, Vernon J, Pilling S, Morris K, Nicholson S, Shearman S, Longshaw C, Wilcox MH, Pan-European Longitudinal Surveillance of Antibiotic Resistance among Prevalent Clostridium difficile Ribotypes Study Group . 2018. The ClosER study: results from a three-year pan-European longitudinal surveillance of antibiotic resistance among prevalent Clostridium difficile ribotypes, 2011–2014. Clin Microbiol Infect 24:724–731. doi: 10.1016/j.cmi.2017.10.008. [DOI] [PubMed] [Google Scholar]
- 8.Thorpe CM, McDermott LA, Tran MK, Chang J, Jenkins SG, Goldstein EJC, Patel R, Forbes BA, Johnson S, Gerding DN, Snydman DR. 2019. U.S.-based national surveillance for fidaxomicin susceptibility of Clostridioides difficile-associated diarrheal isolates from 2013 to 2016. Antimicrob Agents Chemother 63:e00391-19. doi: 10.1128/AAC.00391-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adler A, Miller-Roll T, Bradenstein R, Block C, Mendelson B, Parizade M, Paitan Y, Schwartz D, Peled N, Carmeli Y, Schwaber MJ. 2015. A national survey of the molecular epidemiology of Clostridium difficile in Israel: the dissemination of the ribotype 027 strain with reduced susceptibility to vancomycin and metronidazole. Diagn Microbiol Infect Dis 83:21–24. doi: 10.1016/j.diagmicrobio.2015.05.015. [DOI] [PubMed] [Google Scholar]
- 10.Lynch T, Chong P, Zhang J, Hizon R, Du T, Graham MR, Beniac DR, Booth TF, Kibsey P, Miller M, Gravel D, Mulvey MR, Canadian Nosocomial Infection Surveillance Program (CNISP) . 2013. Characterization of a stable, metronidazole-resistant Clostridium difficile clinical isolate. PLoS One 8:e53757. doi: 10.1371/journal.pone.0053757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moura I, Monot M, Tani C, Spigaglia P, Barbanti F, Norais N, Dupuy B, Bouza E, Mastrantonio P. 2014. Multidisciplinary analysis of a nontoxigenic Clostridium difficile strain with stable resistance to metronidazole. Antimicrob Agents Chemother 58:4957–4960. doi: 10.1128/AAC.02350-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boekhoud IM, Hornung BVH, Sevilla E, Harmanus C, Bos-Sanders I, Terveer EM, Bolea R, Corver J, Kuijper EJ, Smits WK. 2020. Plasmid-mediated metronidazole resistance in Clostridioides difficile. Nat Commun 11:598. doi: 10.1038/s41467-020-14382-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jeong JY, Mukhopadhyay AK, Dailidiene D, Wang Y, Velapatino B, Gilman RH, Parkinson AJ, Nair GB, Wong BC, Lam SK, Mistry R, Segal I, Yuan Y, Gao H, Alarcon T, Brea ML, Ito Y, Kersulyte D, Lee HK, Gong Y, Goodwin A, Hoffman PS, Berg DE. 2000. Sequential inactivation of rdxA (HP0954) and frxA (HP0642) nitroreductase genes causes moderate and high-level metronidazole resistance in Helicobacter pylori. J Bacteriol 182:5082–5090. doi: 10.1128/jb.182.18.5082-5090.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsugawa H, Suzuki H, Satoh K, Hirata K, Matsuzaki J, Saito Y, Suematsu M, Hibi T. 2011. Two amino acids mutation of ferric uptake regulator determines Helicobacter pylori resistance to metronidazole. Antioxid Redox Signal 14:15–23. doi: 10.1089/ars.2010.3146. [DOI] [PubMed] [Google Scholar]
- 15.Olekhnovich IN, Vitko S, Valliere M, Hoffman PS. 2014. Response to metronidazole and oxidative stress is mediated through homeostatic regulator HsrA (HP1043) in Helicobacter pylori. J Bacteriol 196:729–739. doi: 10.1128/JB.01047-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schapiro JM, Gupta R, Stefansson E, Fang FC, Limaye AP. 2004. Isolation of metronidazole-resistant Bacteroides fragilis carrying the nimA nitroreductase gene from a patient in Washington State. J Clin Microbiol 42:4127–4129. doi: 10.1128/JCM.42.9.4127-4129.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Steffens LS, Nicholson S, Paul LV, Nord CE, Patrick S, Abratt VR. 2010. Bacteroides fragilis RecA protein overexpression causes resistance to metronidazole. Res Microbiol 161:346–354. doi: 10.1016/j.resmic.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Veeranagouda Y, Husain F, Boente R, Moore J, Smith CJ, Rocha ER, Patrick S, Wexler HM. 2014. Deficiency of the ferrous iron transporter FeoAB is linked with metronidazole resistance in Bacteroides fragilis. J Antimicrob Chemother 69:2634–2643. doi: 10.1093/jac/dku219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carlier JP, Sellier N, Rager MN, Reysset G. 1997. Metabolism of a 5-nitroimidazole in susceptible and resistant isogenic strains of Bacteroides fragilis. Antimicrob Agents Chemother 41:1495–1499. doi: 10.1128/AAC.41.7.1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leitsch D, Drinic M, Kolarich D, Duchene M. 2012. Down-regulation of flavin reductase and alcohol dehydrogenase-1 (ADH1) in metronidazole-resistant isolates of Trichomonas vaginalis. Mol Biochem Parasitol 183:177–183. doi: 10.1016/j.molbiopara.2012.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dunn LA, Burgess AG, Krauer KG, Eckmann L, Vanelle P, Crozet MD, Gillin FD, Upcroft P, Upcroft JA. 2010. A new-generation 5-nitroimidazole can induce highly metronidazole-resistant Giardia lamblia in vitro. Int J Antimicrob Agents 36:37–42. doi: 10.1016/j.ijantimicag.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wassmann C, Hellberg A, Tannich E, Bruchhaus I. 1999. Metronidazole resistance in the protozoan parasite Entamoeba histolytica is associated with increased expression of iron-containing superoxide dismutase and peroxiredoxin and decreased expression of ferredoxin 1 and flavin reductase. J Biol Chem 274:26051–26056. doi: 10.1074/jbc.274.37.26051. [DOI] [PubMed] [Google Scholar]
- 23.Samarawickrema NA, Brown DM, Upcroft JA, Thammapalerd N, Upcroft P. 1997. Involvement of superoxide dismutase and pyruvate:ferredoxin oxidoreductase in mechanisms of metronidazole resistance in Entamoeba histolytica. J Antimicrob Chemother 40:833–840. doi: 10.1093/jac/40.6.833. [DOI] [PubMed] [Google Scholar]
- 24.Moura I, Spigaglia P, Barbanti F, Mastrantonio P. 2013. Analysis of metronidazole susceptibility in different Clostridium difficile PCR ribotypes. J Antimicrob Chemother 68:362–365. doi: 10.1093/jac/dks420. [DOI] [PubMed] [Google Scholar]
- 25.Kumar M, Adhikari S, Hurdle JG. 2014. Action of nitroheterocyclic drugs against Clostridium difficile. Int J Antimicrob Agents 44:314–319. doi: 10.1016/j.ijantimicag.2014.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Canfield GS, Schwingel JM, Foley MH, Vore KL, Boonanantanasarn K, Gill AL, Sutton MD, Gill SR. 2013. Evolution in fast forward: a potential role for mutators in accelerating Staphylococcus aureus pathoadaptation. J Bacteriol 195:615–628. doi: 10.1128/JB.00733-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Long H, Miller SF, Strauss C, Zhao C, Cheng L, Ye Z, Griffin K, Te R, Lee H, Chen CC, Lynch M. 2016. Antibiotic treatment enhances the genome-wide mutation rate of target cells. Proc Natl Acad Sci U S A 113:E2498–505. doi: 10.1073/pnas.1601208113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lenhart JS, Pillon MC, Guarne A, Biteen JS, Simmons LA. 2015. Mismatch repair in Gram-positive bacteria. Res Microbiol 167:4–12. doi: 10.1016/j.resmic.2015.08.006. [DOI] [PubMed] [Google Scholar]
- 29.Leeds JA, Sachdeva M, Mullin S, Barnes SW, Ruzin A. 2014. In vitro selection, via serial passage, of Clostridium difficile mutants with reduced susceptibility to fidaxomicin or vancomycin. J Antimicrob Chemother 69:41–44. doi: 10.1093/jac/dkt302. [DOI] [PubMed] [Google Scholar]
- 30.Zhou Y, Mao L, Yu J, Lin Q, Luo Y, Zhu X, Sun Z. 2019. Epidemiology of Clostridium difficile infection in hospitalized adults and the first isolation of C. difficile PCR ribotype 027 in central China. BMC Infect Dis 19:232. doi: 10.1186/s12879-019-3841-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cartman ST, Kelly ML, Heeg D, Heap JT, Minton NP. 2012. Precise manipulation of the Clostridium difficile chromosome reveals a lack of association between the tcdC genotype and toxin production. Appl Environ Microbiol 78:4683–4690. doi: 10.1128/AEM.00249-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ho TD, Ellermeier CD. 2015. Ferric uptake regulator fur control of putative iron acquisition systems in Clostridium difficile. J Bacteriol 197:2930–2940. doi: 10.1128/JB.00098-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hastie JL, Hanna PC, Carlson PE. 2018. Transcriptional response of Clostridium difficile to low iron conditions. Pathog Dis 76:fty009. doi: 10.1093/femspd/fty009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yeom J, Imlay JA, Park W. 2010. Iron homeostasis affects antibiotic-mediated cell death in Pseudomonas species. J Biol Chem 285:22689–22695. doi: 10.1074/jbc.M110.127456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pieulle L, Magro V, Hatchikian EC. 1997. Isolation and analysis of the gene encoding the pyruvate-ferredoxin oxidoreductase of Desulfovibrio africanus, production of the recombinant enzyme in Escherichia coli, and effect of carboxy-terminal deletions on its stability. J Bacteriol 179:5684–5692. doi: 10.1128/jb.179.18.5684-5692.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen PY, Aman H, Can M, Ragsdale SW, Drennan CL. 2018. Binding site for coenzyme A revealed in the structure of pyruvate:ferredoxin oxidoreductase from Moorella thermoacetica. Proc Natl Acad Sci U S A 115:3846–3851. doi: 10.1073/pnas.1722329115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuker M. 2003. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Steffens W, Digby D. 1999. mRNAs have greater negative folding free energies than shuffled or codon choice randomized sequences. Nucl Acids Res 27:1578–1584. doi: 10.1093/nar/27.7.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yeo WS, Lee JH, Lee KC, Roe JH. 2006. IscR acts as an activator in response to oxidative stress for the suf operon encoding Fe-S assembly proteins. Mol Microbiol 61:206–218. doi: 10.1111/j.1365-2958.2006.05220.x. [DOI] [PubMed] [Google Scholar]
- 40.Giel JL, Rodionov D, Liu M, Blattner FR, Kiley PJ. 2006. IscR-dependent gene expression links iron-sulphur cluster assembly to the control of O2-regulated genes in Escherichia coli. Mol Microbiol 60:1058–1075. doi: 10.1111/j.1365-2958.2006.05160.x. [DOI] [PubMed] [Google Scholar]
- 41.Andre G, Haudecoeur E, Courtois E, Monot M, Dupuy B, Rodionov DA, Martin-Verstraete I. 2017. Cpe1786/IscR of Clostridium perfringens represses expression of genes involved in Fe-S cluster biogenesis. Res Microbiol 168:345–355. doi: 10.1016/j.resmic.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 42.Santos JA, Alonso-Garcia N, Macedo-Ribeiro S, Pereira PJ. 2014. The unique regulation of iron-sulfur cluster biogenesis in a Gram-positive bacterium. Proc Natl Acad Sci U S A 111:E2251–E2260. doi: 10.1073/pnas.1322728111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fleischhacker AS, Stubna A, Hsueh KL, Guo Y, Teter SJ, Rose JC, Brunold TC, Markley JL, Munck E, Kiley PJ. 2012. Characterization of the [2Fe-2S] cluster of Escherichia coli transcription factor IscR. Biochemistry 51:4453–4462. doi: 10.1021/bi3003204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem 25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 45.Bruschi M, Guerlesquin Fço. 1988. Structure, function and evolution of bacterial ferredoxins. FEMS Microbiol Rev 54:155–176. doi: 10.1111/j.1574-6968.1988.tb02741.x. [DOI] [PubMed] [Google Scholar]
- 46.Cudmore SL, Delgaty KL, Hayward-McClelland SF, Petrin DP, Garber GE. 2004. Treatment of infections caused by metronidazole-resistant Trichomonas vaginalis. Clin Microbiol Rev 17:783–793, table of contents. doi: 10.1128/CMR.17.4.783-793.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marreddy RKR, Wu X, Sapkota M, Prior AM, Jones JA, Sun D, Hevener KE, Hurdle JG. 2019. The fatty acid synthesis protein enoyl-acp reductase ii (fabk) is a target for narrow-spectrum antibacterials for Clostridium difficile infection. ACS Infect Dis 5:208–217. doi: 10.1021/acsinfecdis.8b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leitsch D, Kolarich D, Wilson IB, Altmann F, Duchene M. 2007. Nitroimidazole action in Entamoeba histolytica: a central role for thioredoxin reductase. PLoS Biol 5:e211. doi: 10.1371/journal.pbio.0050211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leitsch D, Schlosser S, Burgess A, Duchene M. 2012. Nitroimidazole drugs vary in their mode of action in the human parasite Giardia lamblia. Int J Parasitol Drugs Drug Resist 2:166–170. doi: 10.1016/j.ijpddr.2012.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huseby DL, Pietsch F, Brandis G, Garoff L, Tegehall A, Hughes D. 2017. Mutation supply and relative fitness shape the genotypes of ciprofloxacin-resistant Escherichia coli. Mol Biol Evol 34:1029–1039. doi: 10.1093/molbev/msx052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diniz CG, Farias LM, Carvalho MA, Rocha ER, Smith CJ. 2004. Differential gene expression in a Bacteroides fragilis metronidazole-resistant mutant. J Antimicrob Chemother 54:100–108. doi: 10.1093/jac/dkh256. [DOI] [PubMed] [Google Scholar]
- 52.Imlay J, Fridovich I. 1992. Exogenous quinones directly inhibit the respiratory NADH dehydrogenase in Escherichia coli. Arch Biochem Biophys 296:337–346. doi: 10.1016/0003-9861(92)90581-g. [DOI] [PubMed] [Google Scholar]
- 53.Fuangthong M, Jittawuttipoka T, Wisitkamol R, Romsang A, Duang-Nkern J, Vattanaviboon P, Mongkolsuk S. 2015. IscR plays a role in oxidative stress resistance and pathogenicity of a plant pathogen, Xanthomonas campestris. Microbiol Res 170:139–146. doi: 10.1016/j.micres.2014.08.004. [DOI] [PubMed] [Google Scholar]
- 54.Arcay RM, Suárez-Bode L, López-Causapé C, Oliver A, Mena A. 2020. Emergence of high-level and stable metronidazole resistance in Clostridioides difficile. Int J Antimicrob Agents 55:105830. doi: 10.1016/j.ijantimicag.2019.10.011. [DOI] [PubMed] [Google Scholar]
- 55.Fletcher JR, Erwin S, Lanzas C, Theriot CM. 2018. Shifts in the gut metabolome and Clostridium difficile transcriptome throughout colonization and infection in a mouse model. mSphere 3:e00089-18. doi: 10.1128/mSphere.00089-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reese AT, Cho EH, Klitzman B, Nichols SP, Wisniewski NA, Villa MM, Durand HK, Jiang S, Midani FS, Nimmagadda SN, O'Connell TM, Wright JP, Deshusses MA, David LA. 2018. Antibiotic-induced changes in the microbiota disrupt redox dynamics in the gut. Elife 7:e35987. doi: 10.7554/eLife.35987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Janoir C, Deneve C, Bouttier S, Barbut F, Hoys S, Caleechum L, Chapeton-Montes D, Pereira FC, Henriques AO, Collignon A, Monot M, Dupuy B. 2013. Adaptive strategies and pathogenesis of Clostridium difficile from in vivo transcriptomics. Infect Immun 81:3757–3769. doi: 10.1128/IAI.00515-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bolton RP, Culshaw MA. 1986. Faecal metronidazole concentrations during oral and intravenous therapy for antibiotic associated colitis due to Clostridium difficile. Gut 27:1169–1172. doi: 10.1136/gut.27.10.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen WJ, Deshpande A, Hevener KE, Endres BT, Garey KW, Palmer KL, Hurdle JG. 2020. Constitutive expression of the cryptic vanGCd operon promotes vancomycin resistance in Clostridioides difficile clinical isolates. J Antimicrob Chemother 75:859–867. doi: 10.1093/jac/dkz513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zirkle RE, Krieg NR. 1996. Development of a method based on alkaline gel electrophoresis for estimation of oxidative damage to DNA in Escherichia coli. J Appl Bacteriol 81:133–138. doi: 10.1111/j.1365-2672.1996.tb04490.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Genomic data for 491858, 490054, and CD26A54_R are available in GenBank under accession numbers SAMN12323916, SAMN12323915, and NZ_AMDM00000000, respectively.